

Abstract
A transient, sublethal ischemic interval confers resistance to a subsequent, otherwise lethal ischemic insult, in a process termed ischemic preconditioning. Poly(ADP-ribose) polymerase-1 (PARP-1) normally functions in DNA repair, but extensive PARP-1 activation is a major cause of ischemic cell death. Because PARP-1 can be cleaved and inactivated by caspases, we investigated the possibility that caspase cleavage of PARP-1 could contribute to ischemic preconditioning. Murine cortical cultures were treated with glucose deprivation combined with 0.5 mm 2-deoxyglucose and 5 mm azide (“chemical ischemia”) to model the reversible energy failure that occurs during transient ischemia in vivo. Cortical cultures preconditioned with 15 min of chemical ischemia showed increased resistance to subsequent, longer periods of chemical ischemia. These cultures were also more resistant to the PARP-1 activating agent, N-methyl-N′-nitro-N-nitrosoguanidine, suggesting reduced capacity for PARP-1 activation after preconditioning. Immunostaining for the 89 kDa PARP-1 cleavage fragment and for poly(ADP-ribose) formation confirmed that PARP-1 was cleaved and PARP-1 activity was attenuated in the preconditioned neurons. Preconditioning also produced an increase in activated caspase-3 peptide and an increase in caspase-3 activity in the cortical cultures. A cause-effect relationship between caspase activation, PARP-1 cleavage, and ischemic preconditioning was supported by studies using the caspase inhibitor Ac-Asp-Glu-Val-Asp-aldehyde (DEVD-CHO). Cultures treated with DEVD-CHO after preconditioning showed reduced PARP-1 cleavage and reduced resistance to subsequent ischemia. These findings suggest a novel interaction between the caspase- and PARP-1-mediated cell death pathways in which sublethal caspase activation leads to PARP-1 cleavage, thereby increasing resistance to subsequent ischemic stress.
Keywords: apoptosis, brain, culture, cycloheximide, mouse, tolerance
Introduction
A brief period of sublethal ischemia induces resistance to a subsequent, otherwise lethal ischemic insult in a process termed ischemic preconditioning (Nandagopal et al., 2001; Kirino, 2002; Dirnagl et al., 2003). Several mechanisms have been implicated in ischemic preconditioning, and the relative importance of these mechanisms differs among different cell types (Chen and Simon, 1997; Ishida et al., 1997; Nandagopal et al., 2001; Kirino, 2002). Studies in brain suggest involvement of post-translational changes such as Ras activation (Gonzalez-Zulueta et al., 2000), altered phosphorylation state of ERK (extracellular signal-regulated kinase) and NMDA receptor subunits (Shamloo and Wieloch, 1999; Shamloo et al., 1999), and altered NMDA receptor function (Aizenman et al., 2000). Other studies suggest important roles for gene transcription and increased expression of proteins such as hsp70 (Chen et al., 1996; Currie et al., 2000), anti-oxidant enzymes (Hoshida et al., 2002), or anti-apoptosis factors (Shimazaki et al., 1994; Shimizu et al., 2001). In only a few instances, however, has it been possible to establish a cause-effect relationship between changes induced by ischemic preconditioning and increased resistance to subsequent stress.
Activation of poly(ADP-ribose) polymerase-1 (PARP-1) is a major cause of neuronal death after brain ischemia (Szabo and Dawson, 1998). Poly(ADP-ribose) polymerases are enzymes that transfer the ADP-ribose groups from NAD+ to form branched ADP-ribose polymers on acceptor proteins in the vicinity of DNA strand breaks or kinks. Several PARP family members are now recognized, with PARP-1 accounting for >80% of nuclear poly(ADP-ribose) polymerase activity (Heller et al., 1995; D'Amours et al., 1999). Although PARP-1 normally functions to facilitate DNA repair, extensive PARP-1 activation promotes cell death through processes involving energy depletion and the release of apoptosis-inducing factor (Ha and Snyder, 1999; Ying et al., 2002; Yu et al., 2002). Genetic or pharmacological inhibition of PARP-1 activity reduces infarct size by up to 80% in brains subjected to transient or permanent ischemia (Eliasson et al., 1997; Endres et al., 1997; Tokime et al., 1998; Takahashi et al., 1999). Similarly, inhibition of PARP-1 in cultured neurons substantially increases resistance to oxygen-glucose deprivation and to NMDA toxicity (Cosi et al., 1994; Ha and Snyder, 1999; Ying et al., 2001).
