

Abstract
Classically, 17β-estradiol (E2) is thought to control homeostatic functions such as reproduction, stress responses, feeding, sleep cycles, temperature regulation, and motivated behaviors through transcriptional events. Although it is increasingly evident that E2 can also rapidly activate kinase pathways to have multiple downstream actions in CNS neurons, the receptor(s) and the signal transduction pathways involved have not been identified. We discovered that E2 can alter μ-opioid and GABA neurotransmission rapidly through nontranscriptional events in hypothalamic GABA, proopiomelanocortin (POMC), and dopamine neurons. Therefore, we examined the effects of E2 in these neurons using whole-cell recording techniques in ovariectomized female guinea pigs. E2 reduced rapidly the potency of the GABAB receptor agonist baclofen to activate G-protein-coupled, inwardly rectifying K+ channels in hypothalamic neurons. These effects were mimicked by the membrane impermeant E2-BSA and selective estrogen receptor modulators, including a new diphenylacrylamide compound, STX, that does not bind to intracellular estrogen receptors α or β, suggesting that E2 acts through a unique membrane receptor. We characterized the coupling of this estrogen receptor to a Gαq-mediated activation of phospholipase C, leading to the upregulation of protein kinase Cδ and protein kinase A activity in these neurons. Moreover, using single-cell reverse transcription-PCR, we identified the critical transcripts, PKCδ and its downstream target adenylyl cyclase VII, for rapid, novel signaling of E2 in GABA, POMC, and dopamine neurons. Therefore, this unique Gq-coupled estrogen receptor may be involved in rapid signaling in hypothalamic neurons that are critical for normal homeostatic functions.
Keywords: GABAB receptor, GIRK, dopamine, GAD, POMC, SERMs
Introduction
It is becoming increasingly evident that the gonadal steroid hormone 17β-estradiol (E2) imparts a multifaceted influence over synaptic transmission in the mammalian CNS. Not only can estrogen alter synaptic responses via estrogen response element-driven target gene transcription, but E2 can also rapidly modulate cell-to-cell communication via membrane-initiated, rapid signaling events (for review, see Kelly and Wagner, 1999). These synaptic alterations are brought about via changes in the cellular responsiveness to the activation of various receptor systems (both G-protein-coupled and ionotropic) to their respective first messengers. E2 can alter the linkage of G-protein-coupled receptor (GPCR) systems such as opioid (both μ and κ), GABAB, and dopamine D2 receptors to their respective effector systems (Demotes-Mainard et al., 1990; Kelly et al., 1992; Takano et al., 1994; Wagner et al., 1994; Lagrange et al., 1996). In addition, E2 can act as an allosteric modulator of ionotropic receptors, such as 5-HT3 and nicotinic receptors (Wetzel et al., 1998; Paradiso et al., 2001), or by direct binding to subunits of ion channels, such as the β1 subunit of the maxi-K+ channel (Valverde et al., 1999). These fundamentally distinct signaling pathways give rise to a coordinated regulation by estrogen of complex physiological processes to maintain homeostasis in the mammal (McEwen, 2001).
The quintessential role of estrogen in the CNS is to transmit feedback information to gonadotropin-releasing hormone (GnRH) neurons that control the female reproductive cycle. Estrogen can alter GnRH neuronal activity directly (Kelly et al., 1984; Lagrange et al., 1995) or it can act upstream to alter synaptic input to GnRH neurons (Watson et al., 1992; Herbison et al., 1995, 2001; Sullivan et al., 1995; Simonian et al., 1999). Two of the major presynaptic target neurons of estrogen are the proopiomelanocortin (POMC) and GABA neurons, both of which provide a prominent synaptic input onto GnRH neurons (Morrell et al., 1985; Leranth et al., 1992; Herbison, 1997). Both opioid peptides and GABA inhibit GnRH output (Ferin et al., 1984; Mitsushima et al., 1996) and luteinizing hormone release (Ferin et al., 1984; Akema et al., 1990; Seltzer and Donoso, 1992; Jarry et al., 1995) from the anterior pituitary. Another target of the actions of estrogen is the arcuate dopamine (tuberoinfundibular) neurons that are located in the arcuate nucleus and project to the median eminence, in which they release dopamine into the portal circulation, which directly inhibits prolactin secretion from anterior pituitary lactotrophs (Neill, 1980; Björklund and Lindvall, 1984; Hökfelt et al., 1984).
μ-Opioid and GABAB receptors are linked to the same population of G-protein-coupled inwardly rectifying K+ (GIRK) channels in hypothalamic POMC, dopamine, and GABA neurons (Loose et al., 1990, 1991; Wagner et al., 2000). Activation of either μ-opioid or GABAB receptors elicits an outward K+ current that robustly hyperpolarizes hypothalamic neurons. However, maximum activation of either receptor occludes the response of the other receptor (Loose et al., 1991; Wagner et al., 1999, 2001). Interestingly, short-term exposure to estrogen reduces the potency of both μ-opioid and GABAB receptor agonists to activate GIRK channels in hypothalamic neurons (Lagrange et al., 1994, 1996, 1997). The underlying mechanism of this estrogen-induced decrease in the responsiveness of hypothalamic neurons to the μ-opioid and GABAB agonists is not known. Recent experiments have shown that selective protein kinase A (PKA) inhibitors can block the effects of estrogen, and PKA activators mimic the effects of estrogen on the coupling of μ-opioid receptor to GIRK (Lagrange et al., 1997). Therefore, in the present study, we characterized the estrogen-mediated rapid signaling pathway in hypothalamic neurons by using novel estrogen receptor (ER) ligands. In addition, by using specific protein kinase inhibitors and single-cell reverse transcription (scRT)-PCR, we found that this novel estrogen receptor is coupled to Gαq and activates a phospholipase C (PLC)-protein kinase Cδ-protein kinase A pathway. We conclude that stimulation of this pathway by binding of natural estrogen hormone and certain selective estrogen receptor modulators (SERMs) to the novel Gq-coupled estrogen receptor mediates the rapid steroid response in hypothalamic neurons.
Materials and Methods
Animals and treatments. All animal procedures described in this study are in accordance with institutional guidelines based on National Institutes of Health standards. Female Topeka guinea pigs (400-600 gm), bred in our institutional breeding facility, and female multicolor guinea pigs (400-500 gm; Elm Hill Breeding Labs, Chelmsford, MA) were used in these experiments. The guinea pigs were maintained under constant temperature (26°C) and light (on between 6:30 A.M. and 8:30 P.M.). Animals were housed individually, with food and water provided ad libitum. They were ovariectomized under ketamine-xylazine anesthesia (33 and 6 mg/kg, respectively, s.c.) 5-7 d before experimentation, and they were given sesame oil vehicle (0.1 ml, s.c.) 24 hr before experimentation. Serum estrogen concentrations were determined by radioimmunoassay (Wagner et al., 2001) from trunk blood collected on the day of experimentation and were <10 pg/ml. An additional group of animals (n = 6) were ovariectomized and, after 1 week, were injected with oil vehicle, estradiol benzoate (25 μg in oil), or STX (25 μg in oil) 24 hr before they were killed. The uteri were collected, weighed, and fixed in 4% paraformaldehyde for later histological analysis, which is not being reported in this paper.
Wild-type C57BL/6 mice in these studies were obtained from The Jackson Laboratory (Bar Harbor, ME). All animals were maintained under controlled temperature (25°C) and photoperiod conditions (14/10 hr light/dark cycle; lights on between 7:00 A.M. and 9:00 P.M.) with food and water ad libitum. Adult mice were ovariectomized under isoflurane anesthesia and allowed to recover for 1 week. At this time, the animals were injected daily for 2 d with oil vehicle, estradiol benzoate (1 μg), or STX (2 or 5 μg) and anesthetized and killed by decapitation after 24 hr. The uteri were collected, weighed, and fixed in 4% paraformaldehyde for later histological analysis, which is not being reported in this paper.
Drugs. All drugs were purchased from Calbiochem (La Jolla, CA) unless otherwise specified. Tetrodotoxin (TTX) (Alomone Labs, Jerusalem, Israel) was dissolved in Milli-Q H2O and further diluted with 0.1% acetic acid (final concentration, 1 mm), pH 4-5. 17β-Estradiol was purchased from Steraloids (Wilton, NH), recrystallized to ensure purity, and dissolved in 100% ethanol to a stock concentration of 1 mm. 17α-Estradiol (17α-E2; 1 mm; Steraloids), anti-estrogen (ICI 182,780; 10 mm; Tocris Cookson, Ballwin, MO), and the selective estrogen receptor modulators 4-OH-tamoxifen (10 mm; Steraloids), raloxifene (10 mm; Eli Lilly and Company, Indianapolis, IN), and STX (10 mm) were also dissolved in 100% ethanol. 17β-Estradiol 17-hemisuccinate: BSA (E2-BSA) (1 mm; Steraloids) was dissolved in H2O. The protein kinase A inhibitor H-89 dihydrochloride (10 mm), the protein kinase A activator forskolin (50 mm), the protein kinase C inhibitors bisindolylmaleimide I hydrochloride (BIS) (100 μm), Gö6976 (2 mm), and rottlerin (10 mm), the phospholipase C inhibitor U73122 (20 mm), the less active analog U73343 (20 mm), and the MEK1 [mitogen-activated protein (MAP) kinase kinase-1] inhibitor PD98059 (50 mm) were dissolved in DMSO. Protein kinase A inhibitory peptide 6-22 amide (1 mm), the protein kinase A inhibitor Rp-cAMPS (50 mm), and cholera toxin (CTX) A subunit (1 μg/μl) were dissolved in H2O. The Gq-binding protein designed to mimic the C terminus of the Gq α subunit and the Gs α-binding protein designed to mimic the C terminus of the Gs α subunit were synthesized by PeptidoGenic Research (Livermore, CA). The peptide sequence for Gq peptide was Ac-LGLNLKEYNLV-OH, and the peptide sequence for Gs peptide was CRMHLRQYELL. The peptides were also dissolved in H2O. 1,2-Bis-(o-aminophenoxyethane)-N,N,N′,N′-tetra-acetic acid (BAPTA) tetrasodium salt was dissolved in the internal solution at a 10 mm concentration. Aliquots of the stock solutions were stored as appropriate until needed.
