

Abstract
The removal of excess neurons by programmed cell death (PCD) is believed to be critical for the proper development and function of the nervous system. A major role of this neuronal loss is to attain quantitative matching of neurons with their targets and afferents. Because motoneurons (MNs) in Bax knock-out (Bax KO) mice fail to undergo PCD in the face of normal target muscle development, we asked whether the excess rescued neurons in Bax KO mice can develop normally. We observed many small atrophied MNs in postnatal Bax KO mice, and these failed to innervate limb muscle targets. When examined embryonically during the PCD period, however, these excess MNs had initiated target innervation. To examine whether a limitation in trophic factor availability is responsible for postnatal MN atrophy and loss of innervation, we applied glial cell line-derived neurotrophic factor (GDNF) to neonatal mice. GDNF injection for 7-14 d induced the regrowth and reinnervation of muscle targets by atrophic MNs in Bax KO mice and prevented the normal postnatal death of MNs in wild-type mice. These results indicate that, although initially all of the MNs, including those rescued by Bax deletion, are able to project to and innervate targets, because of limited target-derived signals required for maintaining innervation and growth, only a subpopulation can grow and retain target contacts postnatally. Although sensory neurons in the dorsal root ganglia are also rescued from PCD by Bax deletion, their subsequent development is less affected than that of MNs.
Keywords: motoneurons, target dependence, cell death, Bax, mouse, spinal cord, innervation
Introduction
Programmed cell death (PCD) is thought to be an important event in the normal development and maintenance of the nervous system (for review, see Oppenheim, 1991). One of the best-characterized cases of PCD occurs when postmitotic motoneurons (MNs) begin to form synapses with their corresponding skeletal muscle targets. In mice, approximately one-half of spinal MNs initially generated undergo PCD during the 4-6 d period [embryonic day 13 (E13)-E19] after MNs contact their muscle targets (Lance-Jones, 1982; Oppenheim et al., 1986; Yamamoto and Henderson, 1999). Competition among MNs for limiting amounts of target-derived trophic factor (the neurotrophic hypothesis) is thought to be crucial for this type of PCD (Hamburger and Levi-Montalcini, 1949; Hamburger, 1975; Phelan and Hollyday, 1991; Caldero et al., 1998; Grieshammer et al., 1998). Consistent with this hypothesis, the exogenous administration of target-derived neurotrophic factors diminishes, whereas genetic elimination of trophic factors or their receptors augments, the PCD of MNs (Oppenheim et al., 1988, 1991, 1992, 1995, 2000a, 2001a; Sendtner et al., 1990, 1992; Yan et al., 1992, 1995; Henderson et al., 1993, 1994; Koliatsos et al., 1993; DeChiara et al., 1995; Pennica et al., 1996; Novak et al., 2000).
MNs undergoing PCD typically exhibit characteristics of apoptosis, such as cytoplasmic membrane blebbing, nuclear condensation, and DNA fragmentation (Chu-Wang and Oppenheim, 1978; Lo et al., 1995). Because many of the gene products regulating apoptosis have been identified, one approach toward understanding the role of PCD in shaping the number of innervating MNs is to examine mice deficient in proapoptotic and antiapoptotic genes. The genetic alteration of either proapoptotic or antiapoptotic Bcl-2 family members rescues MNs from PCD (Dubois-Dauphin et al., 1994; Martinou et al., 1994; Deckwerth et al., 1996; Lentz et al., 1999), and, in fact, the deletion of the proapoptotic gene Bax results in profound effects on the survival of many kinds of neurons (White et al., 1998). Cultured sympathetic and dorsal root ganglia (DRG) neurons from Bax knockout (Bax KO) mice can live indefinitely in the absence of trophic factor in vitro (Deckwerth et al., 1996; Lentz et al., 1999), and the PCD of facial and spinal MNs in vivo is greatly reduced or absent after Bax deletion (Deckwerth et al., 1996; White et al., 1998).
In the present study, we examined neuromuscular development in Bax KO mice to determine the extent to which rescued MNs can develop normally. Using this model, we determined that the limb-muscle targets of MNs in the Bax KO can only support the normal development of a proportion of all of the surviving MNs, and that treatment with exogenous trophic factor [glial cell line-derived neurotrophic factor (GDNF)] can rescue this phenotype, reversing their atrophy and promoting regrowth of the axons of the excess surviving MNs. Our observations suggest that, even after their developmental role in regulating MN survival is completed, trophic signals continue to be required to promote cell growth and to sustain target innervation. Furthermore, these data show that atrophic Bax KO MNs lacking target contacts can be induced to reinnervate their targets by treatment with exogenous GDNF.
Materials and Methods
Animals and histology. Heterozygous Bax-mutant mice are maintained on a C57BL/6 background. Sibling animals at the indicated embryonic or postnatal stages were collected and individually genotyped by PCR as described previously (Knudson et al., 1995). Embryos or postnatal animals were immersion-fixed in Bouin's solution, processed, embedded in paraffin, sectioned (8-10 μm), and stained with thionin. In a few animals, ventral roots (VRs) and dorsal roots (DRs) and spinal cords were fixed with 4% paraformaldehyde and 2% glutaraldehyde, postfixed in 2% osmium tetroxide, dehydrated, and embedded in plastic (Epon). Semithin (1 μm) sections were made, stained with toluidine blue, and photographed by CCD camera, and thin sections were observed by electron microscopy.
For quantification of neuronal numbers, spinal cords and brains were fixed in Bouin's solution, processed for paraffin embedding, sectioned at 8 -10 μm, and stained with thionin. Spinal and facial MNs and sensory neurons were counted in every 5th or 10th section, according to the criteria described previously (Clarke and Oppenheim, 1995), in which only large Nissl-positive cells with a distinct nucleus and nucleolus are included. Pyknotic neurons were identified and counted by criteria described previously (Clarke and Oppenheim, 1995). For examination of cell size, spinal cord or brainstem sections (every 10th) were captured by CCD camera and examined by NIH Image software. Only neurons with a distinct cell membrane and clear nucleus were included in cell size measurements. In addition to spinal MNs, facial MNs were also included in our analysis for two reasons: (1) to demonstrate that the effects of Bax deletion are not restricted to spinal MNs and (2) because the facial motor nucleus only contains somatic α-MNs (Ashwell and Watson, 1983), whereas the lumbar signal motor nucleus contains γ-MNs and a small population of interneurons that are sometimes difficult to distinguish histologically from the small atrophic MNs in the Bax KO.
For immunohistochemical analysis, animals were fixed in 4% paraformaldehyde, cryoprotected in 30% sucrose, and sectioned (10 -20 μm). Frozen sections of spinal cords were blocked with 5% goat serum and 0.1% Triton X-100 in PBS for 30 min, and a cleaved caspase-3-specific antibody (1:400; Cell Signaling, Beverly, MA) or SMI-32, an MN-specific antibody (Gotow and Tanaka, 1994; Carriedo et al., 1995; Bar-Peled et al., 1999) was applied overnight (1:1000; Sternberger-Meyer, Jarrettsville, MD). After washes with PBS, a Cy3-conjugated anti-rabbit antibody (1:400) or an Alexa 594-conjugated anti-mouse antibody (1:400) was applied for 30 min. Subsequently, sections were washed, counterstained with Hoechst 33342, mounted, and observed with a fluorescence microscope.
Muscle development. For muscle fiber staining, the triceps muscles from 1-month-old mice were dissected and quickly frozen in isopentane cooled in liquid nitrogen. Tissues were then sectioned on a cryostat and stained by acid ATPase histochemistry (Green et al., 1982). To identify the neuromuscular junction, soleus muscles were fixed in 4% paraformaldehyde. After blocking with 5% BSA and 0.3% Triton X-100 in PBS for 1 hr, a mixture of 2H3 (neurofilament; 1:100; Development Studies Hybridoma Bank, Iowa City, IA) and SV2 (presynaptic nerve terminal; 1:200; Development Studies Hybridoma Bank) antibodies were applied overnight at room temperature (RT). After several washes, Cy3-conjugated anti-mouse antibody (1:400; Jackson ImmunoResearch, West Grove, PA) and Alexa 594-conjugated α-bungarotoxin (5 μg/ml; Molecular Probes, Eugene, OR) were applied for 1 hr. The muscles were then washed, mounted, and observed with a fluorescence microscope.
Retrograde labeling. E14.5 mouse embryos were dissected and immersion fixed in 4% paraformaldehyde for 2 hr and a DiI crystal was then deposited at the base of the hindlimb bud. DiI was allowed to transfer to the limb and spinal cord for 10 d at 37°C. Whole mounts of the limbs were examined for orthograde labeling of peripheral nerves, and the thickness (diameter) of the tibial nerve was measured from photomicrographs using NIH Image software. Similarly, postnatal day 14 (P14) spinal cord was dissected and fixed, and DiI was deposited into one ventral root [lumbar segment 4 (L4)]. After 40 d, labeled neurons were examined in 20 μm cryostat sections. For retrograde labeling from limb muscle, 1-month-old mice were anesthetized, and 10 μl of a 2.5 μg/ml DiI solution in 50% ethanol and 50% DMSO was injected into three to five locations within the gastrocnemius muscle. Eight days after injection, animals were killed and labeled neurons were examined in 20 μm sections of the spinal cord. Because the injection of DiI in whisker pads gave strong background staining of blood vessels, Alexa 488-conjugated cholera toxin B subunit (2-4 μl of 100 μg/μl in PBS; Molecular Probes) was instead used for retrograde labeling of facial MNs. Retrograde labeling was used as a measure of innervation (i.e., the presence of MN axons in target muscles) and is not meant to imply the presence of structural or functional synapses.
In situ hybridization. Spinal columns were dissected from neonatal mice (P1-P2), snap frozen in optimal cutting temperature medium (O.C.T. Tissue-Tek), and sectioned at a thickness of 16 μm. Sections were postfixed in 4% paraformaldehyde-PBS for 10 min, rinsed in PBS, acetylated for 10 min, and prehybridized for 2 hr at RT, according to Schaeren-Wiemers and Gerfin-Moser (1993). Templates for the GDNF receptor (c-ret, GFRα1) probes were kindly provided by Dr. Chris Henderson (Institut National de la Santé et de la Recherche Médicale, Marseille, France). Digoxigenin-labeled cRNA probes were diluted in prehybridization solution at a concentration of 200 ng/ml and incubated at 68°C overnight, rinsed in 5× SSC, incubated at 68°Cin0.2× SSC for 2 hr, rinsed in 0.2× SSC, rinsed in maleic acid buffer (mab) (125 mm maleic acid, pH 7.5, 150 mm NaCl), blocked in 2% blocking reagent (Hoffmann-La Roche, Basel, Switzerland)-mab for 1 hr at RT, blocked in 20% normal sheep serum (Jackson ImmunoResearch)-2% blocking reagent-mab for 1 hr at RT, and incubated at 4°C overnight in 1/5000 sheep anti-digoxigenin-blocking solution. Sections were rinsed three times in mab, one time in buffer 3 (100 mm Tris, pH 9.5, 100 mm NaCl, 50 mm MgCl2) containing 240 μg/ml levamisole, and finally incubated in 1/100 nitroblue tetrazolium-5-bromo-4-chloro-3-indolylphosphate (Hoffmann-LaRoche)-levamisole-buffer 3 for 1-8 hr at RT. Sections were rinsed in buffer 3, postfixed, rinsed in PBS, mounted, and photographed. In a subset of animals [n = 4 Bax and 4 wild type (WT)], the number of c-ret-labeled cells in the lumbar ventral horn (VH) was counted in every 15th section through the entire lumbar region. The identity of the small immunolabeled profiles as cells was confirmed by Hoechst labeling of all of the sections (see Results).
