

Abstract
The neurobiological underpinnings of mood modulation, molecular pathophysiology of manic-depressive illness, and therapeutic mechanism of mood stabilizers are largely unknown. The extracellular signal-regulated kinase (ERK) pathway is activated by neurotrophins and other neuroactive chemicals to produce their effects on neuronal differentiation, survival, regeneration, and structural and functional plasticity. We found that lithium and valproate, commonly used mood stabilizers for the treatment of manic-depressive illness, stimulated the ERK pathway in the rat hippocampus and frontal cortex. Both drugs increased the levels of activated phospho-ERK44/42, activated phospho-ribosomal protein S6 kinase-1 (RSK1) (a substrate of ERK), phospho-CREB (cAMP response element-binding protein) and phospho-B cell lymphoma protein-2 antagonist of cell death (substrates of RSK), and BDNF. Inhibiting the ERK pathway with the blood-brain barrier-penetrating mitogen-activated protein kinase (MAP kinase)/ERK kinase (MEK) kinase inhibitor SL327, but not with the nonblood-brain barrier-penetrating MEK inhibitor U0126, decreased immobility time and increased swimming time of rats in the forced-swim test. SL327, but not U0126, also increased locomotion time and distance traveled in a large open field. The behavioral changes in the open field were prevented with chronic lithium pretreatment. SL327-induced behavioral changes are qualitatively similar to the changes induced by amphetamine, a compound that induces relapse in remitted manic patients and mood elevation in normal subjects. These data suggest that the ERK pathway may mediate the antimanic effects of mood stabilizers.
Keywords: mood, affect, bipolar disorder, mania, depression, lithium, valproate, ERK, RSK, CREB, RSK, BAD, BDNF
Introduction
Severe mood alteration as manifested in mood disorders (e.g., manic-depressive illness and unipolar depression) affects one's sense of well being, as well as self esteem, judgment, attention, motivation, learning and memory, sleep, appetite, and overall psychomotor activity (Goodwin and Jamison, 1990). Family, twin, and adoption studies strongly suggest that genetic factors are involved in these disorders. Mood disorders have traditionally been conceptualized as neurochemical disorders of certain neurotransmitter systems (such as serotonergic, adrenergic, and, more recently, glutamatergic and GABAergic systems). However, recent brain imaging and morphometric-postmortem studies demonstrate regional changes in CNS volume, as well as changes in the number and/or size of glia and neurons in discrete brain areas of patients with mood disorders. These recent data suggest neurotrophic function, cellular growth, death, and resilience as possible factors that contribute to these disorders (for review see Manji et al., 2001; Nestler et al., 2002; Coyle and Duman, 2003).
Mood stabilizers are a small group of agents used in the treatment of manic-depressive illness. These agents have diverse chemical structures and different direct targets (Gould et al., 2002; Manji and Chen, 2002). Lithium and valproate are the most commonly used mood stabilizers. Lithium is a monovalent cation, and valproate is a branched chain fatty acid. The full clinical beneficial effects of lithium and valproate require weeks of treatment and blood drug levels maintained within a therapeutic window. Given that the pathophysiology of mood disorders have not been well established (Manji and Chen, 2002), it is difficult to link any given mood stabilizer-produced direct or indirect biological action to their therapeutic effect. To guide our research on the mechanism of action of mood stabilizer, we use five therapeutically relevant biological action(s) validating criteria: (1) identity: the action itself or its downstream effects must be shared by structurally dissimilar mood stabilizers; (2) dosing: the action must occur within the therapeutic concentration window; (3) temporal: the action must occur in a time frame consistent with the clinical effects; (4) regional: the action must occur in one or more brain regions implicated in mood modulation; and (5) behavioral: the functional manipulation of the molecules involved in the action(s) of mood stabilizers must induce mood-like behavioral change or alter mood stabilizer-induced behavioral response in experimental animal models.
Many common (or shared) indirect actions of mood stabilizers have been identified (Gould et al., 2002). Among these common actions, the lithium- and valproate-induced increases in activity of activator protein-1 (AP-1) transcription factors and B cell lymphoma protein-2 (Bcl-2) levels meet three or more therapeutically relevant action(s) validating criteria (Gould et al., 2002; Manji and Chen, 2002). In an effort to elucidate the mechanisms by which lithium and valproate affect AP-1 and Bcl-2, we previously investigated the extracellular signal-regulated kinase (ERK) pathway and found that vaproate not only activated the ERK pathway in cultured cells, but also promotes cellular functions associated with the ERK pathway including neurite outgrowth and cell survival (Yuan et al., 2001).
The ERK pathway is activated by neurotrophins and other neuroactive chemicals and is involved in differentiation, survival, and structural and functional plasticity of neurons (Huang and Reichardt, 2001; Marinissen and Gutkind, 2001; Dawson and Ginty, 2002; Weeber and Sweatt, 2002). It has been established that phosphorylation of ERK44 at Thr202/Tyr204 and ERK42 at Thr183/Tyr185 by MAP/ERK kinase (MEK) results in ERK activation; in turn, phospho-ERKs activate ribosomal protein S6 kinase-1 (RSK) by phosphorylation (Frodin and Gammeltoft, 1999). Phospho-RSKs phosphorylate Bcl-2 antagonist of cell death (BAD) at Ser112, thereby inactivating its proapoptotic activity. Phospho-RSKs also phosphorylate cAMP response element-binding protein (CREB) at Ser 133, which increases CREB transcriptional activity. CREB regulates Bcl-2 and BDNF gene expression. The ERK pathway also regulates AP-1 transcription factors directly or through other kinases, such as glycogen synthase kinase-3 (Karin, 1996).