Tissue preparation. On the day of experimentation, the animal was decapitated, its brain was removed from the skull, and the hypothalamus was dissected. The resultant hypothalamic block was mounted on a plastic cutting platform that was then secured in a vibratome well filled with ice-cold, oxygenated (95% O2, 5% CO2) artificial CSF (aCSF) (in mm: 124 NaCl, 26 NaHCO3, 10 dextrose, 10 HEPES, 5 KCl, 2.6 NaH2PO4, 2 MgSO4, and 1 CaCl2). Four coronal slices (350 μm) through the arcuate were cut. The slices were transferred to a multiwell auxiliary chamber containing oxygenated aCSF and kept there until electrophysiological recording after ∼2 hr.
Electrophysiology. Whole-cell patch recordings in voltage clamp were performed as described previously (Wagner et al., 2001). Slices were maintained briefly in a chamber perfused with warmed (35°C), oxygenated aCSF containing the same constituents and respective concentrations, except for CaCl2, which was raised to 2 mm. aCSF and all drug solutions were perfused via a peristaltic pump at a rate of 1.5 ml/min. Drug solutions were prepared in 20 ml syringes by diluting the appropriate stock solution with aCSF, and the flow was controlled via a three-way stopcock.
For whole-cell recordings, electrodes were fabricated from borosilicate glass (1.5 mm outer diameter; World Precision Instruments, Sarasota, FL). Resultant electrodes were then filled with an internal solution containing 0.5% biocytin and consisting of the following (in mm): 128 K+ gluconate, 10 NaCl, 1 MgCl2, 11 EGTA, 10 HEPES, 1.2 ATP, and 0.4 GTP (pH was adjusted to 7.3-7.4 with 1N KOH, 272-315 mOsm). Voltage pulses were amplified and passed through the electrode using an Axo-patch 1D preamplifier (Axon Instruments, Union City, CA). The resultant current deflections were monitored using a digital oscilloscope (Tektronix 2230; Tektronix, Beaverton, OR). After the reduction of the current deflection, negative pressure was applied via a 5 ml syringe connected by polyethylene tubing to the electrode to form a seal (>1 GΩ). After formation of a seal, intracellular access was achieved by suction, followed by perfusion with 1 μm TTX for at least 4-6 min to block spontaneous firing and synaptic potentials before applying the GABAB receptor agonist baclofen (see Fig. 1). All of the responses to baclofen were measured in voltage clamp as outward currents (Vhold = -60 mV), and only those cells that showed <10% change in access resistance (access resistances ranged from 20 to 30 MΩ) throughout the recording were included in this study. Membrane currents underwent analog-to-digital conversion via a Digidata 1200 interface coupled to pClamp 7.0 (Axon Instruments). Low-pass filtering of the currents was conducted at a frequency of 2 kHz. The liquid junction potential was -10 mV and was corrected for in subsequent data analysis.
Figure 1.

17β-Estradiol and SERMs attenuate the GABAB response in hypothalamic neurons. A, Schematic showing the protocol for drug administration in the whole-cell patch voltage-clamp experiments (Vhold, -60 mV). After seals were formed and the whole-cell configuration was obtained, slices were perfused with TTX (1 μm) for 5 min. The first GABAB receptor-mediated response was generated by perfusing baclofen (at EC50 concentration of 5 μm) until a steady-state outward current was obtained (R1). After drug washout, the current returned to its predrug resting level. The cells were then treated with E2 and/or other drugs for 15 min, baclofen (5 μm) was perfused again, and R2 was measured. The effects of estrogen or other drugs on the baclofen response are expressed as a percentage of R2 over R1. B, An example of a biocytin-filled neuron after whole-cell patch-clamp recording for 13 min, illustrating the extent of the biocytin labeling. The slice was fixed immediately after the recording. C, The prebaclofen and postbaclofen (5 μm) I/V relationships from a dopamine neuron. The reversal potential for the outward current was close to the predicted equilibrium potential for potassium. D, The prebaclofen and postbaclofen I/V relationships after E2 treatment in another cell, illustrating the same reversal potential for the baclofen response. I/Vs were done before and after each drug treatment, and none of the drug treatments had any effect on the reversal potential for the baclofen response.
Post hoc identification of hypothalamic arcuate neurons. After electrophysiological recording, the slices were fixed with 4% paraformaldehyde in Sorensen's phosphate buffer, pH 7.4, for 120 min, immersed overnight in 20% sucrose dissolved in Sorensen's buffer, and then frozen in OCT embedding medium and prepared for immunocytochemistry as described previously (Kelly and Rønnekleiv, 1994). Briefly, coronal sections (20 μm) were cut on a cryostat (model 1720 Digital Cryostat; Leitz, Wetzlar, Germany) and mounted on Fisher SuperFrost Plus slides. Sections were washed for 5 min with 0.1 m sodium phosphate buffer, pH 7.4, and then streptavidin-Cy2 (1:1000; Jackson ImmunoResearch, West Grove, PA) was applied for 2 hr. The reaction was terminated by washing with buffer. The slices were scanned for the injected neuron with a Nikon (Melville, NY) Eclipse 800 fluorescence microscope. After localization of the biocytin-filled neurons, the slides containing the appropriate sections were processed for the presence of tyrosine hydroxylase (TH) or β-endorphin using fluorescence immunohistochemistry as described previously (Kelly and Rønnekleiv, 1994). Briefly, the sections with the biocytin-identified neurons were incubated overnight with a monoclonal TH antibody at 1:10,000 (Diasorin, Stillwater, MN) or with a polyclonal β-endorphin antibody at 1:5000 (Dave et al., 1985) and washed in 0.1 m phosphate buffer, followed by incubation with a goat anti-mouse IgG-Cy3 at 1:500 or donkey anti-rabbit IgG-Cy3 at 1:500, respectively (Jackson ImmunoResearch). The sections were washed with sodium phosphate buffer, and coverslips were applied using a glycerolglycine buffer containing 5% N-propylgallate. Immunostained cells were photographed using a Nikon microscope.
Estrogen receptor binding assays. The relative binding affinity of compounds for ERα and ERβ was determined using a spin column assay with commercially available full-length forms of both ERα and ERβ (PanVera, Madison, WI). Receptor was added to a final concentration of 15 nm to a solution containing 10 mm Tris, pH 7.5, 10% glycerol, 2 mm DTT, 1 mg/ml BSA, and 3 nm [2,4,6,7,16,17-3H]estradiol at 4°C. Next, 100 μl of the solution was added to 1 μl of the ligand in ethanol, mixed gently by pipetting, and incubated at 4°C overnight. The mixture was then applied to a micro spin column containing G-25 Sephadex (Harvard Apparatus, Holliston, MA) equilibrated in binding buffer (minus tritiated estradiol) according to the instructions of the manufacturer. Bound estradiol was separated from free ligand by spinning at 2000 × g for 4 min at room temperature. The filtrate was then added to 2.5 ml of scintillant and counted in a liquid scintillation counter. A binding curve was fitted using a single binding site competition model with the Prism statistical analysis software package (GraphPad Software, San Diego, CA). The SD was determined to be <0.2 log units from the EC50 value. Percentage relative binding affinity was then determined by dividing the IC50 determined for unlabeled estradiol by the ligand IC50 and multiplying by 100.
Dispersed single-cell RT-PCR. Guinea pig 350 μm coronal hypothalamic slices were cut on a vibratome from caudal to rostral and placed in an auxiliary chamber containing oxygenated aCSF. The slices were allowed to recover for 1-2 hr in the chamber before dispersion. The arcuate nucleus of the hypothalamus was microdissected and incubated in 2-3 ml of HBSS containing the following: 1.26 mm CaCl2, 1 mm MgSO4, 5.37 mm KCl, 0.44 mm KH2PO4, 136.89 mm NaCl, 0.34 mm Na2HPO4, 5.55 mmd-glucose, and 15 mm HEPES in DEPC-treated water, pH 7.3 (300 mOsm), containing 1 mg/ml protease XIV (Sigma, St. Louis, MO) for ∼15 min at 37°C. The tissue was then washed four times in 1 vol of low-calcium aCSF and two times in HBSS. The cells were isolated by trituration with flame-polished Pasteur pipettes, dispersed on a dish, and perfused continuously with HBSS at a rate of 1.5 ml/min. Cells were visualized using a Nikon inverted microscope, and individual neurons were patched and harvested into the patch pipette by applying negative pressure. The content of the pipette was expelled into a siliconized microcentrifuge tube containing 5 μl of the following solution: 0.5 μl of 10 × buffer (100 mm Tris-HCl, 500 mm KCl, 1% Triton X-100; Promega, Madison, WI), 15 U of RNasin (Promega), 0.5 μl of 100 mm DTT, and DEPC-treated water.
In addition, hypothalamic tissue was homogenized, and total RNA was extracted using the RNeasy kit (Qiagen, Valencia, CA) according to the protocol of the manufacturer. The harvested cell solution and 25 ng of hypothalamic total RNA in 1 μl were denatured for 5 min at 65°C and cooled on ice for 5 min, and then single-stranded cDNA was synthesized from cellular RNA by adding 50 U of murine leukemia virus reverse transcriptase (Applied Biosystems, Foster City, CA), 1.5 μl of 10 × buffer, 2 mm MgCl2, 0.2 μl of deoxynucleotide triphosphates (dNTPs), 15 U of RNasin, 10 mm DTT, 100 ng f random hexamers, and DEPC-treated water to a final volume of 20 μl. Cells and tissue RNA used as negative controls were processed as described above but without reverse transcriptase. The reaction mixtures were incubated at 42°C for 60 min, denatured at 99°C for 5 min, and cooled on ice for 5 min.