PCR. Total RNA was extracted from E14.5 mouse hindlimb muscles using a total RNA extraction kit (SV total RNA extraction kit; Promega, Madison, WI). After quantification, 1 μg of total RNA was reverse transcribed by Moloney murine leukemia virus reverse transcriptase (Promega) and subsequently PCR amplified using specific PCR primer sets. Ciliary neurotrophic factor (CNTF), GDNF, and brain-derived neurotrophic factor (BDNF) cDNAs were amplified by PCR for 33 cycles at 95°C for 45 sec, 58°C for 45 sec, and 72°C for 1 min. As a control, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) cDNA was amplified by PCR for 28 cycles at 95°C for 45 sec, 58°C for 45 sec, and 72°C for 1 min. Primers were as follows: for CNTF, 5′-ATG GCT TTC GCA GAG CAA-3′ and 5′-CTA CAT TTG CTT GGC CCC-3′ (GenBank #U05342); for GDNF, 5′-CTG ACC AGT TTG ATG ACG TC-3′ and 5′-TCT AAA AAC GAC AGG TCG TC-3′ (Inoue et al., 1999); for BDNF, 5′-AGC TGA GCG TGT GTG ACA GT-3′ and 5′-TCC ATA GTA AGG GCC CGA AC-3′ (Tirassa et al., 2000); and for GAPDH, 5′-CAC CAC CAT GGA GAA GGC C-3′ and 5′-GAT GGA TGC CTT GGC CAG G-3′ (Tirassa et al., 2000). Amplified PCR products were resolved in 2.5% agarose gel, stained by ethidium bromide, and visualized under UV illumination.
GDNF injection. Human recombinant BDNF and GDNF were gifts kindly provided by Amgen (Thousand Oaks, CA). Factors were injected daily subcutaneously at a dose of 1 μg/gm of body weight for 7-8 consecutive days beginning on postnatal day 0. Vehicle (saline) was injected as a control. For some experiments, GDNF (1 μg) was injected daily directly into the whisker pad for 14 d, and a retrograde tracer (Alexa 488-cholera toxin B subunit) was applied on the 12th day, and animals were allowed to survive for an additional 48 hr.
Results
Absence of PCD in Bax KO mice
We first reexamined PCD in the spinal cord of Bax KO mice. In a previous study, it was reported that TUNEL+ cells were dramatically reduced or absent in the LMC and DRG during development of Bax KO mice (White et al., 1998). We also observed that active-caspase-3 immunoreactivity was absent in the LMC and DRG of E14.5 Bax KO mice, whereas many neurons of wild-type littermates exhibited active-caspase-3 immunoreactivity (Fig. 1A,B,E,F). However, because neurons may degenerate without caspase-3 activation and DNA degradation (Oppenheim et al., 2001b), we also counted the number of pyknotic cells (Clarke and Oppenheim, 1995) after Nissl staining (Fig. 1C,D,G,H). Although we frequently found typical pyknotic bodies in the LMC and DRG of E14.5 wild-type embryos, these were never observed in Bax KO mice.
Figure 1.
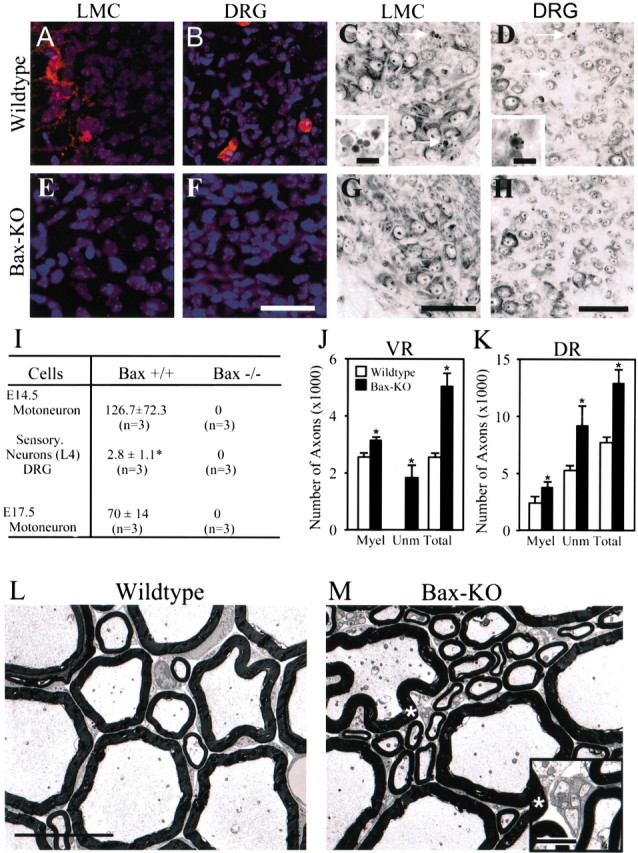
A, B, E, F, Immunostaining of activated caspase-3 in LMC (A, E) or DRG (B, F) at E14.5 in WT (A, B) and Bax KO (E, F) mice. Activated caspase-3 immunoreactivity was never observed in Bax KO motoneurons or DRG neurons. C, D, G, H, Nissl staining of E14.5 lateral motor column (LMC) (C,G) or L3 DRG (D,H) of WT (C,D) and Bax KO (G, H) mice. Arrows indicate pyknotic cells in WT mice (see also insets in C, D). Scale bars: F-H (for A-H), 75 μm; insets, 10 μm. I, Quantification of pyknotic cells in WT and Bax KO mice. For MNs, values are mean (±SD) numbers of pyknotic cells per 1000 surviving cells. J, K, Quantification of axon numbers in 5-month-old WT versus BaxKO L5 ventral (J) and dorsal (K) roots. Values are means ± SDs (n = 3). *p < 0.05. Myel, Myelinated; Unm, unmyelinated. L, M, Morphology of L5 ventral root axons in WT (L) and Bax KO (M) mice. Note that only Bax KO mice exhibit unmyelinated axons (inset in M is an enlargement of the region indicated by the asterisk). Scale bars: L (for L, M), 10 μm; inset, 1 μm.
To determine whether spinal neurons are permanently rescued from PCD in Bax KO mice, we counted the total number of axons (myelinated and unmyelinated in the VRs and DRs of 5-month-old mice). When examined in the electron microscope, the DRs in both Bax KO and WT mice contained bundles of unmyelinated axons (data not shown), whereas unmyelinated axons were only observed in the VRs of Bax KO mice (Fig. 1, compare L, M). The total numbers of Bax KO DR and VR axons were 173 and 204% of WT controls, respectively (Fig. 1J,K). Because ∼40-50% of sensory and motor neurons degenerate during the normal PCD period (Lance-Jones, 1982; Oppenheim et al., 1986; Grieshammer et al., 1998; Yamamoto and Henderson, 1999), the increase in VR and DR axon numbers in the adult Bax KO indicates a virtual total absence of PCD in the Bax KO mice, and suggests that cell death is prevented and not merely delayed.
Neuromuscular development is normal in Bax KO mice
We examined the muscles of 1-month-old mice to determine whether the deletion of Bax modified muscle or neuromuscular junction development. The wet weights of gastrocnemius and triceps muscles in Bax KO mice were comparable with those of wild-type littermates (Fig. 2G). In addition, the fiber type distribution, the numbers of each fiber type, and the diameter of individual fibers were comparable in soleus, gastrocnemius, and triceps muscles of wild-type and Bax KO mice (data not shown) (Fig. 2D-F). Examination of neuromuscular junctions in the soleus muscle indicates that each neuromuscular junction was singly innervated in both the wild-type and Bax KO mice (Fig. 2, compare B, C). Finally, the size and morphology of postsynaptic end plates examined by α-bungarotoxin labeling was found to be similar in Bax KO and WT mice (data not shown).
Figure 2.
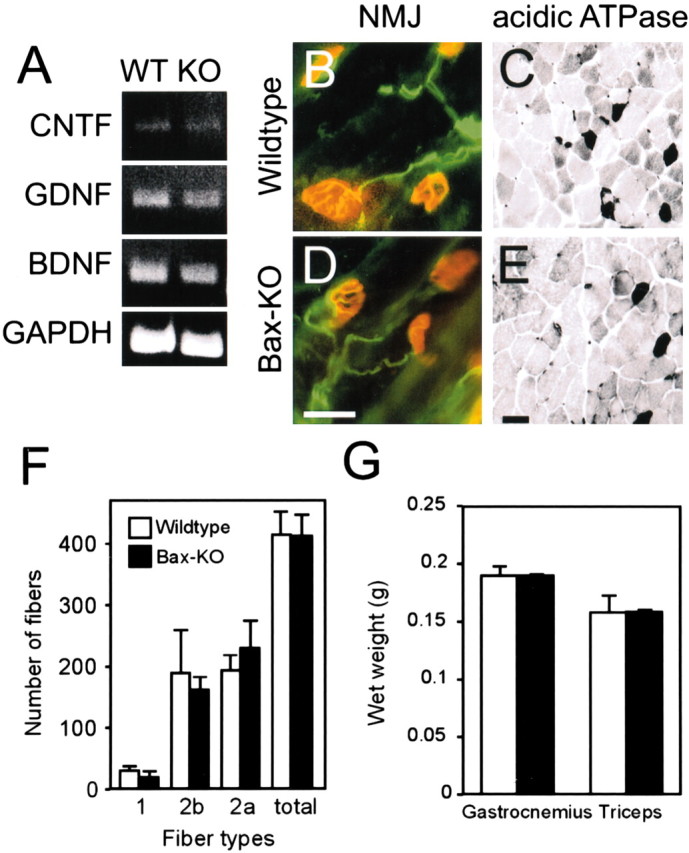
A, Expression levels of neurotrophic factors in E14.5 hindlimbs measured by semiquantitative reverse transcription-PCR. B, D, Neuromuscular junctions (NMJ) in the soleus muscle of 1-month-old WT (B) and Bax KO (D) mice. Postsynaptic acetylcholine receptors (AChRs) were labeled with Alexa 594-α-bungarotoxin (red), and presynaptic axons-nerve terminal are labeled green. Nerve terminals overlying AChR appear yellow. Both genotypes show focal innervation of end plates by single axons, and end plate size and morphology are similar. Scale bar: D (for B, D), 20 μm. C, E, Examination of muscle fiber subtype composition using acidic ATPase staining in triceps muscles of WT (C) and Bax KO (E) mice. In this staining procedure, different muscle fibers are stained at different intensities (type 1 < type 2b < type 2a). Scale bar: E (for C, E), 20 μm. F, Quantification of muscle fiber numbers in adult triceps. Five different fields were chosen and different fiber types were counted in each field and then summated. G, The wet weight of adult gastrocnemius and triceps muscles in Bax KO mice and WT littermates. Values are means ± SDs; n = 3 for F and G.
