

Abstract
The temporally specific nature of neurotrophic factor-induced responses is a general feature of mammalian nervous system development, the mechanisms of which remain to be elucidated. We characterized a mechanism underlying the temporal specificity by which BDNF selectively promotes the survival of newly generated, but not mature, granule neurons of the mammalian cerebellum. We found that BDNF specifically induces the extracellular signal-regulated kinase 5 (ERK5)-myocyte enhancer factor (MEF2) signaling pathway in newly generated granule neurons and thereby induces transcription of neurotrophin-3 (NT-3), a novel gene target of MEF2. Inhibition of endogenous ERK5, MEF2, or NT-3 in neurons by several approaches including disruption of the NT-3 gene in mice revealed a requirement for the ERK5-MEF2-NT-3 signaling pathway in BDNF-induced survival of newly generated granule neurons. These findings define a novel mechanism that underlies the antiapoptotic effect of neurotrophins in a temporally defined pattern in the developing mammalian brain.
Keywords: BDNF, ERK5, MEF2, CREB, NT-3, neuron, apoptosis, survival, transcription
Introduction
The neurotrophins comprise a family of secreted proteins that promote the survival of distinct neuronal populations in the developing mammalian nervous system (Bothwell, 1995; Ip and Yancopoulos, 1996; Lewin and Barde, 1996). Because of the established importance of the neurotrophins in neuronal survival during development and their potential to promote recovery of neurons after injury in the adult nervous system (Jelsma and Aguayo, 1994; Lindsay, 1994; Yuen and Mobley, 1996), there has been a great deal of interest in characterizing the intracellular mechanisms by which the neurotrophins suppress neuronal apoptosis (Kaplan and Miller, 2000).
The granule neurons of the cerebellar cortex represent an attractive cell type for characterizing the molecular control of neuronal survival and death, because these processes have been extensively studied at a cellular and developmental level in these neurons (Williams and Herrup, 1988). Cerebellar granule neurons are generated within the external granular layer (EGL) and then migrate into the internal granule layer (IGL) (Altman and Bayer, 1997; Hatten and Heintz, 1995). After their migration into the IGL, granule neurons undergo maturation that includes the elaboration of dendrites within the IGL and the formation of synapses with Purkinje neurons in the molecular layer. Newly generated granule neurons in the EGL and IGL undergo developmentally regulated apoptosis (Wood et al., 1993).
Early investigations into the role of the neurotrophins in neuron survival in the developing cerebellum revealed that BDNF suppresses apoptosis of cultured primary granule neurons (Segal et al., 1992; Lindholm et al., 1993). In contrast, the addition of neurotrophin-3 (NT-3) to cultured cerebellar granule neurons does not suppress apoptosis (Segal et al., 1992; Lindholm et al., 1993), an observation that cast doubt on a pro-survival role for NT-3 in granule neurons until the gene knock-out data became available. Genetic evidence in mice indicates that both BDNF and NT-3 play important roles in regulating the survival of cerebellar granule neurons (Schwartz et al., 1997; Bates et al., 1999). Still, significant questions remain on the relative roles and relationship of the actions of BDNF and NT-3 in promoting the survival of granule neurons. On the one hand, the BDNF-/- and CNS-specific NT-3-/- mice have very similar cerebellar phenotypes, each displaying a phenocopy of the morphological abnormality of the vermis and increased apoptosis of newly generated granule neurons (Schwartz et al., 1997; Bates et al., 1999). In contrast, inactivation of the major BDNF and NT-3 receptors, TrkB and TrkC, respectively, shows a small increase in apoptosis of granule neurons but a dramatic increase in apoptosis in mice with inactivation of three of the four TrkB and TrkC alleles (TrkB-/-, TrkC+/-, or TrkB+/-, TrkC-/-), suggesting that these neurotrophin receptors collaborate to promote granule neuron survival (Minichiello and Klein, 1996).
Closer examination of the genetic evidence in mice indicates that BDNF suppresses apoptosis of newly generated granule neurons in the EGL and IGL before their maturation (Schwartz et al., 1997). However, beyond this developmental time window, BDNF continues to exert biological effects on developing granule neurons that contribute to their maturation (Gao et al., 1995). These observations raise the important question of what intracellular mechanisms confer on BDNF the ability to promote the survival of granule neurons in a temporally specific manner. The temporal specificity of the biological effects of a neurotrophic factor is a general feature of mammalian brain development, whose underlying mechanisms remain to be elucidated.
Primary granule neurons cultured from the rodent or murine cerebellum recapitulate many aspects of their development in vivo, including the temporally specific profile of granule neuron maturation (Powell et al., 1997). Recent microarray analyses of cultured granule neurons and the developing cerebellum reveal that the temporal changes of gene expression profiles in cultured granule neurons faithfully mimic those observed in vivo, validating cultured primary granule neurons as a good model of developmentally regulated events in the intact cerebellum (Diaz et al., 2002). Consistent with this interpretation, BDNF suppression of apoptosis of cultured granule neurons declines with maturation (Skaper et al., 1998). These results suggest that investigation of BDNF-induced survival of granule neurons should yield insight into the mechanisms underlying the temporal specificity of the pro-survival effect of BDNF.
The intracellular mechanisms by which BDNF promotes the survival of neurons are beginning to be characterized. BDNF enhances the survival of cerebellar granule neurons via the extra-cellular signal-regulated kinase1/2 (ERK1/2) and Akt signaling pathways (Meyer-Franke et al., 1998; Bonni et al., 1999). Akt and the ERK1/2-activated kinases, the Rsks, promote cell survival in part by a transcription-independent mechanism by phosphorylating and thereby inactivating the apoptotic protein BAD (Bonni et al., 1999). In addition to direct inactivation of the cell death machinery, transcription-dependent mechanisms also seem to play a prominent role in neurotrophin-induced cell survival (Bonni et al., 1999; Riccio et al., 1999). In particular, the ERK1/2-Rsk signaling pathway promotes neuronal survival by activating the transcription factor cAMP response element-binding protein (CREB) (Bonni et al., 1999). Whether the temporal specificity of the pro-survival effect of BDNF-induced granule neuron survival is explained by a difference in BDNF activation of the ERK1/2 or Akt signaling pathway or if it is conferred by additional BDNF-induced signals remained to be characterized.
Neurotrophins activate multiple signals downstream of the Trk receptors in addition to the ERK1/2 and Akt signaling cascades (Kaplan and Miller, 2000). However, the biological roles of many of the neurotrophin-induced signals remain to be elucidated. Among these signals, the ERK1/2-related kinase ERK5 has been reported in one study to mediate NGF-induced survival of PNS sensory neurons (Watson et al., 2001), but its role in the survival of neurons in the developing mammalian brain remained unknown. A major substrate of ERK5 is the transcription factor myocyte enhancer factor 2 (MEF2), a member of the superfamily of transcription factors containing a MADS box (Black and Olson, 1998). The MEF2 proteins have established roles in myogenesis, but their high expression in neurons suggests that these transcription factors also play important roles in the developing brain (Lyons et al., 1995; Lin et al., 1996; Mao et al., 1999). Consistent with this conclusion, MEF2 mediates neuronal activity-dependent cell survival (Mao et al., 1999; Gaudilliere et al., 2002; Okamoto et al., 2002). However, the role of MEF2, if any, in neurotrophin-induced cell survival and how MEF2 might mediate neurotrophin-induced neuronal survival remained to be characterized.
In this study, we investigated the mechanisms by which BDNF promotes the survival of granule neurons in an age-specific manner. BDNF robustly promoted the survival of newly generated granule neurons but failed to promote the survival of mature granule neurons. Surprisingly, BDNF induced the ERK1/2 and Akt signaling pathways in both populations of granule neurons, suggesting that additional signals confer on BDNF the temporal specificity of the survival response. We found that BDNF selectively induces the ERK5-MEF2 signaling pathway in newly generated granule neurons. We also identified the NT-3 gene as a novel direct target of MEF2 that is induced by BDNF in granule neurons in an age-specific manner. Finally, activation of the endogenous ERK5-MEF2-NT-3 signaling module in granule neurons was found to mediate BDNF-induced survival of newly generated granule neurons. Together, our findings define a novel mechanism that confers on BDNF the ability to promote the survival of granule neurons in a temporally defined manner. In addition, our results illuminate the relationship of two major neurotrophic factors in the developing cerebellum, suggesting that NT-3 functions downstream of BDNF in the promotion of cerebellar granule neuron survival during the time window of peak developmentally regulated granule neuron apoptosis.
Materials and Methods
Plasmids and mutagenesis. The expression plasmids that contain MEF2CR24L, MEF2A-TA, CREBM1, and MEK1KA97 have been described previously. The pCEFL expression plasmids containing ERK5, MEK5AA, and MEK5DD were kindly provided by Dr. S. Gutkind (National Institute of Dental and Craniofacial Research, Bethesda, MD). The NT-3 promoter-luciferase reporter genes were constructed by inserting fragments of the NT-3 promoter in pGL-2. Mutation of the NT-3 promoter was performed using QuikChange Site Directed Mutagenesis Kit (Stratagene, La Jolla, CA) according to the manufacturer's protocol, and mutations were confirmed by sequencing and restriction analysis. Granule neuron cultures were prepared from cerebella of postnatal day 6 rat pups, as described previously (Bonni et al., 1999). Neurons were placed on polyornithine-coated plates and grown in BME (Sigma, St. Louis, MO) supplemented with 10% calf serum (Hyclone Laboratories, Logan, UT), 25 mm KCl, 2 mm glutamine, penicillin, and streptomycin. One day after cultures were prepared (P6 + 1DIV), they were treated with the antimitotic agent cytosine-β-d-arabinofuranoside (10 μm) to prevent the proliferation of non-neuronal cells. Genes were introduced into cerebellar granule neurons using a calcium phosphate transfection method, as described previously (Konishi et al., 2002).