The present study investigated the hypothesis that the ERK pathway is a common therapeutically relevant target of the mood stabilizers.
Materials and Methods
Animal and drug treatments. Male rats (weight, 150-250 gm) were housed three to four per cage with ad libitum access to water and food, maintained under a 12 hr light/dark cycle, and allowed a 1 week accommodation period before initiation of experiments. All experiments were performed during the light phase of the light/dark cycle and were approved by the National Institutes of Health (NIH) Animal Care and Use Committee in accordance with NIH guidelines on the care and use of animals. Rats were fed with regular rodent chow (control) or lithium carbonate (2.4 gm/kg) or sodium valproate (20 gm/kg) containing rodent chow (custom produced by Bio-Serv, Frenchtown, NJ) for 9 d or 28 d. In addition to tap water, a bottle of saline was available for rats receiving lithium chow to minimize any electrochemical imbalance that may occur because of the diuretic properties of the drug. At the end of treatments, rats were euthanized between 9:00 A.M. and 12:00 P.M., trunk blood samples were collected for monitoring the drug concentration, and frontal cortex and hippocampus were removed, frozen in liquid nitrogen, and stored at -80°C. The MEK inhibitors SL327 (Bristol-Myers Squibb, Princeton, NJ) and U0126 (LC Laboratories, Woburn, MA) were dissolved in DMSO and injected intraperitoneally at an injection volume of 1 ml/kg and at doses ranging from 25 to 100 mg/kg. On the basis of a previous report (Selcher et al., 1999) as well as our initial dose-finding experiments of SL327-induced inhibition of ERK and RSK1 phosphorylation in hippocampus and frontal cortex, the 50 mg/kg dose was chosen for all behavioral experiments. After the injection, rats were kept in their home cage for 1 hr before the behavioral experiments. Amphetamine sulfate was dissolved in saline and injected intraperitoneally at an injection volume of 1 ml/kg and at a dose of 0.5 mg/kg just before the behavioral tests.
Immunoblotting. Frontal cortex and hippocampus tissues were homogenized by brief sonication in an extraction buffer containing 20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1% Triton X-100, 2.5 mm sodium pyrophosphate, 1 mm β-glycerophosphate, pro-tease inhibitor mixture (Sigma, St. Louis, MO), and phosphatase inhibitor mixture I and II (Sigma). Homogenates were centrifuged at 14,000 × g for 15 sec to remove undissolved debris. For immunoblots, total protein contents were adjusted to the same level for all samples, and the amount of total protein loaded on the gel was at levels within the linear range of detection. The blots were first probed with antibodies against the phosphorylated forms of the protein (except for BDNF) and then stripped and probed with antibodies against total proteins of same type. Antibodies were from Cell Signaling (Beverly, MA) (ERK, RSK1, BAD and CREB, phospho- and total-protein except total RSK1) and Santa Cruz Biotechnology [Santa Cruz, CA; BDNF and RSK1 (total protein)] and were used according the manufacturer's instructions. The immuno-complexes were detected using the ECL method (Amersham Biosciences, Piscataway, NJ). Quantitation of the immunoblot was by densitometric scanning using an image analysis system with NIH Image 1.55 software or a Kodak Image Station.
Large open field test. The large open field is a 120 × 120 cm transparent Plexiglas platform, without walls, elevated 80 cm above the ground. This type of arena is effective in the evaluation of exploratory behavior in rats (Decker et al., 2000). Rats were introduced to the center of the open field for a 30 min session. Behavior was videotaped for later analysis with the Ethovision (Noldus, VA) video tracking system. The locomotion time, distance traveled, center time, and number of rearings were recorded.
Forced swim test. The forced swim test was conducted according to previous reports (Porsolt, 1979; Einat et al., 2001). In brief, the test involves two exposures to a cylindrical tank of water in which rats cannot touch the bottom of the tank or escape. The tank is made of transparent Plexiglas, 80 cm tall, 30 cm in diameter, and filled with water (22-23°C) to a depth of 40 cm. Water in the tank was changed after each rat. For the first exposure, rats were placed in the water for 10 min. Twenty-four hours later and 1 hr after SL327, U0126, or DMSO injection, rats were placed in the water again for a 5 min session (test session). Behavior was videotaped for later analysis, and the periods of immobility, swimming, and struggle time were recorded.
Statistical analysis. Data for all experiments were analyzed using parametric statistics with Student's t test, ANOVA, or multiple factors ANOVA, as appropriate. ANOVA analysis was followed by post hoc tests or planned comparisons as projected from the design of each experiment.