To determine whether muscle-peripheral nerve-derived neurotrophic factors were normally expressed, we compared the mRNA expression of CNTF, GDNF, and BDNF in the Bax KO and WT hindlimb muscles on E14.5 (Fig. 2A). Semiquantitative reverse transcription-PCR demonstrated that mRNA levels were similar in the hindlimb muscles of the two groups, indicating that comparable amounts of target- or peripheral nerve-derived trophic support were potentially available to limb-innervating neurons in the Bax KO. Together, these results suggest that, despite the elimination of neuronal PCD, neuromuscular development was normal in Bax KO mice.
A proportion of MNs, but not DRG neurons, are markedly atrophied in Bax KO mice
By counting neurons in Nissl-stained paraffin sections, we found a large increase in DRG sensory neurons in Bax KO mice at all of the time points examined (Table 1). In contrast, when spinal and cranial MNs were counted on P0 (spinal) or P7-P10 (cranial), after the completion of PCD, MN numbers were similar in Bax KO and WT mice. However, when cell counts were done during the PCD period (i.e., E14.5 for spinal MNs and P0 for cranial MNs), there were significantly more cells in the Bax KO mice. This apparent reduction of MN numbers between embryonic and postnatal stages in the Bax KO suggests that either the embryonically rescued MNs subsequently degenerate or are postnatally no longer recognizable as MNs in Nissl-stained sections. Several lines of evidence indicate that the latter alternative is true. In semithin sections of P1 spinal cord, we found many small neurons (Fig. 3F, arrows) in the VH of Bax KO mice, whereas similar neurons were rarely observed in the VH of WT mice; the cross-sectional area occupied by the ventral horn also appeared consistently larger in the Bax KO (Fig. 3, compare E, F). In Nissl-stained sections, the small neurons within the VH of the Bax KO exhibited scant cytoplasm and hence little Nissl labeling, and accordingly, we refer to them as Nissl negative (Fig 3B, arrows) (for spinal MNs, compare A, B) (for facial MNs, compare C, D). Although these Nissl-negative neurons are morphologically distinct from glia (Fig. 3B,F, asterisks), because their soma size and morphology are difficult to distinguish from small spinal interneurons, they were excluded from our postnatal MN counts. However, as described below, these are most likely MNs rescued from PCD after Bax deletion. Similar small atypical MNs were also present in the VH of adult Bax KO mice (data not shown).
Table 1.
Number of neurons in Bax-deficient mice during development
|
|
E14.5 |
E17.5 |
P0 |
P7-P10 |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
Bax+/+
|
Bax−/−
|
Bax+/+
|
Bax−/−
|
Bax+/+
|
Bax−/−
|
Bax+/+
|
Bax−/−
|
||||
| Motoneurons | ||||||||||||
| Hypoglossal | 1833 ± 109 | 2442 ± 271* | 1603 ± 103* | 2058 ± 203* | 1495 | 1970 ± 125 | ||||||
| n = 2 | n = 3 | n = 2 | n = 3 | n = 1 | n = 3 | |||||||
| Facial | 4920 ± 495 | 7038 ± 95* | 3873 ± 244 | 4927 ± 819* | 2540 ± 276 | 2977 ± 225 | ||||||
| n = 2 | n = 3 | n = 2 | n = 3 | n = 3 | n = 3 | |||||||
| Brachial | 3730 | 4990 | 3200 ± 14 | 4194 ± 366* | 3190 ± 181 | 3327 ± 281 | ||||||
| n = 1 | n = 1 | n = 2 | n = 3 | n = 3 | n = 3 | |||||||
| Lumbar | 3693 ± 331 | 4788 ± 513* | 2900 ± 491 | 3620 ± 174* | 2620 ± 643 | 2528 ± 443 | 2187 ± 364 | 2293 ± 126 | ||||
| n = 3 | n = 4 | n = 4 | n = 4 | n = 4 | n = 5 | n = 3 | n = 3 | |||||
| DRG neurons | ||||||||||||
| C7 | 5828 ± 1071 | 8363 ± 979* | ||||||||||
| n = 2 | n = 2 | |||||||||||
| L4 | 5290 ± 382 | 7950 ± 1497* | ||||||||||
| n = 2 | n = 3 | |||||||||||
| L5 | 6288 ± 1404 | 13,880 ± 2835* | 6092 ± 145 | 9386 ± 1424* | 7243 ± 392 | 12,145 ± 3082* | ||||||
|
|
|
|
n = 2
|
n = 2
|
n = 2
|
n = 3
|
n = 2
|
n = 2
|
||||
Data are expressed as mean ± SD. C7, Cervical segment 7; L4, L5, lumbar segments 4 and 5.
p < 0.05.
Figure 3.
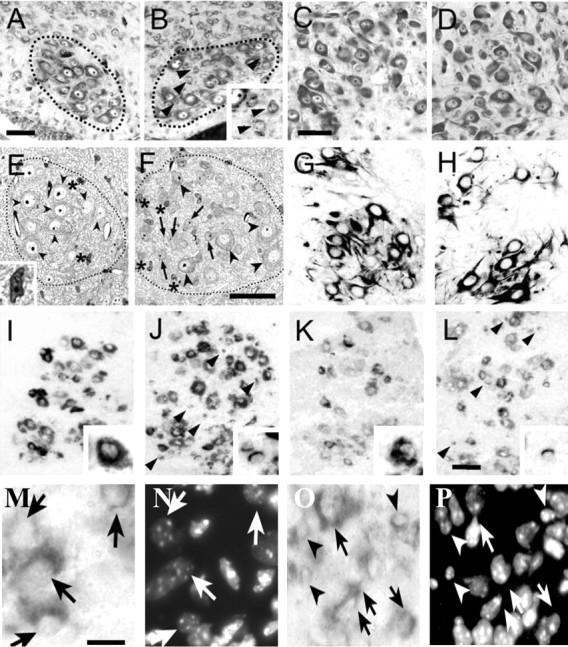
A, B, Nissl-stained lumbar MNs of P0 WT (A) and Bax KO (B) mice (motor column enclosed by dotted lines). Arrowheads in B indicate small neurons within ventral horn motor column (see also inset). Scale bar: A (for A, B), 50 μm. C, D, Nissl-stained facial MNs of P10 WT (C) and Bax KO (D) mice. Scale bar: C (for C, D), 50 μm. E, F, Plastic-embedded (1 μm) sections of lumbar MNs in the ventral horn of P2 WT (E) and Bax KO (F) mice. Arrows indicate small atrophied MNs, arrowheads indicate normal MNs, and asterisks indicate glial cells (inset in E is an enlargement of a glial cell). Aberrantly small neurons were also evident in the adult spinal cord ventral horn of Bax KO (data not shown). Scale bar: F (for E, F), 50 μm. G, H, SMI-32 immunoreactivity in P0 lumbar spinal cord VH of WT (G) and Bax KO (H) mice. I-L, Localization of c-ret (I, J) and GFRα1 (K, L) mRNAs in P2 WT (I, K) and Bax KO (J, L) mice. Scale bar: L (for G-L), 50 μm. Arrowheads in J and L indicate c-ret + atrophic MNs. Insets in I-L are higher magnifications of c-ret+ and GFRα1 + normal and atrophic MNs. M-P, Higher magnification images of c-ret-immunolabeled MNs in the ventral horn of WT (M) and Bax KO (O) together with Hoechst labeling of MN nuclei in the same sections (N, P). Scale bar: M (for M-P), 15 μm. Arrows indicate normal large MNs, and small atrophic MNs are indicated by arrowheads.
Although these small neurons failed to express the putative MN-specific marker SMI-32 (Fig. 3, compare G, H), they retained the MN-specific expression of the GDNF receptors c-ret and GFRα1 (I-L, arrows). In mice, most lumbar MNs express c-ret, whereas only a subset of MNs express GFRα1 (Garces et al., 2000; Oppenheim et al., 2000). Similarly, in the Bax KO, virtually all of the small- as well as normal-sized MNs appear to express c-ret, whereas only a subpopulation of both express GFRα1 mRNA, suggesting that the atrophied MNs retain a typical MN pattern of neurotrophic factor receptor expression. Cell counts of all of the c-ret+ cells in the lumbar ventral horn of WT (n = 4) and Bax KO (n = 4) mice (Fig. 3, M-P) revealed a 45% increase in the Bax KO (p < 0.01; t test). Ultrastructural examination showed that the small neurons in the VH of Bax KO mice contain only a small amount of cytoplasm and cytoplasmic organelles such as mitochondria and rough endoplasmic reticulum compared with the larger, normalsized MNs (Fig. 4). Despite these changes, the atrophic MNs appear otherwise healthy and exhibit no typical signs of degeneration. Furthermore, immature axosomatic and axodendritic synapses were present on these cells as well as on the larger MNs (Fig. 4B,D). These results indicate that, postnatally, the Bax KO-rescued MNs are atrophied, but, on the basis of cytology, these cells appear otherwise healthy. Because the SMI-32 antibody recognizes a nonphosphorylated form of neurofilament protein (neurofilament-H) found only in the cytoplasm of MNs in the spinal cord (Gotow and Tanaka, 1994; Carriedo et al., 1995; Bar-Peled et al., 1999), the scant cytoplasm present in the small Bax KO-rescued MNs may not be sufficient to give a detectable signal. Alternatively, these MNs may not express neurofilament-H. As described above, however, even the small atrophic MNs express mRNA for the MN marker c-ret.
Figure 4.
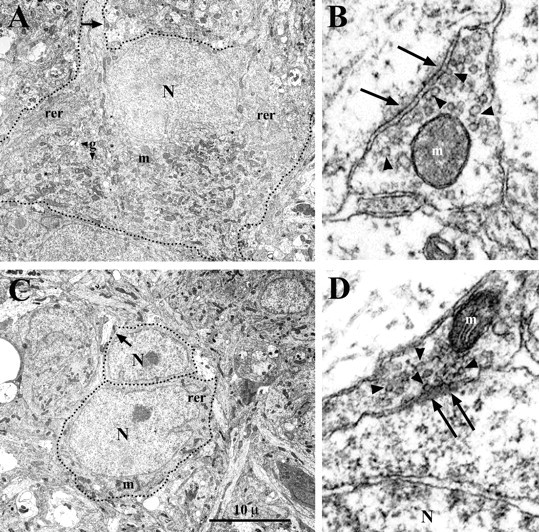
Electron micrographs of MNs in P2 WT (A) and Bax KO (C) lumbar spinal cord. Dotted lines indicate the soma and proximal dendrites. Synapses indicated by arrows in A and C are enlarged in B and D. Although immature, both axodendritic (B) and axosomatic (D) synapses exhibit postsynaptic densities (arrows) and synaptic vesicles (arrowheads). Despite being atrophic, the subpopulation of Bax KO-rescued MNs contain mitochondria (m), rough endoplasmic reticulum (rer), and Golgi apparatus (g). N, MN nuclei.