Indirect immunofluorescence. Indirect immunofluorescence was performed as described previously (Konishi et al., 2002). In transfected cultures, β-galactosidase was detected by incubating fixed cerebellar cultures with a mouse monoclonal antibody to β-galactosidase (dilution, 1:500; Promega, Madison, WI), followed by a goat anti-mouse secondary antibody that was conjugated to Cy3 (dilution, 1:500; Amersham). Cultures were also stained with the DNA dye bisbenzimide (Hoechst 33258). Cell counts were performed by a blinded observer.
Immunoprecipitations and Western blot analyses. Immunoprecipitations and Western blot analyses were performed as described with the following modifications (Bonni et al., 1999). In the experiments in Figure 2 A, lysates of cerebellar granule neurons were immunoblotted using rabbit polyclonal antibodies that recognize the phosphorylated form of ERK5 specifically (dilution, 1:1,000; BioSource, Camarillo, CA) in TBST containing 3% BSA, and 5% milk at 4°C for 12 hr. Antibody binding was detected by ECL with a secondary antibody conjugated to horseradish peroxidase (dilution 1:20,000). Total ERK5 was detected with rabbit polyclonal antibodies (dilution 1:1,000; Calbiochem, La Jolla, CA).
Figure 2.
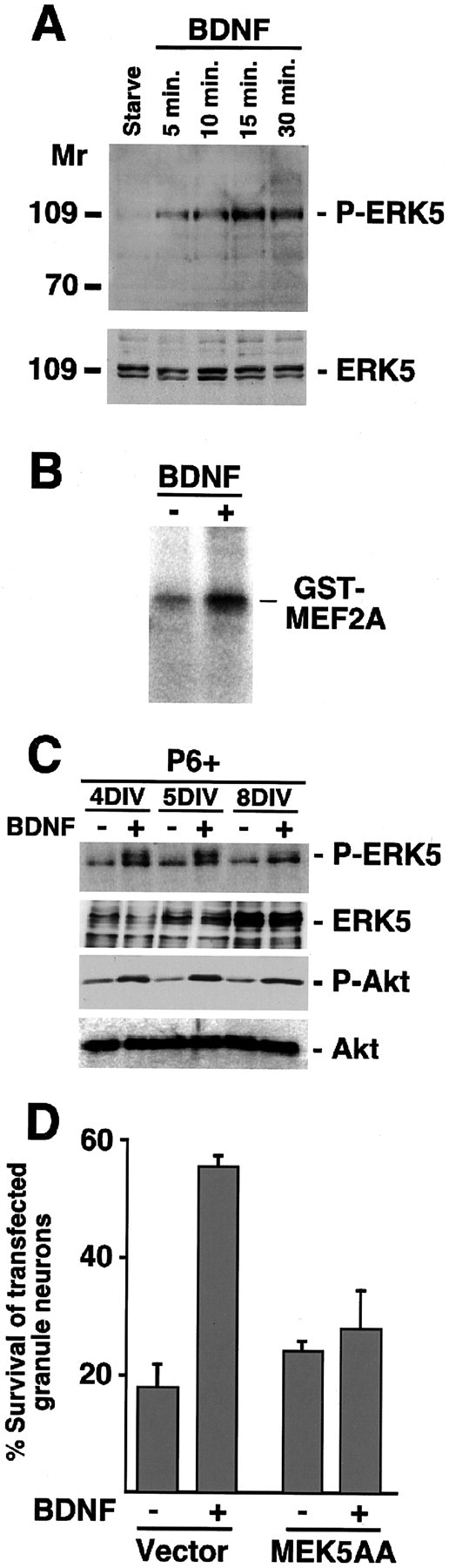
ERK5 is activated in newly generated granule neurons and is necessary for BDNF-mediated survival. A, BDNF activates ERK5 in newly generated granule neurons. Granule neurons were deprived of survival factors and left untreated or treated with BDNF, as in Figure 1 B, for the indicated periods. Immunoblotting was done with a phospho-ERK5 antibody that recognizes ERK5 that is phosphorylated within the TEY motif (top; BioSource) or with an antibody that recognizes ERK5, regardless of phosphorylation state (bottom; Calbiochem). B, BDNF stimulates ERK5 kinase activity. Granule neurons (P6 + 5DIV) were deprived of survival factors and left untreated or treated with BDNF (100 ng/ml) for 15 min. Immunoprecipitated ERK5 was subjected to an in vitro kinase assay using GST-MEF2A as substrate. C, BDNF induces ERK5 phosphorylation in newly generated, but not in mature, cerebellar granule neurons. Newly generated (P6 + 4DIV or + 5DIV) or mature (P6 + 8DIV) granule neurons were treated with BDNF, as in Figure 1 B, for 30 min. Immunoblotting was done with the phospho-ERK5 antibody (top), the ERK5 antibody (second panel), the phospho-Akt antibody (third panel), and the Akt antibody (bottom). D, ERK5 mediates BDNF-induced granule neuron survival. Cerebellar granule neurons (P6 + 5DIV) were transfected with a dominant-negative MEK5 (MEK5AA) or its control vector together with an expression plasmid encoding β-galactosidase. Transfected cultures were deprived of survival factors and left untreated or treated with BDNF (100 ng/ml). After 2 d, cells were fixed and analyzed for cell survival and death. Percentage of survival is shown as mean ± SEM. BDNF significantly increased survival of vector-transfected cerebellar granule neurons (ANOVA; p < 0.001; n = 3) but not MEK5AA-expressing granule neurons. To determine the specificity of MEK5AA in granule neurons, we tested the effect of MEK5AA on BDNF-induced phosphorylation of ERK1/2 immunocytochemically using the phosphoERK1/2 antibody. In two experiments, nearly 80% of vector-transfected, BDNF-treated granule neurons displayed phospho ERK1/2 immunoreactivity. The expression of a dominant interfering form of MEK1 (MEK1KA97) reduced the number of phosphoERK1/2-positive cells to 37%. However, the expression of MEK5AA failed to reduce BDNF activation of ERK1/2, because 77% of MEK5AA-expressing granule neurons were phosphoERK1/2 positive.
In vitro kinase assays. Cerebellar granule neuron cultures maintained in full medium were switched to BME and left untreated or treated with BDNF (100 ng/ml) for 15 min. Lysates were prepared from these cultures in a buffer containing 20 mm Tris, pH 7.5, 5 mm EGTA, 25 mm β-glycerophosphate, 1% Triton X-100, 150 mm NaCl supplemented with 2 mm DTT, 1 mm NaVO3, 1 mm PMSF, and 1% aprotonin. Lysates were subjected to immunoprecipitation by incubating with 10 μl of anti-ERK5 antibody (Calbiochem) for 2 hr at 4°C, followed by 20 μl of protein A-sepharose for 2 hr. Immunoprecpitates were washed three times with a buffer containing 20 mm Tris, pH 7.5, 500 mm NaCl, and 0.05% Tween 20, and 2 mm DTT and then subjected to an in vitro kinase reaction for 30 min at 30°C in 30 μl containing 4 mm Tris, pH 7.5, 2 mm MgCl2, 20 μm cold ATP, 15 μCi [32]-P-γ-ATP, and 1 μg of GST-MEF2A. Reaction products were separated by PAGE and subjected to autoradiography. Immunoprecipitated ERK5 did not induce the phosphorylation of glutathione S-transferase (GST).
Reverse transcription-PCR and Northern blot analyses. Cerebellar granule neurons (15 × 106) were plated on polyornithine-coated 6-cm plates. RNA was extracted from harvested cells using TRIzol reagent (Invitrogen, San Diego, CA) according to the manufacturer's protocol, precipitated, and resuspended in a small volume of DEPC-treated water. Two nanograms of RNA were subjected to reverse transcription (RT)-PCR using the Invitrogen Superscript RT-PCR system. The primers for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were reverse 5′-CATGTCAGATCCACAACGG-3′ and forward 5′-TGCTGGTGCTGAGTATGTCG-3′. The primers for NT-3 were reverse 5′-TCTGAAGTCAGTGCTCGGAC-3′ and forward 5′-GGTCAGAATTTCCAGCCGATG-3′. Reverse transcription was performed at 50°C for 30 min. PCR consisted of an initial 2 min at 95°C, followed by either 33 (NT-3) or 25 (GAPDH) amplification cycles of 95°C for 30 sec, 55°C for 30 sec, and 72°C for 1 min, followed by a final incubation of 72°C for 10 min after the last cycle. PCR products were separated on 3% agarose/Tris-acetate EDTA gels and detected with ethidium bromide.
DNA-binding assays. DNA-binding assays were carried out as described previously (Bonni et al., 1997). Double-stranded oligonucleotide probes corresponding to the wild-type and mutant MEF2-binding site from the NT-3 promoter were labeled with [32]P-γ-ATP using T4 polynucleotide kinase (New England Biolabs, Beverly, MA) and purified. For DNA binding reactions, 2 × 10 5 cpm of probe was incubated with 2 μg of total protein extract from 293T cells that were transfected with vector or a MEF2A expression plasmid, in the presence or absence of cold wild-type or mutant competitors, for 20 min at room temperature in DNA-binding buffer (5 mm HEPES, pH 7.9, 0.5 mm spermidine, 100 μm EGTA, 1 mm DTT, 20 μg/ml poly dIdC, 600 μg/ml BSA, and 7% glycerol). Reaction products were separated by electrophoresis on a nondenaturing 4% acrylamide gel and analyzed by autoradiography.