Results
Effects of lithium on the ERK pathway in rat hippocampus and frontal cortex
We first tested whether lithium stimulates the ERK pathway in regions of brain involved in human mood modulation in the intact animal. Rats were treated with lithium carbonate chow for 9 or 28 d, achieving final blood concentrations within the therapeutic range (∼ 0.8 mEq/l). The activation of the ERK pathway was monitored by measuring the multiple proteins in their functional (phosphorylated) forms along sequential steps of the ERK pathway. The 9 d lithium treatment did not significantly alter the levels of phospho-proteins or proteins (data not shown). The 28 d lithium treatment significantly increased phospho-ERK44/42 levels in hippocampus (Fig. 1a,b) and phospho-ERK42 level in frontal cortex (Fig. 1c,d). The 28 d lithium treatment also significantly increased phospho-RSK1 levels and phospho-BAD levels in hippocampus (Fig. 1a,b) and frontal cortex (Fig. 1c,d), significantly increased phospho-CREB level in hippocampus (Fig. 1a,b), and significantly increased BDNF levels in hippocampus (Fig. 1a,b) and frontal cortex (Fig. 1c,d). Lithium-induced increases in phospho-proteins were not likely attributable to increases in the total amount of the proteins (Fig. 1a,c).
Figure 1.
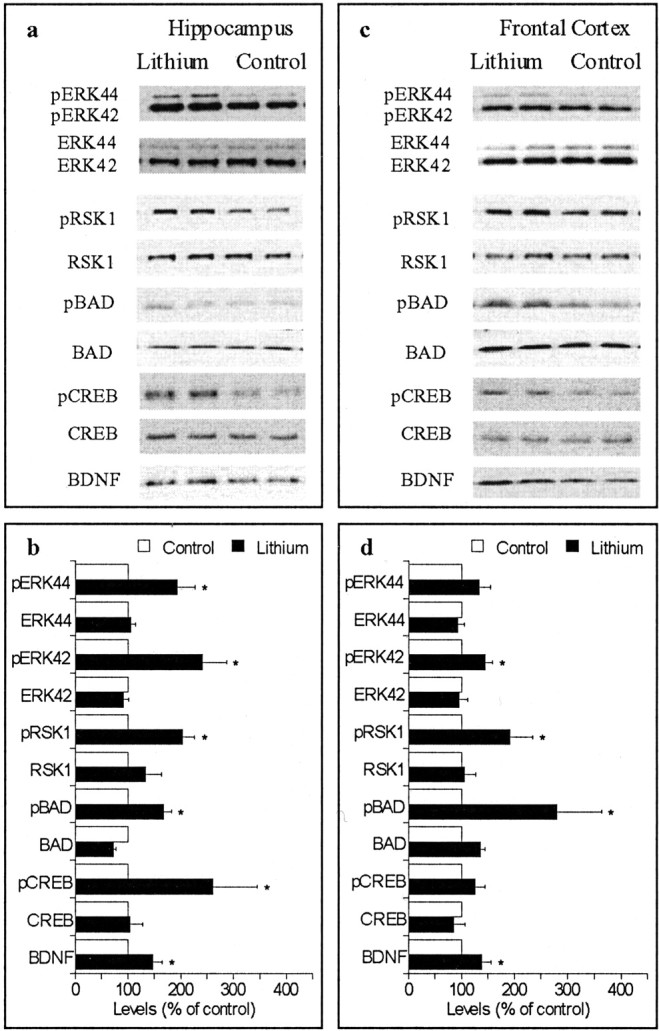
Effects of chronic lithium treatment on the ERK pathway. The photographs are representative immunoblots of the same sample loaded in duplicate. Similar immunoblots were also obtained from eight additional sets of rats. Bar graphs are mean ± SE from nine sets of rats; *p < 0.05. a, b, Results from hippocampal samples. c, d, Results from frontal cortex samples.
Effects of valproate on the ERK pathway in rat hippocampus and frontal cortex
We next tested whether the effects of lithium on the ERK pathway were shared by valproate, one of the most commonly used mood stabilizers in recent years. Rats were treated with sodium valproate chow for 9 or 28 d, achieving final blood concentrations at the low end of the therapeutic range (∼ 40 μg/ml). The 9 d valproate treatment did not significantly alter the levels of phospho-proteins or total protein (data not shown), but, similar to lithium, the 28 d valproate treatment significantly increased phospho-ERK44 levels in the hippocampus (Fig. 2a,b) and frontal cortex (Fig. 2c,d). The valproate treatment significantly increased phospho-RSK1 levels and phospho-BAD levels in hippocampus (Fig. 2a,b) and frontal cortex (Fig. 2c,d). Valproate treatment also significantly elevated phospho-CREB levels in hippocampus (Fig. 2a,b) and frontal cortex (Fig. 2c,d). The treatment-induced increases in phospho-proteins were not likely attributable to increases in total protein levels of these proteins (Fig. 2a,c).
Figure 2.