Axons from developmentally rescued MNs in the postnatal Bax KO do not innervate target muscles
To determine whether the atrophied MNs in postnatal Bax KO mice innervate their targets, we placed DiI either in the L5 ventral root or in target muscles and examined whether the cell bodies were retrogradely labeled. After DiI deposition in the P14 ventral root (L5) of WT mice, we found that only large, Nissl-positive MNs in the ipsilateral VH were brightly labeled (Fig. 5A-C). In contrast, many of the small, Nissl-negative as well as the large, Nissl-positive MNs were labeled in Bax KO mice (Fig. 5D-F, arrowheads and insets). A size-frequency histogram demonstrates that there is a group of small labeled neurons (<100 μm2) in the Bax KO that are absent in WT mice (Fig. 5G). After injection of DiI into adult gastrocnemius muscle, however, small labeled MNs were never observed in either 1-month-old wild-type or Bax KO mice (Fig. 5H,I)(n = 3). Similarly, we never observed retrograde labeling of small Nissl-negative facial MNs at P14; the total number of labeled facial MNs was similar in WT versus Bax KO mice after whisker pad injection of a retrograde tracer (see Fig. 8E). Finally, in a previous study, we observed that injection of DiI into middle to distal regions of the sciatic nerve immediately after axotomy at that site on P2 (Sun and Oppenheim, 2003) fails to label any small atrophic MNs in the Bax KO. From these data, we conclude that, by P2, the Bax KO-rescued MNs have withdrawn their axons to a site between the ventral root and the middle to distal sciatic nerve.
Figure 5.
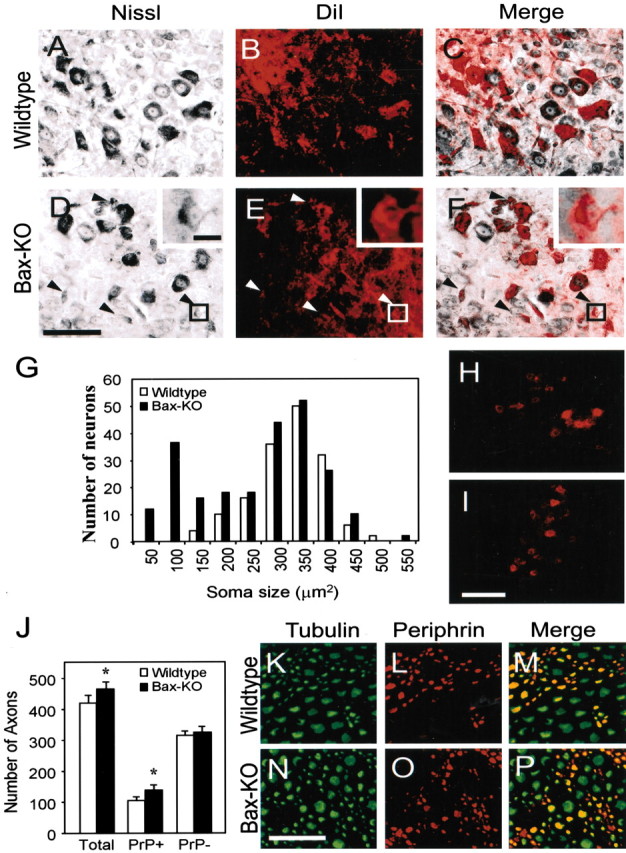
Retrograde labeling by DiI from the ventral root in P14 WT (A-C) or Bax KO (D-F) mice. Comparison of Nissl stained (A, D) and DiI labeled (B, E) (see also merged image in C, F) shows that both small Nissl-negative and large Nissl-positive MNs are DiI labeled in Bax KO mice. Insets show example of a small DiI-labeled neuron in Bax KO. Arrows indicate retrogradely labeled atrophic MNs in Bax KO mice. In A-C, ventral-dorsal is up-down, respectively, and medial-lateral is left-right, respectively. However, to include insets in D-F, the orientation is reversed. G, Size-frequency histogram of DiI-labeled neurons. The number of labeled neurons was counted in every 10th section from three different animals and summated. Bax KO has an additional peak of small neurons in addition to the typical large MNs. H, I, DiI labeling of MNs from muscle in P28 WT (H) and Bax KO (I) mice. No small atrophied neurons were labeled in either group. Scale bars: D (for A-F), I (for H, I), 100 μm; D, inset (for D-F, insets), 10 μm. J-P, Immunostaining of tubulin (K, N) and peripherin (L, O) in the distal sciatic nerve of WT (K, L) and Bax KO (N, O) mice (see also merged images in M and P). Peripherin+ small-sized sensory axons were more abundant in Bax KO mice, whereas the number of large tubulin +, peripherin - axons was comparable in the two genotypes (J). Five different fields in each nerve were examined after double staining in each animal, and the numbers of double- and single-labeled axons were counted. Values are means ± SDs; n = 4; *p < 0.05, t test, WT versus Bax KO. Scale bar, N (for K-P), 50 μm.
Figure 8.
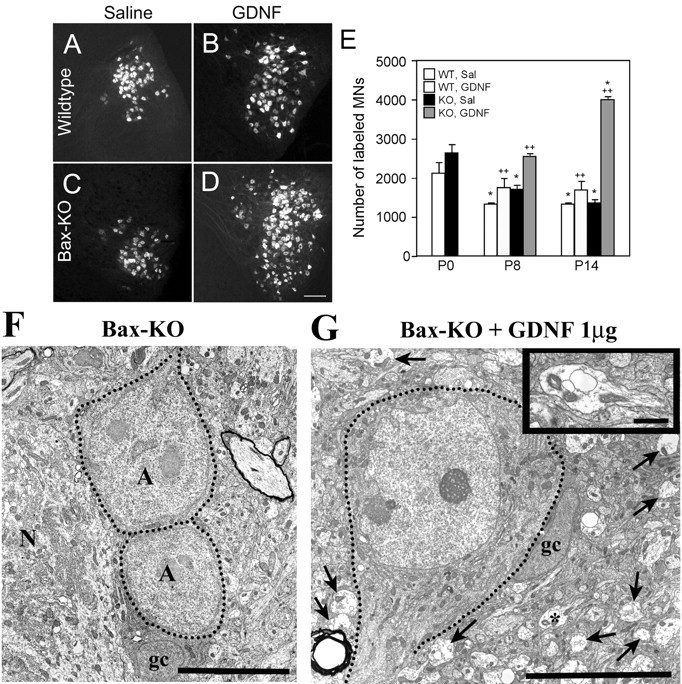
A-D, Retrograde labeling of facial MNs in WT (A, B) and Bax KO (C, D) mice 14 d after intramuscular (whisker pad) injection of saline (A, C) or GDNF (B, D). Scale bar: D (for A-D), 50 μm. E, Quantification of total labeled facial MN numbers after 8 d of subcutaneous injection or 14 d of intramuscular injection of saline (Sal) or GDNF. Values are means ± SD; n = 3; *p < 0.05 compared with P0 value; ++p < 0.05 compared with saline-treated group. F, G, Electron micrographs of lumbar MNs in saline (F)- or GDNF (G)-treated (1 μg/gm of body weight, s.c. injection) Bax KO mice on P7. Dotted lines indicate the soma of atrophic MNs (A) adjacent to a healthy normal (N) MN. gc, Glial cell. Arrows in G indicate growth cones. A growth cone is shown at a higher magnification in the inset to G. Scale bars: F, 10 μm; G, 10 μm; inset, 2 μm.
We also examined the distal sciatic nerve of adult Bax KO mice and wild-type littermates after double immunofluorescence staining with β-tubulin (TuJ-1) and peripherin antibodies. We used a peripherin antibody that only labels sensory axons together with the β-tubulin antibody, which labels both sensory and motor axons, to quantify the number of distal sensory and motor axons in Bax KO mice. Although MNs were never labeled by peripherin, peripherin strongly stained small- to mediumsized DRG neurons and weakly stained large DRG neurons (data not shown). Although the total number of β-tubulin-labeled axons in the Bax KO mice did not quite reach statistical significance (p < 0.10, n = 3) (Fig. 5J), the number of β-tubulin+/peripherin+ sensory axons was significantly increased by 44%, whereas the number of β-tubulin+/peripherin- motor axons was unchanged (n = 3) (Fig. 5K-P), indicating that the number of motor axons reaching the distal sciatic nerve was similar in Bax KO and WT mice. Although we cannot exclude the possibility that small, unmyelinated motor axons were not labeled (or not visible), many of the small unmyelinated, peripherin+ sensory axons in the Bax KO were, in fact, detectable in these preparations. Additionally, the absence of retrograde labeling of small atrophic MNs after either sciatic nerve or muscle injection of DiI (Fig. 5H,I) is consistent with our interpretation that atrophic spinal MNs in the postnatal Bax KO do not innervate target muscles in the hindlimb.
Development of MNs during the PCD period in Bax KO mice
Because the number of Nissl-positive MNs is increased in Bax KO versus WT mice when examined during the embryonic PCD period, we next asked whether these excess MNs are initially able to innervate their targets. The size of MNs in the E14.5 Bax KO appears somewhat smaller than in WT embryos (Fig. 6, compare A, B), and a size-frequency profile is consistent with this impression (C). This is in striking contrast to the situation in postnatal animals in which two MN groups (normal-sized and atrophic MNs) were clearly distinguished (Fig. 5G). A similar developmental profile was also observed for the size of facial MNs in the Bax KO mice (data not shown). To examine limb innervation on E14.5, DiI was placed distal to the plexus region of the hindlimb bud to orthogradely label limb nerves and retrogradely label spinal neurons. Although quantification of axon numbers was not possible, the Bax KO mice appear to have an increased number of nerve fibers innervating the limb (Fig. 6, compare D,F, E,G). For example, when we examined whole mounts from 13 DiI-labeled embryos without previous knowledge of genotype (blinded), the three embryos judged to have the greatest amount (density) of limb innervation were all Bax KO animals; the remaining 10 embryos were either heterozygote or wild-type mice. In higher magnification views of the tibial nerve just distal to the branching point, all of the Bax KO mice exhibit a thicker nerve (Fig. 6, compare E, G). In fact, measures of the thickness (micrometers) of the tibial nerve indicates that it is significantly larger in the Bax KO (18.7 ± 5.6 in WT vs 35.9 ± 6.1 in the Bax KO; n = 3; p < 0.05; t test). In the spinal cord of these same embryos, there was intense and widespread DiI labeling in both the DRG and spinal motor column, suggesting that virtually all of the motor and sensory neurons had projected axons to the limb bud at E14.5 (Fig. 5 H, I). Together, the embryonic and postnatal results indicate that, whereas all of the MNs initially innervate the hindlimb in the Bax KO embryo, a subset of Bax KO MNs fail to maintain innervation postnatally and become atrophied.
Figure 6.