Conditional knock-out mice. All animals were housed and cared for according to Institutional Animal Care and Use Committee guidelines. Mice carrying an allele of NT-3 with exon II flanked by loxP sites have been described and were obtained from Jackson Labs. Nestin-Cre recombinase transgenic mice were generously provided by Dr. Philip Hinds (Harvard Medical School, Boston, MA). Female homozygous floxed NT-3 mice were intercrossed with male heterozygous floxed NT-3 mice carrying a single copy of the Nestin-Cre recombinase transgene. Genotypes were confirmed by PCR analysis of total genomic DNA. Primary cerebellar granule neurons were cultured from postnatal day 5 mice and plated at a density of 1 × 10 5/well in polyornithine-coated 96-well plates in BME (Sigma) supplemented with 10% calf serum (Hyclone), 25 mm KCl, 2 mm glutamine, penicillin, and streptomycin. One day after cultures were prepared (P6 + 1DIV), they were switched to BME supplemented with 25 mm glucose, 2 mm glutamine, penicillin, and streptomycin and 10 μm of the mitotic inhibitor cytosine-β-d-arabinofuranoside, in the presence or absence of neurotrophic factors as detailed in the legend for Figure 6.
Figure 6.

NT-3 is required for BDNF-induced survival of newly generated cerebellar granule neurons. A, Newly generated cerebellar granule neurons (P6 + 5DIV) were deprived of survival factors and left untreated or treated with BDNF (100 ng/ml; Control) or together with a mouse monoclonal neutralizing antibody specific to NT-3 (anti-NT-3; 200 ng/ml; R&D) or with a control neutralizing antibody specific to ciliary neurotrophic factor (anti-CNTF; 200 ng/ml; R&D). Two days later, cultures were fixed and analyzed for cell survival and death. Percentage of survival is shown as mean ± SEM. BDNF significantly promoted the survival of granule neurons in control or anti-CNTF-treated cultures (ANOVA; p < 0.001; n = 3) but not in the presence of the anti-NT-3 antibody. B, C, NT-3 is necessary for BDNF-mediated neuron survival. Cultures of newly generated cerebellar granule neurons (P5 + 1DIV) from mice with disruption of the NT-3 gene generated by CRE-mediated recombination of floxed NT-3 alleles were deprived of survival factors and left untreated or supplemented with increasing amounts of BDNF (0, 25, or 50 ng/ml). After 3 d, cultures were fixed and analyzed for cell survival and death. Shown in B are percentages of apoptosis for matched wild-type littermates (nt3+/+; closed symbols) and knock-out littermates (nt3-/-; open symbols). Shown in C is mean percentage of reduction in apoptosis ± SEM for 25 ng/ml (25) or 50 ng/ml (50) BDNF relative to untreated cultures. The survival effect of BDNF is significantly reduced in NT-3 knock-out (nt3-/-) cultures relative to wild-type controls (nt3+/+) (ANOVA; p < 0.01; n = 3). D, Exogenous NT-3 rescues BDNF-mediated neuron survival in NT-3 knock-out cells. Cultures of newly generated cerebellar granule neurons (P5 + 1DIV) were prepared from nt3+/+ or nt3-/- mice and were deprived of survival factors or treated with 50 ng/ml BDNF alone (BDNF) or 50 ng/ml BDNF plus 50 ng/ml NT-3 (BDNF+NT-3). After 3 d, granule neuron cultures were fixed and analyzed for cell survival and death. Shown is mean percentage of reduction in apoptosis ± SEM relative to untreated cultures. The addition of NT-3 together with BDNF led to a significant increase in survival of NT-3 knock-out (nt3-/-) granule neurons relative to BDNF alone (ANOVA; p < 0.001; n = 4) but not of nt3+/+ granule neurons.
Results
BDNF promotes the survival of cerebellar granule neurons in an age-specific manner
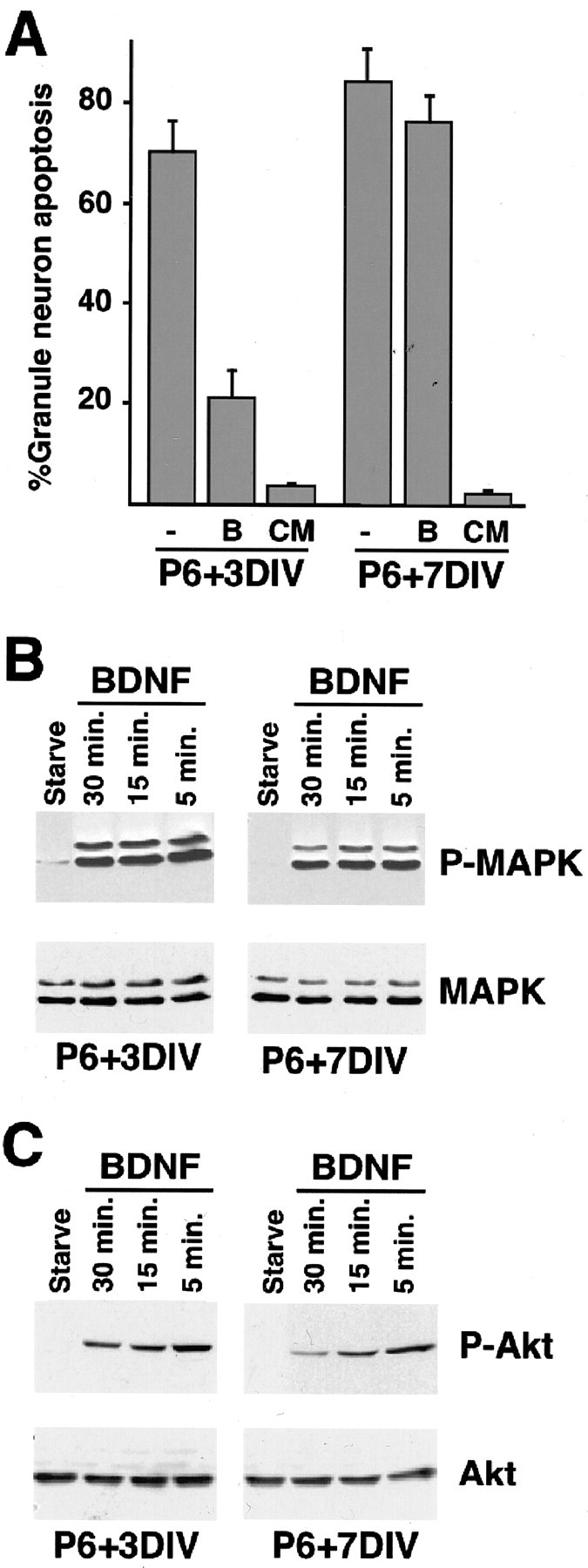
To investigate the mechanisms that might confer temporal specificity to the BDNF-induced survival response in granule neurons of the developing cerebellum, we first characterized BDNF enhancement of survival of newly generated granule neurons isolated from postnatal day 6 rat pups and cultured for 3 d in vitro (P6 + 3DIV) or mature granule neurons (P6 + 7DIV). As expected, BDNF robustly reduced the rate of apoptosis of newly generated granule neurons that had been deprived of full survival medium (Fig. 1A). However, BDNF failed to promote the survival of mature granule neurons (Fig. 1A). The difference in the survival response of newly generated and mature granule neurons to BDNF was not because of the nonspecific failure of mature granule neurons to respond to survival factors, because both populations of neurons underwent little apoptosis in full survival medium (Fig. 1A). These data suggest that BDNF enhancement of granule neuron survival in primary cerebellar cultures occurs in a temporally defined manner that mimics the pattern of BDNF-induced granule neuron survival in vivo.
Figure 1.
BDNF promotes survival of cerebellar granule neurons in a temporally defined manner. A, Newly generated (P6 + 3DIV) or mature (P6 + 7DIV) cerebellar granule neurons were deprived of survival factors and left untreated (control) or treated with BDNF (100 ng/ml) or with full survival medium containing 30 mm KCl and 10% serum (conditioned medium, CM). After 48 hr, cultures were fixed and subjected to immunofluorescence. Percentage of apoptosis is shown as mean ± SEM. BDNF significantly reduced apoptosis in newly generated cerebellar granule neurons (ANOVA; p < 0.0005; n = 3) but had no effect on apoptosis of mature granule neurons. Full survival medium reduced apoptosis of both newly generated and mature cerebellar granule neurons (ANOVA; p < 0.0001; n = 3). B,C, BDNF induces the phosphorylation of ERK1/2 and Akt in both newly generated and mature cerebellar granule neurons. Newly generated (P6 + 3DIV) and mature (P6 + 7DIV) cultures of cerebellar granule neurons were deprived of survival factors for 1 hr and then treated with BDNF (100 ng/ml) for the indicated periods. In B, immunoblotting was performed with an antibody that specifically recognizes ERK1/2 when phosphorylated within the TEY motif (B, top; Promega) or with an antibody that recognizes ERK1/2, regardless of the phosphorylation state (B, bottom; New England Biolabs). In C, immunoblotting was performed with an antibody that recognizes Akt when phosphorylated at serine 478 (C, top; New England Biolabs) or with an antibody that recognizes Akt, regardless of phosphorylation state (C, bottom; New England Biolabs).