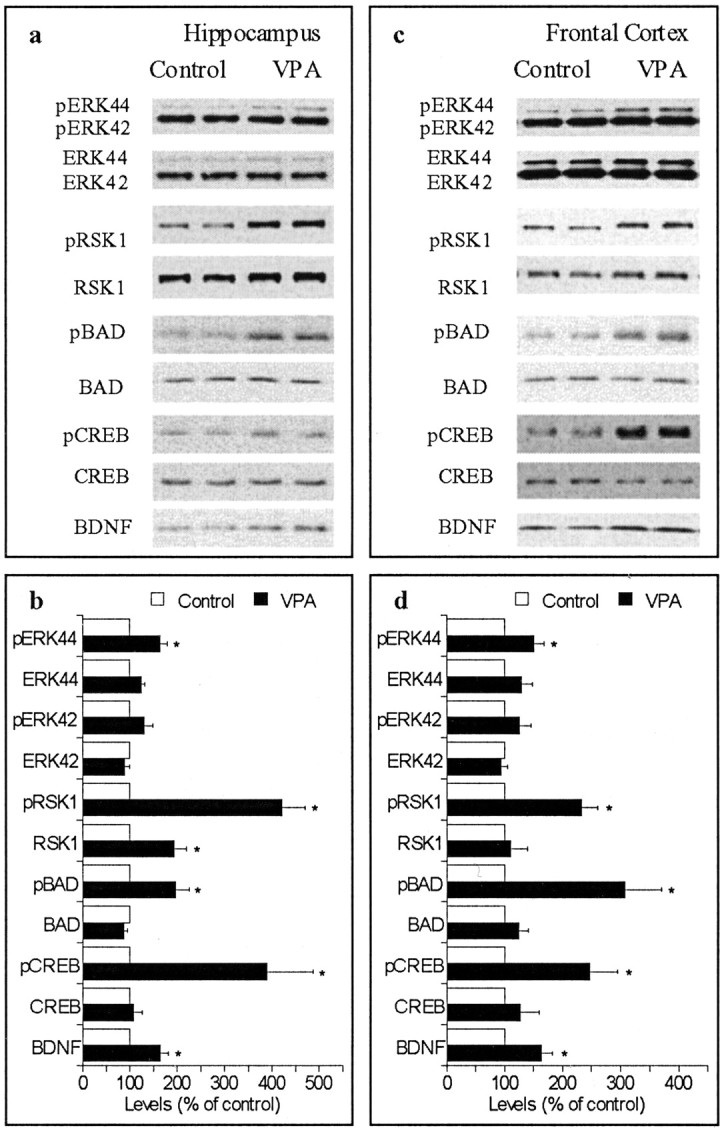
Effects of chronic valproate (VPA) treatment on the ERK pathway. The photographs are representative immunoblots of the same sample loaded in duplicate. Similar immunoblots were also obtained from eight additional sets of rats. Bar graphs are mean ± SE from nine sets of rats; *p < 0.05. a, b, Results from hippocampal samples. c, d, Results from frontal cortex samples.
Effects of the CNS blockade of the ERK pathway on animal behavior in forced swim test
SL327 is a specific MEK inhibitor that penetrates the blood-brain barrier. U0126 is the parent compound of SL327 that does not cross the blood-brain barrier. As reported previously (Selcher et al., 1999), we found that a peripheral injection of SL327, but not U0126, dose dependently (0-100 mg/kg) lowered activated ERK44/42 levels in rat hippocampus and frontal cortex, effects that reached a plateau at the 50 mg/kg dose (data not shown). Injection of SL327 (50 mg/kg) (Fig. 3a,b), but not U0126 (Fig. 3c,d), significantly decreased immobility time and increased swim time in the forced swim test. These data suggest that the blockade of the ERK pathway in the CNS induces antidepressant-like and/or amphetamine-like behavioral effects (see Discussion).
Figure 3.
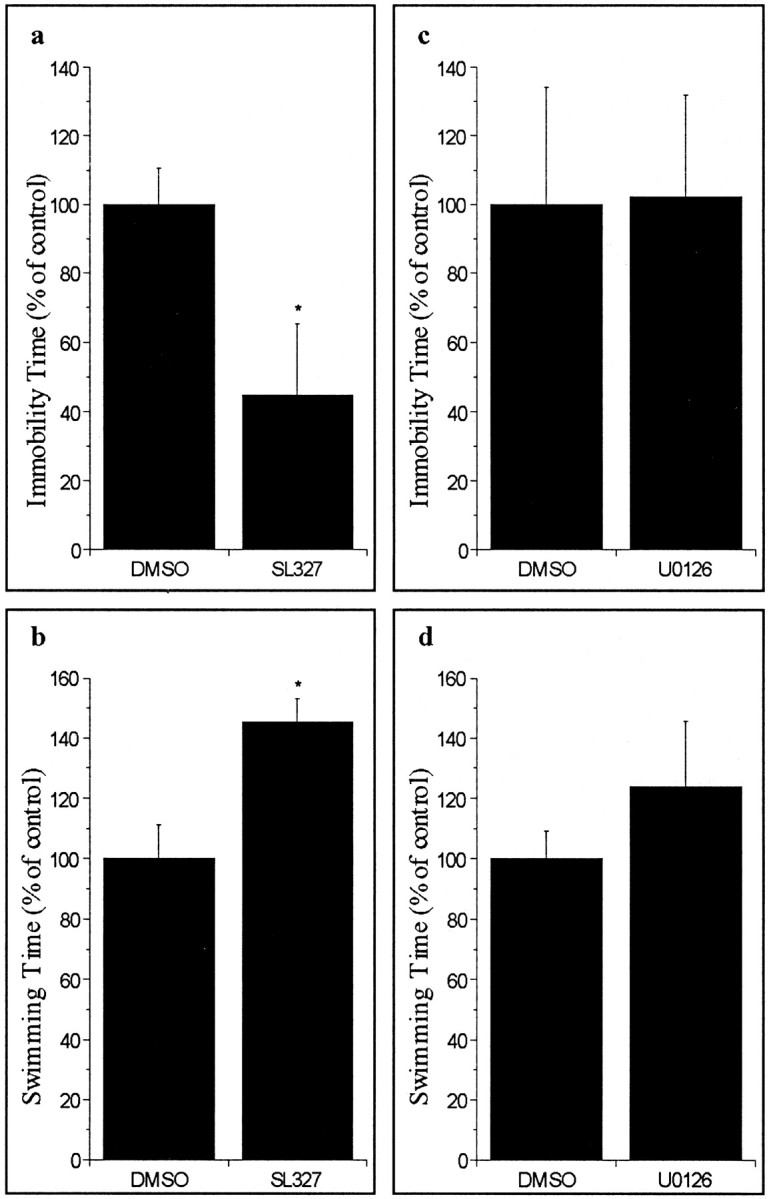
Effects of SL327 and U0126 on animal behavior in the forced swim test. Bar graphs represent mean ± SE from seven to eight rats; *p < 0.05. a, Forced swim test immobility time for SL327/DMSO comparison [t(14) = 2.84; p < 0.02]. b, Forced swim test swimming time for SL327/DMSO comparison [t(14) = 3.9; p < 0.01]. Forced swim test immobility time (c) and forced swim test swimming time (d) for U0126/DMSO comparison (NS).
