

Abstract
During neurite initiation, cells surrounded by a flattened, actin-rich lamellipodium transform to produce thin, microtubule-filled neurite shafts tipped by actin-rich growth cones, but little is known about this transformation. Our detailed time-lapse analyses of cultured hippocampal neurons, a widely used model system for neuronal development, revealed that neurites emerge from segmented lamellipodia, which then gradually extend from the cell body to become nascent growth cones. This suggests that actin- and microtubule-rich structures are reorganized in a coordinated manner. We hypothesized that proteins such as microtubule-associated protein 2 (MAP2), which can interact with both cytoskeletal components, might be critically involved in neurite initiation. Live-cell video and fluorescence microscopy in Neuro-2a cells showed that expression of MAP2c triggers neurite formation via rapid accumulation and bundling of stable, MAP2c-bound microtubules, concurrent with a gradual transformation of lamellipodia into nascent growth cones. The microtubule-stabilizing agent Taxol did not mimic this effect, suggesting that the ability of MAP2c to stabilize microtubules is not sufficient for neurite initiation. However, combination of Taxol treatment with actin disruption induced robust process formation, suggesting that inhibitory effects of F-actin need to be overcome as well. Neurite initiation by MAP2c required its microtubule-binding domain and was enhanced by its binding domain for cAMP-dependent protein kinase (PKA). MAP2c mutants defective in both PKA and microtubule binding acted as dominant negative inhibitors of neurite initiation in neuroblastoma cells and primary hippocampal neurons. Together, these data suggest that MAP2c bears functions that both stabilize microtubules and directly or indirectly alter actin organization during neurite initiation.
Keywords: microtubule-associated proteins, MAP2, growth cone, neurite, cell motility, actin, microtubules, lamellipodia
Introduction
During or shortly after migrating to their final destinations, neurons begin to form processes called neurites. These neurites eventually differentiate to form functionally and structurally distinct axons and dendrites. Several key steps in the morphological differentiation of neurons have been analyzed, and many of the mechanisms controlling these steps have emerged in recent years (Letourneau, 1996; Luo, 2002). However, relatively little is known about either the molecular events or sequence of morphological changes that underlie neurite initiation, the very first step in neuritogenesis. Early imaging studies showed that, shortly after plating, cultured hippocampal neurons first extend one or more actin-rich lamellipodia surrounding the cell body. Later, short microtubule-rich neurites emerge, which normally terminate in actin-rich growth cones (Dotti et al., 1988). These steps clearly involve significant rearrangement of both microtubules and actin-based structures within the lamellipodia.
One candidate to coordinate such rearrangements during neurite initiation is microtubule-associated protein 2 (MAP2), which can interact with both microtubules (Kim et al., 1979) and actin filaments (Selden and Pollard, 1983, 1986; Sattilaro, 1986). MAP2 is an abundant protein expressed during all stages of neuromorphogenesis (Caceres et al., 1986). In primary hippocampal neurons, it is first expressed in neuroblasts before neurite initiation. Later it is present in minor processes, one of which eventually differentiates into an axon. At later stages, MAP2 disappears from axons but is retained in dendrites (Bernhardt and Matus, 1984; Caceres et al., 1986). This expression pattern suggests that MAP2 plays a role in both early and late morphological events. In particular, the embryonic isoform MAP2c, which is highly expressed during early neuronal development, might be involved in neurite initiation. Overexpression of MAP2c in Sf9 cells leads to the formation of protrusions that resemble neurites in their thin diameter and extended length (LeClerc et al., 1993). Dendrite outgrowth is inhibited in map2-/- animals, and growth cones exhibit altered morphology (Teng et al., 2001; Harada et al., 2002). Moreover, acute inhibition of MAP2 expression in cultured cerebellar macroneurons using antisense oligonucleotides inhibited neurite formation (Caceres et al., 1992), suggesting that MAP2 is necessary for neurite initiation in this system.
Here we present a detailed series of time-lapse studies that document the sequence of events underlying microtubule and lamellipodia reorganization during neurite initiation. We show for the first time that the smallest known MAP2 isoform, MAP2c, is sufficient to induce neurites in undifferentiated neuronal cells, and we characterize the molecular basis for this action. Finally, we show that mutant forms of MAP2c can interfere with neurite initiation in primary neurons expressing endogenous MAP2. Our data suggest a model in which MAP2c coordinates rearrangements of microtubules with forward protrusion of lamellipodia to establish a newly formed growth cone in front of a bundle of stabilized microtubules.
Materials and Methods
Constructs. Plasmids pEGFP-MAP2c and pEGFP-MAP2c-EEE coding for rat MAP2c containing a C-terminal fusion of green fluorescent protein (GFP-MAP2c) and the mutant GFP-MAP2c-EEE were described previously (Ozer and Halpain, 2000). To delete the conserved regulatory subunit II (RII)-binding site (86-103; Vijayaraghavan et al., 1999), DNA coding for amino acids 1-85 was amplified using the primers 5′-AAAAAAGCTTATGGCTGACGAGAGG-3′ and 5′-GGAATTCTCTGTCAGCTGAGG-3′, which introduce HindIII and EcoRI sites. Nucleotides coding for amino acids 104-467 were amplified using primers 5′-GGAATTCGCAGTCCTGAAAGG-3′ and 5′-AAAAGGATCCAAGCCCTGCTTAGCGAG-3′ containing EcoRI and BamHI sites. Both fragments were ligated into pEGFP-N1 digested with HindIII and BamHI. The deletion mutants contain one additional phenylalanine replacing the RII-binding site. Plasmids coding fot red fluorescent MAP2c RED2-MAP2c (dsRED2-MAP2c) were generated by ligating the pEGFP-MAP2c insert digested with BamHI and HindIII into similarly digested pDsRED2 (Clontech). All constructs were fully sequenced. A plasmid coding for GFP-γ-actin was a kind gift from A. Matus (Friedrich Miescher Institute, Basel, Switzerland).
Cell culture, transfection, and drug treatments. Neuro-2a cells (American Type Culture Collection, Manassas, VA) were cultured using standard conditions (Wu et al., 1998). Cells were seeded at a density of 2.5 × 104 cells/cm2 onto 12-well plates for morphological quantification and onto poly-l-lysine-coated glass coverslips for immunocytochemistry and live cell imaging. Cells were rested for 4 hr at 37°C in 5% CO2 before treatment. Transfection was performed using the FuGENE 6 transfection reagent (3 μl/μg of DNA; Roche Diagnostic Corp., Indianapolis, IN). In some experiments, serum was reduced to 5%, and 40 μm retinoic acid (Sigma, St. Louis, MO) was added to the cells 4 hr after transfection. Treatment with Taxol (paclitaxel; Cytoskeleton, Denver, CO), cytochalasin D (cytoD; Sigma), latrunculin A (latA), and jasplakinolide (jasp) (both from Molecular Probes, Eugene, OR) was performed 4 hr after plating for an additional 4 hr. Human embryonic kidney 293 (HEK-293) cells were maintained using standard conditions. Primary hippocampal neurons were prepared as described (Goslin et al., 1998) with minor modifications. Transfection of neurons was performed using Ca/PO4 precipitation (Kohrmann et al., 1999) 1 d after plating, and neurons were fixed 24 hr after transfection (transfection efficiencies were usually ∼10%). In some experiments, freshly prepared dissociated neurons were frozen at -80°C as described previously (Ruthel and Hollenbeck, 2000). Neurons were maintained in the absence of glia for the short observation times during live cell imaging. Instead, glial conditioned medium was used to provide survival factors. These modifications did not cause discernable alterations in normal neuron development.
Immunocytochemistry. To localize MAP2c or βIII-tubulin, cells were fixed in 0.5% glutaraldehyde in PHEM buffer (60 mm PIPES, 25 mm HEPES, 10 mm EGTA, and 2 mm MgCl2, pH 6.9) for 10 min at 37°C (Marsden et al., 1996). For α-tubulin and F-actin localization, cells were fixed in 4% paraformaldehyde in PBS for 20 min at 37°C. Cells were permeabilized with 0.25% Triton X-100 in PBS, treated with 0.1% NaBH4 to minimize autofluorescence, and incubated 1 hr at 37°C with 10% bovine serum albumin (BSA) in PBS to block nonspecific binding. As indicated, cultures were stained using mouse monoclonal anti-βIII-tubulin antibody TUJ1 at 1:1000 (a gift from A. Frankfurter, University of Virginia, Charlottesville, VA), monoclonal anti-α-tubulin antibody DM1A at 1:1000 (Sigma), rabbit polyclonal anti-MAP2 antibody 4170 at 1:5000 (Ozer and Halpain, 2000), Alexa-568-phalloidin at 1:1000, or a combination thereof in 2% BSA and PBS for 2 hr at room temperature. Anti-IgG antibodies conjugated to Alexa-488 (1:1000; Molecular Probes) were used as secondary antibodies.