In recent studies, the phosphatidylinositol 3-kinase (PI3K)-Akt and the ERK-Rsk kinase pathways have emerged as critical mediators of BDNF-dependent neuron survival (Meyer-Franke et al., 1998; Bonni et al., 1999). To determine whether a difference in the activation of these signaling pathways in newly generated and mature granule neurons might account for the differential BDNF survival response at the two ages, we examined the activation of ERK1/2 and Akt in the two populations of granule neurons. Surprisingly, immunoblotting of cerebellar granule neuron lysates using phospho-specific antibodies revealed little difference in the ability of BDNF to induce phosphorylation of ERK1/2 and Akt on sites that reflect their activation in mature and newly generated granule neurons (Fig. 1B,C). These data indicate that although BDNF selectively promotes the survival of newly generated granule neurons, both newly generated and mature granule neurons are responsive to BDNF. In addition, these data suggest that other BDNF-induced signals, in addition to the PI3K-Akt and ERK-Rsk kinase cascades, are required for BDNF to promote neuron survival in a temporally defined manner.
The ERK5-MEF2 signaling pathway mediates BDNF-induced survival of newly generated granule neurons
We next considered the possibility that a signaling pathway that is distinct from the ERK1/2-Rsk and PI3K-Akt cascades might confer on BDNF the ability to selectively promote the survival of newly generated granule neurons. Recent reports suggest that the ERK1/2-related kinase ERK5 mediates neurotrophin-induced responses, including BDNF-induced transcription in cortical neurons and NGF-induced survival of sensory neurons in the PNS (Kamakura et al., 1999; Cavanaugh et al., 2001; Watson et al., 2001). However, the role of ERK5 in the pro-survival function of neurotrophins in the developing mammalian brain remained to be characterized.
To determine whether the ERK5 signaling pathway plays a role in regulating BDNF-induced survival, we first asked whether BDNF stimulates the activity of ERK5 in newly generated granule neurons. BDNF induced the rapid phosphorylation of ERK5 on sites that reflect its activation as determined by immunoblotting of granule neuron lysates using phospho-specific ERK5 antibodies (Fig. 2A). In other experiments, BDNF stimulated the kinase activity of ERK5 in granule neurons as determined by in vitro kinase assays of ERK5 immunoprecipitated from granule neuron lysates (Fig. 2B). BDNF triggered ERK5 phosphorylation in newly generated granule neurons but failed to effectively induce ERK5 phosphorylation in mature granule neurons, suggesting that BDNF stimulates the ERK5 signaling pathway in granule neurons in an age-specific manner (Fig. 2C). Interestingly, the total level of ERK5 increased with age in granule neurons (Fig. 2C), suggesting an even more striking age-associated difference in the ability of BDNF to induce ERK5 phosphorylation in granule neurons.
We next determined the role of BDNF-activated ERK5 in BDNF-induced survival of newly generated granule neurons. We tested the effect of a dominant interfering form of the ERK5 activator MEK5, in which two key regulatory residues have been replaced by alanines (MEK5AA) (Marinissen et al., 1999), on BDNF-induced survival. Cultures of newly generated cerebellar granule neurons were transfected with an expression plasmid encoding MEK5AA or a control vector together with an expression plasmid encoding β-galactosidase. Transfected cultures were deprived of survival factors and left untreated or treated with BDNF. After 2 d, cultures were fixed and subjected to indirect immunofluorescence using an antibody to β-galactosidase and the DNA dye bisbenzimide (Hoechst 33258) to label the nuclei. A determination of cell death in transfected neurons was made on the basis of the presence or absence of nuclear pyknosis and fragmentation and the integrity of the neurites as visualized by β-galactosidase. We found that the expression of MEK5AA reduced significantly BDNF-induced survival of newly generated granule neurons (Fig. 2D). MEK5AA specifically blocks the activation of ERK5 (Marinissen et al., 1999), and in granule neurons the expression of MEK5AA failed to inhibit BDNF activation of ERK1/2, as determined by immunocytochemistry experiments using an antibody that specifically recognizes the activated form of ERK1/2 (see legend to Fig. 2D). Together, these results suggest that BDNF activates ERK5 in granule neurons in a temporally defined manner and, once activated, the ERK5 signaling pathway confers on BDNF the ability to mediate the survival of newly generated granule neurons.
We next examined the mechanism by which ERK5 mediates BDNF-induced granule neuron survival. Members of the MEF2 family of transcription factors are thought to be physiological substrates of ERK5 (Marinissen et al., 1999). In neurons, electrical activity induces MEF2-dependent survival via the p38MAPK signaling pathway (Mao et al., 1999). In contrast, BDNF-induced granule neuron survival occurs independently of the p38MAPK signaling pathway. The expression of a dominant interfering form of the p38MAPK activator MKK6 (MKK6KA) in granule neurons that reduces activity-dependent cell survival (Mao et al., 1999) failed to inhibit BDNF-induced cell survival (data not shown). Nevertheless, we considered the possibility that BDNF might induce MEF2-dependent survival of newly generated granule neurons owing to the ability of BDNF to activate ERK5 in these neurons.
We expressed two dominant interfering forms of MEF2, MEF2CR24L and MEF2A-TA, in cerebellar granule neurons (Fig. 3A) to block endogenous MEF2 function. MEF2CR24L is a mutant MEF2C protein that contains a mutation within its conserved DNA-binding region (Molkentin et al., 1996). MEF2A-TA is a mutant MEF2A protein in which threonines 312 and 319 have been replaced with alanine (Zhao et al., 1999). MEF2CR24L and MEF2A-TA act as dominant-negative MEF2 proteins by distinct mechanisms. Whereas MEF2CR24L forms dimers with endogenous MEF2 proteins that then fail to bind to the MEF2 response element (MRE) within MEF2-responsive genes, MEF2A-TA competes with endogenous MEF2s for binding to the MRE within MEF2-responsive genes. However, once bound to DNA, MEF2A-TA fails to activate transcription because MEF2A-TA cannot undergo phosphorylation at the critical regulatory sites of threonines 312 and 319 in response to extracellular stimuli. We found that the expression of both dominant interfering forms of MEF2 reduced significantly BDNF-induced survival of newly generated granule neurons (Fig. 3B). These results suggest that MEF2-dependent transcription is required for BDNF-induced granule neuron survival.
Figure 3.
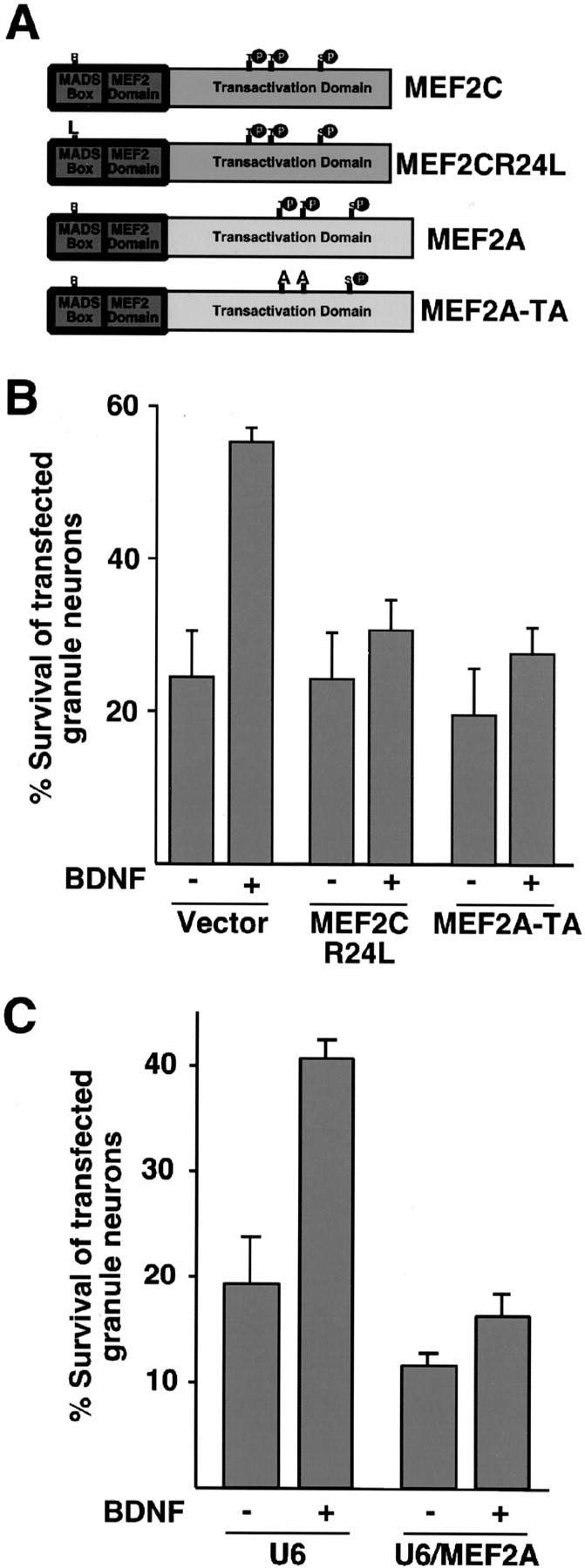
MEF2 mediates BDNF-induced survival of newly generated cerebellar granule neurons. A, Schematic representation of dominant-negative MEF2s. In MEF2CR24L, arginine 24 is substituted to lysine within the conserved DNA-binding domain. In MEF2A-TA, the key regulatory sites of phosphorylation threonines 312 and 319 are replaced by alanines. B, MEF2 mediates BDNF-induced granule neuron survival. Granule neurons were transfected with an expression plasmid encoding the dominant interfering forms of MEF2 (see Fig.3A) or the control vector together with the β-galactosidase expression plasmid. Transfected cultures were analyzed as in Figure 2 D. BDNF significantly increased survival of vector-transfected cerebellar granule neurons (ANOVA; p < 0.001; n = 3) but not of MEF2CR24L- or MEF2A-TA-expressing granule neurons. C, Knockdown of MEF2A inhibits BDNF-induced granule neuron survival. Granule neurons (P6 + 2DIV) were transfected with the U6 control vector or the U6/MEF2A plasmid encoding MEF2A hairpin RNAs, together with the β-galactosidase expression plasmid. After 2 d, cells were changed to medium without survival factors or were supplemented with BDNF (100 ng/ml). After 1 d, cells were fixed and subjected to immunofluorescence. Percentage of survival is shown as mean ± SEM. BDNF significantly increased survival of U6-transfected cerebellar granule neurons (ANOVA; p < 0.0005; n = 3) but not of MEF2A hpRNA-expressing transfected granule neurons. BDNF-induced survival was significantly reduced in MEF2A hpRNA-expressing neurons (percentage of survival, 16.3 ± 1.8; p < 0.01; ANOVA; n = 3) but not in Cdk2 hpRNA-expressing neurons (percentage of survival, 32.3 ± 4.7; n = 3) when compared with U6-transfected granule neurons (percentage of survival, 40.7 ± 1.5; n = 3).