Effects of the CNS blockade of the ERK pathway on locomotion in an open field
Amphetamine-induced hyperactivity in the open field test has been used as a model of mania (Einat et al., 2000). To further explore the effects of SL327, we studied its effects on behavior in a large open field. Injection of SL327 (Fig. 4a,b), but not U0126 (Fig. 4c,d), significantly increased locomotion time and distance traveled in a large open field. Interestingly, co-injection of SL327 with amphetamine did not produce additive effects (data not shown).
Figure 4.
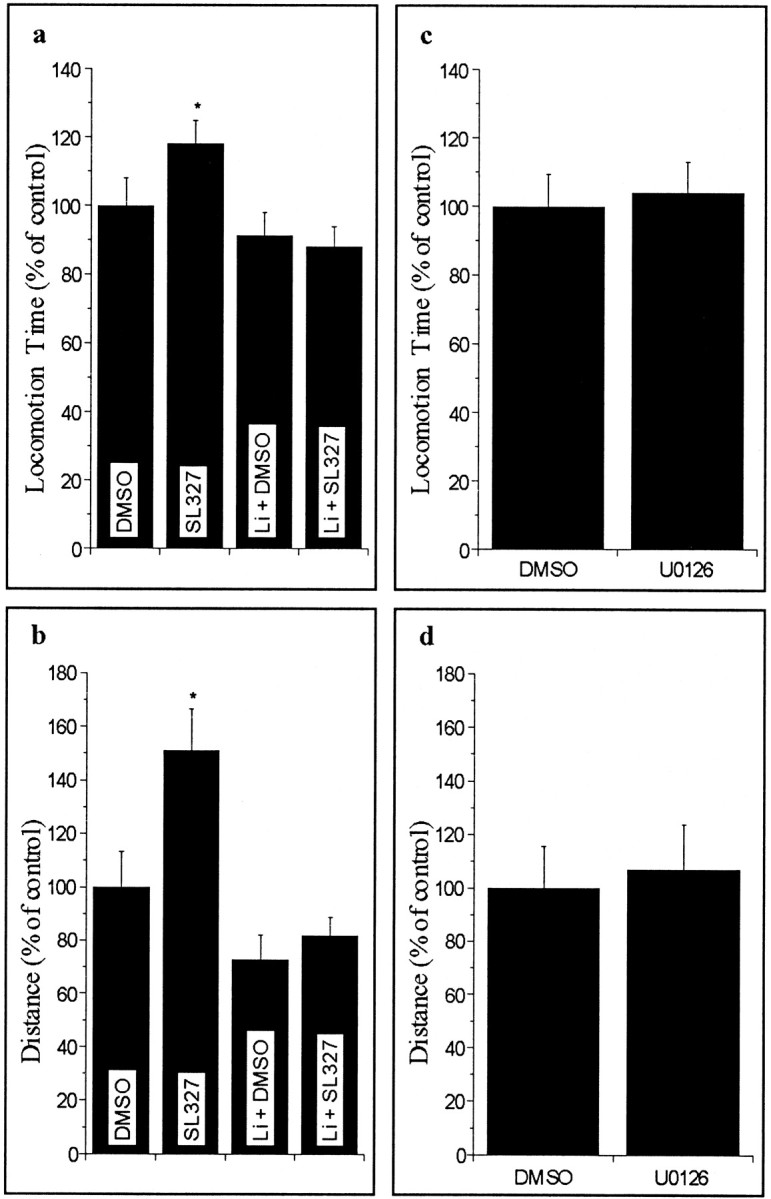
Effects of SL327 and U0126 on animal behavior in a large open field. a, Large open field locomotion time for the lithium (Li)/SL327 interaction experiment (F(3,28) = 3.7; p < 0.03). b, Large open field distance traveled for the Li/SL327 interaction experiment (F(3,28) = 6.15; p < 0.01; post hoc test, SL327 group different than all other groups). Large open field locomotion time (c) and large open field distance (d) traveled for the U0126/DMSO comparison (NS).