PARP-1 can be cleaved and inactivated by caspase-3, caspase-7, caspase-8, and calpains (D'Amours et al., 1998; Germain et al., 1999; Wang, 2000). Because caspases and calpains are activated by cerebral ischemia (Lipton, 1999; Chen et al., 2002), we hypothesized that cleavage and inactivation of PARP-1 could be an effector mechanism of ischemic preconditioning. Caspase-3 can be activated by low levels of oxidative stress or excitotoxicity (Bonfoco et al., 1995). Moreover, caspase-3 activation has recently been shown to be important for ischemic preconditioning in brain (McLaughlin et al., 2003). Here we show that sublethal activation of caspase-3 in cultured neurons by a preconditioning ischemic stimulus leads to PARP-1 cleavage, and this renders neurons resistant to insults, such as subsequent ischemia, that would otherwise induce PARP-1-mediated cell death.
Materials and Methods
Materials. Chemicals were purchased from Sigma-Aldrich (St. Louis, MO), except where noted otherwise.
Astrocyte-neuron cocultures. The animal use protocol was approved by the Animal Studies Committee of the San Francisco Veterans Affairs Medical Center. Cortical astrocyte-neuron cocultures were prepared by seeding neurons onto a preexisting astrocyte layer, as described previously (Ying et al., 1999). Wild-type (WT) neurons were prepared from fetal Swiss-Webster mice (Simonsen, Gilroy, CA). PARP-1 gene deficient (PARP-1 -/-) neurons were prepared from fetal mice of the inbred PARP-1 -/- strain developed by Wang et al. (1995) on a mixed 129/Sv x C57BL/6 background. Cocultures were maintained in glial-conditioned medium prepared by placing MEM with 2 mm glutamine, 50 μg/ml streptomycin, and 2.5% FBS supplemented with 100 nm sodium selenate and 200 nm α-tocopherol (Leist et al., 1996) into a flask of confluent cortical astrocytes for 72 hr. Experiments were conducted when neurons were 14 -15 d in vitro.
Experimental procedures. Experiments were performed using a balanced salt solution (BSS) composed of (in mm): 3.1 KCl, 134 NaCl, 1.2 CaCl2, 1.2 MgSO4, 0.25 KH2PO4, 15.7 NaHCO3, as described previously (Ying et al., 2001). The pH was adjusted to 7.2 while the solution was equilibrated with 5% CO2 at 37°C. Osmolarity was verified at 280-310 mOsm with a Wescor vapor pressure osmometer (Logan, UT). Concentrated drug stocks were prepared in BSS and likewise adjusted to pH 7.2 and 280-320 mOsm. Chemical ischemia (CI) was induced by incubating the cultures in glucose-free BSS containing 0.5 mm 2-deoxyglucose and 5 mm sodium azide in a 37°C, 5% CO2 incubator (Swanson and Benington, 1996) and terminated by washing in BSS and replacement of glial-conditioned medium.
Assessment of neuronal injury. Neurons were distinguished from the underlying astrocyte layer by their phase-bright, process-bearing morphology (Ying et al., 1999). Dead neurons were identified 24, 48, or 72 hr after chemical ischemia exposures by propidium iodide (PI) fluorescence. PI was added at 0.04 mg/ml to each well, and both the PI-fluorescing dead neurons and nonfluorescing live neurons were counted in four randomly selected optical fields using a Nikon fluorescence microscope. At least 300 neurons were counted in each well, and results from each well were expressed as “% neuronal death.”
Immunostaining. Immunostaining for poly(ADP-ribose) (PAR) was performed according to the method of Burkle et al. (1993) with modifications. After one wash in cold PBS, the cultures were fixed in 10% trichloroacetic acid on ice for 15 min, dehydrated by sequential washes in 70, 90, and 100% ethanol, and air dried. The fixed cultures were preincubated with blocking buffer (PBS), 10% goat serum, 0.1% Triton X-100) for 60 min at room temperature, and anti-PAR monoclonal antibody (Trevigen, Gaithersburg, MD) was added at a 1:2000 dilution for incubation overnight at 4°C. After washing with PBS containing 0.1% Triton X-100, the cells were incubated with Alexa-fluor 488-conjugated goat anti-mouse IgG (Molecular Probes, Eugene, OR) at 1:500 dilution for 1 hr at room temperature and then washed again to remove excess antibody. Immunostaining for the 89 kDa PARP-1 cleavage product was performed in cultures fixed in 4% paraformaldehyde for 45 min at room temperature. After preincubation in blocking buffer, rabbit polyclonal anti-cleaved PARP-1 antibody (#9544, Cell Signaling Technology, Beverly, MA) was added at a 1:500 dilution and incubated overnight at 4°C. Antibody visualization was achieved with Alexa-fluor 488-conjugated goat anti-mouse IgG.