Because MEF2A is the most highly expressed member of the MEF2 family in newly generated granule neurons (Mao et al., 1999), we assessed the specific role of MEF2A in the BDNF-induced survival response. Using a U6 promoter-based DNA template method expressing MEF2A hairpin RNAs (hpRNAs) that specifically reduced endogenous MEF2A expression in cerebellar granule neurons (Gaudilliere et al., 2002), we found that the MEF2A hpRNAs significantly reduced BDNF enhancement of neuronal survival (Fig. 3C). This was not due to a non-specific effect of productive hpRNAs because the expression of hpRNAs targeted to the unrelated gene cyclin dependent kinase 2 (Cdk2) was found to have little effect on BDNF-induced neuronal survival (see legend of Fig. 3C). Together, these results suggest that endogenous MEF2A mediates BDNF enhancement of the survival of newly generated granule neurons.
NT-3: a novel target of the ERK5-MEF2 signaling pathway
The results suggesting that MEF2-dependent transcription is required for BDNF-induced granule neuron survival led us next to investigate the mechanism by which the ERK5-MEF2 signaling pathway mediates BDNF-induced neuron survival. As a first step toward identifying the set of MEF2-induced genes that might promote the survival of neurons, we performed computer-aided searches to identify genes that contain within their promoters conserved potential MEF2-binding sites (MREs). By sequence gazing, we found that the promoter of the NT-3 gene contains a potential MRE at nucleotide -1026 upstream of the transcriptional start site that is conserved in the human and rat NT-3 genes. Consistent with the possibility that NT-3 might represent a novel target of MEF2, BDNF has been reported to induce the expression of NT-3 in granule neurons (Leingartner et al., 1994).
Because BDNF induces the activation of ERK5 in granule neurons in a temporally defined manner and the ERK5-MEF2 signaling pathway mediates BDNF-induced survival of newly generated granule neurons, we determined the temporal profile of BDNF induction of NT-3 gene expression. We found that BDNF stimulated the expression of the late response gene encoding NT-3 in newly generated granule neurons but failed to induce NT-3 expression in mature granule neurons (Fig. 4A). In contrast, BDNF induced transcription of the immediate early gene c-fos in both newly generated and mature granule neurons (Fig. 4A). The temporal profiles of BDNF-induced granule neuron survival, BDNF-induced NT-3 expression, and BDNF-induced ERK5 activation correlated tightly (Fig. 1A, 2C, 4A,B), suggesting that NT-3 may be a direct target of the ERK5-MEF2 signaling pathway.
Figure 4.

NT-3: a novel target of the BDNF-induced ERK5-MEF2 signaling pathway in newly generated granule neurons. A, B, BDNF induces the expression of NT-3 mRNA in newly generated, but not in mature, cerebellar granule neurons. In A, P6 + 3DIV and P6 + 7DIV cerebellar granule neurons were deprived of survival factors for 4 hr and treated with BDNF for the indicated periods. In B, P6 + 5DIV, P6 + 7DIV, or P6 + 9DIV granule neurons were treated with 10 μg/ml insulin in the presence or absence of BDNF for 24 hr. RNA was isolated and subjected to RT-PCR with primers specific for c-fos (Fig. 4 A only), NT-3, or GAPDH. C, BDNF activates NT-3 promoter-dependent transcription. NT-3-luciferase reporter constructs containing nucleotides -1087 to +91 or nucleotides -838 to +91 relative to the NT-3 transcription start site of the NT-3 promoter were transfected in newly generated cerebellar granule neurons (P6 + 3DIV). After 6 hr, cells were treated without or with BDNF (100 ng/ml) for 20 hr, and luciferase activity was determined. Shown are mean ± SEM luciferase values normalized relative to uninduced -1087/+91 reporter. BDNF significantly induced the -1087 NT-3-luciferase reporter gene (ANOVA; p < 0.0001; n = 5) but not the -838 NT-3-luciferase reporter. D, The NT-3 promoter contains a MEF2-binding site. Extracts from cerebellar granule neurons (CGN; P6 + 3DIV) were incubated in the presence or absence a 30 bp oligonucleotide containing the putative NT-3 MRE, with or without a 10-fold excess of an oligonucleotide containing a consensus MRE (10x WT MRE), an oligonucleotide containing a mutant MRE does not bind MEF2 (10x Mut MRE), or a polyclonal antibody against MEF2A (left). Similar reactions were performed with extracts from 293T cells overexpressing MEF2A (right). FP, Free oligonucleotide probe. E, The MRE is required for BDNF induction of NT-3 transcription in response to BDNF. A NT-3-luciferase reporter gene containing nucleotides -1117 to +91 of the NT-3 promoter with either the wild-type MRE (WT MRE) or a point mutation of the MRE (Mut MRE) that abolished MEF2 binding in vitro were introduced to P6 + 3DIV granule neurons, as in C. After transfection, cells were analyzed as in C. BDNF induced the wild-type -1117 NT-3-luciferase reporter significantly (ANOVA; p < 0.0001; n = 5) but not the Mut MRE -1117 NT-3-luciferase reporter gene. F, Granule neurons were transfected with the -1087 NT-3-luciferase reporter gene together with an expression plasmid encoding MEF2A-TA or MEF2CR24L or the control plasmid. Transfected cultures were analyzed as in Figure 4C. BDNF induced the NT-3 promoter significantly in control-transfected cultures (ANOVA; p < 0.0005; n = 3) but not in MEF2A-TA- or MEF2CR24L-expressing granule neurons. G, Activation of the ERK5 pathway is required for BDNF-induced NT-3 transcription. Newly generated granule neurons (P6 + 3DIV) were transfected, as in C, with the -1087 NT-3-luciferase reporter gene and with an expression vector encoding dominant-negative MEK5 (MEK5AA) or its control vector and analyzed as in C. BDNF significantly induced the NT-3 reporter in vector-transfected cultures (ANOVA; p < 0.005; n = 4) but not in MEK5AA-expressing granule neurons.
In transient transfection assays in cerebellar granule neurons, we found that BDNF stimulates the expression of a luciferase reporter gene controlled by ∼1 kb of the NT-3 promoter in newly generated granule neurons but not in mature neurons, suggesting that BDNF induces NT-3 transcription in a temporally defined manner (Fig. 4C) (data not shown). Deletion mapping revealed that a region of ∼250 bp between -1087 and -838 bp 5′ to the transcription start site that contains the potential MEF2-binding site (MRE) is necessary for BDNF-induced expression of a NT-3 promoter-luciferase reporter construct (Fig. 4C).
In DNA-binding assays, the MEF2 protein MEF2A, when expressed in 293T cells, formed a complex with a radiolabeled NT-3 promoter, MRE (Fig. 4D). The protein-DNA complex was disrupted by competition with a consensus MRE but not with a mutant MRE, and the complex was supershifted with an anti-MEF2A antibody (Fig. 4D). Likewise, we found that the radiolabeled NT-3 promoter MRE formed a specific protein-DNA complex when incubated with lysates of cerebellar granule neurons. Formation of the granule neuron-derived protein-DNA complex was competed by wild-type MRE but not mutant MRE and was supershifted by the anti-MEF2A antibody (Fig. 4D). Together, these results suggest that endogenous MEF2A in granule neurons binds the NT-3 promoter MRE.
We next determined the role of endogenous MEF2 in BDNF-induction of NT-3 transcription in granule neurons. We first tested the effect of a mutation of the NT-3 MRE known to disrupt MEF2 binding in vitro on the ability of BDNF to stimulate NT-3 promoter-mediated transcription. Whereas BDNF stimulated the expression of a reporter gene controlled by the wild-type NT-3 promoter, BDNF failed to effectively induce the NT-3 promoter containing the mutant MRE, suggesting that MEF2 binding to the NT-3 promoter MRE is critical for BDNF induction of NT-3 transcription (Fig. 4E). We also tested the effect of the two dominant interfering forms of MEF2 (Fig. 3A) on the BDNF-induced NT-3 response. We found that both MEF2CR24L and MEF2A-TA significantly reduced the ability of BDNF to stimulate NT-3 promoter-mediated transcription (Fig. 4F). These results suggest that MEF2 binds to the NT-3 MRE and thereby mediates BDNF-induced NT-3 transcription in cerebellar granule neurons.