PCR was performed using 3 μl of cDNA template from each RT reaction in a 30 μl of PCR reaction volume containing the following: 3 μl of 10 × buffer, 2.4 μl of MgCl2 [2 mm final concentration for TH, POMC, GABAB receptor 2 (GABAB R2), PKCδ, adenylyl cyclase VII (AC VII), and glyceraldehyde-3-phospate dehydrogenase (GAPDH)], or 3.6 μl of MgCl2 (3 mm final concentration for GAD), 0.2 mm dNTPs, 0.2 μm forward and reverse primers, 2 U of Taq DNA polymerase (Promega), and 0.22 μg of TaqStart antibody (Clontech, Palo Alto, CA). Taq DNA polymerase and TaqStart antibody were combined and incubated at room temperature for 5 min, and the remainder of the reaction contents were added to the tube and incubated at 94°C for 2 min. Then, each reaction went through 60 cycles (35 cycles for GAPDH) of amplification according to the following protocols: 94°C, 45 sec; 55°C (GAD), 57°C (PKCδ), 58°C (GABAB-R2), 60°C (TH and adenylyl cyclase VII), 61°C (POMC), 63°C (GAPDH) 45 sec; 72°C, 1 min 10 sec; with a final 72°C extension for 5 min. Ten microliters of the PCR products were visualized with ethidium bromide on a 1.5% agarose gel.
All of the primers were synthesized by Invitrogen (Carlsbad, CA) and were as follows: guinea pig GAD65, 207 bp product, forward primer 5′-GGCTCTGGTGATGGAATA-3′, reverse primer 5′-CAGAATCACGCTGTCTGTT-3′; guinea pig TH, 223 bp product, forward primer 5′-TCCACGTTATACTGGTTCAC-3′, reverse primer 5′-TTGCATCACTGAAGCTCTC-3′; guinea pig GABAB-R2, 241 bp product, forward primer 5′-TGTTTGTGCCAAAGCTCATC-3′ reverse primer 5′-GTGTCTTGCAGTTGCATAGT-3′; guinea pig POMC (GenBank accession number S78260), 344 bp product, forward primer (bases 40-60) 5′-CTGGCCTTGCTGCTTCAGAT-3′ reverse primer (bases 383-363) 5′-ATGGAGTAGGAGCGCTTGTC-3′; guinea pig GAPDH, 212 bp product (GenBank accession number CPU51572), forward primer 5′CATCCACTGGTGCTGCCAAG-3′, reverse primer 5′-GTCCTCGGTGTAGCCCAAGA-3′; human protein kinase Cδ, 251 bp product (GenBank accession number L07861) forward primer (bases 1127-1147) 5′-AAAGGCAGCTTCGGGAAGGT-3′, reverse primer (bases 1377-1357) 5′-TGGATGTGGTACATCAGGTC-3′; and guinea pig adenylyl cyclase VII, 235 bp product, forward primer 5′-CTGTTCGGCAAGTTTGACCAG-3′, reverse primer 5′-TGACGCCACACAGCACATT-3′.
Statistical analyses. Comparisons between groups were performed using a one-way ANOVA [with post hoc (Newman-Keuls) paired analysis]. Differences were considered statistically significant if the probability of error was <5%.
Results
E2 and SERMs rapidly attenuate the GABAB response in hypothalamic dopamine and POMC neurons
Whole-cell recordings were made in arcuate neurons (n = 195) from ovariectomized female guinea pigs (Fig. 1). A subgroup of these neurons (n = 55) was identified using dual-labeling immunocytochemistry (Fig. 2). This revealed that 41% of the cells were TH positive (i.e., dopamine neurons) and 39% were β-endorphin positive (i.e., POMC neurons). Moreover, on the basis of dual-immunocytochemical staining and in situ hybridization for GAD65, a subgroup of arcuate dopamine neurons coexpress GABA (O. K. Rønnekleiv, unpublished findings), which was substantiated by the scRT-PCR data (see below). For the electrophysiology analysis, only cells with gigaohm or better seals were included in this study. The mean resting membrane potential was -54.3 ± 0.4 mV at a 0 pA holding current, and the mean input resistance was 1.9 ± 0.3 GΩ. Moreover, 50% of A12 dopamine neurons exhibited a T-type Ca2+ current and a hyperpolarization-activated cation current (Ih), as we described previously (Loose et al., 1990). Seventy-one percent of the POMC neurons exhibited Ih and a transient outward K+ current (IA), as we described previously (Kelly et al., 1990). Therefore, the passive membrane properties measured with whole-cell patch recording are similar to what we described using single-electrode voltage-clamp recordings (Kelly et al., 1990; Loose et al., 1990).
Figure 2.

Double labeling of dopamine and POMC neurons. Representative dopamine (A, B) and POMC (C, D) neurons that responded to estrogen. Arcuate neurons were filled with biocytin during the whole-cell recording. A, Biocytin-streptavidin-Cy2 labeling of fusiform arcuate neuron. Scale bar, 20 μm. B, Immunocytochemical staining of TH in the same neuron (arrow). C, Biocytin-streptavidin-Cy2 labeling of a small pyramidal arcuate neuron. Scale bar, 20 μm. D, Immunocytochemical staining of β-endorphin in the same neuron (arrow).
We used the whole-cell recording method to measure the rapid effects of E2 on the activation of the GIRK conductance by the GABAB receptor agonist baclofen. We showed previously that E2 rapidly attenuates both μ-opioid and GABAB receptor-mediated responses in hypothalamic arcuate neurons (Lagrange et al., 1994, 1996, 1997). Therefore, for measuring E2 modulation of the GABAB response, we used an EC50 concentration (5 μm) of baclofen and the protocol depicted in Figure 1. A robust outward current was measured in response to baclofen that subsided after washout (Fig. 3A,H). The application of baclofen 20 min later elicited the same robust response, suggesting that desensitization and rundown were not occurring in response to successive applications of 5 μm baclofen. However, if E2 (100 nm) was applied during the interim period (i.e., after the washout of the first application of baclofen), there was a significant (p < 0.005) decrease of 41% in the response to a second application of baclofen (Fig. 3B,H). Current-voltage relationships generated before and during the application of 100 nm E2 showed that this steroid did not change the reversal potential for the baclofen-mediated response: control Ebaclofen, -88.8 ± 3.6 mV, n = 13; versus after E2Ebaclofen, -85.4 ± 3.9 mV, n = 12 (Fig. 1C,D). The effects of E2 were stereospecific such that the biologically inactive stereoisomer 17α-estradiol (100 nm) had no effect on the baclofen response (Fig. 3C,H). Furthermore, the effects of E2 were blocked by the anti-estrogen ICI 182,780 when coperfused with E2 (Fig. 3H). Treatment with ICI 182,780 alone had no effect on the baclofen response (data not shown).
Figure 3.

Estrogen and SERMs rapidly attenuate the GABAB response. A-F, Representative traces of the GABAB responses before and after steroid treatment. Experiments were conducted as described in Figure 1. G, The chemical structures of E2 and the SERMs. H, Bar graphs summarizing the effects of E2 and SERMs on the baclofen response. E2 (100 nm) attenuated the GABAB receptor-mediated outward current by 41%. The inhibitory effects of E2 on the baclofen response were blocked by the estrogen receptor antagonist ICI 182,780 (1 μm). E2-BSA (100 nm), 4-OH tamoxifen (1 μm), raloxifene (1 μm), GW-5638 (1 μm), and STX (10 nm) also inhibited the baclofen response; however, 17α-E2 (1 μm) had no effect. Error bars represent the mean ± SEM of 4-11 cells tested per group. *p < 0.05, **p < 0.01, and ***p < 0.005, versus vehicle control group.
We next investigated the cellular localization of this estrogen receptor using the membrane-impermeable estrogen conjugate E2-BSA. Interestingly, E2-BSA (100 nm) was fully efficacious in inhibiting the baclofen response, indicating that this estrogen receptor-mediated response is initiated at the plasma membrane (Fig. 3D,H). We checked the integrity of the E2-BSA preparation by performing an E2 radioimmunoassay of the slice perfusate. We found no unbound E2 in the media (data not shown), indicating that E2-BSA conjugate did not contain contamination-free E2.
We also characterized the estrogen receptor by using several SERMs. Tamoxifen (1 μm) was inactive (p > 0.05 vs control) and did not attenuate the effects of E2 on the baclofen activation of GIRK (R2/R1 for E2, 58.6 ± 3.4%, n = 10; vs tamoxifen plus E2, 60.4 ± 6.6%, n = 5). However, 4-OH tamoxifen (1 μm) did partially mimic the actions of E2 by blocking the baclofen response by 25% (Fig. 3E,H). Because 4-OH tamoxifen always exists as an E/Z mixture of olefin isomers (Katzenellenbogen et al., 1985), we suspect that only one of the isomers is active at mediating this novel estrogen response. Raloxifene (1 μm), another SERM with a hydroxylated aromatic ring, mimicked completely the actions of E2 in terms of efficacy in the suppression of the baclofen response (Fig. 3H). In contrast, the nonhydroxylated SERM GW-5638, which structurally resembles the triphenylethylene core of tamoxifen, was found to be significantly more efficacious than E2 at inhibiting GIRK channel activation by baclofen (Fig. 3H).
STX is a SERM devoid of nuclear ER activity that selectively attenuates rapid responses
All of the above-mentioned compounds are high-affinity ligands for the nuclear estrogen receptors (Table 1), which complicates the interpretation of the observed pharmacology and makes it difficult to exclude unequivocally a role for nuclear ERs. Therefore, we next tested a novel diphenylacrylamide compound (STX) (Fig. 3G), which is a close structural analog of 4-OH tamoxifen. However, unlike 4-OH tamoxifen, STX is geometrically stable and does not exist as a mixture of E/Z olefin isomers. An additional distinction between STX and other SERMs is that STX has an ∼1 million-fold reduced binding affinity for the nuclear ERα or ERβ compared with that of E2 (Table 1). In addition, this compound has no uterotropic actions, even at five times the dose of E2 (Fig. 4), confirming in vivo that STX has no 4-OH tamoxifen-like estrogenic activity mediated by the nuclear ERs. However, in the whole-cell electrophysiological assay, 10 nm STX was as efficacious as 100 nm E2 in attenuating the GABAB response (Fig. 3F,H).
Table 1.