Western blots. Cells were lysed at 4°C in buffer containing 1% SDS, 1 mm Na-vanadate, 0.1 mm phenylmethylsulfonylfluoride, 2.5 μg/ml pepstatin, 10 μg/ml aprotinin, 5 μg/ml leupeptin, and 10 mm Tris-HCl, final pH 7.4. Viscosity of the samples was reduced by brief sonication. Aliquots containing 20 μg of protein were mixed with Laemmli buffer, boiled for 5 min, and electrophoresed on a SDS-polyacrylamide gel. Proteins were transferred to a polyvinylidene difluoride membrane (Amersham Biosciences, Piscataway, NJ), incubated overnight at 4°C in Tris-buffered saline containing 5% nonfat dry milk and 0.1% Tween 20, and then incubated for 4 hr at room temperature with the primary antibody. After three washes, the membrane was incubated for 90 min with a 1:10,000 dilution of anti-mouse or anti-rabbit IgG horseradish peroxidase antibody (Vector Laboratories, Burlingame, CA). The membrane was washed three additional times, and the signal was detected with the Amersham ECL system (Amersham Biosciences). After antibody stripping, membranes were immunostained for β-actin to confirm consistent protein loading in each lane. The primary antibody dilutions were as follows: 1:1500 for rabbit polyclonal antibody to PARP-1 (#9544, Cell Signaling Technology); 1:1000 for rabbit polyclonal antibody to cleaved caspase-3 (#9661, Cell Signaling Technology); and 1:10,000 for the mouse monoclonal antibody to β-actin (Sigma-Aldrich). Bands were quantified using the SCION Image system. The analysis of β-actin expression showed negligible variation in protein loading.
Caspase-3 activity. Caspase-3 activity was measured as described by Sordet et al. (2002), with minor modifications. After lysing, cell samples were centrifuged at 10,000 × g for 20 min. The protein concentration of the resulting supernatant was measured by the bicinchonic acid method (Smith et al., 1985), and 25 μg protein aliquots were loaded into 96-well plates. A reaction mixture containing 100 μm fluorogenic peptide substrate Ac-Asp-Glu-Val-Asp-7-amino-4-methyl coumarin (Calbiochem, San Diego, CA), 1 mm EDTA, 0.1% 3-[(3-cholamidopropyl)d:methyl amino]-1-propane sulfonate, 10% glycerol, 20 mm dithiothreitol, and 100 mm HEPES, final pH 7.0 was added to the wells. After 1 hr at 37°C, fluorescence was measured using an Fmax fluorescence plate reader with Softmax Pro software (Molecular Devices, Sunnyvale, CA) at an excitation wavelength of 355 nm and an emission wavelength of 460 nm. Fluorescence values were expressed as increase over control and normalized to the increase induced by staurosporine.
Caspase inhibition. Effects of caspase inhibition on PARP-1 cleavage and ischemic preconditioning were tested by adding 25 μm of Ac-Asp-Glu-Val-Asp-aldehyde (DEVD-CHO) (Calbiochem) to the culture medium 2 hr before the preconditioning and during the subsequent 24 hr. Control experiments were performed in parallel by adding only the vehicle, 0.25% dimethylsulfoxide, to the culture medium.
Statistical analyses. Data are presented as means ± SE. Statistical significance was assessed using ANOVA and the Student-Newman-Keuls post hoc test to compare the indicated experimental groups.
Results
A model of ischemic preconditioning in mouse cortical cultures was established using inhibitors of energy metabolism (Rajdev and Reynolds, 1994; Aizenman et al., 2000). Oxidative ATP production was blocked with 5 mm azide, and glycolytic ATP production was blocked by removing glucose and adding 0.5 mm 2-deoxyglucose to prevent metabolism of retained intracellular glucose (Swanson and Benington, 1996). A 15 min period of CI was sublethal, resulting in neuronal death no greater than that caused by medium exchanges alone. In contrast, CI of ≥30 min killed >75% of the neurons (Fig. 1A,B). Cell death was assessed 24 hr after CI (Fig. 1B). The neuronal death resulting from 30 or 45 min of CI was substantially reduced in cultures that had been preconditioned with a sublethal, 15 min interval of CI 24 hr previously. Assessment of neuronal death at 48 or 72 hr after CI gave nearly identical results. This model of ischemic preconditioning was used for all subsequent studies in this report.