To determine whether BDNF-activated ERK5 is required for BDNF-induced NT-3 expression in newly generated granule neurons, we tested the effect of the dominant interfering form of MEK5, MEK5AA, on the BDNF-induced response. We found that the expression of MEK5AA in newly generated granule neurons inhibited BDNF-induction of NT-3 promoter-mediated transcription (Fig. 4G). Together, these findings implicate the ERK5-MEF2 pathway as a developmentally regulated signaling module that mediates BDNF-induced NT-3 expression in newly generated granule neurons.
MEF2 and CREB cooperate to mediate BDNF-induced NT-3 transcription
Although the activation of MEF2 is required for BDNF-induced NT-3 transcription (Fig. 4), in transient expression assays, we found that MEF2A, when targeted to the DNA-binding site of the heterologous transcription factor GAL4 just upstream of the TATA box of a reporter gene, mediated BDNF-induced reporter transcription only to a modest level (data not shown). These results suggested that MEF2 might cooperate with other transcription factors to mediate BDNF-induced NT-3 transcription.
The MEF2A transcriptional response to neurotrophins in the absence of other transcription factors is similar to the neurotrophin-induced CREB response under similar circumstances (Ginty et al., 1994; Bonni et al., 1995). Interestingly, to mediate neurotrophin-induced transcription of the immediate early gene c-fos, CREB cooperates with serum response factor (SRF), a member of the MADS superfamily of transcription factors that includes the MEF2 proteins (Bonni et al., 1995). These observations raised the intriguing possibility that MEF2, in an analogous manner to SRF at the c-fos promoter, might cooperate with CREB or a related protein at the NT-3 promoter to mediate BDNF-induced NT-3 transcription.
Remarkably, the 250-bp region of the NT-3 promoter that is required for BDNF-induced NT-3 transcription contains, in addition to the MRE, a conserved potential CREB-binding sequence (CRE). In DNA-binding assays, extracts of cerebellar granule neurons formed two protein-DNA complexes when incubated with the putative NT-3 CRE. The appearance of the two complexes was disrupted by competition with a 10-fold excess of unlabeled probe containing the wild-type CRE or on incubation with an anti-CREB antibody. In contrast, competition with an excess of unlabeled probe containing a mutant CRE or incubation with an anti-MEF2A antibody had no effect on formation of the protein-DNA complexes (Fig. 5A). Together, these data suggest that endogenous CREB in granule neurons binds the NT-3 promoter CRE.
Figure 5.

MEF2 cooperates with CREB to mediate BDNF-induced NT-3 transcription. A, The NT-3 promoter contains a CREB-binding sequence. Extracts from cerebellar granule neurons (CGN; P6 + 3DIV) were incubated in the presence or absence a 30 bp oligonucleotide containing the putative NT-3 CRE, with or without a 10-fold excess of the wild-type oligonucleotide (10x WT CRE), an oligonucleotide containing a mutant CRE (10x Mut CRE), or a polyclonal antibody against either CREB or MEF2A. FP, Free oligonucleotide probe. B, The CRE is required for BDNF-induction of NT-3 transcription in response to BDNF. An NT-3-luciferase reporter gene containing nucleotides -1087 to +91 of the NT-3 promoter with either the wild-type CRE (WT) or a point mutation of the CRE (Mut CRE) that abolished CREB binding in vitro was introduced to P6 + 3DIV granule neurons and analyzed as in Figure 4C. BDNF induced the wild-type -1087 NT-3-luciferase reporter significantly (ANOVA; p < 0.005; n = 6) but not the Mut CRE -1087 NT-3-luciferase reporter gene. C, Dominant-negative CREB prevents BDNF induction of NT-3. Granule neurons were transfected with the -1087 NT-3-luciferase reporter gene together with an expression plasmid encoding CREBM1 or a control plasmid. Transfected cultures were analyzed as in Figure 4C. BDNF induced the NT-3 promoter significantly in control-transfected cultures (ANOVA; p < 0.0005; n = 3) but not in CREBM1-expressing granule neurons. D, The ERK1/2 pathway is necessary for BDNF-induced NT-3 transcription. Newly generated granule neurons (P6 + 3DIV) were transfected as in Figure 4C with the -1087 NT-3-luciferase reporter gene and an expression vector encoding dominant-negative MEK1 (MEK1KA97) or its control vector and analyzed as in Figure 4C. BDNF significantly induced the NT-3 reporter in vector-transfected cultures (ANOVA; p < 0.05; n = 3) but not in MEK1KA97-expressing granule neurons.
To determine the role of endogenous CREB in BDNF induction of NT-3 transcription, we tested the effect of a point mutation in the NT-3 promoter CRE known to disrupt CREB binding in vitro on the ability of BDNF to stimulate NT-3 promoter-mediated transcription. Whereas BDNF induced the activity of the wild-type NT-3 promoter, BDNF failed to effectively stimulate the NT-3 promoter containing a mutant CRE, suggesting that, in addition to MEF2, the binding of CREB to the NT-3 promoter is necessary for BDNF-induced NT-3 transcription (Fig. 5B). In other experiments, a dominant interfering form of CREB, in which the key regulatory site of serine 133 is replaced with an alanine (CREBM1), significantly inhibited BDNF induction of NT-3 promoter-mediated transcription (Fig. 5C).
Neurotrophins are thought to induce the phosphorylation of CREB at serine 133 via the ERK1/2-Rsk signaling pathway (Shaywitz and Greenberg, 1999; Finkbeiner, 2000; Lonze and Ginty, 2002). We found that expression of the dominant interfering form of the ERK1/2 activator MEK1 (MEK1KA97) also reduced the ability of BDNF to induce the NT-3 promoter (Fig. 5D). Together, our results suggest that on BDNF stimulation of newly generated granule neurons, the ERK1/2 and ERK5 signaling pathways trigger the activation of CREB and MEF2. Once activated, MEF2 and CREB cooperate at the promoter of the NT-3 gene and thereby induce NT-3 transcription.
NT-3 mediates BDNF-induced survival of newly generated cerebellar granule neurons
Our results suggest that BDNF triggers the activation of the ERK5-MEF2 signaling pathway in newly generated granule neurons and thereby stimulates NT-3 transcription and granule neuron survival in a temporally specific manner. Although NT-3 alone is not sufficient to promote the survival of cultured granule neurons (Segal et al., 1992; Lindholm et al., 1993), mice with a CNS-specific knock-out of the NT-3 gene display a phenocopy of the cerebellar defects observed in the BDNF knock-out mice, including the increased apoptosis of newly generated granule neurons (Bates et al., 1999). Together, these observations raised the possibility that NT-3 might mediate the age-specific BDNF survival response.
We first examined the effect of a mouse monoclonal antibody that specifically neutralizes NT-3 on BDNF-dependent survival of cerebellar granule neurons. We found that BDNF failed to effectively promote the survival of granule neurons in the presence of the neutralizing antibody to NT-3 (Fig. 6A). A control mouse monoclonal antibody that neutralizes ciliary neurotrophic factor failed to inhibit the BDNF survival response (Fig. 6A). In other experiments, we confirmed that the NT-3 neutralizing antibody acts specifically as this antibody failed to block the BDNF-triggered immediate responses in neurons including BDNF-induced ERK1/2 phosphorylation (data not shown). Together, these results suggest that NT-3 is required for BDNF-induced survival of granule neurons.
To establish the importance of NT-3 in the developmentally regulated BDNF enhancement of granule neuron survival, we tested the ability of BDNF to promote the survival of newly generated granule neurons obtained from mice in which the NT-3 gene is disrupted in a CNS-specific pattern. BDNF robustly promoted the survival of newly generated granule neurons cultured from nt3+/+ mice (Fig. 6B,C). In contrast, we found that BDNF failed to effectively support the survival of newly generated granule neurons cultured from nt3-/- littermates (Fig. 6B,C). Whereas BDNF treatment of the nt3+/+ granule neurons led to a reduction of apoptosis of over 50%, BDNF reduced apoptosis of nt3-/- granule neurons by just over 20% (Fig. 6B,C). In contrast to the relative inability of BDNF to promote the survival of nt3-/- granule neurons, neuronal activity promoted the survival of granule neurons from nt3-/- mice as effectively as that of granule neurons from wild-type mice (data not shown). In other experiments, we found that exogenous NT-3 almost completely rescued the ability of BDNF to promote the survival of the nt3-/- granule neurons (Fig. 6D). Together, these findings demonstrate the specific requirement for NT-3 in BDNF-induced survival of newly generated granule neurons.
Discussion
We have characterized a signaling mechanism that mediates the survival of neurons in a temporally defined manner in the developing mammalian brain. The neurotrophin BDNF promotes the survival of newly generated, but not mature, cerebellar granule neurons. Although activation of the ERK1/2-Rsk and PI3K-Akt signaling cascades is required for BDNF-induced neuron survival (Meyer-Franke et al., 1998; Bonni et al., 1999), these two pathways alone do not seem to contain the temporally specific signal necessary for BDNF to promote the survival of granule neurons. Rather, we found that BDNF induces the ERK5-MEF2 pathway and thereby stimulates transcription of the neurotrophin NT-3 specifically in newly generated granule neurons. Disruption of each endogenous component of the ERK5-MEF2-NT-3 signaling pathway was found to abrogate the ability of BDNF to promote the survival of newly generated granule neurons. We also found that the NT-3 gene represents a convergence point for the ERK5-MEF2 and ERK1/2-Rsk-CREB signaling pathways, because CREB and MEF2 cooperate to mediate BDNF-induced NT-3 transcription. Together, our findings suggest that the ERK5-MEF2 signaling pathway acts a switch to confer temporal specificity to the pro-survival function of BDNF and links the sequential actions of the two major neurotrophic factors, BDNF and NT-3, in developing cerebellar granule neurons.