Microscopy. Confocal fluorescence images were collected from an Olympus Optical (Tokyo, Japan) BX-50 microscope equipped with an Ultraview spinning disk scan head (PerkinElmer Life Sciences, Boston, MA) using a 60× 1.4 numerical aperture plan APO oil immersion objective and a krypton-argon laser. Images were collected using an Orca 100 camera (Hamamatsu, Bridgewater, NJ). Live cell imaging of Neuro-2a cells was performed using an Olympus BX-50 microscope equipped with a C4742-95 camera (Hamamatsu) and Nomarski differential interference contrast (DIC) optics. Imaging of primary hippocampal neurons used an Olympus IX-70 microscope equipped with an environmentally controlled chamber (Solent Scientific, Portsmouth, UK) and a C4742-98 camera (Hamamatsu). For combined DIC and fluorescence live cell imaging, an Olympus Fluoview 500 confocal microscope was used. DIC and fluorescence images were simultaneously collected by illumination using the 488 line of an argon laser. Images were processed using Metamorph (Universal Imaging Corporation, West Chester, PA) and Photoshop (Adobe, San Jose, CA).
Neurite initiation. To synchronize MAP2c-induced neurite initiation, Neuro-2a cells were transfected with pEGFP-MAP2c and trypsinized after overnight expression of GFP-MAP2c. Cells were replated onto poly-l-lysine-coated coverslips. After 1 hr, coverslips were mounted into closed aluminum slide chambers. Imaging was performed using a Biostage 600 heated stage (20/20 Technologies, Wilmington, NC) set to 36.5°C. Laser intensity was set to minimize phototoxicity and bleaching. More than 10 MAP2c-induced neurite initiation events were recorded at low spatial resolution (10× objective), and four were recorded at high resolution (40-60× objective). For primary hippocampal neurons, 30 spontaneous neurite initiation events were acquired at medium spatial resolution (20× objective), and six events were acquired at high resolution (40×, 6.6 frames/min). For live cell recording of drug-induced neurite formation, cells were plated onto poly-l-lysine-coated coverslips. After attachment overnight, cells were mounted into a heat-controlled perfusion system (20/20 Technologies) and imaged using DIC optics. More than 100 initiation events were observed. The presented examples are representative of the combined data.
Morphological measurements. Neuro-2a cells were analyzed for the presence of neurites on a Nikon (Melville, NY) TE200 microscope using a 20× phase objective. Transfected cells were identified in epifluorescence (typically 50% of cells display detectable green fluorescence; 30% of transfected cells reach high expression levels 24 hr after transfection). To assess neurite initiation induced by the expressed constructs, all cells with detectable fluorescence were analyzed for the presence of neurites, which in this study were defined as thin protrusions that were at least one cell diameter in length. To assess dominant negative effects of GFP constructs on retinoic acid-induced neurite initiation, only strongly expressing cells (>50% of maximal intensity observed) were selected for analysis. Each experiment was repeated at least three times, and the number of cells analyzed was at least 100 per experiment. Neurite initiation was assayed by at least two independent observers and always yielded similar results. Retrograde flow was measured as the slope of diagonal lines in kymographs, acquired perpendicular to the lamellipodial edge using DIC optics. For analysis of hippocampal neurons, processes of >10 μm were counted as neurites. Neurite length and growth rates were measured by manual tracking of neurites using Metamorph (Universal Imaging Corporation). All statistical analyses were performed using the one-way ANOVA and Dunnett's post hoc test built into Prizm (Graph Pad, San Diego, CA), unless otherwise noted.
Results
Neurite induction results in a substantial reorganization of cellular morphology and must therefore require specific changes in the underlying actin and microtubule cytoskeletons. A recent review postulates that actin plays a leading role in this process and that microtubules only play a secondary role (da Silva and Dotti, 2002). Actin-based processes are clearly important in neurite initiation, but microtubules are increasingly implicated in aspects of cell motility previously thought to be purely actin-mediated (Wittmann and Waterman-Storer, 2001). To gain mechanistic insight into neurite initiation, we first characterized the detailed sequence of events during this process using primary hippocampal neurons. We then extended our analyses using Neuro-2a neuroblastoma cells as a more tractable model system in which the timing of neurite induction can be synchronized and controlled by incubation with retinoic acid (Shea et al., 1985; Wu et al., 1998).
Coordinated reorganization of microtubule- and actin-rich structures during spontaneous neurite initiation in hippocampal neurons
Before neurite initiation, primary hippocampal neurons exhibit specialized peripheral, actin-rich structures called lamellipodia (Dotti et al., 1988) (Fig. 1A). After neurite initiation, similar lamellipodial structures, called growth cones, are located at the tips of neurites. At this stage, microtubules, which were previously concentrated in the cell soma, become organized into bundled arrays inside neurites (Fig. 1B). Previously, it was reported that lamellipodia disappear before neurite initiation and that nascent neurites emerge as pointed bud-like structures that lack recognizable growth cones (Dotti et al., 1988; da Silva and Dotti, 2002). Such a sequence of events suggests that growth cones emerge as distinct entities and thus might be fundamentally different from lamellipodia.
Figure 1.

Hippocampal neurons initiate neurites by coordinated rearrangements of F-actin- and microtubule-rich structures. A, B, Cytoskeletal organization of cultured neurons before and after neurite initiation. Primary hippocampal neurons were prepared according to the method of Goslin et al. (1998), fixed 20 hr after plating, and stained for F-actin (red) and βIII-tubulin (green). Neurons that have not yet formed neurites are characterized by fairly uniform actin-rich lamellipodia (A). After neurite formation (B), microtubules become bundled inside elongated, thin neurite shafts. The original actin-rich lamellipodia transform into smaller structures, called growth cones, on the tips of these neurites. At both stages, microtubules are concentrated in the cell body and neurites. However, note that single microtubules invade the actin-rich structures and are frequently seen to align with F-actin cables in filopodia (insets). C, Morphological changes in actin-rich lamellipodia are coordinated with the growth of microtubule-rich neurites. DIC images were acquired beginning 1 hr after plating. Time points (hours:minutes) were selected to illustrate the appearance of representative intermediates of neurite initiation (also see Fig 1.mov). Initially, most neurons display one or few fairly uniform lamellas (00:00, arrowhead). Then these veil-like structures segregate (04:10, small arrows) to form multiple individual lamellipodia, which begin to resemble growth cones. After a delay, neurite elongation proceeds, and a thin shaft condenses between cell body and nascent growth cone (04:44, 06:23, large arrows). During this time, some lamellipodia continue to segment still further (06:23, small arrows). The delay is highly variable, ranging from minutes to hours. In the example shown, after a second delay, one minor process elongated rapidly and eventually became the axon (19:59, asterisk), as is typical of hippocampal neurons.
We reinvestigated this issue using high-resolution time-lapse imaging with DIC optics to document the sequence of cellular events preceding and during neurite initiation. Contrary to previous suggestions, we observe that neurite initiation usually starts from one or more large lamellipodia, which segment and later become growth cones on the tips of neurites. As in the example in Figure 1C (also see Fig. 1.mov, available at www.jneurosci.org), a broad lamellipodium (00:00, arrowhead) first becomes segmented at several positions to form localized, growth cone-like structures that are initially adjacent to the cell body (04:10, small arrows). After a variable delay, these lamellipodia either segment further to form multiple neurite initiation sites (06:23, small arrows), or migrate outward as a condensed neurite shaft forms behind (06:23, 9:06, large arrows).
Throughout the entire sequence of initiation, lamellipodia rarely collapse completely but instead become the tip of nascent neurites. In the rare cases of collapse, such events usually accompany neurite retraction and never directly precede neurite initiation. In single images, some protrusions can occasionally resemble bud-like membrane protrusions. However, such small protrusions are transient, and time-lapse imaging using DIC optics reveals that they exhibit typical lamellipodial behavior, including membrane ruffling and retrograde flow. For example, in 15 representative cells recorded throughout neurite initiation, 33 of 36 neurites clearly emerged from existing lamellipodia; three initiation events were ambiguous. Thus, for primary hippocampal neurons, >90% of neurites emerge from lamellipodia. However, we cannot exclude the possibility that different initiation structures might exist in vivo or in different neuronal model systems.
Examination of actin filament organization using phalloidin staining suggests that growth cones and their lamellipodial precursors are very similar in containing both F-actin meshwork and filopodia-like F-actin bundles (Fig. 1A,B) (cf. Schaefer et al., 2002). Furthermore, single microtubules are frequently seen in alignment with actin bundles in both lamellipodia and growth cones. Despite little apparent change in the overall organization of actin filaments, we observe that lamellipodial structures become spatially relocated to positions more distant from the cell center, whereas the membrane condenses around a nascent microtubule-rich neurite shaft. This implies that the underlying actin and microtubule cytoskeletons are locally and dynamically regulated. Proteins such as MAP2c, which can interact with both microtubules and F-actin, are strong candidates to coordinate the reorganization of microtubule- and actin-rich structures during neurite initiation.