Figure 1.

Preconditioning (PC) increases neuronal resistance to subsequent chemical ischemia. Cultures were preconditioned with 15 min of chemical ischemia (15 min PC) or sham washes only (0 min PC). Twenty-four hours later, the cultures were exposed to 0, 30, or 45 min of CI. Cell death was assessed by propidium iodide fluorescence 24 hr after the CI. A, Representative cell fields photographed with phase-contrast and epifluorescence optics show normal, phase-bright neurons and dead neurons with nuclear propidium iodide fluorescence. Scale bar, 100 μm. B, Effect of preconditioning on neuronal resistance to CI. C, Neuronal resistance to CI at sequential time points after a 15 min ischemic preconditioning. D, Effects of CHX on ischemic preconditioning. Cultures were incubated with 10 μg/ml CHX for the 24 hr interval between the 15 min ischemic preconditioning and the onset of chemical ischemia. *p < 0.05; **p < 0.01; representative of five independent experiments each with n = 7-11.
Previous studies have reported that ischemic preconditioning requires several hours or days to become fully manifest (Kirino, 2002; Dirnagl et al., 2003). Consistent with these reports, we found that significant resistance to subsequent ischemia in this model first developed between 3 and 6 hr after the preconditioning ischemia and increased through at least 24 hr (Fig. 1C). As shown in Figure 1D, the preconditioning effect was substantially reduced in cultures treated with the protein synthesis inhibitor cycloheximide (CHX). Cycloheximide alone (in the absence of preconditioning) also had a modest neuroprotective effect, as reported previously (Lobner and Choi, 1996).
Ischemic preconditioning attenuates PARP-1-mediated neuronal death
Neuronal death caused by CI was substantially reduced by the PARP inhibitor 3,4-dihydro-5-[4-(1-piperidinyl)butoxy]-1(2H)-isoquinolinone (DPQ) and by PARP-1 genetic deletion (Fig. 2), consistent with a major role for PARP-1 activation in ischemic neuronal death. These results suggested the possibility that the resistance to ischemia conferred by ischemic preconditioning might likewise result from a block in PARP-1 activation. We tested this possibility by examining the effect of ischemic preconditioning on PARP-1 activation and neuronal death induced by N-methyl-N′-nitro-N-nitrosoguanidine (MNNG), a DNA alkylating agent widely used to trigger PARP-1 activation and PARP-1-mediated cell death (Virag and Szabo, 2002; Ying et al., 2002; Yu et al., 2002). MNNG-induced neuronal death was substantially reduced by ischemic preconditioning (Fig. 3A), as well as by the PARP inhibitor DPQ (data not shown). PARP-1 activation was also assessed by immunostaining for poly(ADP-ribose), the enzymatic product of PARP-1. In the absence of preconditioning, poly(ADP-ribose) immunoreactivity was evident in most neurons after 40 min of either CI or MNNG. These treatments produced almost no detectable poly(ADP-ribose) in cultures that had been treated with ischemic preconditioning, suggesting a reduced capacity for PARP-1 activation in the preconditioned neurons. The caspase inhibitor DEVD-CHO negated this effect of preconditioning, whereas CHX had no appreciable effect (Fig. 3B).
Figure 2.

Ischemic neuronal death is mediated by PARP-1 activation. PARP-1 -/- neurons and WT neurons in cultures pretreated with the PARP inhibitor DPQ showed markedly reduced neuronal death after CI. **p < 0.01; representative of three independent experiments each with n = 3-4.
Figure 3.