The ERK5-MEF2 signaling pathway joins the ERK1/2-Rsk-CREB and PI3K-Akt signaling pathways as important mediators of BDNF-induced survival of neurons in the developing CNS (Kaplan and Miller, 2000; Meyer-Franke et al., 1998). Our results indicate that the activation of each of these three major pathways is required for BDNF-promoted survival of newly generated cerebellar granule neurons, suggesting that these pathways act in a nonadditive manner. Consistent with this idea, we have found common points of convergence on which these signals act to enhance neuronal survival. For example, our results suggest that both ERK5 and ERK1/2 induce the Rsk-mediated phosphorylation of BAD at serine 112 and thereby inhibit a component of the cell death machinery directly (Bonni et al., 1999) (data not shown). In addition, we have found that the transcription factors MEF2 and CREB cooperate to mediate BDNF induction of NT-3 transcription in newly generated granule neurons leading to their survival, suggesting that the ERK5-MEF2 and ERK1/2-Rsk-CREB signaling pathways converge on the NT-3 promoter. The possibility also remains that both the ERK5 and ERK1/2 signaling cascades collaborate to induce the activation of CREB in granule neurons. In future studies, it will be important to determine the extent of the dependence of BDNF responses on the cooperativity of CREB and MEF2 by characterizing the set of genes that require both CREB and MEF2 activation in BDNF-treated neurons.
Our findings illustrate the possibility of how the participation of several signaling cascades in mediating the pro-survival function of a neurotrophic factor might allow these signals to mediate distinct neurotrophic factor-induced responses. In the case of granule neurons, the activation of the ERK5-MEF2-NT-3 signaling module occurs selectively in newly generated but not in mature granule neurons and may, thus, act as a switch to confer on BDNF the ability to specifically promote the survival of newly generated granule neurons. The subsequent developmental un-coupling of ERK5 activation from BDNF may allow BDNF to exert distinct biological effects at the later stages of granule neuron development. Therefore, it will be interesting in future studies to determine the role of the ERK1/2 and PI3K-Akt signaling pathways in the biological responses of mature granule neurons to BDNF.
In this study, we have characterized NT-3 as the first identified pro-survival target gene of MEF2 in neurons. Our study establishes a requirement for NT-3 in BDNF-induced granule neuron survival. However, although critical, NT-3 is neither the sole nor sufficient pro-survival target of the ERK5-MEF2 signaling pathway in neurons. The addition of NT-3 did not protect neurons against apoptosis induced by the expression of the dominant interfering form of MEK5 (MEK5AA) (data not shown). Therefore, additional antiapoptotic targets of the ERK5-MEF2 pathway that remain to be identified are required to function together with NT-3 to effectively mediate BDNF-induced neuronal survival.
Although a critical role for BDNF in the enhancement of cerebellar granule neuron survival has been established by both in vitro and genetic evidence in mice (Lindholm et al., 1993; Schwartz et al., 1997), the role of NT-3 in the regulation of cerebellar granule neuron survival in early studies was less clear. The addition of NT-3 to cultured cerebellar granule neurons does not promote the survival of the granule neurons, indicating that NT-3 is not sufficient to promote granule neuron survival (Segal et al., 1992; Gao et al., 1995). However, the results of the CNS-specific knock-out of the NT-3 gene in mice (Bates et al., 1999) and our finding that NT-3 is required for BDNF-induced granule neuron survival suggest that, although not sufficient, NT-3 plays a critical role in the survival of granule neurons in the developing cerebellum by mediating BDNF enhancement of neuronal survival.
Our results provide new insights into the understanding of the relationship of the two major neurotrophic factors, BDNF and NT-3, in the developing cerebellum. Supporting our results of a requirement for NT-3 in BDNF enhancement of granule neuron survival are the observations that disruption of the BDNF gene or the CNS-specific disruption of the NT-3 gene in mice leads to a very similar phenotype in the developing cerebellum, including an increase in apoptosis of newly generated granule neurons (Schwartz et al., 1997; Bates et al., 1999). However, knock-out studies of the major BDNF and NT-3 receptors TrkB and TrkC, respectively, reveal that loss of TrkB or TrkC leads to a modest increase in apoptosis of granule neurons, but combined loss of three of the four TrkB and TrkC alleles (TrkB-/-, TrkC+/- or TrkB+/-, TrkC-/-) leads to a greater rate of apoptosis of granule neurons (Minichiello and Klein, 1996), suggesting that TrkB and TrkC act synergistically to promote granule neuron survival.
Disparities in the cerebellar phenotypes of the neurotrophin ligand and receptor knock-outs may be explained in part by the observation that a small fraction of the TrkB and TrkC knock-out mice reach maturity and may, thus, reflect the existence of modifier genes that alter neurotrophin function in the CNS. In addition, neurotrophins are known to interact with multiple Trk receptor subtypes with varying affinities, leading to some discordance in the phenotype of ligand and receptor knock-outs (Bothwell, 1995; Farinas et al., 1998). Importantly, closer examination of these genetic studies reveals that the increase in apoptosis in the receptor knock-outs was measured at P12 (Minichiello and Klein, 1996), whereas in the ligand knock-outs the increase in apoptosis is maximal at P8 (Schwartz et al., 1997; Bates et al., 1999) and in the BDNF -/- mice there is little evidence of increased apoptosis at P14 (Schwartz et al., 1997). These observations raise the possibility that differences in the ligand and receptor knock-out phenotypes may reflect distinct relationships between BDNF and NT-3 in suppressing granule neuron apoptosis at different developmental time points. Together with the genetic data of both the ligand and receptor knock-outs, our findings suggest the following model: NT-3 acts downstream of BDNF to mediate the ability of BDNF to promote the survival of newly generated granule neurons in a developmentally specific time window, one that occurs during the peak of developmentally regulated granule neuron apoptosis. However, the relationship between BDNF and NT-3 evolves with further development of the cerebellum. BDNF and NT-3, and perhaps other factors, subsequently act in a synergistic manner to promote the survival of granule neurons at these later stages of development.
Beyond the enhancement of neuronal survival, the neurotrophins regulate multiple aspects of the maturation of neurons. A recurring theme in brain development is that distinct neurotrophic factors act on a particular population of developing neurons in a sequential manner (Verdi and Anderson, 1994; Davies, 1997). In the developing cerebellum, the biological effects of BDNF and NT-3 on granule neuron development are thought to occur sequentially (Segal et al., 1992). Our results indicating that NT-3 is also required for the survival of cerebellar granule neurons on exposure to BDNF suggests that the transcription-dependent mechanism of BDNF-induced survival mediated by MEF2 might couple the process of neuronal survival to neuronal differentiation.
Footnotes
This work was supported by a Burroughs Wellcome Career Development Award (A.B.), National Institutes of Health Grant R01-NS41021-01 (A.B.), an MPM scholar award (A.S.), an Albert J. Ryan Foundation award (A.S.), and a National Science Foundation graduate research fellowship (M.L.). A.B. is the recipient of a fellowship from the Alfred P. Sloan Foundation, a Robert H. Ebert Clinical Scholar Award from the Esther A. and Joseph Klingenstein Fund, an EJLB Foundation award, and a Sidney Kimmel Foundation Award. We thank S. Gutkind for providing the ERK5 and MEK5 plasmids, E. Olson for the MEF2 plasmids, A. Shintani for the NT-3 promoter plasmids, and S. Vasquez for technical assistance.
Correspondence should be addressed to Dr. Azad Bonni, Department of Pathology, Harvard Medical School, 200 Longwood Avenue, Boston, MA 02115. E-mail: azad_bonni@hms.harvard.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/237326-11$15.00/0
References
- Altman J, Bayer S ( 1997) Development of the cerebellar system. In: Relation to its evolution, structure, and functions. New York: CRC.