MAP2c initiates neurites in Neuro-2a cells
We showed previously that the small embryonic isoform MAP2c can associate with either microtubules or the actin cytoskeleton in HeLa cells (Ozer and Halpain, 2000), making it a plausible candidate for coordination of microtubule and actin reorganization during neurite initiation. Indeed, transfection of MAP2c into Neuro-2a cells led to a 2.4-fold increase in the percentage of cells bearing neurites compared with GFP-transfected controls (Fig. 2A,B). For purposes of quantification, we defined a neurite as a narrow protrusion that contains microtubules and has a length of at least one cell body diameter. Cells expressing both high and low levels of MAP2c displayed robust neurite formation. MAP2c induced 1.6 ± 0.1 (mean ± SEM) neurites per cell (n = 60). MAP2c-induced neurites often terminated in wide, flat growth cone-like structures that morphologically resembled those seen on minor processes of primary hippocampal neurons (Fig. 2A; see Fig. 6D).
Figure 2.
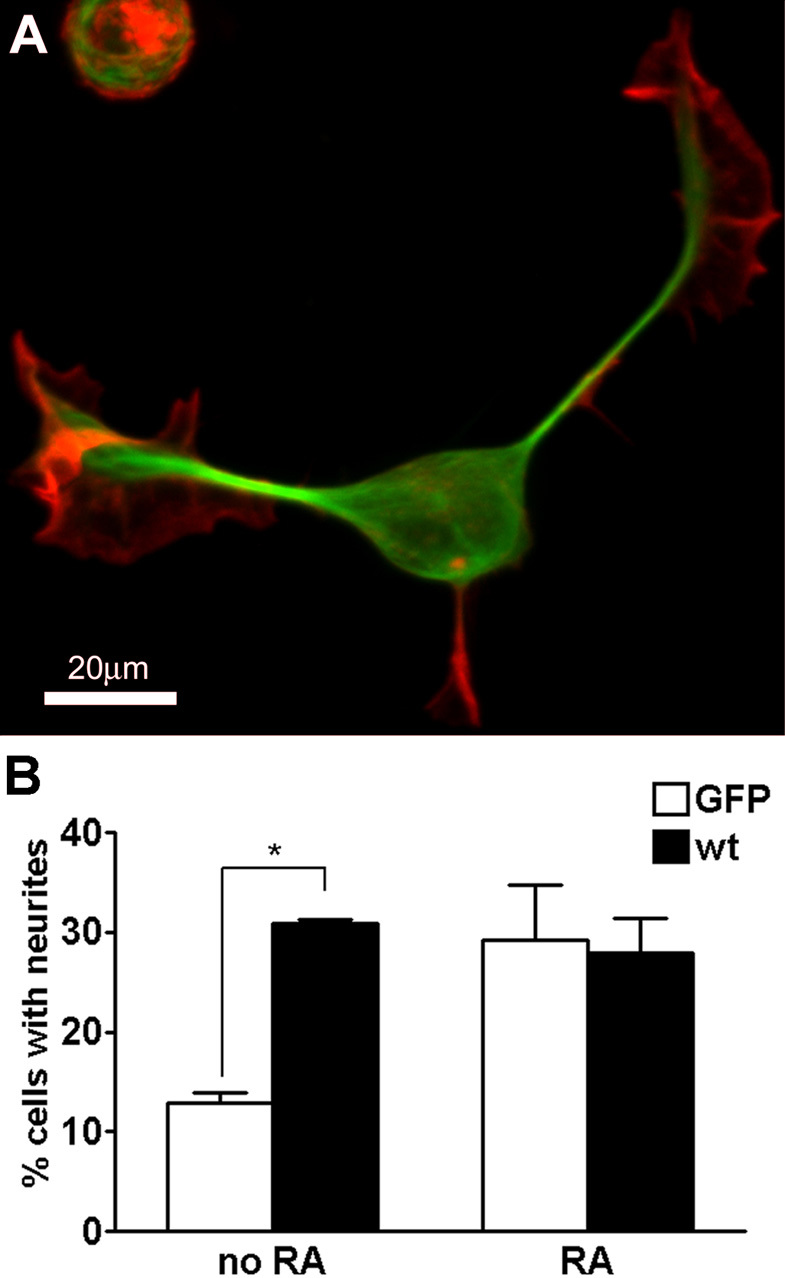
MAP2c is sufficient to induce neurite formation. A, Neuro-2a cell transfected with GFP-MAP2c (green), fixed with glutaraldehyde after 24 hr, and stained for F-actin (red). B, Quantification of neurite initiation by MAP2c. Neuro-2a cells were transfected with GFP-MAP2c or control plasmids in the absence or presence of 40 μm retinoic acid (RA) and evaluated for neurite formation as described in Materials and Methods. MAP2c induced neurites to a similar extent as retinoic acid treatment. Overexpression of MAP2c had no additional effect on retinoic acid-induced neurite induction. Data represent three independent experiments each with >100 cells evaluated per treatment condition (*p < 0.05, one-way ANOVA). wt, Wild type.
Figure 6.

Neuro-2a cells form neurite-like processes in response to the combined action of Taxol and inhibitors of actin dynamics. A, Four hours after application of 1 μm Taxol alone or combined with 4 μm cytoD, 1 μm latA, or 40 nm jasp, cells were fixed and stained for α-tubulin (green) and F-actin (red) and imaged using confocal microscopy. B, Concentration dependence of process induction in Neuro-2a cells after 4 hr of incubation with the indicated actin-disrupting drug in the presence of 1 μm Taxol. Processes were identified as described in Materials and Methods, and >100 cells were evaluated per data point. C, Representative kymograph obtained for measurement of retrograde flow (rf) before and after application of 1 μm Taxol and 40 nm jasp (for the DIC time-lapse recording used to produce this kymograph, see Fig 6.mov). The image represents a depiction of pixel intensity values measured along the yellow line indicated in the inset at the right, plotted as distal to proximal in the horizontal dimension and elapsed time in the vertical dimension. Diagonal lines represent the retrograde movement of particles within the lamellipodium. These were used to measure the velocity of retrograde flow. The two panels depict the first and second 30 min of the time-lapse sequence, respectively. D, Comparison of MAP2c-induced neurites (green, EGFP-MAP2c) with drug-induced processes (enlargements from A; green, α-tubulin) and minor processes of primary hippocampal neurons (HN; green, endogenous MAP2). F-actin is stained using phalloidin (red). All processes contained a microtubule array in the shaft. MAP2c-induced neurites usually terminated in growth cones, whereas neurites induced by Taxol plus jasp were devoid of detectable growth cones, and neurites induced by Taxol plus cytoD terminated in small F-actin-containing structures. Note that jasp competes with phalloidin binding, leading to underestimation of F-actin content in the image. However, DIC images confirm the lack of normal growth cones (see Fig 6.mov).
Incubation of control Neuro-2a cells with retinoic acid led to a similar increase in neurite induction (Fig. 2B). However, transfection of retinoic acid-treated cells with MAP2c did not further enhance neurite initiation, suggesting that both treatments share a common mechanism. Indeed, differentiation of Neuro-2a cells with retinoic acid leads to upregulation of MAP2 (Fischer et al., 1986; Wu et al., 1998).
MAP2c induces coordinated reorganization of microtubule- and actin-rich structures
Using time-lapse video microscopy, we observed that MAP2c induced neurites in Neuro-2a cells by invasion of multiple microtubules into existing lamellipodia, similar to the case in hippocampal neurons. In the example shown in Figure 3 (see Fig 3.mov), the cell was first characterized by a large, uncondensed lamellipodium (time point 00:00), into which single or a few MAP2c-decorated microtubules invaded at random for several minutes without inducing a neurite (time point 00:12). The broad lamellipodium was highly dynamic, and it quickly became segmented into multiple smaller lamellipodia. At a later stage (00:55), a prominent MAP2c-decorated microtubule bundle accumulated in the lamellipodium. Subsequently, this process elongated, and the neurite persisted for at least 2 hr.
Figure 3.

Lamellipodia segregate and condense during MAP2c-induced neurite initiation. Neuro-2a cells were transfected with GFP-MAP2c and imaged using confocal microscopy as described in Materials and Methods. The extent of the plasma membrane of the cell (white line) was manually traced from images in which low-level signals were maximized. At the beginning of the time sequence, the cell exhibited a broad lamellipodium (00:00, 00:12) surrounding a large portion of the cell body. MAP2c-decorated microtubules were readily visualized during the time course. During the first minutes, single microtubules or small microtubule bundles emerged from the soma and penetrated into the lamellipodium without inducing neurites (00:12). At a later stage (00:55), the lamellipodium became segmented, and shortly thereafter, a thick microtubule bundle rapidly formed (00:55, 01:28, 01:34). This neurite remained stable for at least 2 hr (also see Fig 3.mov; time points represent hours:minutes). Fluorescent images represent either confocal planes or three-dimensional reconstructions (3d Recon).
Using simultaneous time-lapse recording of Neuro-2a cells doubly transfected with GFP-γ-actin and dsRed2-MAP2c, we observed that single MAP2c-decorated microtubules frequently aligned with actin bundles (Fig. 4A, arrowheads), similar to previous studies in Aplysia growth cones (Schaefer et al., 2002) or newt lung epithelial cells (Salmon et al., 2002). During the time-lapse recording, one of these actin bundles breaks, and the microtubule is seen to break in parallel, suggesting that the two structures are mechanically coupled.