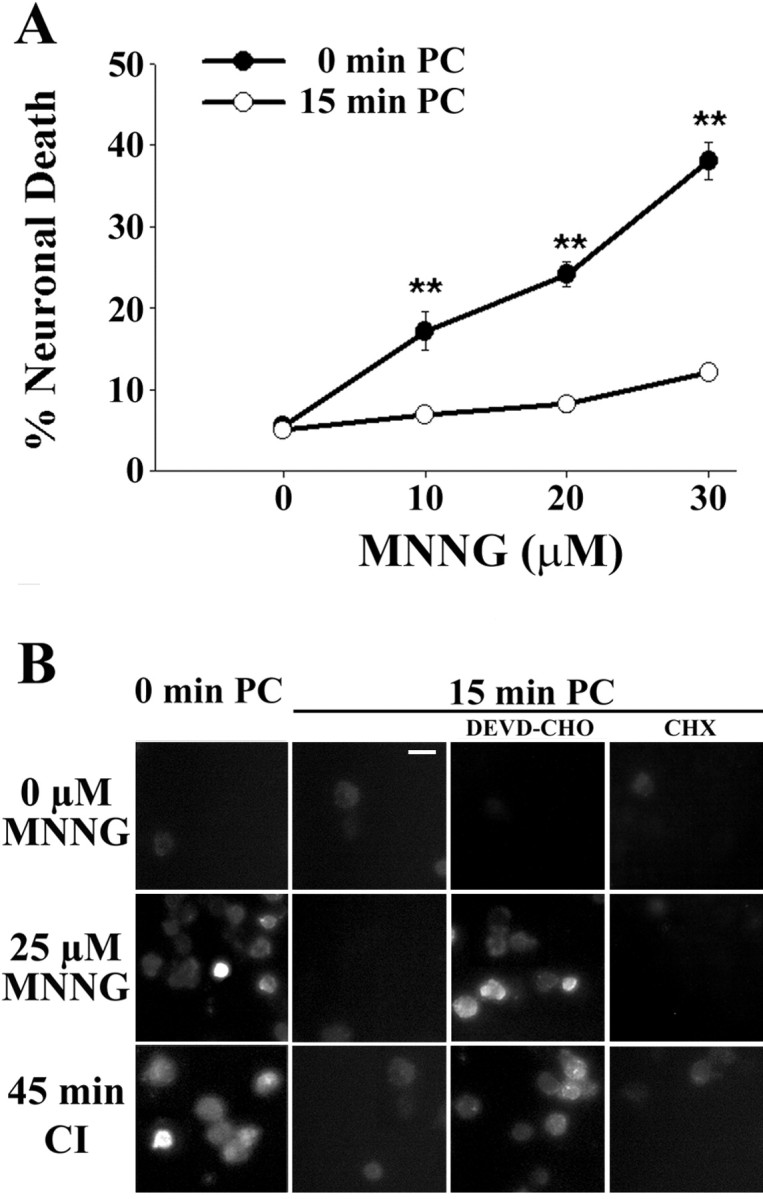
Preconditioning increases neuronal resistance to PARP-1-mediated cell death and blocks PARP-1 activation. Cultures were treated with 15 min of ischemic preconditioning (15 min PC) or sham washes only (0 min PC). Twenty-four hours later, the cultures were exposed to MNNG to induce PARP-1 activation. A, Neuronal death assessed 24 hr after MNNG exposures. **p < 0.01; representative of three independent experiments each with n = 3-4. B, Effects of PC on immunostaining for poly(ADP-ribose), the enzymatic product of PARP-1. The poly(ADP-ribose) immunoreactivity induced by either MNNG or CI was confined to neurons and attenuated in preconditioned cultures. The effect of preconditioning was negated by the addition of DEVD-CHO (25 μm) but not by CHX (10 μg/ml) added immediately after PC. Cells were fixed 40 min after onset of CI or MNNG incubation. Similar results were observed in four independent experiments. Scale bar, 25 μm.
Ischemic preconditioning causes caspase activation and PARP-1 cleavage
Because PARP-1 can be irreversibly inactivated by caspase cleavage, we examined whether this process could account for the reduced PARP-1 activity and PARP-mediated cell death observed in preconditioned cultures. Cultures were harvested 24 hr after ischemic preconditioning and assessed by Western blots and immunocytochemistry for the 89 kDa PARP-1 fragment produced by caspase cleavage (Lazebnik et al., 1994; Le et al., 2002). As shown in Figure 4A, ischemic preconditioning caused a significant increase in the 89 kDa fragment, and this increase was attenuated in cultures treated with the caspase inhibitor, DEVD-CHO. Cultures immunostained with the same antibody used in the Western blots showed the presence of PARP-1 cleavage product in most neurons (and no astrocytes) after preconditioning (Fig. 4B). Consistent with the Western blots, appearance of the 89 kDa PARP-1 cleavage product was significantly reduced in cultures treated with DEVD-CHO.
Figure 4.