Effects of lithium pretreatment on SL327-induced increase in locomotion
Lithium pretreatment for 28 d prevented the SL327-induced increase in locomotor activity (Fig. 4a,b), further supporting the notion that SL327-induced hyperactivity may be relevant to mania.
Discussion
Lithium and valproate activate the ERK pathway and facilitate ERK pathway function at therapeutic doses
MEK phosphorylates ERK44 at Thr202/Tyr204 and ERK42 at Thr183/Tyr185; this phosphorylation results in severalfold increases in ERK44/42 activity (Frodin and Gammeltoft, 1999). We found that chronic treatments with therapeutic concentrations of lithium and valproate increased the levels of activated phospho-ERK44/42 in rat hippocampus and frontal cortex (Figs. 1, 2), suggesting that these treatments stimulated ERK activity. We also found that phosphorylation of the ERK substrate RSK1 was also increased concurrently with phospho-ERK44/42 (Figs. 1, 2). Taken together, these data suggest lithium and valproate stimulate the ERK pathway.
Bcl-2, BAD, CREB, and BDNF are targets downstream of ERK/RSK and play key roles in neuronal development, neuronal survival, and long-term neuronal plasticity (Huang and Reichardt, 2001; Dawson and Ginty, 2002; Weeber and Sweatt, 2002). Impairments of neuroplasticity and cellular resilience have been postulated to play a role in the pathophysiology of manic-depressive illness (Manji et al., 2001; Coyle and Duman, 2003). Chronic lithium and valproate treatments increased phospho-BAD and phospho-CREB levels (Figs. 1, 2). Consistent with an earlier report (Fukumoto et al., 2001), both treatments also increased BDNF levels in the rat brain (Figs. 1, 2). The neuroprotective ability of lithium has been well documented (Chuang et al., 2002). In a preliminary study, we found that valproate activates the ERK pathway in cortical neurons and protects cortical neurons against β-amyloid toxicity; furthermore, this protection is dependent on activation of the ERK pathway activation (our unpublished observations). These cell signaling and neurobiological data support the hypothesis that lithium and valproate activate the ERK/RSK pathway and enhance associated functions.
In addition to hippocampus and prefrontal cortex, clinical and postmortem studies also reveal abnormalities in other brain regions of patients with mood disorders (Manji et al., 2001). The possible effects of mood stabilizers on the ERK pathway in other potentially relevant areas of the brain, including the nucleus accumbens and amygdala, are yet to be investigated.
Blockade of ERK pathway in CNS-induced hyperactivity that may be relevant to mania
Lithium and valproate are primarily used for treatment and prophylaxis of mania. We found that both drugs activate the ERK pathway in the rat frontal cortex and hippocampus. In line with these findings, we found that blockade of the ERK pathway induced behavioral changes similar to the change induced by amphetamine in the forced swim test (Fig. 3) and in the open field test (Fig. 4). Antidepressants relieve depression and can result in a switch to mania in some depressed or euthymic patients with manic-depressive illness. Amphetamine can induce manic relapse in recently remitted patients and various degrees of mood elevation in normal subjects (for review, see Goodwin and Jamison, 1990). Both agents induce reduction of immobility in the forced swim test, and amphetamine, but not most antidepressants, induces hyperactivity in the open field in rats. Amphetamine-induced hyperactivity in the open field is attenuated by chronic pretreatment with lithium (Lerer et al., 1984). Similar to this phenomenon, we found that chronic pretreatment with lithium attenuated SL327-induced hyperactivity (Fig. 4). Taken together, the data supports the notion that blockade of the ERK pathway in the CNS results in behavioral changes that are relevant to facets of mania.
Our results need to be discussed in the context of the body of data demonstrating that antidepressants increase BDNF expression (Nestler et al., 2002; Coyle and Duman, 2003). Because BDNF is one upstream regulator of the ERK cascade, those findings may appear inconsistent with our results at first glance. However, it is clear that BDNF and ERK bring about distinct biochemical effects, and a growing body of data suggests that region-specific manipulation of these plasticity cascades may bring about quite distinct affective behavioral phenotypes (Newton et al., 2002).
Lithium and valproate do not cause depression in patients or healthy subjects. The effects of acute and subchronic (15 d) treatments of lithium have been studied in the forced swim test in rats, finding that neither treatment has significant effects (Kitamura et al., 2002; Wegener et al., 2003). Whether these two agents block the SL327-induced effects in the forced swim test is yet to be investigated.