Relative binding affinities of ligands to full-length ERα or ERβ
|
|
RBA |
||
|---|---|---|---|
| Ligand
|
ERα
|
ERβ
|
|
| 17β-Estradiol | 100 | 100 | |
| 4-Hydroxytamoxifen | 36 | 43 | |
| Raloxifene | 34 | 76 | |
| GW-5638
|
5
|
8
|
|
| STX | 4.3E-6 | 9.0E-6 | |
Relative binding affinities (RBA) are expressed as a percentage of the potency of 17β-estradiol. Under the experimental conditions described in Materials and Methods, 17β-estradiol was found to have an IC50 of 5 nm for ERα and 3 nm for ERβ.
Figure 4.

Uteri are enlarged after estradiol treatment but not after STX or oil vehicle treatment. After a 48 hr treatment period, the uteri of wild-type C57BL/6 mice were collected and examined. A, In E2-treated mice, there was a noticeable increase in uterine size after estradiol benzoate (EB) compared with oil vehicle or STX treatment. B, Bar graph shows the uterine weights. **p < 0.01 versus oil-treated females (n = 3-5 mice per group).
The rapid effect of E2 on the GABAB response involves protein kinase A
We next examined the involvement of specific signaling proteins in the E2-mediated modulation of GABAB. If activation of the PKA pathway is involved, then the effect of E2 on GABAB responses should be blocked by inhibiting PKA and mimicked by stimulating PKA. To test this, we applied selective PKA activators and inhibitors. As shown in Figure 5, A and E, forskolin (10 μm) could mimic the actions of E2 to attenuate the GABAB response. However, the specific PKA inhibitor H-89 (10 μm) blocked the E2-induced suppression of the GABAB response (Fig. 5B,E). To confirm the involvement of PKA in E2 modulation of GABAB responses further, we dialyzed neurons with the specific PKA-inhibitory peptide PKI (PKA inhibitor 6-22 amide, 20 μm) or the nonhydrolyzable cAMP analog Rp-cAMPS (200 μm) that blocks PKA activation. After ∼15 min of dialysis with PKI or Rp-cAMPS, the E2-induced reduction of the GABAB response was abolished (Fig. 5C,E). CTX, which is a bacterial exotoxin secreted by vibrio cholerae, elevates intracellular cAMP levels in a variety of tissues by ADP ribosylating the G-protein Gs, thereby stimulating adenylyl cyclase activity in an apparently irreversible manner. Intracellular dialysis with the active unit of CTX into individual cells occluded the rapid inhibition of GABAB response by estrogen (Fig. 5D,E). These results indicate that the suppression of the GABAB response by E2 requires the activation of PKA.
Figure 5.
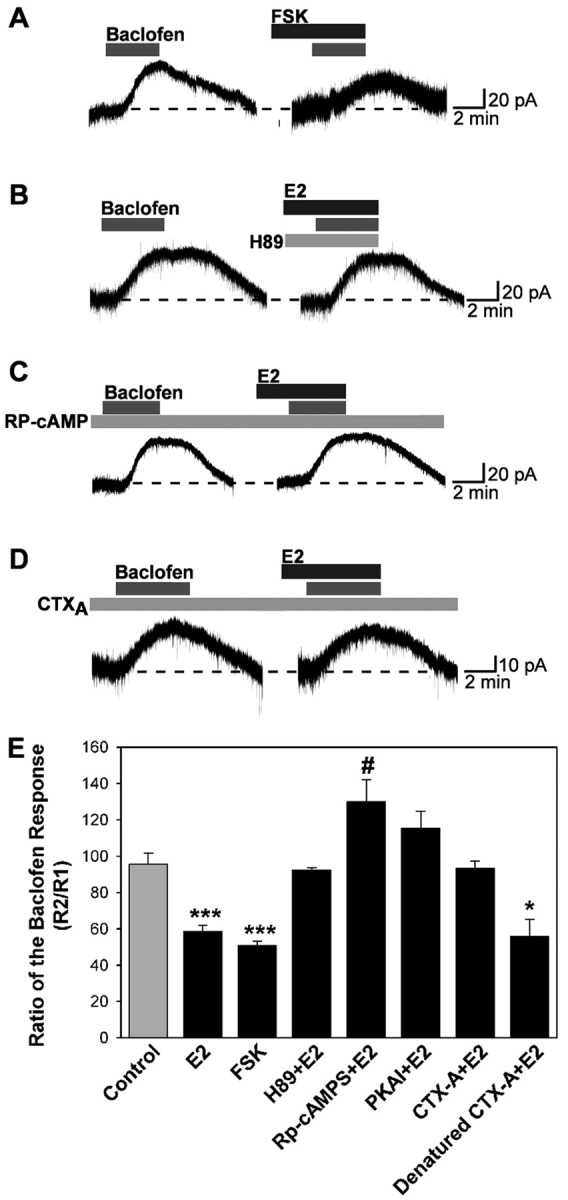
E2 attenuation of the GABAB response involves protein kinase A. A-D, Representative traces of the baclofen responses in the presence of PKA activators or inhibitors. Experiments were conducted as described in Figure 1, except that cells were dialyzed for 15 min before the first baclofen application with Rp-cAMPS (200 μm), PKA inhibitor 6-22 amide (PKAI; 20 μm), CTX A subunit (CTX-A; 10 μg/ml), or denatured CTX A subunit (10 μg/ml, 100°C, 10 min). H-89 (10 μm) and forskolin (FSK; 10 μm) were bath perfused. E, Bar graphs summarizing the effects of PKA drugs. The PKA activator forskolin could mimic the effects of E2, and the specific PKA inhibitors H-89 (10 μm), Rp-cAMPS, and PKAI could block the effects. CTX A subunit could occlude the attenuation of the baclofen response by E2; however, the denatured CTX A subunit could not. Error bars represent the mean ± SEM of 4-11 cells tested per group. *p < 0.05, #p < 0.05, and ***p < 0.005 versus vehicle control; CTX A subunit plus E2 versus denatured CTX A subunit plus E2, p < 0.05.
Attenuation of the GABAB response involves protein kinase Cδ
We next examined whether activation of PKC is also critical for E2 modulation of the GABAB response using several selective PKC inhibitors. The first, bisindolymaleimide, is a selective inhibitor of PKC that does not distinguish among the conventional, novel, and atypical isoforms of PKC. The second, Gö6976, is a selective inhibitor of the conventional PKC isoforms (Martiny-Baron et al., 1993; Way et al., 2000). Treatment of neurons with BIS nearly eliminated the effects of E2 (Fig. 6A,E). In contrast, Gö6976 treatment was without effect (Fig. 6E). Indeed, when similar experiments were performed after replacing intracellular EGTA with 10 mm BAPTA, a calcium buffer with similar Ca2+ affinity as EGTA but a much faster on rate, the estrogen inhibition of the GABAB response was still observed (Fig. 6B,E). Because conventional isoforms of PKC are unlikely to be active with this level of calcium buffering, these results also support a role for a Ca2+-independent, novel PKC isoform in mediating the effects of estrogen. Finally, the selective PKCδ inhibitor rottlerin (5 μm) completely blocked the ability of E2 to inhibit the GABAB response in hypothalamic neurons (Fig. 6C,E).
Figure 6.
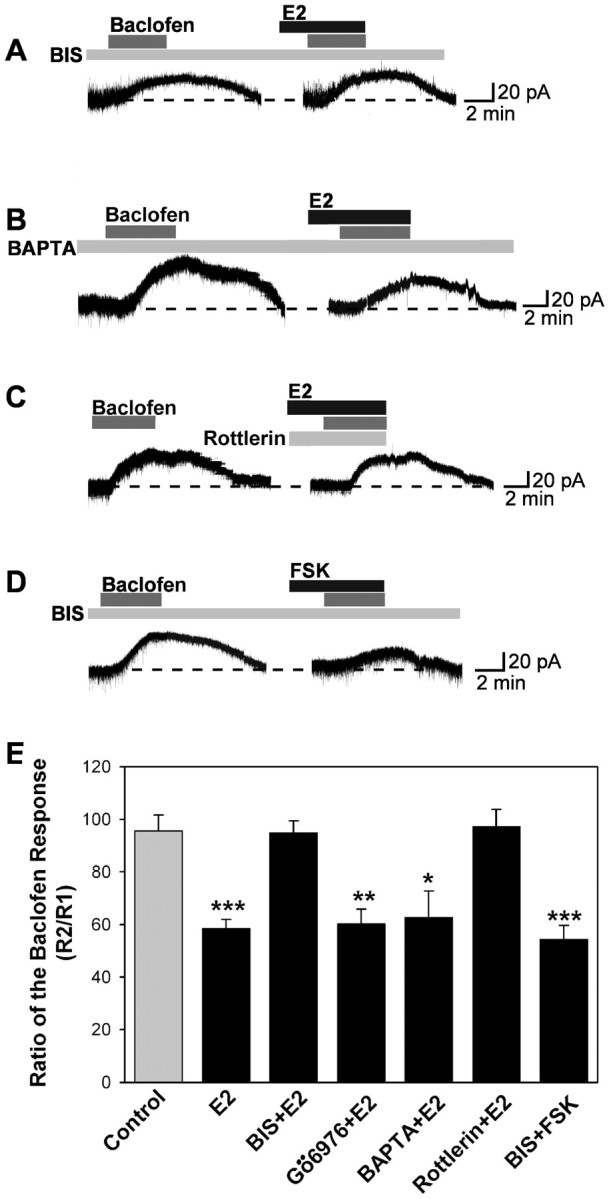
E2 attenuation of the GABAB response involves protein kinase Cδ. A-D, Representative traces of the baclofen responses in the presence of PKC inhibitors. Experiments were conducted as described in Figure 1. For the BIS or fast Ca2+ chelator BAPTA, cells were dialyzed for 15 min before baclofen application with BAPTA (replacing the 11 mm EGTA with 10 mm BAPTA) or BIS (100 nm), which were included in the patch pipette solution. E, Bar graph summarizing the effects of PKC inhibitors. The broad PKC inhibitor BIS and a selective PKCδ inhibitor rottlerin (5 μm) could reverse, but the selective inhibitor of conventional PKC isoforms Gö6976 (2 μm) or BAPTA could not block the attenuation of the baclofen response by E2. BIS could not block the attenuation of the baclofen response by forskolin (FSK; 10 μm). Error bars represent the mean ± SEM of 4-11 cells tested per group. *p < 0.05, **p < 0.01, and ***p < 0.005 versus vehicle control.