Preconditioning produces caspase-mediated cleavage of PARP-1. A, Western blots with antibody to the 89 kDa PARP-1 fragment were prepared from cultures 24 hr after sham or 15 min PC. The increase produced by preconditioning was attenuated in cultures treated with the caspase inhibitor DEVD-CHO (25 μm). Values above the bands denote the increase ± SE relative to the paired control (**p < 0.01; n = 3). B, Immunostaining for the 89 kDa PARP-1 cleavage product showed it to accumulate in neurons after preconditioning. The accumulation was almost totally blocked by 25 μm DEVD-CHO, as observed in the Western blots. Similar results were observed in three independent experiments. Scale bar, 100 μm.
Caspase-3 is the major effector of PARP-1 cleavage during apoptosis (Lazebnik et al., 1994; Le et al., 2002). Procaspase-3 becomes activated by cleavage to a 17 kDa fragment (Cohen, 1997). To assess procaspase-3 cleavage in preconditioning, Western blots were prepared at serial time points after preconditioning and probed with an antibody specific to the active 17 kDa caspase-3 fragment. These studies showed an increase in active caspase-3 fragment after an interval of 3-6 hr (Fig. 5A). We also assessed the time course of PARP-1 cleavage. Western blots for the 89 kDa PARP-1 cleavage product showed a significant increase by 12 hr after preconditioning ischemia (Fig. 5A). Direct measures of caspase-3 enzymatic activity in lysed cultures showed a pattern similar to that observed in the Western blots (Fig. 5B). An increase in caspase-3 enzymatic activation was significant by 3 hr and maximal by 6 hr after preconditioning ischemia. The preconditioning-induced caspase-3 activity was blocked by the caspase inhibitor DEVD-CHO, whereas CHX had no effect. Of note, the magnitude of caspase-3 activation was modest relative to that induced by staurosporine, a classic inducer of apoptosis (Fig. 5B). Staurosporine caused extensive neuronal death at 24 hr (data not shown).
Figure 5.

Time course of caspase-3 activation and PARP-1 cleavage after preconditioning. A, Western blots with antibody to the cleaved (activated) 17 kDa caspase-3 fragment or the 89 kDa (inactivated) PARP-1 fragment were prepared from cultures 0-24 hr after a sham or 15 min ischemic preconditioning. Values above the bands denote the fold increase ± SE, relative to the 0 hr time point (*p < 0.05; **p < 0.01; n = 3-4). B, Caspase-3 enzymatic activity was measured in cultures lysed at sequential time points after sham washes only (0 min PC) or 15 min of ischemic preconditioning (15 min PC). PC produced a significant increase in caspase-3 within 3 hr of PC. The increase was blocked in cultures treated with the caspase inhibitor DEVD-CHO after PC, but not in cultures treated with CHX. Staurosporine (150 nm) was used as a positive control. Fluorescence values are expressed as increase over control, normalized to the increase induced by staurosporine. *p < 0.05; **p < 0.01; representative of three independent experiments each with n = 3-4.
Caspase inhibition attenuates ischemic preconditioning
To further evaluate the role of caspase activation in ischemic preconditioning, cultures were incubated with DEVD-CHO for a 24 hr interval after the preconditioning stimulus. As shown in Figure 6, cultures treated with 25 μm DEVD-CHO acquired less resistance to a second, longer ischemic challenge, whereas the 24 hr incubation with DEVD-CHO did not itself affect cell viability. DEVD-CHO reduced the extent of the preconditioning-induced resistance by ∼40% when tested with a 30 min ischemic challenge and by ∼100% when tested with a 45 min ischemic challenge. These differences cannot be compared directly because the degree of preconditioning-induced resistance also differed at the two ischemic time points in the absence of DEVD-CHO, but they suggest that the relative contribution of caspase-mediated processes to ischemic tolerance may vary with the severity of the ischemic challenge.
Figure 6.

Caspase inhibition reduces the resistance to CI conferred by ischemic preconditioning. Cultures were treated sham washes only (0 min PC), with 15 min of chemical ischemia (15 min PC), or with 15 min of chemical ischemia in the presence of 25 μm DEVD-CHO. After 24 hr the cultures were exposed to CI for durations of 0, 30, or 45 min. DEVD-CHO was added 2 hr before onset of PC and maintained until onset of CI. Neuronal death was assessed 24 hr after CI. **p < 0.01; representative of three independent experiments each with n = 3-4.