Genetic alterations in rodents of the ERK pathway and its downstream targets cause behavioral changes
Genetic disruptions within the ERK pathway and the downstream targets demonstrate behavioral alterations and drug responses that support the hypothesis that the ERK pathway modulates manic-depressive illness-related behavior in rodent. BDNF-deficient mice exhibit increased spontaneous locomotion (Kernie et al., 2000), increased sensitivity to acute amphetamine, altered response to chronic cocaine, increased aggression, an increase in risk-taking behavior, and changes in eating patterns (Horger et al., 1999; Lyons et al., 1999; Kernie et al., 2000; Dluzen et al., 2001). ERK-1 knock-out mice also exhibit an increased locomotor activity (Selcher et al., 2001; Mazzucchelli et al., 2002). CREB-deficient mice exhibit reduced immobility in the forced swim and tail suspension tests (Conti et al., 2002). cAMP response element modulator-deficient mice are hyperactive and show less anxiety-related behavior (Maldonado et al., 1999). Direct injection of viral vector encoding mutant CREB (HVS-mCREB) into nucleus accumbens results in decreased anxiety-related behavior in the elevated plus maze and open field test (Barrot et al., 2002) and reduction in depression-related behavior in the forced swim test (Pliakas et al., 2001) and in the learned helplessness paradigm (Newton et al., 2002). Overexpression of CREB in the region produces opposite effects. These data appear to support our current findings and implicate the involvement of the ERK pathway in the modulation of behaviors relevant to manic-depressive illness.
In summary, the present study demonstrates that two structurally distinct mood stabilizers stimulate the ERK pathway at therapeutic concentrations, in a clinically relevant time frame, and in brain regions implicated in mood modulation. The study further demonstrates that inhibiting the ERK pathway produces behavioral effects in rats that are normalized by mood stabilizer treatment. These pharmacological, intracellular cell signaling, and behavioral data are consistent with data from the genetic manipulations of the ERK pathway. Together, these data strongly suggest that the ERK pathway is a common therapeutically relevant target of antimanic mood stabilizers.
Footnotes
This work was supported by the Intramural Research Program of the National Institute of Mental Health, a grant from Stanley Medical Research Institute, and the National Alliance for Research on Schizophrenia and Depression.
Correspondence should be addressed to Dr. Guang Chen, Laboratory of Molecular Pathophysiology, Mood and Anxiety Disorders Program, National Institute of Mental Health, National Institutes of Health, Department of Health and Human Services, Building 49, Room B1EE16, 49 Convent Drive, MSC 4405, Bethesda, MD 20892-4405. E-mail: cheng@intra.nimh.nih.gov.
Copyright © 2003 Society for Neuroscience 0270-6474/03/237311-06$15.00/0
References
- Barrot M, Olivier JD, Perrotti LI, DiLeone RJ, Berton O, Eisch AJ, Impey S, Storm DR, Neve RL, Yin JC, Zachariou V, Nestler EJ ( 2002) CREB activity in the nucleus accumbens shell controls gating of behavioral responses to emotional stimuli. Proc Natl Acad Sci USA 99: 11435-11440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang DM, Chen R, Chalecka-Franaszek E, Ren M, Hashimoto R, Senatorov V, Kanai H, Hough C, Hiroi T, Leeds P ( 2002) Neuroprotective effects of lithium in cultured cells and animal model of diseases. Bipolar Disord 4: 129-136. [DOI] [PubMed] [Google Scholar]
- Conti AC, Cryan JF, Dalvi A, Lucki I, Blendy JA ( 2002) cAMP response element-binding protein is essential for the upregulation of brain-derived neurotrophic factor transcription, but not the behavioral or endocrine responses to antidepressant drugs. J Neurosci 22: 3262-3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle JT, Duman RS ( 2003) Finding the intracellular signaling pathways affected by mood disorder treatments. Neuron 38: 157-160. [DOI] [PubMed] [Google Scholar]
- Dawson TM, Ginty DD ( 2002) CREB family transcription factors inhibit neuronal suicide. Nat Med 8: 450-451. [DOI] [PubMed] [Google Scholar]
- Decker S, Grider G, Cobb M, Li XP, Huff MO, El-Mallakh RS, Levy RS ( 2000) Open field is more sensitive than automated activity monitor in documenting ouabain-induced hyperlocomotion in the development of an animal model for bipolar illness. Prog Neuropsychopharmacol Biol Psychiatry 24: 455-462. [DOI] [PubMed] [Google Scholar]
- Dluzen DE, Gao X, Story GM, Anderson LI, Kucera J, Walro JM ( 2001) Evaluation of nigrostriatal dopaminergic function in adult +/+ and +/- BDNF mutant mice. Exp Neurol 170: 121-128. [DOI] [PubMed] [Google Scholar]
- Einat H, Kofman O, Belmaker RH ( 2000) Animal models of bipolar disorder: from a single episode to progressive cycling models. In: Contemporary issues in modeling psychopharmacology (Myslobodsky M, Weiner I, eds), pp 165-180. Boston: Kluwer Academic Publishers.
- Einat H, Clenet F, Shaldubina A, Belmaker RH, Bourin M ( 2001) The antidepressant activity of inositol in the forced swim test involves 5-HT(2) receptors. Behav Brain Res 118: 77-83. [DOI] [PubMed] [Google Scholar]
- Frodin M, Gammeltoft S ( 1999) Role and regulation of 90 kDa ribosomal S6 kinase (RSK) in signal transduction. Mol Cell Endocrinol 151: 65-77. [DOI] [PubMed] [Google Scholar]
- Fukumoto T, Morinobu S, Okamoto Y, Kagaya A, Yamawaki S ( 2001) Chronic lithium treatment increases the expression of brain-derived neurotrophic factor in the rat brain. Psychopharmacology (Berl) 158: 100-106. [DOI] [PubMed] [Google Scholar]
- Goodwin FK, Jamison KR ( 1990) Manic-depressive illness. New York: Oxford UP.