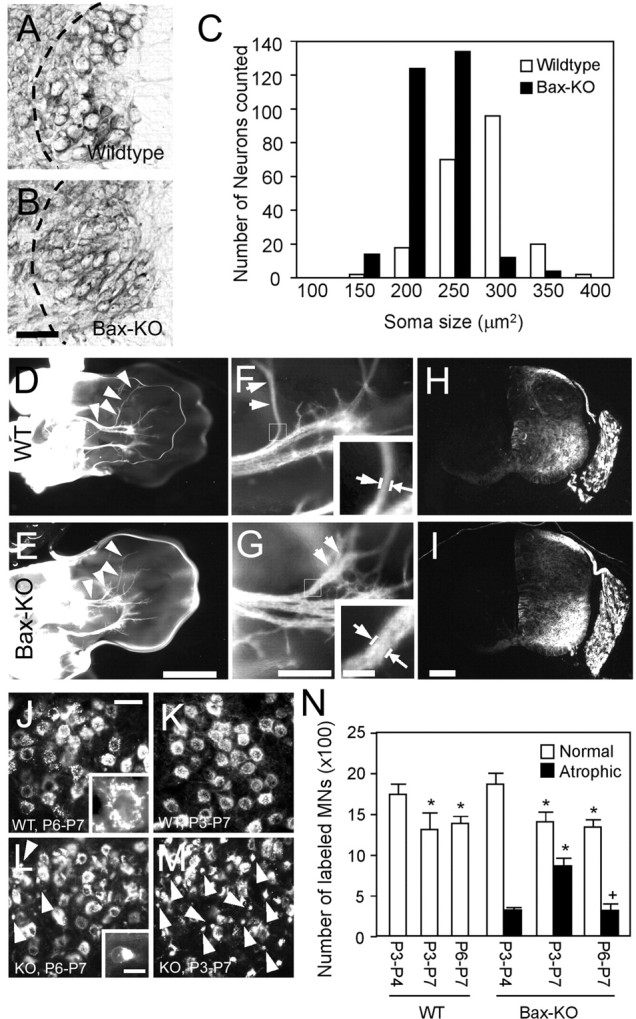
A-C, Comparison of MN size in lumbar VH of E14 WT (A) and Bax KO (B) mice. Dashed lines in A and B indicate medial border of the ventral horn. The size-frequency histogram shows that Bax KO MNs (n = 4) are smaller than WT MNs (n = 3) (C). Ventral (D-G) views of orthogradely labeled developing nerves in hindlimb whole mounts of E14 WT (D, F) and Bax KO (E, G). Arrowheads and arrows in D-G indicate tibial nerve. Arrows in insets indicate where tibial nerve diameter is measured. Higher magnification of the tibial nerve branch shows that the nerve in Bax KO (G) appears thicker and more arborized than in WT (F). H, I, Retrograde labeling of neurons in the VH and DRG of WT (H) and Bax KO (I). Scale bars: B (for A, B), 50 μm; E (for D, E), 1 mm; G (for F, G), 100 μm; I (for H, I), 100 μm; G, inset (for F, G, insets), 20 μm. J-M, Retrograde labeling of facial MNs from the whisker pad for P6-P7 (J, L) or P3-P7 (K, M) in WT (J, K) and Bax KO (L, M) mice (see Results). Arrows indicate a subpopulation of atrophic MNs retrogradely labeled in Bax KO mice (see also insets J vs L showing individual labeled normal vs atrophic MNs). Scale bars: J (for J-M), 50 μm; L, inset (for J, L, insets), 10 μm. N, Quantification of retrogradely labeled healthy (open bars) or atrophic (closed bars) MNs (n = 3). *p < 0.05, compared with P3-P4 value; +p < 0.05, compared with P3-P7 value; t tests.
To directly examine the retraction of axons and the atrophy of Bax KO MNs, we applied a retrograde tracer (Alexa 488-conjugated cholera toxin B subunits) into the whisker pad of P3 or P6 WT and Bax KO mice, and counted the number of labeled MNs (Fig. 6J-N). Because the PCD of facial MNs occurs between E16 and P7, postnatal labeling of innervating MNs provides a means of assessing the extent of atrophy and axon retraction by excess Bax KO-rescued MNs. In Bax KO mice, we found many very small (<100 μm2) retrogradely labeled MNs (Fig. 6L, inset) on P3 through P8, whereas by P14 such atrophic neurons could no longer be retrogradely labeled (Figs. 6L, 8C). In contrast, labeled atrophic MNs were never observed at any age in WT mice. To quantify the cellular atrophy and axon retraction, healthy and atrophic (<100 μm2) retrogradely labeled MNs were counted separately after different labeling schedules. In WT mice, there were fewer retrogradely labeled facial MNs on P7 after tracer injection on either P3 (4 d survival) or P6 (1 d survival) compared with the number present on P4 after tracer injection on P3 (Fig. 6N). This reduction between P3 and P7 most likely reflects the normal postnatal PCD of facial MNs during this period (Ashwell and Watson, 1983). In contrast, a similar comparison in the Bax KO indicates that, between P3 and P7, there is a reduced number of labeled normal MNs and an increased number of labeled atrophic MNs, resulting in no net change in the total number of labeled MNs (2190 ± 151 vs 2267 ± 30; n = 3). This is consistent with a subpopulation of MNs becoming atrophic between P3 and P7 in the Bax KO mice. Many Bax KO facial MNs remain atrophic into adult stages (White et al., 1998). A comparison of labeled MNs on P7 after tracer injection on P3 versus P6 in the Bax KO shows similar numbers of normal large MNs in the two groups but a reduction in the number of labeled atrophic MNs present on P7 in the P3-versus-P6 injected group (Fig. 6). This is consistent with the occurrence of axon retraction and target denervation by a subpopulation of facial MNs between P3 and P6 in the Bax KO mice.
Postnatal injection of GDNF rescues late dying WT MNs from normal PCD and induces growth of the atrophic Bax KO-rescued MNs
Our examination of MN development in Bax KO mice indicates that a proportion of MNs are unable to differentiate normally and fail to maintain target innervation beyond the embryonic period (spinal MNs) or the early postnatal period (facial MNs). We reasoned that a plausible explanation for this failure could be insufficient trophic support from target muscles; that is, although because of Bax deletion, these MNs can survive independent of target-derived survival signals, they may still require trophic support for cellular growth and maintenance of innervation, and this putative trophic support may in some sense be limiting. To test this, we applied exogenous trophic factor (GDNF) to postnatal Bax KO mice and observed whether such treatment could prevent or reverse MN atrophy (Fig. 7). We chose GDNF for this study, because it is a potent survival factor for embryonic MNs (Henderson et al., 1994; Oppenheim et al., 2000a), and because we recently found that neonatal axotomy increased GDNF expression in the proximal nerve, resulting in regrowth of the subpopulation of atrophic Bax KO MNs (Sun and Oppenheim, 2003). We injected GDNF subcutaneously daily (1 μg/gm of body weight) from P0. Both wild-type and Bax KO mice treated with GDNF exhibited a retardation of the normal increase in body weight and a severe motor tremor (data not shown), as has been reported previously (Keller-Peck et al., 2001). Because many animals died after 9 -10 d of subcutaneous GDNF treatment, we examined animals after 7 d of daily injections when all of the animals appeared healthy and viable. Wild-type mice treated with GDNF exhibited 22% more lumbar MNs on P7 compared with saline-injected control animals, whereas Bax KO mice treated with GDNF had 47% more discernible (i.e., large Nissl-positive) lumbar MNs compared with saline-treated Bax KO mice (Fig. 7A). In contrast, BDNF treatment (daily subcutaneous injection; 1 μg/gm of body weight) did not affect the number of discernible MNs in either group. Examination of the distribution of lumbar spinal MNs along the rostrocaudal axis after GDNF treatment showed that the number of caudal lumbar MNs was selectively increased in GDNF-injected WT mice, whereas more MNs were present in both rostral and caudal regions in Bax KO mice (Fig. 7B). Because the small amount of normal postnatal PCD of lumbar MNs occurs primarily in caudal segments (Yamamoto and Henderson, 1999; Oppenheim et al., 2000a), these data suggest that prevention of postnatal PCD is responsible for the increase in MN number in WT animals, whereas the marked increase in MNs in Bax KO mice is attributable to the growth of the atrophic Bax KO-rescued MNs, which are located in all segments of the lumbar spinal cord.
Figure 7.

The effect of postnatal GDNF treatment in WT and Bax KO mice. GDNF and BDNF were injected subcutaneously from P0 for 7 consecutive days, and the number of MNs (mean ± SD) in spinal cord (A, right) and facial nucleus (C, right) were counted at P7 (n = 3 for A-C) (*p < 0.01). The left side of A and C shows the number of MNs at different ages in untreated animals. B, Number (mean ± SD) of MNs in the rostral or caudal one-fifth of lumbar spinal cord after 1 week of GDNF injections (*p < 0.01). D, Size-frequency histogram of facial MNs in WT and Bax KO mice after GDNF treatment. The number of labeled neurons was counted in every 10th section from three different animals and summated. Because atrophic MNs in saline-treated Bax KO mice did not meet counting criteria, only Nissl-stained, healthy MNs were included in this analysis (see Results).
The effect of GDNF treatment was even more profound for facial MNs. There were 59% more MNs in WT and 130% more MNs in Bax KO mice on P7 after GDNF treatment (Fig. 7C). The number of facial MNs in P7 GDNF-treated WT mice is close to the P0 value for WT facial MN number, whereas the number of facial MNs in GDNF-treated Bax KO mice is similar to the number of MNs present before the embryonic onset of PCD on E16 (Ashwell and Watson, 1983). Therefore, GDNF appears to completely prevent the postnatal PCD of facial MNs in WT mice and rescues cell size in all of the atrophied facial MNs in Bax KO mice.
The soma size of facial MNs was markedly affected by GDNF (Fig. 6D). Soma size-frequency profiles of saline-treated WT and Bax KO facial MNs were similar. In GDNF-treated WT mice, MNs >250 μm2 were greatly increased, whereas the number of MNs in GDNF-treated Bax KO mice was increased in all of the cell size categories (Fig. 7D). Because we excluded the small atrophic (Nissl-negative) neurons in the saline-treated Bax KO group from the soma size analyses, the increase in the number of small neurons in GDNF-treated Bax KO mice is attributable to the presence of previously atrophic neurons that were now increased in size, so that they met our criteria (e.g., Nissl-positive) for inclusion in both the cell counts and MN soma size measures (see Materials and Methods). These results indicate that GDNF increased the soma size of both normal and atrophic MNs in WT and Bax KO mice.