Discussion
A major role for PARP-1 activation in ischemic neuronal death is well established (Virag and Szabo, 2002). The increased resistance to ischemic insults resulting from genetic or pharmacological inhibition of PARP-1 activity, as confirmed in the cell culture model used here, led us to propose that a block in PARP-1 activation might similarly contribute to the protective effect of ischemic preconditioning. This proposal was supported by the finding that preconditioning blocks the poly(ADP-ribose) formation and cell death that otherwise result from exposure to the PARP-1 activating agent MNNG. PARP-1 can be irreversibly inactivated by cleavage of the catalytic site from the DNA binding domain, through the actions of caspase-3 and other proteolytic enzymes (Pieper et al., 1999). The present studies provide evidence for both PARP-1 cleavage and caspase-3 activation in preconditioned neurons. Additionally, treatment with the caspase inhibitor DEVD-CHO blocked PARP-1 cleavage and reduced the development of ischemic tolerance. Taken together, these results suggest that preconditioning provides increased resistance to ischemia at least in part by inducing caspase-mediated PARP-1 cleavage and thereby blocking the PARP-1-mediated cell death pathway.
Ischemic tolerance developed over a period of 6 -24 hr after the preconditioning stimulus, similar to observations made in vivo and in other cell culture systems (Nandagopal et al., 2001; Kirino, 2002; Dirnagl et al., 2003). This time course can be compared with the time course of procaspase-3 cleavage, increased caspase enzymatic activity, and PARP-1 cleavage presented in Figure 5. Procaspase-3 cleavage and caspase activity both reached maximum values at ∼6 hr after ischemic preconditioning, and PARP-1 cleavage reached a maximum ∼12 hr after preconditioning. These events correlate well with the development of ischemic tolerance, which first became apparent between 3 and 6 hr after preconditioning and neared a maximum at 12 hr after preconditioning. These time course studies suggest that procaspase-3 cleavage, caspase activation, and PARP-1 cleavage occur sequentially and precede the development of ischemic tolerance. A causative link between these events is supported by the effects of the caspase inhibitor DEVD-CHO. Cultures incubated with DEVD-CHO after ischemic preconditioning showed no inactivation of PARP-1 activity, as judged by lack of poly(ADP-ribose) formation in response to CI or MNNG, and showed an attenuated resistance to a subsequent ischemic challenge.
The extent of caspase-3 activation induced by ischemic preconditioning was small relative to that induced by staurosporine. The relatively modest activation of caspase-3 is consistent with the observation that preconditioning, unlike staurosporine, did not lead to neuronal death. Although caspase-3 activation is an effector mechanism of cell death during apoptosis, caspases can also be activated in processes that do not lead to cell death. For example, caspase-3 cleavage of the transcription factor GATA-1 regulates differentiation of erythroid cells (De Maria et al., 1999). Reversible, sublethal activation of caspase-3 has been demonstrated previously in neurons (Francois et al., 2001), and caspase-3 cleavage of glutamate receptors has been proposed as a mechanism for synaptic plasticity (Glazner et al., 2000). The present results suggest that PARP-1 cleavage is an additional mechanism by which caspase-3 may act outside of its classical role as an effector of cell death.
A comparison of Figure 1C and Figure 2 shows that the degree of neuroprotection achieved by ischemic preconditioning was substantial, approaching that achieved with the PARP inhibitor DPQ and with PARP-1 gene deletion. If the neuroprotective effect of preconditioning effect is caused primarily by PARP-1 cleavage, a substantial fraction of neuronal PARP-1 must be cleaved after preconditioning. It is difficult to quantify the fraction of neuronal PARP-1 cleaved after preconditioning because of the presence of astrocytes in the cocultures. However, the studies of PARP-1 function showed that preconditioning markedly decreased poly(ADP-ribose) formation in both MNNG- and CI-treated neurons (Fig. 4B), suggesting that substantial PARP-1 inactivation does occur after ischemic preconditioning.
Modest ischemic insults can trigger apoptosis (Bonfoco et al., 1995), and it is likely that the sublethal insults that induce preconditioning activate the caspase cascade by similar mechanisms. Several studies support NMDA receptor activation and nitric oxide production as important upstream components of neuronal preconditioning (Grabb and Choi, 1999; Gonzalez-Zulueta et al., 2000; Nandagopal et al., 2001). Although not investigated here, these upstream events may be coupled to caspase-3 activation in several ways. One well established sequence involves mitochondrial depolarization and cytochrome c release, which triggers the intrinsic pathway of caspase activation (Lemasters et al., 1999; Kroemer and Reed, 2000). Evidence also suggests that the Fas pathway of caspase activation can be induced by transient ATP depletion (Feldenberg et al., 1999) and that NMDA receptor activation and nitric oxide production can trigger caspase activation through the endoplasmic reticulum stress response (Rao et al., 2002).