Inhibition of the GABAB response by E2 involves Gαq
Although the specific PKC inhibitor BIS blocked the E2 effect, forskolin (10 μm) was found to mimic the effects of estrogen in the presence of BIS blockade (Fig. 6D,E). This indicated that the action of PKC is upstream of the activation of PKA. Therefore, we focused on pathways upstream of PKC to elucidate further the E2-mediated signaling pathway. To examine whether the estrogen receptor-mediated inhibition of the GABAB response depended on the activation of Gαq, arcuate neurons were dialyzed with a peptide (11 amino acids) that mimics the C-terminal binding site of Gαq (Akhter et al., 1998). This peptide blocks the interaction between G-protein-coupled receptors and Gαq proteins. In cells dialyzed with this peptide (200 μm), the E2-mediated reduction of the GABAB response was blocked significantly (Fig. 7A,E) compared with cells dialyzed with a control peptide (11 amino acids) that mimics the C-terminal domain of Gαs (Fig. 7B,E).
Figure 7.
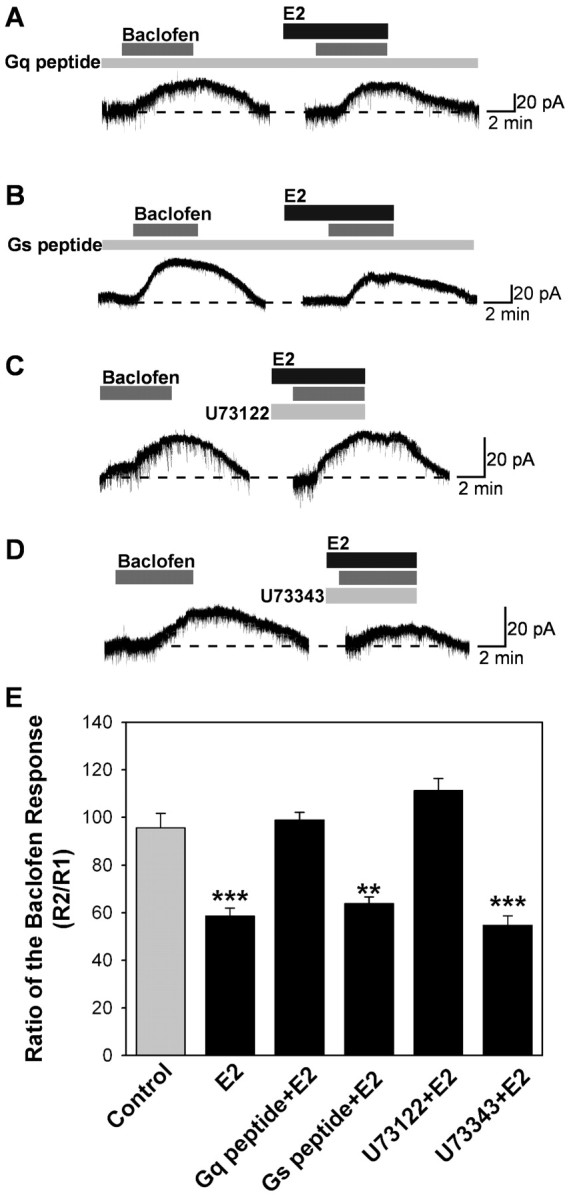
E2 attenuation of the GABAB response depends on activation of Gαq, and PKC activation is in the upstream of PKA. A-D, Representative traces of the baclofen responses in the presence of PLC and Gαq inhibitors. Experiments were conducted as described in Figure 1. Cells were dialyzed for 15 min before baclofen application with Gαq (200 μm) or Gαs peptide (200 μm). E, Bar graphs summarizing the effects of PLC and Gq peptide. The PLC inhibitor U73122 (10 μm) and Gαq peptide could inhibit, but the inactive analog of U73122, U73343 (10 μm), and Gαs peptide could not block the attenuation of the baclofen response by E2. Error bars represent the mean ± SEM of 4-11 cells tested per group. **p < 0.01 and ***p < 0.005 versus vehicle control; U73122 plus E2 versus U73343 plus E2, p < 0.05; Gq peptide plus E2 versus Gs peptide plus E2, p < 0.05.
In light of these results for a primary role for Gαq in E2-mediated rapid inhibition, we tested whether the activation of PLC, a well known Gαq effector, might also play a role. To determine whether the activation of PLCβ is required for the estrogen-induced inhibition of GABAB response, neurons were treated with the broad-spectrum PLC inhibitor U73122 (10 μm). U73122 (10 μm) was perfused in the extracellular bathing media. Under these conditions, the estrogen-mediated reduction of GABAB response was blocked (Fig. 7C,E), whereas the less active PLC inhibitor U73343 at the same concentration had no effect (Fig. 7D,E).
The attenuation of the GABAB response does not involve MAP kinase
Recent studies have shown that 17β-E2 rapidly activates the MAP kinase pathway in primary neuronal cortical cultures and in organotypic cerebrocortical explant cultures (Watters et al., 1997; Singh et al., 1999, 2000). We therefore tested whether inhibition of MAP kinase activity could prevent estrogen modulation of the baclofen response. Treatment with MAP kinase inhibitors PD98059 (10 μm, in the pipette) or U0126 (5 μm) did not affect E2 inhibition of baclofen responses (R2/R1 for E2, 58.6 ± 3.4%, n = 10; vs PD98059 plus E2, 66.1 ± 11.8%, n = 5).
Expression of GABAB receptor PKCδ and adenylate cyclase VII transcripts in arcuate (GABA, dopamine, and POMC) neurons
Using single-cell RT-PCR from 75 acutely dispersed arcuate neurons, we found that 90% of the neurons expressed GAD65 transcripts, including TH-expressing and POMC-expressing neurons (data not shown). Most importantly, 92% of the neurons expressed GABAB-R2 transcripts, which correlates with the 90% response rate to baclofen. Furthermore, we also determined that dopamine and POMC neurons express PKCδ and adenylyl cyclase VII transcripts using single-cell RT-PCR. In one group of cells (n = 22), we found that PKCδ and adenylyl cyclase VII transcripts are expressed in the majority (70%) of TH neurons (Fig. 8A), including those that coexpress GAD65. TH and GAD were colocalized in 60% of this population of neurons because of a limited amount of cDNA from individual neurons, POMC expression was determined in another group of cells (n = 29), and we found that PKCδ and adenylyl cyclase VII transcripts were expressed in the majority (75%) of POMC neurons, including those that coexpress GAD65 (Fig. 8B). POMC and GAD were colocalized in 28% of this population of neurons. Therefore, the single-cell RT-PCR data support the electrophysiological findings that dopamine and POMC neurons express the critical transcripts for rapid estrogen signaling.
Figure 8.

Expression of GAD, TH, POMC, PKCδ, and AC VII mRNA in arcuate neurons. Two representative gels illustrating that single arcuate neurons express mRNA for TH, GAD, PKCδ, AC VII, and GAPDH (A) or mRNA for POMC, GAD, PKCδ, AC VII, and GAPDH (B). The expected size of the PCR products is as follows (in bp): GAD, 207; TH, 223; POMC, 344; PKCδ, 251; AC VII, 235; and GAPDH, 212. GAPDH transcripts were analyzed in the same cells as an internal control for the RT reaction. One cell contained no RT as a negative control (-RT). Basal hypothalamic tissue RNA was also reverse transcribed in the presence of RT (tissue controls, +). A tissue control without RT (-) was included in each experiment. In addition, the following controls were included: HBSS from the dispersed cellular milieu and a water blank, both of which were negative after RT-PCR (data not shown).
Discussion
A unique membrane estrogen receptor mediates the rapid effects of E2
Estrogen suppresses the action of the GABAB receptor agonist baclofen to activate GIRK channels in GABA, POMC, and dopamine neurons. This E2 effect is rapid, with measurable suppression occurring within minutes after addition of E2. The kinetics of this response support the notion that a membrane E2 receptor is mediating the response and argue against the involvement of the classical nuclear estrogen receptors operating by transcription regulation.
The pharmacology we observed for this rapid estrogen response further supports the involvement of a novel transmembrane estrogen receptor. The membrane-impermeable E2-BSA conjugate gives an identical response to free E2, suggesting that the hormone-binding site of this receptor is accessible from the extracellular surface of the plasma membrane. The E2 response is stereospecific with respect to the configuration of the D-ring hydroxyl group; 17β-estradiol elicits the rapid response, whereas 17α-estradiol is inactive. This is notable because 17α-estradiol functions as an agonist of the nuclear ERs, albeit with slightly reduced potency compared with 17β-estradiol (Barkhem et al., 1998). The SERMs 4-OH tamoxifen, raloxifene, and GW-5638 all behave like E2 in mediating this response, whereas the steroidal anti-estrogen ICI 182,780 antagonizes the E2 response. Most importantly, the novel SERM STX that is devoid of estrogen (or anti-estrogen) activity with the nuclear ERs is a stronger activator of this rapid E2 response than E2, even at a 10-fold lower concentration. Moreover, we know that this membrane ER has a subnanomolar affinity for estrogen on the basis of our pharmacological (Schild) analysis (Lagrange et al., 1997). These results demonstrate that the pharmacology of this rapid response is different and, in the case of STX, separable from that of the nuclear ERs (Razandi et al., 1999; Levin, 2001; Chambliss and Shaul, 2002).