Figure 4.
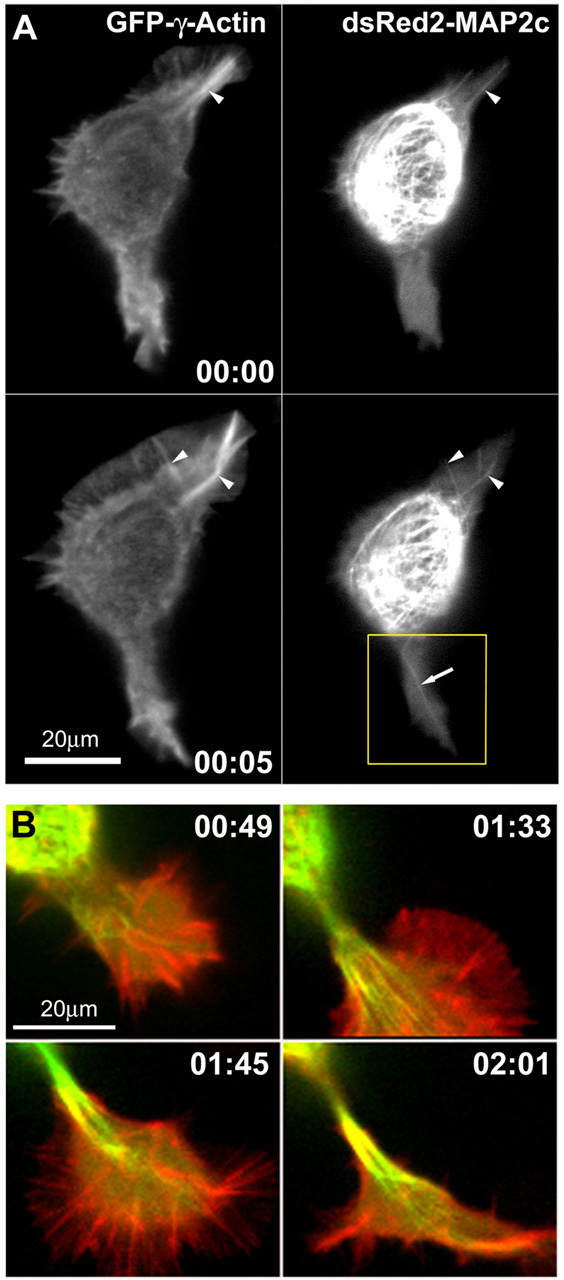
Changes in microtubules and F-actin during neurite initiation. A, MAP2c-decorated microtubules frequently align with actin cables in lamellipodia of Neuro-2a cells (also see Fig 4.mov; time points represent hours:minutes). Neuro-2a cells were cotransfected with plasmids encoding GFP-γ-actin and dsRED2-MAP2c and imaged using a spinning disk confocal microscope. Red and green fluorescence signals were acquired at intervals of 10 sec, with a time delay of 3.5 sec between the two channels. Single MAP2c-decorated microtubules (arrow) frequently invade deeply into the lamellipodia and align with actin cables (arrowheads). At time 0:05, a large actin bundle in the lamellipodium at the top right breaks, and an associated microtubule bundle appears to break in concert. The boxed region is shown in B at later time points, as this protrusion goes on to produce a neurite. B, Disordered microtubule arrays become ordered and aligned into bundles to form a neurite shaft. These panels depict four later time points in observations of the lower lamellipodium shown in A. Images are digital overlays of the GFP-actin fluorescence shown here in red and the dsRed2-MAP2c fluorescence shown here in green. Initially, the process is invaded by multiple microtubules, which are disorganized (0:44) but eventually become aligned into quasiparallel arrays (1:33). Thereafter, the microtubule array becomes quickly condensed into a compact bundle (1:45). The process elongates as microtubules continue to advance distally into the growth cone, and they become tightly packed into bundles behind the growth cone core (02:01).
As before, single MAP2c-decorated microtubules often invaded actin-rich lamellipodia without inducing a neurite (see Fig 4.mov). In our observations neurite formation was always preceded by the invasion of multiple MAP2c-decorated microtubules; however, because of limitations in optical resolution, we were not able to determine the threshold of microtubules necessary to induce a neurite. Microtubule behavior is exemplified in the lower lamellipodial protrusion seen in Figure 4A, which, as shown in Figure 4B, goes on to develop into a neurite. First, multiple MAP2c-decorated microtubules accumulate in the base of the lamellipodium (Fig. 4B, 00:49). These microtubules are dynamic and initially oriented in multiple directions; however, they subsequently become aligned into parallel arrays to form a bundle. Concurrent with microtubule bundle formation, the lamellipodium condenses at its base near the cell body to form a nascent, actin-rich growth cone (Fig. 4B, 00:49, 01:33). The end result is a protrusion having a narrow neck, which contains bundled microtubules and is tipped by a growth cone. This transition, from initial microtubule invasion to microtubule alignment to bundling and lamellipodial condensation, usually occurs rapidly (in the example shown in Fig. 4, over ∼1 hr and in other examples as fast as 15 min; e.g., Fig. 5).
Figure 5.

Neurites in Neuro-2a cells exhibit dynamic behaviors. A, Growth cone dynamics in MAP2c-transfected Neuro-2a cells. Simultaneous DIC and fluorescence imaging of a GFP-MAP2c-transfected cell revealed the rapid formation of a growth cone-like structure (large arrows) at the tip of a newly formed neurite (also see Fig 5.mov; time points represent hours:minutes). On encountering a nonpermissive substrate (a scratch in the surface of the coverslip), the growth cone structure collapsed, and the neurite retracted rapidly, showing morphological characteristics of those in primary neurons, including a retraction bulb (arrowhead) and trailing membrane remnants (small arrow). B, Rapid bundling of MAP2c-decorated microtubules during process formation. Shown is a magnified view of the boxed portion of A. During the first 5 min, a short protrusion containing a few unbundled MAP2c-decorated microtubules was formed (arrowhead). During the next 10 min, the microtubules quickly formed a packed array to produce a transient, neurite-like structure.
MAP2c-induced neurites behave similarly to spontaneously formed neurites in hippocampal neurons
During both extension and retraction processes, MAP2c-induced neurites exhibit morphology and dynamic behaviors that closely resemble those of primary neurons. Figure 5A demonstrates the rapid transition of a condensed lamellipodium at a neurite initiation site to form the growth cone at the tip of the neurite (Fig. 5A, arrows; see Fig 5.mov). Ultimately, in this example, the growth cone collapsed, and the neurite retracted after encountering a scratch on the glass coverslip (presumably a surface nonpermissive for growth). During this process, a retraction bulb and trailing membrane remnants formed (Fig. 5A, arrowhead, small arrow respectively), similar to such retraction events in primary neurons (Griffin and Letourneau, 1980).
We did note some differences between hippocampal neurons and Neuro-2a cells. In particular, neurite growth rates were lower in hippocampal neurons, which exhibited initial net growth rates of 0.31 ± 0.1 μm/min, reaching instantaneous speeds of up to 0.93 μm/min (n = 10). By comparison, the initial net growth rate of MAP2c-induced neurites was higher (1.33 ± 0.2 μm/min; n = 10). MAP2c-induced processes also retracted more frequently over greater distances and spent less time pausing. Figure 5B shows a magnified view of the smaller, transient neurite shown in Figure 5A (box), again demonstrating the rapid alignment of initially splayed MAP2c-decorated microtubules to form a bundle.
Microtubule stabilization is not sufficient for neurite initiation
A simple explanation for MAP2c-induced neurite initiation might be that stabilization of microtubules, a well known effect of MAP2c, is sufficient to induce neurites in neuroblastoma cells. However, we observe that stabilization of microtubules alone by incubation for 4 hr with the drug Taxol (1 μm) was not sufficient to induce neurite-like protrusions (Fig. 6A). Under control conditions, Neuro-2a cells were round, with ∼30% exhibiting broad, actin-rich lamellipodia at their peripheries. Less than 1% of control or Taxol-treated cells exhibited neurite-like protrusions (n > 100 cells per group). These observations indicate that stable microtubules per se are not sufficient to induce neurite-like processes.