- Gould TD, Chen G, Manji HK ( 2002) Mood stabilizer psychopharmacology. Clin Neurosci Res 2: 193-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horger BA, Iyasere CA, Berhow MT, Messer CJ, Nestler EJ, Taylor JR ( 1999) Enhancement of locomotor activity and conditioned reward to cocaine by brain-derived neurotrophic factor. J Neurosci 19: 4110-4122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF ( 2001) Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci 24: 677-736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M ( 1996) The regulation of AP-1 activity by mitogen-activated protein kinases. Philos Trans R Soc Lond B Biol Sci 351: 127-134. [DOI] [PubMed] [Google Scholar]
- Kernie SG, Liebl DJ, Parada LF ( 2000) BDNF regulates eating behavior and locomotor activity in mice. EMBO J 19: 1290-1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura Y, Araki H, Gomita Y ( 2002) Influence of ACTH on the effects of imipramine, desipramine and lithium on duration of immobility of rats in the forced swim test. Pharmacol Biochem Behav 71: 63-69. [DOI] [PubMed] [Google Scholar]
- Lerer B, Globus M, Brik E, Hamburger R, Belmaker RH ( 1984) Effect of treatment and withdrawal from chronic lithium in rats on stimulant-induced responses. Neuropsychobiology 11: 28-32. [DOI] [PubMed] [Google Scholar]
- Lyons WE, Mamounas LA, Ricaurte GA, Coppola V, Reid SW, Bora SH, Wihler C, Koliatsos VE, Tessarollo L ( 1999) Brain-derived neurotrophic factor-deficient mice develop aggressiveness and hyperphagia in conjunction with brain serotonergic abnormalities. Proc Natl Acad Sci USA 96: 15239-15244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado R, Smadja C, Mazzucchelli C, Sassone-Corsi P, Mazucchelli C ( 1999) Altered emotional and locomotor responses in mice deficient in the transcription factor CREM. Proc Natl Acad Sci USA 96: 14094-14099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manji HK, Chen G ( 2002) PKC, MAP kinases and the bcl-2 family of proteins as long-term targets for mood stabilizers. Mol Psychiatry 7 [Suppl 1]: S46-S56. [DOI] [PubMed] [Google Scholar]
- Manji HK, Drevets WC, Charney DS ( 2001) The cellular neurobiology of depression. Nat Med 7: 541-547. [DOI] [PubMed] [Google Scholar]
- Marinissen MJ, Gutkind JS ( 2001) G-protein-coupled receptors and signaling networks: emerging paradigms. Trends Pharmacol Sci 22: 368-376. [DOI] [PubMed] [Google Scholar]
- Mazzucchelli C, Vantaggiato C, Ciamei A, Fasano S, Pakhotin P, Krezel W, Welzl H, Wolfer DP, Pages G, Valverde O, Marowsky A, Porrazzo A, Orban PC, Maldonado R, Ehrengruber MU, Cestari V, Lipp HP, Chapman PF, Pouyssegur J, Brambilla R ( 2002) Knockout of ERK1 MAP kinase enhances synaptic plasticity in the striatum and facilitates striatal-mediated learning and memory. Neuron 34: 807-820. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM ( 2002) Neurobiology of depression. Neuron 34: 13-25. [DOI] [PubMed] [Google Scholar]
- Newton SS, Thome J, Wallace TL, Shirayama Y, Schlesinger L, Sakai N, Chen J, Neve R, Nestler EJ, Duman RS ( 2002) Inhibition of cAMP response element-binding protein or dynorphin in the nucleus accumbens produces an antidepressant-like effect. J Neurosci 22: 10883-10890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pliakas AM, Carlson RR, Neve RL, Konradi C, Nestler EJ, Carlezon Jr WA ( 2001) Altered responsiveness to cocaine and increased immobility in the forced swim test associated with elevated cAMP response element-binding protein expression in nucleus accumbens. J Neurosci 21: 7397-7403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porsolt RD ( 1979) Animal model of depression. Biomedicine 30: 139-140. [PubMed] [Google Scholar]
- Selcher JC, Atkins CM, Trzaskos JM, Paylor R, Sweatt JD ( 1999) A necessity for MAP kinase activation in mammalian spatial learning. Learn Mem 6: 478-490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selcher JC, Nekrasova T, Paylor R, Landreth GE, Sweatt JD ( 2001) Mice lacking the ERK1 isoform of MAP kinase are unimpaired in emotional learning. Learn Mem 8: 11-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeber EJ, Sweatt JD ( 2002) Molecular neurobiology of human cognition. Neuron 33: 845-848. [DOI] [PubMed] [Google Scholar]
- Wegener G, Bandpey Z, Heiberg IL, Mork A, Rosenberg R ( 2003) Increased extracellular serotonin level in rat hippocampus induced by chronic citalopram is augmented by subchronic lithium: neurochemical and behavioural studies in the rat. Psychopharmacology (Berl) 166: 188-194. [DOI] [PubMed] [Google Scholar]
- Yuan PX, Huang LD, Jiang YM, Gutkind JS, Manji HK, Chen G ( 2001) The mood stabilizer valproic acid activates mitogen-activated protein kinases and promotes neurite growth. J Biol Chem 276: 31674-31683. [DOI] [PubMed] [Google Scholar]