- Bates B, Rios M, Trumpp A, Chen C, Fan G, Bishop JM, Jaenisch R ( 1999) Neurotrophin-3 is required for proper cerebellar development. Nat Neurosci 2: 115-117. [DOI] [PubMed] [Google Scholar]
- Black BL, Olson EN ( 1998) Transcriptional control of muscle development by myocyte enhancer factor-2 (MEF2) proteins. Annu Rev Cell Dev Biol 14: 167-196. [DOI] [PubMed] [Google Scholar]
- Bonni A, Ginty DD, Dudek H, Greenberg ME ( 1995) Serine 133-phosphorylated CREB induces transcription via a cooperative mechanism that may confer specificity to neurotrophin signals. Mol Cell Neurosci 6: 168-183. [DOI] [PubMed] [Google Scholar]
- Bonni A, Sun Y, Nadal-Vicens M, Bhatt A, Frank DA, Rozovosky I, Stahl N, Yancopoulos GD, Greenberg ME ( 1997) Regulation of gliogenesis in the central nervous system by the JAK-STAT signaling pathway. Science 278: 477-483. [DOI] [PubMed] [Google Scholar]
- Bonni A, Brunet A, West A, Datta SR, Takasu M, Greenberg ME ( 1999) Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science 286: 1358-1362. [DOI] [PubMed] [Google Scholar]
- Bothwell M ( 1995) Functional interactions of neurotrophins and neurotrophin receptors. Annu Rev Neurosci 18: 223-253. [DOI] [PubMed] [Google Scholar]
- Cavanaugh JE, Ham J, Hetman M, Poser S, Yan C, Xia Z ( 2001) Differential regulation of mitogen-activated protein kinases ERK1/2 and ERK5 by neurotrophins, neuronal activity, and cAMP in neurons. J Neurosci 15: 434-443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies AM ( 1997) Neurotrophin switching: where does it stand? Curr Opin Neurobiol 7: 110-118. [DOI] [PubMed] [Google Scholar]
- Diaz E, Ge Y, Yang YH, Loh KC, Serafini TA, Okazaki Y, Hayashizaki Y, Speed TP, Ngai J, Scheiffele P ( 2002) Molecular analysis of gene expression in the developing pontocerebellar projection system. Neuron 36: 417-434. [DOI] [PubMed] [Google Scholar]
- Farinas I, Wilkinson GA, Backus C, Reichardt LF, Patapoutian A ( 1998) Characterization of neurotrophin and Trk receptor functions in developing sensory ganglia: direct NT-3 activation of TrkB neurons in vivo Neuron 21: 325-334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkbeiner S ( 2000) CREB couples neurotrophin signals to survival messages. Neuron 25: 11-14. [DOI] [PubMed] [Google Scholar]
- Gao W, Zheng L, Karihaloo M ( 1995) Neurotrophin-4/5 (NT-4/5) and brain-derived neurotrophic factor (BDNF) act at later stages of cerebellar granule cell. J Neurosci 15: 2656-2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudilliere B, Shi Y, Bonni A ( 2002) RNA interference reveals a requirement for myocyte enhancer factor 2A in activity-dependent neuronal survival. J Biol Chem 277: 46442-46446. [DOI] [PubMed] [Google Scholar]
- Ginty DD, Bonni A, Greenberg ME ( 1994) NGF activates a Ras-dependent protein kinase that stimulates c-fos transcription via phosphorylation of CREB. Cell 77: 713-725. [DOI] [PubMed] [Google Scholar]
- Hatten ME, Heintz N ( 1995) Mechanisms of neural patterning and specification in the developing cerebellum. Annu Rev Neurosci 18: 385-408. [DOI] [PubMed] [Google Scholar]
- Ip NY, Yancopoulos GD ( 1996) The neurotrophins and CNTF: Two families of collaborative neurotrophic factors. Annu Rev Neurosci 19: 491-515. [DOI] [PubMed] [Google Scholar]
- Jelsma TN, Aguayo AJ ( 1994) Trophic factors. Curr Opin Neurobiol 4: 717-725. [DOI] [PubMed] [Google Scholar]
- Kamakura S, Moriguchi T, Nishida E ( 1999) Activation of the protein kinase ERK5/BMK1 by receptor tyrosine kinases. J Biol Chem 274: 26563-26571. [DOI] [PubMed] [Google Scholar]
- Kaplan DR, Miller FD ( 2000) Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol 10: 381-391. [DOI] [PubMed] [Google Scholar]
- Konishi Y, Lehtinen M, Donovan N, Bonni A ( 2002) Cdc2 phosphorylation of BAD links the cell cycle to the cell death machinery. Mol Cell 9: 1005-1016. [DOI] [PubMed] [Google Scholar]
- Leingartner A, Heisenberg C-P, Kolbeck R, Thoenen H, Lindholm D ( 1994) Brain derived neurotrophic factor increases neurotrophin-3 expression in cerebellar granule neurons. J Biol Chem 269: 828-830. [PubMed] [Google Scholar]
- Lewin GR, Barde Y-A ( 1996) Physiology of the neurotrophins. Annu Rev Neurosci 19: 289-317. [DOI] [PubMed] [Google Scholar]
- Lin X, Shah S, Bulleit RF ( 1996) The expression of MEF2 genes is implicated in CNS neuronal differentiation. Brain Res Mol Brain Res 42: 307-316. [DOI] [PubMed] [Google Scholar]
- Lindholm D, Dechant G, Heisenberg CP, Thoenen H ( 1993) Brain-derived neurotrophic factor is a survival factor for cultured rat cerebellar granule neurons and protects them against glutamate-induced neurotoxicity. Eur J Neurosci 5: 1455-1464. [DOI] [PubMed] [Google Scholar]
- Lindsay RM ( 1994) Neurotrophic growth factors and neurodegenerative diseases: therapeutic potential of the neurotrophins and ciliary neurotrophic factor [review]. Neurobiol Aging 15: 249-251. [DOI] [PubMed] [Google Scholar]
- Lonze BE, Ginty DD ( 2002) Function and regulation of CREB family transcription factors in the nervous system. Neuron 35: 605-623. [DOI] [PubMed] [Google Scholar]
- Lyons GE, Micales BK, Schwarz J, Martin JF, Olson EN ( 1995) Expression of mef2 genes in the mouse CNS suggests a role in neuronal maturation. J Neurosci 15: 5727-5738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Z, Bonni A, Xia F, Nadal-Vicens M, Greenberg ME ( 1999) Neuronal activity-dependent cell survival mediated by transcription factor MEF2. Science 286: 785-790. [DOI] [PubMed] [Google Scholar]
- Marinissen MJ, Chiariello M, Pallante M, Gutkind JS ( 1999) A network of mitogen-activated protein kinase links G. protein-coupled receptors to the c-jun promoter: a role for c-Jun NH2-terminal kinase, p38s, and extracellular signal-regulated kinase 5. Mol Cell Biol 19: 4289-301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Franke A, Wilkinson GA, Kruttgen A, Hu M, Munro E, Hanson Jr MG, Reichardt LF, Barres BA ( 1998) Depolarization and cAMP elevation rapidly recruit TrkB to the plasma membrane of CNS neurons. Neuron 21: 681-693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minichiello L, Klein R ( 1996) TrkB and TrkC neurotrophin receptors cooperate in promoting survival of hippocampal and cerebellar granule neurons. Genes Dev 15: 2849-2858. [DOI] [PubMed] [Google Scholar]
- Molkentin JD, Black BL, Martin JF, Olson EN ( 1996) Mutational analysis of the DNA binding, dimerization, and transcriptional activation domains of MEF2C. Mol Cell Biol 16: 2627-2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto S, Li Z, Ju C, Scholzke MN, Mathews E, Cui J, Salvesen GS, Bossy-Wetzel E, Lipton SA ( 2002) Dominant-interfering forms of MEF2 generated by caspase cleavage contribute to NMDA-induced neuronal apoptosis. Proc Natl Acad Sci USA 99: 3974-3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell SK, Rivas RJ, Rodriguez-Boulan E, Hatten ME ( 1997) Development of polarity in cerebellar granule neurons. J Neurobiol 32: 223-236. [DOI] [PubMed] [Google Scholar]
- Riccio A, Ahn S, Davenport CM, Blendy JA, Ginty DD ( 1999) Mediation by a CREB family transcription factor of NGF-dependent survival of sympathetic neurons. Science 286: 2358-2361. [DOI] [PubMed] [Google Scholar]
- Schwartz PM, Borghesani PR, Levy RL, Pomeroy SL, Segal RA ( 1997) Abnormal cerebellar development and foliation in BDNF-/- mice reveals a role for neurotrophins in CNS patterning. Neuron 19: 269-281. [DOI] [PubMed] [Google Scholar]
- Segal RA, Takahashi H, McKay RD ( 1992) Changes in neurotrophin responsiveness during the development of cerebellar granule neurons. Neuron 9: 1041-1052. [DOI] [PubMed] [Google Scholar]
- Shaywitz AJ, Greenberg ME ( 1999) CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular factors. Annu Rev Biochem 68: 821-861. [DOI] [PubMed] [Google Scholar]
- Skaper SD, Floreani M, Negro A, Facci L, Giusti P ( 1998) Neurotrophins rescue cerebellar granule neurons from oxidative stress-mediated apoptotic death: selective involvement of phosphatidylinositol 3-kinase and the mitogen-activated protein kinase pathway. J Neurochem 70: 1859-1868. [DOI] [PubMed] [Google Scholar]
- Verdi JM, Anderson DJ ( 1994) Neurotrophins regulate sequential changes in neurotrophin receptor expression by sympathetic neuroblasts. Neuron 13: 1359-1372. [DOI] [PubMed] [Google Scholar]
- Watson FL, Heerssen HM, Bhattacharyya A, Klesse L, Lin MZ, Segal RA ( 2001) Neurotrophins use the Erk5 pathway to mediate a retrograde survival response. Nat Neurosci 4: 981-988. [DOI] [PubMed] [Google Scholar]
- Williams RW, Herrup K ( 1988) The control of neuron number. Annu Rev Neurosci 11: 423-453. [DOI] [PubMed] [Google Scholar]
- Wood KA, Dipasquale B, Youle RJ ( 1993) In situ labeling of granule cells for apoptosis-associated DNA fragmentation reveals different mechanisms of cell loss in developing cerebellum. Neuron 11: 621-632. [DOI] [PubMed] [Google Scholar]
- Yuen EC, Mobley WC ( 1996) Therapeutic potential of neurotrophic factors for neurological disorders. Ann Neurol 40: 346-354. [DOI] [PubMed] [Google Scholar]
- Zhao M, New L, Kravchenko VV, Kato Y, Gram H, di Padova F, Olson EN, Ulevitch RJ, Han J ( 1999) Regulation of the MEF2 family of transcription factors by p38. Mol Cell Biol 19: 21-30. [DOI] [PMC free article] [PubMed] [Google Scholar]