Requirement for F-actin alterations in neurite initiation
MAP2c can also interact with actin, resulting in bundle formation in vitro (Selden and Pollard, 1983); however, the effects of MAP2c on actin in vivo are not known. In an attempt to mimic actin-related cell properties that might be involved in neurite induction, we treated Neuro-2a cells with compounds that affect polymerization or depolymerization of actin through distinct mechanisms. cytoD stops addition of monomers to the fast-growing barbed end (Cooper, 1987); latA sequesters actin monomers (Spector et al., 1989); and jasp stabilizes and nucleates actin filaments (Bubb et al., 2000). Each of these three drugs induced the loss of lamellipodia in Neuro-2a cells, confirming their effects on actin dynamics. However, none led to a significant induction of neurite-like processes. Interestingly, however, when cells were simultaneously incubated with the actin-perturbing drugs and Taxol together, we observed consistent formation of neurite-like extensions (Fig. 6A). Treatment with Taxol plus cytoD, latA, or jasp yielded an average of 5.1 ± 0.8, 7.1 ± 0.6, or 6.0 ± 0.5 processes per cell, respectively (n = 40). This effect was maximal within 4 hr and was dose-dependent over a narrow concentration range of actin-perturbing drugs (Fig. 6B). Furthermore, this morphological change was induced by a wide range of Taxol concentrations (0.1-20 μm) and was not specific for Neuro-2a cells because we observed a similar effect in non-neuronal HEK-293 cells (see supplementary Fig. S1). This contrasts with previous observations using similarly treated human hepatoma PLC cells (Edson et al., 1993), where cytoD plus Taxol failed to induce processes. In that study, numerous small microtubule bundles were detected inside the cells. Such bundles are indicative of frequent breakage of microtubules; thus microtubules might not have grown sufficiently long to promote process formation.
Although drug-induced processes are different from MAP2c-induced or spontaneously formed neurites in certain respects (number per cell, dynamic behavior, and F-actin organization), these processes also share many similarities (diameter, length, bundled microtubules in the shaft, and active outgrowth from the cell body). Thus, although drug-induced processes do not precisely phenocopy MAP2c-induced neurites, they nevertheless reinforce the notion that alterations in both microtubules and F-actin are required for initiation of neurite-like protrusions. In fact, because all three actin compounds induced collapse of the lamellipodia, it suggests that some aspect of F-actin is strongly inhibitory to neurite induction. Previous experiments suggested that alterations in both microtubules and F-actin are important for the related but distinct phenomenon of neurite elongation as well. Similar to our results in neurite initiation, application of either cytochalasin B or Taxol to cultured dorsal root ganglion neurons inhibited neurite elongation, whereas this inhibition was reversed by simultaneous application of both drugs (Letourneau et al., 1987).
Cessation of retrograde flow correlates with drug-induced outgrowth of neurite-like protrusions in Neuro-2a cells
There are several potential mechanisms whereby microtubule stabilization combined with actin disruption could elicit growth of neurite-like processes. Previous studies in Aplysia growth cones and migrating epithelial cells have suggested that retrograde flow, which is defined as a centripetal movement of the actin meshwork from the cell periphery toward the center, acts to inhibit the advance of microtubules toward the leading edge (Waterman-Storer and Salmon, 1997; Schaefer et al., 2002). Using time-lapse video microscopy, we observed a strong temporal correlation between the cessation of retrograde flow and initiation of neurite-like processes after combined jasp and Taxol treatment (Fig. 6C; see Fig 6.mov). We constructed kymographs (see Materials and Methods) and measured the rate of retrograde flow as the speed of endogenous particle movement in lamellipodia, as shown in Figure 6C.
Under control conditions, we observed retrograde flow rates of 5.2 ± 0.3 μm/min (n = 12) in lamellipodia of Neuro-2a cells, similar to rates of retrograde flow reported for neuronal growth cones (Schaefer et al., 2002). Shortly after combined drug application, a partial collapse of lamellipodia was typically observed. In the example shown in Figure 6C, complete lamellipodial collapse and cessation of flow occurred ∼24 min after drug application. Within seconds to minutes thereafter, thin processes began to emerge. These protrusions typically terminated in small, bulbous, semiadherent structures and underwent repeated extension and retraction as they elongated rapidly (see Fig 6.mov). Staining for F-actin revealed that drug-induced neurites do not form normal growth cones, although F-actin is often detectable at the tip (Fig. 6D).
In contrast to these observations, we have so far not detected reductions in retrograde flow during the process of initiation of MAP2c-induced neurites or spontaneously formed neurites in primary hippocampal neurons. This suggests that the mechanism by which MAP2c overcomes the inhibitory effects of F-actin may be different from the action of drugs that inhibit retrograde flow.
MAP2c binding to microtubules is necessary for neurite initiation; binding to cAMP-dependent protein kinase is modulatory
We used mutated MAP2c variants (Fig. 7A) to analyze the significance of the two best-characterized interaction partners for MAP2c: microtubules and the RII regulatory subunit of cAMP-dependent protein kinase (PKA). To inhibit binding to PKA, we deleted the relevant binding site between amino acids 85-104 (Vijayaraghavan et al., 1999). Previously, we described MAP2c pseudo-phosphorylated at the three KXGS motifs within the microtubule-binding domain. At these sites, serines 319, 350, and 382 were mutated to glutamic acid (MAP2c-EEE), which strongly impairs the ability of MAP2c to bind and stabilize microtubules (Ozer and Halpain, 2000). Finally, we generated the double mutant (MAP2c-ΔRII-EEE), which is impaired in both microtubule and PKA interaction. Transfection of these mutants into Neuro-2a cells revealed that the capacity for neurite initiation by MAP2c-ΔRII is reduced by 43% compared with wild-type MAP2c (p < 0.05, one-way ANOVA; n = 3; Fig. 7B,D). This suggests that targeting of PKA by MAP2c is modulatory but not completely essential for neurite formation. In contrast, MAP2c-EEE completely failed to induce neurites (Fig. 7C,D), indicating that binding of MAP2c to microtubules is necessary for neurite initiation.
Figure 7.

Functional significance of MAP2c-interacting proteins during neurite initiation. Neuro-2a cells were transfected to express variants of GFP-MAP2c (green), fixed, and stained for F-actin (red). A, Domain diagram depicting MAP2c mutants used in this study. B, Cells transfected with GFP-MAP2c-ΔRII were able to induce neurites; however, the efficiency was significantly reduced. C, Pseudophosphorylated MAP2c (GFP-MAP2c-EEE), which is strongly impaired in microtubule binding, failed to induce neurites. D, Quantification of neurite formation. Data represent three independent experiments, each with >100 cells evaluated per treatment condition (*p < 0.05; ***p < 0.001, one-way ANOVA). MT, Microtubules; MTBR, microtubule binding repeats; wt, wild type.
MAP2c mutants are dominant negative inhibitors of neurite initiation
Retinoic acid-induced neurite formation is correlated with upregulation of MAP2 expression (Fischer et al., 1986; Wu et al., 1998). Treatment of control Neuro-2a cells with 40 nm retinoic acid induced a twofold to threefold increase in the percentage of cells exhibiting neurites after 24 hr (Fig. 8A,D). Surprisingly, expression of the mutant MAP2c-EEE completely inhibited this increase (Fig. 8B,D). This inhibition does not appear to reflect merely a delay in neurite formation because this effect was also evident after 72 hr. Because MAP2c-EEE is strongly inhibited in microtubule binding and should therefore not interfere with microtubule stabilization by any endogenous MAP induced by retinoic acid, this dominant negative effect seems to point to a different function of MAP2c. It was not a consequence of mislocalization of the regulatory subunit II of PKA from sites important for neurite formation and elongation because a double mutant impaired in binding both PKA and microtubules (MAP2c-EEEΔRII) was equally effective in inhibiting retinoic acid-induced neurite formation (Fig. 8C,D).
Figure 8.

Expression of MAP2c pseudophosphorylated at KXGS sites blocks retinoic acid-induced neurite formation. Neuro-2a cells were treated with retinoic acid 4 hr after transfection for a total time of 24 or 72 hr. Cells were fixed and analyzed for the presence of neurites using combined phase contrast and fluorescence microscopy (transfected cells are marked by asterisks). A, The expression of GFP alone did not inhibit retinoic acid-induced neurite formation. B, C, Expression of the mutants GFP-MAP2c-EEE (B) and GFP-MAP2c-EEEΔRII (C) significantly reduced neurite initiation. D, Quantification of neurite formation. Retinoic acid (RA) induced a significant increase in neurite number, which was inhibited by the EEE mutants of GFP-MAP2c to control levels. Data represent three independent experiments, each with >100 cells evaluated per treatment condition (**p < 0.01 for 24 hr RA; p < 0.001 for 72 hr RA, one-way ANOVA).
This dominant negative effect was also observed in primary hippocampal neurons (Fig. 9A,B). Because of low transfection efficiency at the day of plating, we analyzed neurons transfected 1 d after plating and fixed 24 hr thereafter. Although this time frame is somewhat later than the period of maximal neurite initiation, we nonetheless observed a statistically significant decrease in neurite number in GFP-MAP2c-EEE-transfected neurons compared with control neurons transfected with GFP alone (Fig. 9C). Such a reduction could be an indirect effect resulting from inhibition of neurite elongation. However, this appears to be not the case because the mean length of neurites was not altered significantly during this period (Fig. 9D).
Figure 9.