GDNF induces regrowth and reinnervation of axons of the Bax KO-rescued MNs
Because previous studies have demonstrated that GDNF promotes MN survival but fails to prevent axonal degeneration in a mouse model of MN disease (Sagot et al., 1996, 1998), we examined whether GDNF can induce axonal growth and target muscle reinnervation by the excess Bax KO-rescued MNs. After 6 d of daily subcutaneous GDNF treatment, we injected a retrograde tracer into the whisker pad, the target of a subset of facial MNs, and assessed the number of retrogradely labeled MNs 2 d after tracer injection (Fig. 8). Although there was a significant increase in the number of labeled MNs in the GDNF-versus saline-treated Bax KO mice [2549 ± 77 (n = 3) vs 1690 ± 139 (n = 3)], labeled MN numbers in the GDNF-treated Bax KO mice were similar to the number of labeled MNs in P1 Bax KO mice [2549 ± 77 (n = 3) vs 2635 ± 220 (n = 3)]. These results suggest that, whereas 8 d of GDNF injection completely inhibited additional postnatal axon retraction, such treatment failed to induce target reinnervation by already atrophied and denervated MNs. Because most mice injected subcutaneously with GDNF from P0 died after 9 -10 d, we used an alternative method of injection to address the question of whether a longer duration of GDNF treatment could promote reinnervation. Mice that received intramuscular injections of GDNF (1 μg/mice) daily into the whisker pad were viable for >2 weeks, with only minor side effects such as weak tremor and a slight retardation of body growth. Accordingly, we examined the number of retrogradely labeled facial MNs on P14 after 2 weeks of GDNF treatment (Fig. 8A-E). After saline treatment, the number of labeled MNs in Bax KO mice was not significantly different from that in WT mice, indicating that all of the atrophic Bax KO-rescued MNs failed to innervate their targets by 2 weeks. In contrast, when compared with the Bax KO saline group, GDNF treatment for 2 weeks resulted in a 307% increase in the number of labeled MNs in the Bax KO (Fig. 8E). Because these animals also had significantly more labeled MNs than did the P1 Bax KO group, these data indicate that 2 weeks of daily GDNF treatment (P0 -P13) induced most of the Bax KO-rescued subpopulation of facial MNs to regrow axons to their whisker pad targets. Although it is possible that these increases in retrograde labeling after GDNF treatment could reflect effects that are independent of regrowth and reinnervation (e.g., terminal sprouting, resulting in increased uptake of tracer or enhanced retrograde transport), we consider this unlikely. First, atrophic MNs were never retrogradely labeled in the Bax KO mice. Second, we also observed that, after only 7 d of subcutaneous injection of GDNF, there appear to be increased numbers of well defined growth cone-like profiles (for similar examples of spinal cord growth cones, see Foelix and Oppenheim, 1973; Skoff and Hamburger, 1974; Vaughn et al., 1977) in the ventral spinal cord of Bax KO mice, suggesting that GDNF, acting either directly or indirectly, also induces neurite growth and synaptogenesis within the spinal cord of Bax KO mice (Fig. 8, compare F, G). Finally, results from retrograde labeling with DiI, which acts by passive diffusion in the cell membrane and not by active fast axoplasmic transport (Honig and Hume, 1989), were similar to that after cholera toxin labeling (data not shown).
Discussion
Since its discovery almost a decade ago as a proapoptotic member of the Bcl-2 gene family (Oltvai et al., 1993), Bax has been shown to be essential for the PCD of several populations of developing neurons (Deckwerth et al., 1996; Vekrellis et al., 1997; White et al., 1998; Lentz et al., 1999; Patel et al., 2000; Fan et al., 2001; Li et al., 2001). In addition to its role in the execution pathway during neuronal PCD, a related question that has received much less attention is the extent to which neurons rescued from PCD by Bax deletion can grow, differentiate, and function normally in vivo.
Absence of PCD but normal neuromuscular development in Bax KO mice
Using a variety of methods, we confirmed and extended a previous report that the PCD of sensory (DRG) and motor neurons is absent in Bax KO mice (White et al., 1998). Because the TUNEL technique used by White et al. (1998) may not detect neurons undergoing PCD by a nonapoptotic pathway (Oppenheim et al., 2001b; Yaginuma et al., 2001), we used additional criteria of cell death including morphological indices at the light- and electron-microscopic levels as well as caspase activation. Regardless of the technique used, we were unable to detect PCD of sensory or motor neurons. In addition, we found that the number of axons in the VR and DR of adult Bax KO mice animals was greatly increased, consistent with the complete rescue of these populations from PCD.
On the basis of a number of quantitative and histological criteria, the limb muscles of WT and Bax KO mice were indistinguishable, suggesting that neither endogenous Bax within muscle cells nor the excess number of proprioceptive DRG sensory neurons or MNs in the Bax KO mice altered muscle development. Similarly, when examined during the PCD period, comparable amounts of mRNAs for GDNF, CNTF, and BDNF were expressed in the limb muscles of Bax KO mice and WT littermates. Therefore, it is unlikely that the rescue of sensory or motor neurons in the Bax KO mice can be explained by an indirect effect on their targets. Although we did not attempt to quantify the number of neuromuscular synapses in WT versus Bax KO muscles, because we did not find any alterations in the size or shape of end plates and because, despite the presence of excess MNs, individual end plates were innervated by a single axon in both WT and Bax KO mice, neuromuscular innervation appears to be normal after Bax deletion. This was our first indication that the subpopulation of MNs rescued from PCD in the Bax KO may be unable to sustain target innervation.
Atrophic changes in the excess MNs rescued from PCD in Bax KO mice
We found that a subpopulation of the MNs in Bax KO mice apparently undergo a gradual atrophy between embryonic and postnatal stages. Because these atrophied MNs have little rough endoplasmic reticulum, which is the substrate for Nissl stains (Lieberman, 1974), they appear as Nissl-negative profiles in histological sections. Because the atrophy of cultured sympathetic neurons from Bax KO can be reversed by treatment with NGF (Deckwerth et al., 1996), we reasoned that the atrophy of a subpopulation of MNs in the Bax KO mice may also be attributable to trophic factor deprivation. Our observation that the treatment of Bax KO mice with GDNF can reduce MN atrophy is consistent with this idea. Furthermore, we found that axotomy-induced target deprivation in neonatal Bax KO mice fails to induce PCD, but increases the number of MNs that are atrophied (Sun and Oppenheim, 2003). Together, these data indicate that the atrophied MNs in the Bax KO may be unable to obtain sufficient target-derived trophic support to maintain normal growth and development. In fact, we find that, although all of the MNs in the Bax KO appear to initiate target innervation in the embryo, by the end of the PCD period on E19-P1 for spinal MNs and by P7-P14 for facial MNs, the subpopulation of atrophied MNs fail to maintain target innervation.
On the face of it, the atrophy and loss of innervation by the excess MNs in the Bax KO mice may appear inconsistent with the situation after the rescue of MNs by activity blockade. In both chick and mouse embryos, the loss of neuromuscular activity rescues virtually all of the MNs from PCD (Oppenheim et al., 2000b; Banks et al., 2001; Terrado et al., 2001; Misgeld et al., 2002; Brandon et al., 2003), yet the excess MNs grow and differentiate normally and maintain target innervation. However, in the case of activity blockade, MN axons branch profusely intramuscularly at the very onset of both innervation and PCD, resulting in increased access of MNs to target-derived trophic support (Oppenheim et al., 2000b), which promotes growth and differentiation and maintains innervation. It is likely this difference between MNs rescued by activity blockade versus by Bax deletion that is crucial for the occurrence of atrophy and loss of innervation in the Bax KO mice.
In striking contrast to MNs, the growth of other populations of spinal neurons appears to be less affected by Bax deletion. For example, DRG neurons remain Nissl positive, and although moderately reduced in size, they retain the cytological criteria for inclusion in cell counts at all stages of development. Whereas postnatally a subpopulation of Bax KO MNs are atrophied and Nissl negative, and therefore excluded from our cell counts (thus, MN numbers are similar in WT and Bax KO), we consistently found 150 -200% more DRG neurons in Bax KO mice at all of the time points examined. Therefore, although, as a population, Bax KO-rescued DRG neurons are somewhat smaller than normal, they appear considerably less affected than MNs. Recently, Patel et al. (2000) reported increased numbers of sensory fibers innervating central and peripheral targets at P0 in Bax KO mice, and they showed that the peripheral innervation is NGF dependent (Patel et al., 2000). We also find that, even in the adult, there are increased numbers of peripherin+ (sensory) axons in the distal sciatic nerve of Bax KO mice. Therefore, the excess sensory neurons in Bax KO mice may be able to maintain target innervation, which is likely to be responsible for the reduced atrophic phenotype of DRG neurons.
Postnatal application of GDNF rescues MNs from death and atrophy
Our observations indicate that neuromuscular development in Bax KO mice is normal, and that, despite the presence of excess MNs, hindlimb muscles can only support innervation by control numbers of MNs (i.e., approximately one-half of the MNs present after Bax deletion). From this perspective, it appears that the remaining MNs are unable to maintain innervation and thus undergo atrophy. We reasoned that, if these deficits were caused by insufficient target-derived trophic support, then treatment with exogenous trophic factors might be able to reverse the deficits. We chose GDNF, because it is a potent survival factor for embryonic MNs and is present in embryonic muscles and Schwann cells (Henderson et al., 1994; Oppenheim et al., 1995; Garces et al., 2000; Oppenheim et al., 2000a). In addition, GDNF appears to be involved in the regulation of neuromuscular development at multiple stages, including MN survival, axonal growth, and postnatal remodeling of innervation (Nguyen et al., 1998; Costantini and Isacson, 2000; Widmer et al., 2000; Keller-Peck et al., 2001; Linnarsson et al., 2001). Furthermore, in a separate study, we found that the loss of proximal axons from the atrophied MNs in the Bax KO could be prevented by putative endogenous regeneration signals after neonatal axotomy, and that the expression of GDNF is upregulated in the proximal sciatic nerve after axotomy (Sun and Oppenheim, 2003). For these reasons, GDNF appeared to be a good candidate as a trophic signal responsible for the regrowth of axons in this situation.
The postnatal injection of GDNF completely inhibited the late stage of normal PCD of facial and spinal MNs in WT mice. Because the PCD of mouse facial MNs occurs between E16 -P7, the application of GDNF was begun in the middle of the PCD period for facial MNs (P0). We found a 47% increase in MN number in WT mice, indicating that nearly all of the postnatal MN death was prevented by GDNF. This was surprising in that neither single nor combinations of exogenous neurotrophic factor(s) have been shown previously to completely block PCD (Oppenheim, 1996). Part of the reason for this striking effect may be that GDNF appears to be more effective during late stages of PCD. In the chick, application of GDNF in ovo at early stage (E5-E9) rescued 25% of spinal MNs from PCD, whereas treatment from E9 completely prevented PCD during this late stage in the cell death period (Oppenheim et al., 1995). GDNF has also been shown to completely prevent postnatal MN death in an in vitro spinal cord slice model (Rakowicz et al., 2002). We are currently examining the mechanisms by which GDNF has such a striking effect on postnatal MN survival.