Recently, Toran-Allerand et al. (2002) identified a high-affinity, saturable estrogen receptor, ERX, that is associated with caveolar-like microdomains in developing neocortical neurons. This membrane-associated receptor is coupled to the activation of MAPKs (MAP kinases), ERK1 (extracellular-signal related kinase 1), and ERK2, which appear to be important for the development and survival of neurons (Watters et al., 1997; Singh et al., 1999, 2000; Fitzpatrick et al., 2002). Interestingly, ERX also has a distinct pharmacology in that 17α-estradiol is equipotent as E2 in activating the MAP kinase pathway (Wade et al., 2001; Toran-Allerand et al., 2002). However, we did not see any effects of 17α-estradiol on the GABAB response (present findings) or the μ-opioid response, which is coupled to the same family of GIRK channels in hypothalamic neurons (Lagrange et al., 1997). Similarly, Gu and Moss (1996) found that 17α-estradiol did not mimic the actions of E2 in the hippocampus to potentiate the glutamate (kainate)-mediated currents in CA1 pyramidal neurons. Likewise, Mermelstein et al. (1996) found that 17α-estradiol was much less efficacious than E2 in reducing L-type calcium currents in neostriatal neurons. Therefore, it appears that the membrane estrogen receptor that modulates channel activity in neurons via the PKC-PKA pathway is pharmacologically distinct from the receptor that is coupled to activation of ERK1 and ERK2 that promotes growth and survival.
Although the GABAA ionotropic receptor has been identified as a target for 5α-reduced progesterone metabolites in CNS neurons (Harrison et al., 1987; Lambert et al., 1995; Rupprecht and Holsboer, 1999), the nature of the transmembrane receptor for estrogen that uncouples GABAB (μ-opioid) receptors from GIRK is not known. In sea trout oocytes, progestins inhibit adenylyl cyclase via a pertussis toxin-sensitive mechanism to initiate oocyte maturation, and recently, a high-affinity progestin, GPCR, has been cloned from spotted seatrout oocytes (Zhu et al., 2003). However, whether there is an estrogen GPCR with similar homology needs to be determined.
E2 activates PKCδ and PKA to alter the coupling of GPCRs to K+ channels in hypothalamic neurons
A number of studies have shown that protein kinase pathways affect GABAB receptor-mediated signaling in CNS neurons. Activation of protein kinase C suppresses the GABAB receptor activation of GIRK channels in the hippocampal CA1 pyramidal neurons (Dutar and Nicoll, 1988) and attenuates the GABAB receptor-mediated inhibition of norepinephrine release from cerebellar slices (Taniyama et al., 1992).
Currently, there are 12 known members of the PKC family (Way et al., 2000). The family is divided into three groups on the basis of sequence homology and biochemical regulation. Class A, or conventional PKCs (PKCα, βI, βII, and γ) are the well known, Ca2+-dependent PKCs. Class B, or novel PKCs (PKCδ, ϵ, θ, and η), are Ca2+-independent. Finally, class C PKCs, or atypical PKCs (PKCζ and τ/λ), are the most divergent class. Atypical PKCs are also Ca2+ independent and do not require diacylglycerol for activation (Way et al., 2000). We found that the rapid GABAB-suppressing effects of estrogen in hypothalamic neurons were sensitive to the broad-spectrum PKC inhibitor BIS but not to Gö6976, suggesting the involvement of a PKC not belonging to the conventional PKC class. In addition, the inhibition of estrogen of the GABAB response was not altered by inclusion of 10 mm BAPTA in the intracellular recording patch pipette, providing additional evidence that the Ca2+-dependent conventional PKCs are not involved. However, the selective PKCδ inhibitor rottlerin blocked the actions of E2, suggesting that this novel-class PKC is a mediator of the rapid E2 response. Moreover, our scRT-PCR data on the expression of PKCδ transcripts in arcuate neurons support the involvement of PKCδ in the E2-mediated inhibition of the GABAB response. Likewise, PKCδ is involved in the estrogen-mediated inhibition of K+ channels and fluid retention in female distal colonic epithelial cells, although the upstream signaling pathway is not known (Doolan et al., 2000).
PKC activation is in the upstream of PKA activation
In our study, internal perfusion of BIS could completely block the inhibition of the baclofen response by E2 but could not attenuate the inhibition of the baclofen response by forskolin applied via bath perfusion. PKC is known to activate adenylyl cyclases (Jacobowitz et al., 1993; Yoshimura and Cooper, 1993; Lin and Chen, 1998); moreover, when AC is activated by PKC instead of by Gαs or forskolin, it is resistant to inhibition by Gαi (Pieroni et al., 1993). To date, nine AC isozymes have been cloned (AC types I-IX). Notably, AC VII has a potential binding site for PKCδ that is not present in the sequences of the other adenylyl cyclases, which would allow PKCδ to directly phosphorylate AC VII (Nelson et al., 2003). Interestingly, GABA neurons in the cortex, hippocampus, striatum, and cerebellum are immunoreactive for AC VII (Mons et al., 1998), and, in the present study, we show that hypothalamic GABA, TH, and POMC neurons express AC VII transcripts.
Gαq mediates the inhibition of the GABAB response by E2 through PLC
Most PKCs are activated by diacylglycerol, and some require the presence of Ca2+. Thus, PKCs are downstream of the PLC-inositol triphosphate-diacylglycerol signaling cascade. Because different forms of PLC can be activated by various messengers, including Gαq, Gβγ (PLCβ), and tyrosine kinases (PLCγ), the PKC family is involved in a diverse array of signaling cascades (Tanaka and Nishizuka, 1994; Battaini, 2001). Our results show that a membrane ER is specifically coupled to Gαq protein. This conclusion is based on experiments in which intracellular dialysis with a peptide fragment of Gαq blocked the receptor interaction with G-protein. This Gαq peptide has been used to block Gαq signaling pathways in cortical pyramidal neurons (Carr et al., 2002). In addition, the estrogen-mediated reduction of the GABAB response was significantly reduced by the phospholipase C inhibitor U73122 compared with cells perfused with the less active inhibitor U73343.
Therefore, from the collective results of this study, we formulate the signal transduction pathway for the rapid response to estrogen in hypothalamic neurons depicted in Figure 9. The sequence of events in this model are as follows: (1) E2 binds to a novel transmembrane estrogen receptor; (2) ligand binding activates Gαq; (3) activated Gαq in turn activates PLC; (4) activated PLC liberates DAG; (5) free DAG stimulates PKCδ; (6) PKCδ activates adenylyl cyclase (VII); (7) cAMP levels are elevated; (8) cAMP stimulates PKA; and (9) PKA phosphorylates membrane targets critical for K+ channel function.
Figure 9.

A cellular model of the rapid signaling of estrogen in hypothalamic neurons. Schematic overview showing the rapid versus delayed ER-mediated modulation of neurotransmitter-regulated G-protein-coupled receptors via a membrane-associated ER in hypothalamic neurons. Rapid signaling: E2 activates a membrane-associated ER that is Gαq-coupled to activation of phospholipase C that catalyzes the hydrolysis of membrane-bound phosphatidylinositol 4,5-biphosphate (PIP2) to inositol 1,4,5 triphosphate (IP3) and DAG. Calcium is released from intracellular stores (endoplasmic reticulum) by IP3, and DAG activates PKCδ. Through phosphorylation, AC VII activity is upregulated by PKCδ. The generation of cAMP activates PKA, which can rapidly uncouple GABAB and μ-opioid (μ) receptors from their effector system through phosphorylation of a downstream effector molecule (e.g., the inwardly rectifying K+ channel, or GIRK). ER-mediated modulation of kinase pathways either reduces the capacity of neuromodulators such as GABA and β-endorphin (βEnd) to inhibit hypothalamic neuronal excitability, or augments the ability of neurotransmitters such as glutamate to increase neuronal excitability (data not shown). Delayed signaling: rapid ER-mediated activation of PKA can lead to phosphorylation of cAMP-responsive element-binding protein (pCREB), which can then alter gene transcription through its interaction with the cAMP-responsive element (CRE). Moreover, other isoforms of PKC can phosphorylate raf-1 (Raf), leading to activation of the MAP kinase pathway and new gene transcription and protein (e.g., GPCRs) synthesis in an estrogen-response element-independent manner.
Functional significance of rapid membrane effects of E2 in CNS neurons
It was discovered previously that E2 could rapidly modulate synaptic efficacy via activation of PKA (Gu and Moss, 1996, 1998; Lagrange et al., 1997; Kelly et al., 1999). Presently, we delineated the upstream components of this signaling pathway that includes Gq, phospholipase C, and PKCδ activation (Fig. 9). This is a novel signaling pathway for E2 to rapidly modulate hypothalamic neuronal excitability, and there is most likely a similar E2 signaling pathway in hippocampal CA1 neurons (Gu and Moss, 1996, 1998). Therefore, we believe that this pathway is important for increasing synaptic efficacy not only in hypothalamic neurons but also in other neurons in the CNS. In addition, we identified a specific ligand (STX) that is selective for activating this pathway. The consequences of STX effects in hypothalamic neurons are evident in that these neurons are involved in controlling the ovulatory cycle, lactation, stress responses, temperature, and energy balance, all of which require rapid feedback regulation by estrogen. Furthermore, having a selective E2 agonist for rapid signaling is critical because SERMs such as tamoxifen and raloxifene increase the incidence of hot flashes in women, suggesting that they act as E2 antagonists in the hypothalamus (Stearns et al., 2002; Sherwin, 2003). In addition, raloxifene treatment is no better than placebo treatment in maintaining cognitive performance of postmenopausal women (Sherwin, 2003), which suggests that raloxifene is not an E2 agonist in hippocampus. Most importantly, raloxifene and tamoxifen bind to ERα and ERβ with high affinity (Barkhem et al., 1998). In contrast, the STX (E2) receptor is similarly coupled as the serotonin 5HT2A,C receptor (Carr et al., 2002) in CNS neurons, which may explain the ability of serotonin uptake inhibitors (SSRIs) to prevent hot flashes in postmenopausal women (Stearns et al., 2002). Hence, we would predict that STX would prevent hot flashes, maintain sleep cycles, elevate mood, etc. Therefore, this rapid PLC-PKCδ-PKA signaling of E2 may synergize with CNS transmitter systems to enhance synaptic efficacy in brain circuits that are critical for maintaining homeostatic functions.