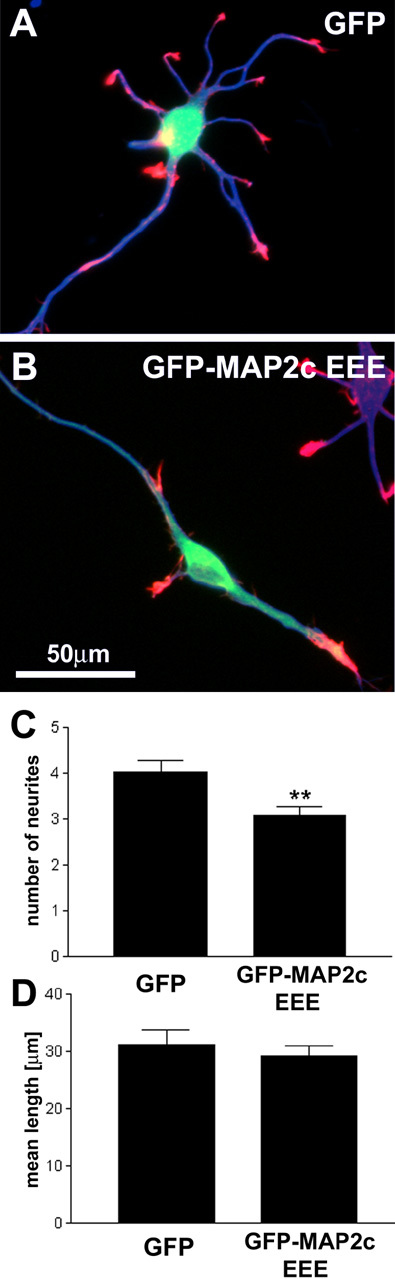
MAP2c EEE inhibits neurite initiation in primary hippocampal neurons. Primary hippocampal neurons were transfected with GFP or GFP-MAP2c EEE (green) 1 d after plating. Neurons were fixed 24 hr after transfection and stained for the neuron-specific marker βIII-tubulin TUJ1 (blue) and F-actin (red). A, Transfection of GFP alone does not induce significant alterations in the development of hippocampal neurons. B, Transfection of GFP-MAP2c-EEE induces a significant reduction of neurite number. Other aspects of neuromorphogenesis, such as polarization and axon elongation and branching, appear normal during this time frame. C, Quantification of neurite number per cell (defined as protrusions of length >10 μm). Data represent three independent experiments with a total of 40 neurons evaluated per treatment condition (**p < 0.01, Student's t test). Axons were excluded from the analysis. D, Quantification of mean neurite length per cell (total neurite arbor/number of neurites). Data represent three independent experiments with a total of 40 neurons evaluated per treatment condition (p > 0.05, Student's t test). Axons were excluded from the analysis.
Discussion
These results provide novel observations on neurite initiation and specifically advance our understanding of MAP2 function in this context. First, we show what, to our knowledge, is the first complete recording of neurite initiation in primary neurons. We demonstrate that neurites are initiated by a gradual transformation and outward migration of actin-rich lamellipodia to form growth cones, and that this occurs concurrently with extension and narrowing of the microtubule-rich neurite shaft. Second, we present new information regarding the function of the microtubule-associated protein MAP2 in very early stages of neuromorphogenesis. Using Neuro-2a cells, we demonstrate that MAP2c triggers neurite initiation. This activity depends critically on its ability to bind microtubules and is enhanced by its ability to bind PKA. Unexpectedly, we find that mutant forms of MAP2c lacking microtubule- and PKA-binding domains act in a dominant negative manner to inhibit neurite initiation in both neuroblastoma cells and primary neurons. Finally, by comparing neurite formation induced by MAP2c with that induced by cytoskeletal disrupters, we deduce key principles concerning the roles of microtubules, actin filaments, and MAPs in neurite initiation. We conclude, contrary to earlier suggestions, that neurite initiation is equally dependent on reorganization in both microtubules and actin-rich lamellipodia, and that MAP2c must be capable of effecting both changes, either directly or indirectly, to account for its ability to induce neurites in neuroblastoma cells.
Sequence of neurite initiation
Our observations suggest the following sequence of events for neurite initiation: (1) segmentation of the peripheral lamellipodium into smaller lamellipodia; (2) invasion of multiple microtubules into lamellipodia; (3) alignment of multiple microtubules into parallel arrays; (4) condensation of the lamellipodial membrane at the base of the microtubule bundle, resulting in a nascent neurite shaft proximal to the cell body and a growth cone distal from the cell body; and (5) elongation and further condensation of the nascent neurite. Clearly, microtubules play important roles in steps 2-5; it will be of interest to determine whether microtubules, MAP2c, or both also actively participate in the first step, lamellipodial segmentation.
Earlier studies observed bundles of microtubules associated with sites of process initiation or branching; however, the relationship of such bundles to neurite initiation versus neurite elongation is unclear. Tang and Goldberg (2000) reported that formation of microtubule bundles precedes laminin-induced outgrowth of new branches from existing growth cones. In contrast, Dent and Kalil (2001) emphasized that a loop of bundled microtubules splays apart (i.e., “de-bundles”) before invasion of individual microtubules into lamellipodia to establish nascent axon branches. Our observations in hippocampal neurons and similar ones by Yu et al. (2001) in sympathetic neurons lead us to postulate that the invasion of many individual microtubules into a lamellipodium initiates the neurite, and at a later time point, the bundling of those microtubules serves to elongate and stabilize the new process.
A recent review suggests common mechanisms between neurite formation and yeast budding (da Silva and Dotti, 2002). Indeed, some structures that we observed in single images appear to resemble buds. However, using DIC time-lapse microscopy, we obtained more details regarding neurite and lamellipodial morphology. We nearly always observe a seamless conversion of lamellipodia into growth cones, rather than emergence of growth cones from buds or other small membrane protrusions.
Drug-induced process formation
The finding that disruption of actin in the presence of Taxol induces robust formation of neurite-like processes suggests that microtubule stabilization promotes neurite formation, whereas one or more properties of actin inhibit neurite formation. One of these properties could be retrograde flow because we find a strong temporal correlation between its inhibition and the initiation of neurite-like processes. We frequently observe MAP2c-decorated microtubules getting swept back by retrograde flow as they invade the distal portions of lamellipodia, confirming this strong inhibitory influence. Tensile forces in the cortical actin cytoskeleton might also play a role in suppressing process formation (Edson et al., 1993). Indeed, elasticity measurements using atomic force microscopy showed that the rigidity of cells decreases after application of jasp, latA, or cytoD (Rotsch and Radmacher, 2000) at concentrations similar to those effective in promoting neurite-like processes in our study.
Role of MAP2 in neurite initiation
In contrast to drug-induced processes in Neuro-2a cells, cultured hippocampal neurons form neurites without collapse of lamellipodia or detectable inhibition of retrograde flow. Here, the inhibitory effects of tension, retrograde flow, or both are presumably overcome by coordinated growth of the microtubule-rich neurite shaft and advance of the actin-rich lamellipodium. Expression of MAP2c in Neuro-2a cells induces a similar behavior, suggesting that MAP2c is sufficient either directly or indirectly to promote microtubule growth in the neurite shaft and to alter actin behavior to promote lamellipodial advance. Such apparent cytoskeletal coordination has also been observed in phenomena such as growth cone steering (Zhou et al., 2002) and neurite branching (Dent and Kalil, 2001). Our study uniquely attributes such coordination to the expression of a specific molecule, MAP2. It remains to be seen whether MAP2 plays a similar role in either neurite branching or pathfinding.
The well characterized property of MAP2 to stabilize microtubules is likely responsible for enhanced growth of the microtubule-rich neurite shaft. How, however, might MAP2 facilitate advance of the actin-rich lamellipodium? We suggest three nonexclusive mechanisms through which MAP2c could alter lamellipodium behavior to initiate neurite formation. First, MAP2c might recruit signaling complexes. Second, MAP2c might alter the equilibrium of forces at neurite initiation sites. Third, MAP2c might promote microtubule-F-actin interactions.
MAP2c and microtubules might affect F-actin by recruiting signaling complexes
Targeting of signaling proteins along microtubules or to microtubule plus ends gained recent attention as a possible mechanism for regulation of cell motility (Wittmann and Waterman-Storer, 2001; Fukata et al., 2002). A positive feedback loop has been proposed (Wittmann and Waterman-Storer, 2001) whereby microtubules promote actin-based cell protrusion by activating the small GTPase Rac (Waterman-Storer et al., 1999), and Rac in turn promotes microtubule stabilization via inhibition of the microtubule-destabilizing MAP stathmin/OP18 (Daub et al., 2001). Such a feedback loop might also underlie neurite initiation. Nevertheless, our results suggest that stabilization of microtubules per se is not sufficient to initiate such a feedback loop because treatment of Neuro-2a cells with Taxol alone does not induce neurite formation. However, by linking signaling molecules such as PKA or other effectors to microtubules, MAP2c might promote positive feedback between microtubules and actin. Consistent with this idea, we find that deletion of the binding site for PKA diminishes the neurite initiation activity of MAP2c.