In addition to preventing postnatal PCD in WT mice, GDNF also reverses the atrophy of excess MNs in the Bax KO. Approximately 47% more spinal MNs were Nissl positive and thus were now included in the cell counts of GDNF-treated Bax KO mice. The effect of GDNF on facial MNs in the Bax KO mice was even more profound in that virtually all of the MNs now met the criteria for inclusion in cell counts. Accordingly, the number of facial MNs in GDNF-treated Bax KO mice was similar to the number of MNs present before the onset of PCD on E15-E16. The soma size of GDNF-treated MNs was significantly larger than MNs in the saline-treated group of both WT and Bax KO mice, indicating that GDNF can induce the growth of both normal and atrophied MNs. Consistent with these observations, a growth-promoting effect of GDNF on injured MNs has also been reported (Oppenheim et al., 1995; Yan et al., 1995; Hottinger et al., 2000). We also observed many growth cone-like profiles in the ventral horn of the Bax KO spinal cord after treatment with GDNF, suggesting that GDNF may induce axonal or dendritic growth and synaptogenesis in the spinal cord. These data indicate that the subpopulation of MNs that would have normally been lost by PCD, but are rescued by Bax deletion, retain the capacity to regrow and reinnervate peripheral targets in response to trophic signals such as GDNF.
A model for explaining the development of excess MNs in Bax KO mice
At the peak period of PCD for spinal (E14.5) and facial MNs (P0), when the excess MNs are Nissl positive and can be readily counted in Bax KO mice, the overall size of MNs appears to be only moderately reduced compared with normal WT MNs. In contrast, by the end of the PCD period, the excess MNs become severely atrophied, and only control numbers of MNs that retain a normal size and morphology are present. A comparison of the smallest cell sizes in embryonic versus postnatal Bax KO mice suggests that the MNs actually atrophy versus exhibit arrested growth. We postulate that, in WT mice, as MNs undergo normal PCD and are eliminated from the competitive pool, there would be more target-derived trophic factors available for the survival and growth of the remaining MNs. However, because the subpopulation of MNs that would normally undergo PCD are retained after Bax deletion, competition among MNs for access to limiting amounts of trophic support from the target is increased, resulting in the retarded growth of a proportion of the surviving MNs and a gradual loss of innervation. A similar hypothesis has been proposed to explain the atrophy and reduced neurite growth of cultured Bax-deficient sympathetic neurons (Deckwerth et al., 1996) and the reduced cell size of mouse spinal MNs in vivo rescued from developmental PCD by overexpression of the antiapoptotic gene Bcl-2 (Zup et al., 2003).
What are initially only small differences in the access to target-derived trophic signals between MNs may affect the growth rate of their axons, which could further increase differences in their capacity to grow and acquire target-derived trophic support. The ability of GDNF to reverse this process in postnatal Bax KO animals (Fig. 9) suggests that GDNF affects primarily terminal growth and extension of axons, although we cannot exclude an effect on intercalary axonal growth (Goldberg, 2003). Whether the Bax KO MNs that reinnervate their targets after GDNF treatment can form functional synaptic contacts and contribute to motor behaviors is currently under investigation.
Figure 9.

A schematic model of the development of Bax KO neurons after the elimination of PCD. In wild-type animals (A), excess MNs are eliminated from competition during the PCD period, so that the remaining MNs can now obtain sufficient trophic support for normal growth and survival. In contrast, because excess MNs in Bax KO mice (B) survive and compete for trophic support, the growth of all of the MNs is initially retarded. However, as a result of increasing limb size, the excess MNs whose axonal growth is slower than that of normal MNs lose contact with targets, which causes them to atrophy. As a consequence of the loss of competition from the excess MNs, the remaining MNs grow, differentiate normally, and maintain synaptic contact with the target. Postnatal treatment with GDNF (C) is able to rescue MNs from atrophy and loss of innervation.
Footnotes
This work was supported by National Institutes of Health Grant NS20402 to R.W.O. We thank Carol Mansfield for her assistance.
Correspondence should be addressed to Dr. Ronald W. Oppenheim, Department of Neurobiology and Anatomy, Wake Forest University School of Medicine, Winston-Salem, NC 27157. E-mail: roppenhm@wfubmc.edu.
W. Sun's present address: Department of Anatomy, College of Medicine, Brain Korea 21, Korea University, 126-1 Anam-Dong, Sungbuk-Gu, Seoul, Korea 136-705.
Copyright © 2003 Society for Neuroscience 0270-6474/03/237298-13$15.00/0
References
- Ashwell KW, Watson CR ( 1983) The development of facial motoneurones in the mouse—neuronal death and the innervation of the facial muscles. J Embryol Exp Morphol 77: 117-141. [PubMed] [Google Scholar]
- Banks GB, Chan TNP, Barlett SE, Noakes PG ( 2001) Promotion of motoneuron survival and branching in rapsyn-deficient mice. J Comp Neurol 429: 156-165. [DOI] [PubMed] [Google Scholar]
- Bar-Peled O, Knudson M, Korsmeyer SJ, Rothstein JD ( 1999) Motoneuron degeneration is attenuated in Bax-deficient neurons in vitro. J Neurosci Res 55: 542-556. [DOI] [PubMed] [Google Scholar]
- Brandon EP, Lin W, D'Amour KA, Pizzo DP, Dominquez B, Sugiure Y, Thode S, Ko CP, Thal LJ, Gage FH, Lee KR ( 2003) Aberrant patterning of neuromuscular synapses in choline acetyltransferase-deficient mice. J Neurosci 23: 539-549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderó J, Prevette D, Mei X, Oakley RA, Li L, Milligan C, Houenou L, Burek M, Oppenheim RW ( 1998) Peripheral target regulation of the development and survival of spinal sensory and motor neurons in the chick embryo. J Neurosci 18: 356-370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carriedo SG, Yin H, Lamberta R, Weiss JH ( 1995) In vitro kainite injury to large SMI-32 + spinal neurons is Ca 2+ dependent. NeuroReport 6: 945-948. [DOI] [PubMed] [Google Scholar]
- Chu-Wang IW, Oppenheim RW ( 1978) Cell death of motoneurons in the chick embryo spinal cord. I. A light and electron microscopic study of naturally occurring and induced cell loss during development. J Comp Neurol 177: 33-57. [DOI] [PubMed] [Google Scholar]
- Clarke PG, Oppenheim RW ( 1995) Neuron death in vertebrate development: in vivo methods. Methods Cell Biol 46: 277-321. [PubMed] [Google Scholar]
- Costantini LC, Isacson O ( 2000) Immunophilin ligands and GDNF enhance neurite branching or elongation from developing dopamine neurons in culture. Exp Neurol 164: 60-70. [DOI] [PubMed] [Google Scholar]
- DeChiara TM, Vejsada R, Poueymirou WT, Acheson A, Suri C, Conover JC, Friedman B, McClain J, Pan L, Stahl N, Yip NY, Kato A, Yancopoulas GD ( 1995) Mice lacking the CNTF receptor, unlike mice lacking CNTF, exhibit profound motor neuron deficits at birth. Cell 83: 313-322. [DOI] [PubMed] [Google Scholar]
- Deckwerth TL, Elliott JL, Knudson CM, Johnson Jr EM, Snider WD, Korsmeyer SJ ( 1996) BAX is required for neuronal death after trophic factor deprivation and during development. Neuron 17: 401-411. [DOI] [PubMed] [Google Scholar]
- Dubois-Dauphin M, Frankowski H, Tsujimoto Y, Huarte J, Martinou JC ( 1994) Neonatal motoneurons overexpressing the bcl-2 protooncogene in transgenic mice are protected from axotomy-induced cell death. Proc Natl Acad Sci USA 91: 3309-3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan H, Favero M, Vogel MW ( 2001) Elimination of Bax expression in mice increases cerebellar Purkinje cell numbers but not the number of granule cells. J Comp Neurol 436: 82-91. [PubMed] [Google Scholar]
- Foelix RF, Oppenheim RW ( 1973) Synaptogenesis in the avian embryo. In: Studies on the development of the nervous system and behavior, Vol 1 (Gottlieb G, ed), pp 103-139. New York: Academic. [Google Scholar]
- Garces A, Haase G, Airaksinen MS, Livet J, Filippi P, deLapeyriere O ( 2000) GFRα1 is required for development of distinct subpopulations of motoneuron. J Neurosci 20: 4992-5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg JL ( 2003) How does an axon grow? Genes Dev 17: 941-958. [DOI] [PubMed] [Google Scholar]
- Gotow T, Tanaka J ( 1994) Phosphorylation of neurofilament-H subunits as related to arrangements of neurofilaments. J Neurosci Res 37: 691-713. [DOI] [PubMed] [Google Scholar]
- Green HJ, Reichmann H, Pette D ( 1982) A comparison of two ATPase based schemes for histochemical muscle fibre typing in various mammals. Histochemistry 76: 21-31. [DOI] [PubMed] [Google Scholar]
- Grieshammer U, Lewandoski M, Prevette D, Oppenheim RW, Martin GR ( 1998) Muscle-specific cell ablation conditional upon Cre-mediated DNA recombination in transgenic mice leads to massive spinal and cranial motoneuron loss. Dev Biol 197: 234-247. [DOI] [PubMed] [Google Scholar]
- Hamburger V ( 1975) Cell death in the development of the lateral motor column of the chick embryo. J Comp Neurol 160: 535-546. [DOI] [PubMed] [Google Scholar]
- Hamburger V, Levi-Montalcini R ( 1949) Proliferation, differentiation and degeneration in the spinal ganglia of the chick embryo under normal and experimental conditions. J Exp Zool 111: 457-501. [DOI] [PubMed] [Google Scholar]
- Henderson CE, Camu W, Mettling C, Gouin A, Poulsen K, Karihaloo M, Rullamas J, Evans T, McMahon SB, Armanini MP, Berkemeier L, Phillips HS, Rosenthal A ( 1993) Neurotrophins promote motor neuron survival and are present in embryonic limb bud. Nature 363: 266-270. [DOI] [PubMed] [Google Scholar]
- Henderson CE, Phillips HS, Pollock RA, Davies AM, Lemeulle C, Armanini M, Simmons L, Moffet B, Vandlen RA, Simpson LC, Moffet B, Vandlen RA, Koliatsor VE, Rosenthal A ( 1994) GDNF: a potent survival factor for motoneurons present in peripheral nerve and muscle. Science 266: 1062-1064. [DOI] [PubMed] [Google Scholar]
- Honig MG, Hume RI ( 1989) DiI and DiO: versatile fluorescent dyes for neuronal labeling and pathway tracing. Trends Neurosci 12: 333-341. [PubMed] [Google Scholar]
- Hottinger AF, Azzouz M, Deglon N, Aebischer P, Zurn AD ( 2000) Complete and long-term rescue of lesioned adult motoneurons by lentiviral-mediated expression of glial line-derived neurotrophic factor in the facial nucleus. J Neurosci 20: 5587-5593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue T, Tsui J, Wong N, Wong SY, Suzuki F, Kwok YN ( 1999) Expression of glial cell line-derived neurotrophic factor and its mRNA in the nigrostriatal pathway following MPTP treatment. Brain Res 826: 306-308. [DOI] [PubMed] [Google Scholar]
- Keller-Peck CR, Feng G, Sanes JR, Yan Q, Lichtman JW, Snider WD ( 2001) Glial cell line-derived neurotrophic factor administration in postnatal life results in motor unit enlargement and continuous synaptic remodeling at the neuromuscular junction. J Neurosci 21: 6136-6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudson CM, Tung KS, Tourtellotte WG, Brown GA, Korsmeyer SJ ( 1995) Bax-deficient mice with lymphoid hyperplasia and male germ cell death. Science 270: 96-99. [DOI] [PubMed] [Google Scholar]
- Koliatsos VE, Clatterbuck RE, Winslow JW, Cayouette MH, Price DL ( 1993) Evidence that brain-derived neurotrophic factor is a trophic factor for motor neurons in vivo. Neuron 10: 359-367. [DOI] [PubMed] [Google Scholar]
- Lance-Jones C ( 1982) Motoneuron cell death in the developing lumbar spinal cord of the mouse. Brain Res 256: 473-479. [DOI] [PubMed] [Google Scholar]
- Lentz SI, Knudson CM, Korsmeyer SJ, Snider WD ( 1999) Neurotrophins support the development of diverse sensory axon morphologies. J Neurosci 19: 1038-1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Oppenheim RW, Milligan CE ( 2001) Characterization of the execution pathway of developing motoneurons deprived of trophic support. J Neurobiol 46: 249-264. [DOI] [PubMed] [Google Scholar]
- Lieberman AR ( 1974) Some factors affecting retrograde neuronal responses to axonal lesions. In: Essays on the nervous system (Bellairs R and Gray EG, eds), pp 71-105. Clarendon, Oxford UP.