Footnotes
This research was supported by United States Public Health Service Grants NS 35944 (Office of Research on Women's Health), NS 38809, DA 05158, DA 10703, and DK57574. We thank Drs. D. James Surmeier and Tania Tkatch for their help and advice in establishing the single-cell RT-PCR technique in our laboratory. We also recognize Barry R. Naylor and Rebecka D. Amodei for their expert technical contribution.
Correspondence should be addressed to Dr. Martin J. Kelly, Department of Physiology and Pharmacology, L334, Oregon Health and Science University, Portland, OR 97239-3098. E-mail: kellym@ohsu.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/239529-12$15.00/0
References
- Akema T, Chiba A, Kimura F ( 1990) On the relationship between noradendritic stimulatory and GABAergic inhibitory systems in the control of luteinizing hormone secretion in female rats. Neuroendocrinology 52: 566-572. [DOI] [PubMed] [Google Scholar]
- Akhter SA, Luttrell LM, Rockman HA, Iaccarino G, Lefkowitz RJ, Koch WJ ( 1998) Targeting the receptor-Gq interface to inhibit in vivo pressure overload myocardial hypertrophy. Science 280: 574-577. [DOI] [PubMed] [Google Scholar]
- Barkhem T, Carlsson B, Nilsson Y, Enmark E, Gustafsson JÅ, Nilsson S ( 1998) Differential response of estrogen receptor α and estrogen receptor β to partial estrogen agonists/antagonists. Mol Pharmacol 54: 105-112. [DOI] [PubMed] [Google Scholar]
- Battaini F ( 2001) Protein kinase C isoforms as therapeutic targets in nervous system disease states. Pharmacol Res 44: 353-361. [DOI] [PubMed] [Google Scholar]
- Björklund A, Lindvall O ( 1984) Dopamine-containing systems in the CNS. In: Handbook of chemical neuroanatomy: classical transmitters in the CNS, Pt 1 (Björklund A, Hökfelt T, eds), pp 55-122. Amsterdam: Elsevier.
- Carr DB, Cooper DC, Ulrich SL, Spruston N, Surmeier DJ ( 2002) Serotonin receptor activation inhibits sodium current and dendritic excitability in prefrontal cortex via a protein kinase C-dependent mechanism. J Neurosci 22: 6846-6855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambliss KL, Shaul PW ( 2002) Estrogen modulation of endothelial nitric oxide synthase. Endocr Rev 23: 665-686. [DOI] [PubMed] [Google Scholar]
- Dave JR, Rubinstein N, Eskay RL ( 1985) Evidence that β-endorphin binds to specific receptors in rat peripheral tissues and stimulates the adenylate cyclase-adenosine 3′, 5′-monophosphate system. Endocrinology 117: 1389-1396. [DOI] [PubMed] [Google Scholar]
- Demotes-Mainard J, Arnauld E, Vincent JD ( 1990) Estrogens modulate the responsiveness of in vivo recorded striatal neurons to iontophoretic application of dopamine in rats: role of D1 and D2 receptor activation. J Neuroendocrinol 2: 825-832. [DOI] [PubMed] [Google Scholar]
- Doolan CM, Condliffe SB, Harvey BJ ( 2000) Rapid non-genomic activation of cytosolic cyclic AMP-dependent protein kinase activity and [Ca2+]i by 17β-oestradiol in female rat distal colon. Br J Pharmacol 129: 1375-1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutar P, Nicoll RA ( 1988) Pre- and postsynaptic GABA(B) receptors in the hippocampus have different pharmacological properties. Neuron 1: 585-591. [DOI] [PubMed] [Google Scholar]
- Ferin M, Van Vugt D, Wardlaw S ( 1984) The hypothalamic control of the menstrual cycle and the role of endogenous opioid peptides. Recent Prog Horm Res 40: 441-485. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick JL, Mize AL, Wade CB, Harris JA, Shapiro RA, Dorsa DM ( 2002) Estrogen-mediated neuroprotection against β-amyloid toxicity requires expression of estrogen receptor α or β and activation of the MAPK pathway. J Neurochem 82: 674-682. [DOI] [PubMed] [Google Scholar]
- Gu Q, Moss RL ( 1996) 17β-Estradiol potentiates kainate-induced currents via activation of the cAMP cascade. J Neurosci 16: 3620-3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Q, Moss RL ( 1998) Novel mechanism for non-genomic action of 17β-estradiol on kainate-induced currents in isolated rat CA1 hippocampal neurones. J Physiol (Lond) 506: 745-754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison NL, Majewska MD, Harrrington JW, Barker JL ( 1987) Structure-activity relationships for steroid interaction with the gamma-aminobutyric acidA receptor complex. J Pharmacol Exp Ther 241: 346-353. [PubMed] [Google Scholar]
- Herbison AE ( 1997) Estrogen regulation of GABA transmission in rat preoptic area. Brain Res Bull 44: 321-326. [DOI] [PubMed] [Google Scholar]
- Herbison AE, Horvath TL, Naftolin F, Leranth C ( 1995) Distribution of estrogen receptor-immunoreactive cells in monkey hypothalamus: relationship to neurones containing luteinizing hormone-releasing hormone and tyrosine hydroxylase. Neuroendocrinology 61: 1-10. [DOI] [PubMed] [Google Scholar]
- Herbison AE, Skynner MJ, Sim JA ( 2001) Lack of detection of estrogen receptor-α transcripts in mouse gonadotropin-releasing hormone neurons. Endocrinology [Erratum] 142: 492-493. [Google Scholar]
- Hökfelt T, Mårtensson R, Björklund A, Kleinau S, Goldstein M ( 1984) Distributional maps of tyrosine-hydroxylase-immunoreactive neurons in the rat brain. In: Handbook of chemical neuroanatomy (Björklund A, Hökfelt T, eds), pp 277-379. Amsterdam: Elsevier.
- Jacobowitz O, Chen J, Premont RT, Iyengar R ( 1993) Stimulation of specific types of Gs-stimulated adenylyl cyclases by phorbol ester treatment. J Biol Chem 268: 3829-3832. [PubMed] [Google Scholar]
- Jarry H, Leonhardt S, Wuttke W ( 1995) The inhibitory effect of β-endorphin on LH release in ovariectomized rats does not involve the preoptic GABAergic system. Exp Clin Endocrinol Diabetes 103: 317-323. [DOI] [PubMed] [Google Scholar]
- Katzenellenbogen JA, Carlson KE, Katzenellenbogen BS ( 1985) Facile geometric isomerization of phenolic non-steroidal estrogens and antiestrogens: limitations to the interpretation of experiments characterizing the activity of individual isomers. J Steroid Biochem 22: 589-596. [DOI] [PubMed] [Google Scholar]
- Kelly MJ, Wagner EJ ( 1999) Estrogen modulation of G-protein-coupled receptors. Trends Endocrinol Metab 10: 369-374. [DOI] [PubMed] [Google Scholar]
- Kelly MJ, Rønnekleiv OK ( 1994) Electrophysiological analysis of neuroendocrine neuronal activity in hypothalamic slices. In: Methods in neurosciences: pulsatility in neuroendocrine systems (Levine JE, ed), pp 47-67. San Diego: Academic.