Force-based interactions between MAP2c-induced microtubule bundles and F-actin
It is generally believed that leading edge protrusion is driven by actin polymerization (Mitchison and Kirschner, 1988; Condeelis, 1993; Suter and Forscher, 2000). However, the leading edge membrane cannot be easily pushed forward against the retrogradely flowing actin meshwork in lamellipodia. The clutch hypothesis proposed by Mitchison and Kirschner (1988) suggests that if a “clutch” in the form of a physical linkage between the extracellular matrix and the actin meshwork is engaged, actin polymerization at the leading edge pushes the membrane against the linked actin meshwork, resulting in forward protrusion (Mitchison and Kirschner, 1988;Jay, 2000).
During MAP2c-induced neurite formation, we observe accumulation of MAP2c-decorated microtubules into arrays that quickly align to form large bundles. Furthermore, in agreement with observations in non-neuronal cells (Salmon et al., 2002) and Aplysia growth cones (Schaefer et al., 2002), we observe that microtubules physically interact with F-actin at neurite initiation sites. Similar to linking F-actin to the relatively rigid extracellular matrix, as in the original clutch hypothesis, physical linkage of actin to stable, cell body-anchored microtubules might act as a clutch as well, pushing the leading edge membrane against the microtubule-linked actin meshwork.
What is the origin of MAP2c-induced microtubule bundles?
Microtubule bundle formation could be either a direct or an indirect effect of MAP2c. Direct cross-linking of microtubules by MAP2c was initially proposed and remains a possibility; however, there is evidence both for and against this notion (Lewis et al., 1989; Burgin et al., 1994). Although high-affinity dimerization seems to be ruled out for MAP2c (Burgin et al., 1994), low-affinity dimerization could be sufficient to align microtubules via the cumulative effects of large numbers of MAP2c molecules bound along the microtubule surface. Indeed, MAP2c can oligomerize, as demonstrated by the formation of homodimers and straight filaments in vitro (Wille et al., 1992; DeTure et al., 1996). Another possibility is that bundle formation indirectly results from stabilization of microtubules inside the confinement of the cell borders (Burgin et al., 1994; Ferralli et al., 1994). However, MAP2c-stabilized microtubules rapidly reform bundles throughout the cell after nocodazole washout, suggesting that spatial constraint is not a major factor (Takemura et al., 1995). Finally, MAP2c expression might alter the association or behavior of motor or adapter proteins on microtubules, which in turn results in microtubule bundling.
Direct effects of MAP2c on F-actin?
MAP2c could also directly affect F-actin behavior because an interaction between MAP2 and F-actin has repeatedly been demonstrated in vitro (Selden and Pollard, 1983; Sattilaro, 1986; Correas et al., 1990; Cunningham et al., 1997); we recently confirmed and extended these results (B. Roger, J. Al-Bassam, L. Dehmelt, R. A. Milligan, S. Halpain, unpublished results). We find that MAP2c exhibits comparable affinities for microtubules and F-actin and show that MAP2c promotes actin bundle formation in vitro. Furthermore, the microtubule-binding domain of MAP2c is both necessary and sufficient for the F-actin binding and bundling activity. Surprisingly, the highly homologous domain in tau does not bind F-actin, and we find that both tau itself and a chimeric MAP2c containing the tau microtubule-binding domain are greatly impaired in initiating neurites in Neuro-2a cells compared with MAP2c despite retaining the ability to bind and stabilize microtubules (Roger, Al-Bassam, Dehmelt, Milligan, Halpain, unpublished results). These data argue that the stabilization of microtubules by MAP2c is not sufficient for neurite initiation and that its interaction with F-actin could play a key role as well.
In cells, MAP2 is mostly localized to microtubules; however, MAP2 can also associate with the actin cytoskeleton (Morales and Fifkova, 1989; Ozer and Halpain, 2000). By binding to both actin and microtubules, MAP2 might promote interactions between these cytoskeletal structures. Such direct interaction could contribute to guidance of microtubule polymerization along actin bundles in filopodia, which channels them along the most efficient path against retrograde flow (Schaefer et al., 2002). By promoting net accumulation of microtubules into the lamellipodial periphery, guidance along F-actin could enhance both signaling cross talk and force transduction between microtubules and F-actin.
Dominant negative effects of MAP2c mutants
Dominant negative inhibition of neurite initiation by MAP2c mutants that can bind neither microtubules nor PKA might be explained by any of the above three mechanisms. First, expression of MAP2c mutants could sequester critical signaling molecules from endogenous MAPs. Here we provide evidence supporting the role of MAP2c as a scaffold for the signaling molecule PKA, but deletion of the PKA-binding site does not abolish the dominant negative effect, so perhaps other effectors have yet to be identified. Second, MAP2c mutants could interfere with microtubule bundle formation (e.g., by dimerizing with endogenous MAP2 or via intermediary molecules). Third, the mutants could interfere with actin-microtubule cross-linking. Additional structure-function studies will ultimately reveal the nature of the dominant negative actions of mutant MAP2c on neurite initiation.
Footnotes
This work was supported by National Institutes of Health Grant MH50861 (S.H.). We thank Dr. Andrew Matus for providing a GFP-γ-actin construct, Dr. A. Frankfurter for supplying antibodies against βIII-tubulin, Dr. Torsten Wittmann, Dr. Benoit Roger, and Dr. Perihan Nalbant for critically reading this manuscript, Dr. Elizabeth Gardiner for technical advice in image acquisition, Dr. Simone Graber for providing cultures of primary hippocampal neurons, Judy Chu for construction of MAP2c deletion constructs, and Julia Braga and Brian Jenkins for technical assistance.
Correspondence should be addressed to Shelley Halpain, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA 92037. E-mail: shelley@scripps.edu.
F. M. Smart's present address: Department of Neurobiology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA 92037.
R. S. Ozer's present address: Department of Psychiatry and Behavioral Sciences, Stanford University Medical Center, Stanford, CA 94304-5485.
Copyright © 2003 Society for Neuroscience 0270-6474/03/239479-12$15.00/0
References
- Bernhardt R, Matus A ( 1984) Light and electron microscopic studies of the distribution of microtubule-associated protein 2 in rat brain: a difference between dendritic and axonal cytoskeleton. J Comp Neurol 226: 203-221. [DOI] [PubMed] [Google Scholar]
- Bubb MR, Spector I, Beyer BB, Fosen KM ( 2000) Effects of jasplakinolide on the kinetics of actin polymerization: an explanation for certain in vivo observations. J Biol Chem 275: 5163-5170. [DOI] [PubMed] [Google Scholar]
- Burgin KE, Ludin B, Ferralli J, Matus A ( 1994) Bundling of microtubules in transfected cells does not involve an autonomous dimerization site on the MAP2 molecule. Mol Biol Cell 5: 511-517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caceres A, Banker GA, Binder L ( 1986) Immunocytochemical localization of tubulin and microtubule-associated protein 2 during the development of hippocampal neurons in culture. J Neurosci 6: 714-722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caceres A, Mautino J, Kosik KS ( 1992) Suppression of MAP2 in cultured cerebellar macroneurons inhibits minor neurite formation. Neuron 9: 607-618. [DOI] [PubMed] [Google Scholar]
- Condeelis J ( 1993) Life at the leading edge: the formation of cell protrusions. Annu Rev Cell Biol 9: 411-444. [DOI] [PubMed] [Google Scholar]
- Cooper JA ( 1987) Effects of cytochalasin and phalloidin on actin. J Cell Biol 105: 1473-1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correas I, Padilla R, Avila J ( 1990) The tubulin-binding sequence of brain microtubule-associated proteins, tau and MAP-2, is also involved in actin binding. Biochem J 269: 61-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham CC, LeClerc N, Flanagan LA, Lu M, Janmey PA, Kosik KS ( 1997) Microtubule-associated protein 2c reorganizes both microtubules and microfilaments into distinct cytological structures in an actin-binding protein-280-deficient melanoma cell line. J Cell Biol 136: 845-857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva JS, Dotti CG ( 2002) Breaking the neuronal sphere: regulation of the actin cytoskeleton in neuritogenesis. Nat Rev Neurosci 3: 694-704. [DOI] [PubMed] [Google Scholar]
- Daub H, Gevaert K, Vandekerckhove J, Sobel A, Hall A ( 2001) Rac/Cdc42 and p65PAK regulate the microtubule-destabilizing protein stathmin through phosphorylation at serine 16. J Biol Chem 276: 1677-1680. [DOI] [PubMed] [Google Scholar]
- Dent EW, Kalil K ( 2001) Axon branching requires interactions between dynamic microtubules and actin filaments. J Neurosci 21: 9757-9769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeTure MA, Zhang EY, Bubb MR, Purich DL ( 1996) In vitro polymerization of embryonic MAP-2c and fragments of the MAP-2 microtubule binding region into structures resembling paired helical filaments. J Biol Chem 271: 32702-32706. [DOI] [PubMed] [Google Scholar]
- Dotti CG, Sullivan CA, Banker GA ( 1988) The establishment of polarity by hippocampal neurons in culture. J Neurosci 8: 1454-1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edson K, Weisshaar B, Matus A ( 1993) Actin depolymerisation induces process formation on MAP2-transfected non-neuronal cells. Development 117: 689-700. [DOI] [PubMed] [Google Scholar]
- Ferralli J, Doll T, Matus A ( 1994) Sequence analysis of MAP2 function in living cells. J Cell Sci 107: 3115-3125. [DOI] [PubMed] [Google Scholar]
- Fischer I, Shea TB, Sapirstein VS, Kosik KS ( 1986) Expression and distribution of microtubule-associated protein 2 (MAP2) in neuroblastoma and primary neuronal cells. Brain Res 390: 99-109. [DOI] [PubMed] [Google Scholar]
- Fukata M, Watanabe T, Noritake J, Nakagawa M, Yamaga M, Kuroda S, Matsuura Y, Iwamatsu A, Perez F, Kaibuchi K ( 2002) Rac1 and Cdc42 capture microtubules through IQGAP1 and CLIP-170. Cell 109: 873-885. [DOI] [PubMed] [Google Scholar]
- Goslin K, Asmussen H, Banker GA ( 1998) Rat hippocampal neurons in low density culture. In: Culturing nerve cells (Banker GA, Goslin K, eds), pp 339-370. Cambridge, MA: MIT.