- Linnarsson S, Mikaels A, Baudet C, Ernfors P ( 2001) Activation by GDNF of a transcriptional program repressing neurite growth in dorsal root ganglia. Proc Natl Acad Sci USA 98: 14681-14686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo AC, Houenou LJ, Oppenheim RW ( 1995) Apoptosis in the nervous system: morphological features, methods, pathology, and prevention. Arch Histol Cytol 58: 139-149. [DOI] [PubMed] [Google Scholar]
- Martinou JC, Dubois-Dauphin M, Staple JK, Rodriguez I, Frankowski H, Missotten M, Albertini P, Talabot D, Catsicas S, Pietra C ( 1994) Overexpression of BCL-2 in transgenic mice protects neurons from naturally occurring cell death and experimental ischemia. Neuron 13: 1017-1030. [DOI] [PubMed] [Google Scholar]
- Misgeld T, Burgess RW, Lewis RM, Cunningham JM, Lichtman JW, Sanes JR ( 2002) Roles of neurotransmitter in synapse formation: development of neuromuscular junctions lacking choline acetyltransferase. Neuron 36: 635-648. [DOI] [PubMed] [Google Scholar]
- Nguyen QT, Parsadanian AS, Snider WD, Lichtman JW ( 1998) Hyperinnervation of neuromuscular junctions caused by GDNF overexpression in muscle. Science 279: 1725-1729. [DOI] [PubMed] [Google Scholar]
- Novak KD, Prevette D, Wang S, Gould TW, Oppenheim RW ( 2000) Hepatocyte growth factor/scatter factor is a neurotrophic survival factor for lumbar but not for other somatic motoneurons in the chick embryo. J Neurosci 20: 326-337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oltvai ZN, Milliman CL, Korsmeyer SJ ( 1993) Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell 74: 609-619. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW ( 1991) Cell death during development of the nervous system. Annu Rev Neurosci 14: 453-501. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW ( 1996) Neurotrophic survival molecules for motoneurons: an embarrassment of riches. Neuron 17: 195-197. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW, Houenou L, Pincon-Raymond M, Powell JA, Rieger F, Standish LJ ( 1986) The development of motoneurons in the embryonic spinal cord of the mouse mutant, muscular dysgenesis (mdg/mdg): survival, morphology, and biochemical differentiation. Dev Biol 114: 426-436. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW, Haverkamp LJ, Prevette D, McManaman JL, Appel SH ( 1988) Reduction of naturally occurring motoneuron death in vivo by a target-derived neurotrophic factor. Science 240: 919-922. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW, Prevette D, Yin QW, Collins F, MacDonald J ( 1991) Control of embryonic motoneuron survival in vivo by ciliary neurotrophic factor. Science 251: 1616-1618. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW, Yin QW, Prevette D, Yan Q ( 1992) Brain-derived neurotrophic factor rescues developing avian motoneurons from cell death. Nature 360: 755-757. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW, Houenou LJ, Johnson JE, Lin LF, Li L, Lo AC, Newsome AL, Prevette DM, Wang S ( 1995) Developing motor neurons rescued from programmed and axotomy-induced cell death by GDNF. Nature 373: 344-346. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW, Houenou LJ, Parsadanian AS, Prevette D, Snider WD, Shen L ( 2000a) Glial cell line-derived neurotrophic factor and developing mammalian motoneurons: regulation of programmed cell death among motoneuron subtypes. J Neurosci 20: 5001-5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheim RW, Prevette D, D'Costa A, Wang S, Houenou LJ, McIntosh JM ( 2000b) Reduction of neuromuscular activity is required for the rescue of motoneurons from naturally occurring cell death by nicotinic-blocking agents. J Neurosci 20: 6117-6124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheim RW, Wiese S, Prevette D, Armanini M, Wang S, Houenou LJ, Holtmann B, Gotz R, Pennica D, Sendtner M ( 2001a) Cardiotrophin-1, a muscle-derived cytokine, is required for the survival of subpopulations of developing motoneurons. J Neurosci 21: 1283-1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheim RW, Flavell RA, Vinsant S, Prevette D, Kuan CY, Rakic P ( 2001b) Programmed cell death of developing mammalian neurons after genetic deletion of caspases. J Neurosci 21: 4752-4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel TD, Jackman A, Rice FL, Kucera J, Snider WD ( 2000) Development of sensory neurons in the absence of NGF/TrkA signaling in vivo Neuron 25: 345-357. [DOI] [PubMed] [Google Scholar]
- Pennica D, Arce V, Swanson TA, Vejsada R, Pollock RA, Armanini M, Dudley K, Phillips HS, Rosenthal A, Kato AC, Henderson CE ( 1996) Cardiotrophin-1, a cytokine present in embryonic muscle, supports long-term survival of spinal motoneurons. Neuron 17: 63-74. [DOI] [PubMed] [Google Scholar]
- Phelan KA, Hollyday M ( 1991) Embryonic development and survival of brachial motoneurons projecting to muscleless chick wings. J Comp Neurol 311: 313-320. [DOI] [PubMed] [Google Scholar]
- Rakowicz WP, Staples CS, Milbrandt J, Brunstrom JE, Johnson Jr EM ( 2002) Glial cell line-derived neurotrophic factor promotes the survival of early postnatal spinal motor neurons in the lateral and medial motor columns in slice culture. J Neurosci 22: 3953-3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagot Y, Tan SA, Hammang JP, Aebischer P, Kato AC ( 1996) GDNF slows loss of motoneurons but not axonal degeneration or premature death of pmn/pmn mice. J Neurosci 16: 2335-2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagot Y, Rosse T, Vejsada R, Perrelet D, Kato AC ( 1998) Differential effects of neurotrophic factors on motoneuron retrograde labeling in a murine model of motoneuron disease. J Neurosci 18: 1132-1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeren-Wiemers N, Gerfin-Moser A ( 1993) A single protocol to detect transcripts of various types and expression levels in neural tissue and cultured cells: in situ hybridization using digoxigenin-labelled cRNA probes. Histochemistry 100: 431-440. [DOI] [PubMed] [Google Scholar]
- Sendtner M, Kreutzberg GW, Thoenen H ( 1990) Ciliary neurotrophic factor prevents the degeneration of motor neurons after axotomy. Nature 345: 440-441. [DOI] [PubMed] [Google Scholar]
- Sendtner M, Holtmann B, Kolbeck R, Thoenen H, Barde YA ( 1992) Brain-derived neurotrophic factor prevents the death of motoneurons in newborn rats after nerve section. Nature 360: 757-759. [DOI] [PubMed] [Google Scholar]
- Skoff RP, Hamburger V ( 1974) Fine structure of dendritic and axonal growth cones in embryonic chick spinal cord. J Comp Neurol 153: 107-148. [DOI] [PubMed] [Google Scholar]
- Sun W, Oppenheim RW ( 2003) Response of motoneurons to neonatal sciatic nerve atotomy in Bax-knockout mice. Mol Cell Neurosci, in press. [DOI] [PubMed]
- Terrado J, Burgess RW, DeChiara T, Yancopoulas G, Sanes JR, Kato AC ( 2001) Motoneuron survival is enhanced in the absence of neuromuscular junction formation in embryos. J Neurosci 21: 3144-3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirassa P, Manni L, Stenfors C, Lundeberg T, Aloe L ( 2000) RT-PCR ELISA method for the analysis of neurotrophin mRNA expression in brain and peripheral tissues. J Biotechnol 84: 259-272. [DOI] [PubMed] [Google Scholar]
- Vaughn JE, Sims T, Nakashims M ( 1977) A comparison of the early development of axodendritic and axosomatic synapses upon embryonic mouse spinal motor neurons. J Comp Neurol 175: 79-100. [DOI] [PubMed] [Google Scholar]
- Vekrellis K, McCarthy MJ, Watson A, Whitfield J, Rubin LL, Ham J ( 1997) Bax promotes neuronal cell death and is downregulated during the development of the nervous system. Development 124: 1239-1249. [DOI] [PubMed] [Google Scholar]
- White FA, Keller-Peck CR, Knudson CM, Korsmeyer SJ, Snider WD ( 1998) Widespread elimination of naturally occurring neuronal death in Bax-deficient mice. J Neurosci 18: 1428-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widmer HR, Schaller B, Meyer M, Seiler RW ( 2000) Glial cell line-derived neurotrophic factor stimulates the morphological differentiation of cultured ventral mesencephalic calbindin- and calretinin-expressing neurons. Exp Neurol 164: 71-81. [DOI] [PubMed] [Google Scholar]
- Yaginuma H, Shiraiwa N, Shimada T, Nishiyama K, Hong J, Wang S, Momoi T, Uchiyama Y, Oppenheim RW ( 2001) Caspase activity is involved in, but is dispensable for, early motoneuron death in the chick embryo cervical spinal cord. Mol Cell Neurosci 18: 168-182. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Henderson CE ( 1999) Patterns of programmed cell death in populations of developing spinal motoneurons in chicken, mouse, and rat. Dev Biol 214: 60-71. [DOI] [PubMed] [Google Scholar]
- Yan Q, Elliott J, Snider WD ( 1992) Brain-derived neurotrophic factor rescues spinal motor neurons from axotomy-induced cell death. Nature 360: 753-755. [DOI] [PubMed] [Google Scholar]
- Yan Q, Matheson C, Lopez OT ( 1995) In vivo neurotrophic effects of GDNF on neonatal and adult facial motor neurons. Nature 373: 341-344. [DOI] [PubMed] [Google Scholar]
- Zup SL, Carrier H, Waters EM, Tabor A, Bengston L, Rosen GJ, Simerly RB, Forger NG ( 2003) Overexpression of Bcl-2 reduces sex differences in neuron number in the brain and spinal cord. J Neurosci 23: 2357-2362. [DOI] [PMC free article] [PubMed] [Google Scholar]