- Kelly MJ, Rønnekleiv OK, Eskay RL ( 1984) Identification of estrogen-responsive LHRH neurons in the guinea pig hypothalamus. Brain Res Bull 12: 399-407. [DOI] [PubMed] [Google Scholar]
- Kelly MJ, Loose MD, Rønnekleiv OK ( 1990) Opioids hyperpolarize β-endorphin neurons via mu-receptor activation of a potassium conductance. Neuroendocrinology 52: 268-275. [DOI] [PubMed] [Google Scholar]
- Kelly MJ, Loose MD, Rønnekleiv OK ( 1992) Estrogen suppresses μ-opioid and GABAB-mediated hyperpolarization of hypothalamic arcuate neurons. J Neurosci 12: 2745-2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly MJ, Lagrange AH, Wagner EJ, Rønnekleiv OK ( 1999) Rapid effects of estrogen to modulate G-protein-coupled receptors via activation of protein kinase A and protein kinase C pathways. Steroids 64: 64-75. [DOI] [PubMed] [Google Scholar]
- Lagrange AH, Rønnekleiv OK, Kelly MJ ( 1994) The potency of μ-opioid hyperpolarization of hypothalamic arcuate neurons is rapidly attenuated by 17β-estradiol. J Neurosci 14: 6196-6204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagrange AH, Rønnekleiv OK, Kelly MJ ( 1995) Estradiol-17β and mu-opioid peptides rapidly hyperpolarize GnRH neurons: a cellular mechanism of negative feedback? Endocrinology 136: 2341-2344. [DOI] [PubMed] [Google Scholar]
- Lagrange AH, Wagner EJ, Rønnekleiv OK, Kelly MJ ( 1996) Estrogen rapidly attenuates a GABAB response in hypothalamic neurons. Neuroendocrinology 64: 114-123. [DOI] [PubMed] [Google Scholar]
- Lagrange AH, Rønnekleiv OK, Kelly MJ ( 1997) Modulation of G-protein-coupled receptors by an estrogen receptor that activates protein kinase A. Mol Pharmacol 51: 605-612. [DOI] [PubMed] [Google Scholar]
- Lambert JJ, Belelli D, Hill-Venning C, Peters JA ( 1995) Neurosteroids and GABAA receptor function. Trends Pharmacol Sci 16: 295-303. [DOI] [PubMed] [Google Scholar]
- Leranth C, MacLusky NJ, Brown TJ, Chen EC, Redmond Jr DE, Naftolin F ( 1992) Transmitter content and afferent connections of estrogen-sensitive progestin receptor-containing neurons in the primate hypothalamus. Neuroendocrinology 55: 667-682. [DOI] [PubMed] [Google Scholar]
- Levin ER ( 2001) Cell localization, physiology, and nongenomic actions of estrogen receptors. J Appl Physiol 91: 1860-1867. [DOI] [PubMed] [Google Scholar]
- Lin W-W, Chen BC ( 1998) Distinct PKC isoforms mediate the activation of cPLA2 and adenylyl cyclase by phorbol ester in RAW264.7 macrophages. Br J Pharmacol 125: 1601-1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loose MD, Rønnekleiv OK, Kelly MJ ( 1990) Membrane properties and response to opioids of identified dopamine neurons in the guinea pig hypothalamus. J Neurosci 10: 3627-3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loose MD, Rønnekleiv OK, Kelly MJ ( 1991) Neurons in the rat arcuate nucleus are hyperpolarized by GABAB and mu-opioid receptor agonists: evidence for convergence at a ligand-gated potassium conductance. Neuroendocrinology 54: 537-544. [DOI] [PubMed] [Google Scholar]
- Martiny-Baron G, Kazanietz MG, Mischak H, Blumberg PM, Kochs G, Hug H, Marme D, Schachtele C ( 1993) Selective inhibition of protein kinase C isozymes by the indolocarbazole Gö6976. J Biol Chem 268: 9194-9197. [PubMed] [Google Scholar]
- McEwen BS ( 2001) Estrogens effects on the brain: multiple sites and molecular mechanisms. J Appl Physiol 91: 2785-2801. [DOI] [PubMed] [Google Scholar]
- Mermelstein PG, Becker JB, Surmeier DJ ( 1996) Estradiol reduces calcium currents in rat neostriatal neurons via a membrane receptor. J Neurosci 16: 595-604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsushima D, Marzban F, Luchansky LL, Burich AJ, Keen KL, Durning M, Golos TG, Terasawa E ( 1996) Role of glutamic acid decarboxylase in the prepubertal inhibition of the luteinizing hormone releasing hormone release in female rhesus monkeys. J Neurosci 16: 2563-2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mons N, Yoshimura M, Ikeda H, Hoffman PL, Tabakoff B ( 1998) Immunological assessment of the distribution of type VII adenylyl cyclase in brain. Brain Res 788: 251-261. [DOI] [PubMed] [Google Scholar]
- Morrell JI, McGinty JF, Pfaff DW ( 1985) A subset of β-endorphin or dynorphin-containing neurons in the medial basal hypothalamus accumulates estradiol. Neuroendocrinology 41: 417-426. [DOI] [PubMed] [Google Scholar]
- Neill JD ( 1980) Neuroendocrine regulation of prolactin secretion. Front Neuroendocrinol 6: 129-155. [Google Scholar]
- Nelson EJ, Hellevuo K, Yoshimura M, Tabakoff B ( 2003) Ethanol-induced phosphorylation and potentiation of the activity of type 7 adenylyl cyclase: involvement of PKC delta. J Biol Chem 278: 4552-4560. [DOI] [PubMed] [Google Scholar]
- Paradiso K, Zhang J, Steinbach JH ( 2001) The C terminus of the human nicotinic α4β2 receptor forms a binding site required for potentiation by an estrogenic steroid. J Neurosci 21: 6561-6568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieroni JP, Jacobowitz O, Chen J, Iyengar R ( 1993) Signal recognition and integration by Gs-stimulated adenylyl cyclases. Curr Opin Neurobiol 3: 345-351. [DOI] [PubMed] [Google Scholar]
- Razandi M, Pedram A, Greene GL, Levin ER ( 1999) Cell membrane and nuclear estrogen receptors (ERs) originate from a single transcript: studies of ERα and ERβ expressed in Chinese hamster ovary cells. Mol Endocrinol 13: 307-319. [DOI] [PubMed] [Google Scholar]
- Rupprecht R, Holsboer F ( 1999) Neuroactive steroids: mechanisms of action and neuropsychopharmacological perspectives. Trends Neurosci 22: 410-416. [DOI] [PubMed] [Google Scholar]
- Seltzer AM, Donoso AO ( 1992) Restraining action of GABA on estradiol-induced LH surge in the rat: GABA activity in brain nuclei and effects of GABA mimetics in the medial preoptic nucleus. Neuroendocrinology 55: 28-34. [DOI] [PubMed] [Google Scholar]
- Sherwin BB ( 2003) Estrogen and cognitive functioning in women. Endocr Rev 24: 133-151. [DOI] [PubMed] [Google Scholar]
- Simonian SX, Spratt DP, Herbison AE ( 1999) Identification and characterization of estrogen receptor α-containing neurons projecting to the vicinity of the gonadotropin-releasing hormone perikarya in the rostral preoptic area of the rat. J Comp Neurol 411: 346-358. [DOI] [PubMed] [Google Scholar]
- Singh M, Setalo Jr G, Guan X, Warren M, Toran-Allerand CD ( 1999) Estrogen-induced activation of mitogen-activated protein kinase in cerebral cortical explants: convergence of estrogen and neurotrophin signaling pathways. J Neurosci 19: 1179-1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M, Setalo GJ, Guan X, Frail DE, Toran-Allerand CD ( 2000) Estrogen-induced activation of the mitogen-activated protein kinase cascade in the cerebral cortex of estrogen receptor-α knock-out mice. J Neurosci 20: 1694-1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stearns V, Ullmer L, Lopez JF, Smith Y, Isaacs C, Hayes D ( 2002) Hot flushes. Lancet 360: 1851-1861. [DOI] [PubMed] [Google Scholar]
- Sullivan KA, Witkin JW, Ferin M, Silverman AJ ( 1995) Gonadotropin-releasing hormone neurons in the rhesus macaque are not immunoreactive for the estrogen receptor. Brain Res 685: 198-200. [DOI] [PubMed] [Google Scholar]
- Takano K, Asano S, Yamashita N ( 1994) Activation of G-protein-coupled K+ channels by dopamine in human GH-producing cells. Am J Physiol 266: E318-E325. [DOI] [PubMed] [Google Scholar]
- Tanaka C, Nishizuka Y ( 1994) The protein kinase C family for neuronal signaling. Annu Rev Neurosci 17: 551-567. [DOI] [PubMed] [Google Scholar]
- Taniyama K, Niwa M, Kataoka Y, Yamashita K ( 1992) Activation of protein kinase C suppresses the gamma-aminobutyric acid B receptor-mediated inhibition of the vesicular release of noradrenaline and acetylcholine. J Neurochem 58: 1239-1245. [DOI] [PubMed] [Google Scholar]
- Toran-Allerand CD, Guan X, MacLusky NJ, Horvath TL, Diano S, Singh M, Connolly Jr ES, Nethrapalli IS, Tinnikov AA ( 2002) ER-X: a novel, plasma membrane-associated, putative estrogen receptor that is regulated during development and after ischemic brain injury. J Neurosci 22: 8391-8401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valverde MA, Rojas P, Amigo J, Cosmelli D, Orio P, Bahamonde MI, Mann GE, Vergara C, Latorre R ( 1999) Acute activation of Maxi-K channels (hSlo) by estradiol binding to the β subunit. Science 285: 1929-1931. [DOI] [PubMed] [Google Scholar]
- Wade CB, Robinson S, Shapiro RA, Dorsa DM ( 2001) Estrogen receptor (ER)alpha and ERbeta exhibit unique pharmacologic properties when coupled to activation of the mitogen-activated protein kinase pathway. Endocrinology 142: 2336-2342. [DOI] [PubMed] [Google Scholar]
- Wagner EJ, Manzanares J, Moore KE, Lookingland KJ ( 1994) Neurochemical evidence that estrogen-induced suppression of kappa-opioid-receptor-mediated regulation of tuberoinfundibular dopaminergic neurons is prolactin-independent. Neuroendocrinology 59: 197-201. [DOI] [PubMed] [Google Scholar]
- Wagner EJ, Bosch MA, Kelly MJ, Rønnekleiv OK ( 1999) A powerful GABAB receptor-mediated inhibition of GABAergic neurons in the arcuate nucleus. NeuroReport 10: 2681-2687. [DOI] [PubMed] [Google Scholar]
- Wagner EJ, Reyes-Vazquez C, Rønnekleiv OK, Kelly MJ ( 2000) The role of intrinsic and agonist-activated conductances in determining the firing patterns of preoptic area neurons in the guinea pig. Brain Res 879: 29-41. [DOI] [PubMed] [Google Scholar]
- Wagner EJ, Rønnekleiv OK, Bosch MA, Kelly MJ ( 2001) Estrogen biphasically modifies hypothalamic GABAergic function concomitantly with negative and positive control of luteinizing hormone release. J Neurosci 21: 2085-2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson RE, Langub MC, Landis JW ( 1992) Further evidence that most luteinizing hormone-releasing hormone neurons are not directly estrogen-responsive: simultaneous localization of luteinizing hormone-releasing hormone and estrogen receptor immunoreactivity in the guinea-pig brain. J Neuroendocrinol 4: 311-317. [DOI] [PubMed] [Google Scholar]
- Watters JJ, Campbell JS, Cunningham MJ, Krebs EG, Dorsa DM ( 1997) Rapid membrane effects of steroids in neuroblastoma cells: effects of estrogen on mitogen-activated protein kinase signaling cascade and c-fos immediate early gene transcription. Endocrinology 138: 4030-4033. [DOI] [PubMed] [Google Scholar]
- Way KJ, Chou E, King GL ( 2000) Identification of PKC-isoform-specific biological actions using pharmacological approaches. Trends Pharmacol Sci 21: 181-187. [DOI] [PubMed] [Google Scholar]
- Wetzel CH, Hermann B, Behl C, Pestel E, Rammes G, Zieglgansberger W, Holsboer F, Rupprecht R ( 1998) Functional antagonism of gonadal steroids at the 5-hydroxytryptamine type 3 receptor. Mol Endocrinol 12: 1441-1451. [DOI] [PubMed] [Google Scholar]
- Yoshimura M, Cooper DM ( 1993) Type-specific stimulation of adenylyl cyclase by protein kinase C. J Biol Chem 268: 4604-4607. [PubMed] [Google Scholar]
- Zhu Y, Rice CD, Pang Y, Pace M, Thomas P ( 2003) Cloning, expression, and characterization of a membrane progestin receptor and evidence it is an intermediary in meiotic maturation of fish oocytes. Proc Natl Acad Sci USA 100: 2231-2236. [DOI] [PMC free article] [PubMed] [Google Scholar]