- Griffin CG, Letourneau PC ( 1980) Rapid retraction of neurites by sensory neurons in response to increased concentrations of nerve growth factor. J Cell Biol 86: 156-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada A, Teng J, Takei Y, Oguchi K, Hirokawa N ( 2002) MAP2 is required for dendrite elongation, PKA anchoring in dendrites, and proper PKA signal transduction. J Cell Biol 158: 541-549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay DG ( 2000) The clutch hypothesis revisited: ascribing the roles of actin-associated proteins in filopodial protrusion in the nerve growth cone. J Neurobiol 44: 114-125. [DOI] [PubMed] [Google Scholar]
- Kim H, Binder LI, Rosenbaum JL ( 1979) The periodic association of MAP2 with brain microtubules in vitro. J Cell Biol 80: 266-276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohrmann M, Haubensak W, Hemraj I, Kaether C, Lessmann VJ, Kiebler MA ( 1999) Fast, convenient, and effective method to transiently transfect primary hippocampal neurons. J Neurosci Res 58: 831-835. [PubMed] [Google Scholar]
- LeClerc N, Kosik KS, Cowan N, Pienkowski TP, Baas PW ( 1993) Process formation in Sf9 cells induced by the expression of a microtubule-associated protein 2C-like construct. Proc Natl Acad Sci USA 90: 6223-6227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letourneau PC ( 1996) The cytoskeleton in nerve growth cone motility and axonal pathfinding. Perspect Dev Neurobiol 4: 111-123. [PubMed] [Google Scholar]
- Letourneau PC, Shattuck TA, Ressler AH ( 1987) “Pull” and “push” in neurite elongation: observations on the effects of different concentrations of cytochalasin B and taxol. Cell Motil Cytoskeleton 8: 193-209. [DOI] [PubMed] [Google Scholar]
- Lewis SA, Ivanov IE, Lee G-H, Cowan NJ ( 1989) Organization of microtubules in dendrites and axons is determined by a short hydrophobic zipper in microtubule-associated proteins MAP2 and tau. Nature 342: 498-505. [DOI] [PubMed] [Google Scholar]
- Luo L ( 2002) Actin cytoskeleton regulation in neuronal morphogenesis and structural plasticity. Annu Rev Cell Dev Biol 18: 601-635. [DOI] [PubMed] [Google Scholar]
- Marsden KM, Doll T, Ferralli J, Botteri F, Matus A ( 1996) Transgenic expression of embryonic MAP2 in adult mouse brain: implications for neuronal polarization. J Neurosci 16: 3265-3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchison T, Kirschner M ( 1988) Cytoskeletal dynamics and nerve growth. Neuron 1: 761-772. [DOI] [PubMed] [Google Scholar]
- Morales M, Fifkova E ( 1989) Distribution of MAP2 in dendritic spines and its colocalization with actin. Cell Tissue Res 256: 447-456. [DOI] [PubMed] [Google Scholar]
- Ozer RS, Halpain S ( 2000) Phosphorylation-dependent localization of microtubule-associated protein MAP2c to the actin cytoskeleton. Mol Biol Cell 11: 3573-3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotsch C, Radmacher M ( 2000) Drug-induced changes of cytoskeletal structure and mechanics in fibroblasts: an atomic force microscopy study. Biophys J 78: 520-535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruthel G, Hollenbeck PJ ( 2000) Growth cones are not required for initial establishment of polarity or differential axon branch growth in cultured hippocampal neurons. J Neurosci 20: 2266-2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon WC, Adams MC, Waterman-Storer CM ( 2002) Dual-wavelength fluorescent speckle microscopy reveals coupling of microtubule and actin movements in migrating cells. J Cell Biol 158: 31-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattilaro W ( 1986) Interaction of microtubule-associated protein 2 with actin filaments. Biochemistry 25: 2003-2009. [DOI] [PubMed] [Google Scholar]
- Schaefer AW, Kabir N, Forscher P ( 2002) Filopodia and actin arcs guide the assembly and transport of two populations of microtubules with unique dynamic parameters in neuronal growth cones. J Cell Biol 158: 139-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selden SC, Pollard TD ( 1983) Phosphorylation of microtubule-associated proteins regulates their interaction with actin filaments. J Biol Chem 258: 7064-7071. [PubMed] [Google Scholar]
- Selden SC, Pollard TD ( 1986) Interaction of actin filaments with microtubules is mediated by microtubule-associated proteins and regulated by phosphorylation. Ann NY Acad Sci 466: 803-12:803-812. [DOI] [PubMed] [Google Scholar]
- Shea TB, Fischer I, Sapirstein VS ( 1985) Effect of retinoic acid on growth and morphological differentiation of mouse NB2a neuroblastoma cells in culture. Brain Res 353: 307-314. [DOI] [PubMed] [Google Scholar]
- Spector I, Shocet NR, Blasberger D, Kashman Y ( 1989) Latrunculins—novel marine macrolides that disrupt microfilament organization and affect cell growth: I. Comparison with cytochalasin D. Cell Motil Cytoskeleton 13: 127-144. [DOI] [PubMed] [Google Scholar]
- Suter DM, Forscher P ( 2000) Substrate-cytoskeletal coupling as a mechanism for the regulation of growth cone motility and guidance. J Neurobiol 44: 97-113. [PubMed] [Google Scholar]
- Takemura R, Okabe S, Umeyama T, Hirokawa N ( 1995) Polarity orientation and assembly process of microtubule bundles in nocodazole-treated, MAP2c-transfected COS cells. Mol Biol Cell 6: 981-996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang D, Goldberg DJ ( 2000) Bundling of microtubules in the growth cone induced by laminin. Mol Cell Neurosci 15: 303-313. [DOI] [PubMed] [Google Scholar]
- Teng J, Takei Y, Harada A, Nakata T, Chen J, Hirokawa N ( 2001) Synergistic effects of MAP2 and MAP1B knockout in neuronal migration, dendritic outgrowth, and microtubule organization. J Cell Biol 155: 65-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayaraghavan S, Liberty GA, Mohan J, Winfrey VP, Olson GE, Carr DW ( 1999) Isolation and molecular characterization of AKAP110, a novel, sperm-specific protein kinase A-anchoring protein. Mol Endocrinol 13: 705-717. [DOI] [PubMed] [Google Scholar]
- Waterman-Storer CM, Salmon ED ( 1997) Actomyosin-based retrograde flow of microtubules in the lamella of migrating epithelial cells influences microtubule dynamic instability and turnover and is associated with microtubule breakage and treadmilling. J Cell Biol 139: 417-434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman-Storer CM, Worthylake RA, Liu BP, Burridge K, Salmon ED ( 1999) Microtubule growth activates Rac1 to promote lamellipodial protrusion in fibroblasts. Nat Cell Biol 1: 45-50. [DOI] [PubMed] [Google Scholar]
- Wille H, Mandelkow EM, Mandelkow E ( 1992) The juvenile microtubule-associated protein MAP2c is a rod-like molecule that forms antiparallel dimers. J Biol Chem 267: 10737-10742. [PubMed] [Google Scholar]
- Wittmann T, Waterman-Storer CM ( 2001) Cell motility: can Rho GTPases and microtubules point the way? J Cell Sci 114: 3795-3803. [DOI] [PubMed] [Google Scholar]
- Wu G, Fang Y, Lu ZH, Ledeen RW ( 1998) Induction of axon-like and dendrite-like processes in neuroblastoma cells. J Neurocytol 27: 1-14. [DOI] [PubMed] [Google Scholar]
- Yu W, Ling C, Baas PW ( 2001) Microtubule reconfiguration during axogenesis. J Neurocytol 30: 861-875. [DOI] [PubMed] [Google Scholar]
- Zhou FQ, Waterman-Storer CM, Cohan CS ( 2002) Focal loss of actin bundles causes microtubule redistribution and growth cone turning. J Cell Biol 157: 839-849. [DOI] [PMC free article] [PubMed] [Google Scholar]