It is generally accepted that delayed-onset ischemic tolerance requires de novo protein synthesis (Kirino, 2002). Consistent with this, we observed that ischemic tolerance was substantially reduced in cultures treated with the protein synthesis inhibitor CHX. Because caspase-3 is abundant in cells in an inactive form, it is unlikely that de novo synthesis of caspase-3 is required for PARP-1 cleavage. Accordingly, we observed no effect of CHX treatment on the preconditioning-induced caspase-3 activation or PARP-1 inhibition. This suggests the possibility that preconditioning may induce synthesis of proteins that influence the PARP-1 cell death pathway at steps downstream of PARP-1 activation. Alternatively, CHX may have cytoprotective effects through mechanisms independent of protein synthesis inhibition, as has been suggested by previous studies (Ratan et al., 1994).
During apoptosis, PARP-1 cleavage by caspase-3 and other enzymes prevents the PARP-1 activation and resultant ATP depletion that would otherwise be triggered during DNA fragmentation (Oliver et al., 1998; Boulares et al., 1999; Le et al., 2002). Support for caspase-3 as a mediator of PARP-1 cleavage after ischemic preconditioning was provided in the present study by biochemical measurements showing increased caspase-3 activity in cell lysates, by Western blots showing the active caspase-3 fragment in preconditioned cultures, and by the block of PARP-1 cleavage produced by 25 μm DEVD-CHO, a concentration at which DEVD-CHO is a relatively specific caspase-3 inhibitor (Margolin et al., 1997). These findings are in agreement with a study published during the preparation of this manuscript that reports widespread caspase-3 activation in a rat brain model of ischemic preconditioning (McLaughlin et al., 2003). It is possible, however, that other proteolytic enzymes may also be involved. PARP-1 can also be cleaved by caspase-7, caspase-8, and calpains (Germain et al., 1999; Wang, 2000; Le et al., 2002), and because DEVD-CHO is not entirely specific for caspase-3, the results obtained with DEVD-CHO do not exclude the possibility that other caspases or calpains may contribute to PARP-1 inactivation in ischemic preconditioning. Conversely, the possibility that DEVD-CHO does not entirely block PARP-1 cleavage means that the observed effect of DEVD-CHO on ischemic preconditioning may underestimate the true contribution of PARP-1 cleavage to this process.
Footnotes
This work was supported by the Department of Veterans Affairs and by National Institutes of Health Grants NS41421 (R.A.S.) and NS11048 (W.Y.). We thank Drs. Z. Q. Wang and B. Zingarelli for the PARP-1 -/- mouse strain and Elizabeth Gum for technical assistance.
Correspondence should be addressed to Raymond A. Swanson, (127) Neurology, Veterans Affairs Medical Center, 4150 Clement Street, San Francisco, CA 94121. E-mail: ray@itsa.ucsf.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/237967-07$15.00/0
References
- Aizenman E, Sinor JD, Brimecombe JC, Herin GA (2000) Alterations of N-methyl-d-aspartate receptor properties after chemical ischemia. J Pharmacol Exp Ther 295:572-577. [PubMed]
- Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton SA (1995) Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N-methyl-d-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci USA 92:7162-7166. [DOI] [PMC free article] [PubMed]
- Boulares AH, Yakovlev AG, Ivanova V, Stoica BA, Wang G, Iyer S, Smulson M (1999) Role of poly(ADP-ribose) polymerase (PARP) cleavage in apoptosis. Caspase 3-resistant PARP mutant increases rates of apoptosis in transfected cells. J Biol Chem 274:22932-22940. [DOI] [PubMed]
- Burkle A, Chen G, Kupper JH, Grube K, Zeller WJ (1993) Increased poly(ADP-ribosyl)ation in intact cells by cisplatin treatment. Carcinogenesis 14:559-561. [DOI] [PubMed]
- Chen J, Simon R (1997) Ischemic tolerance in the brain. Neurology 48:306-311. [DOI] [PubMed]
- Chen J, Graham SH, Zhu RL, Simon RP (1996) Stress proteins and tolerance to focal cerebral ischemia. J Cereb Blood Flow Metab 16:566-577. [DOI] [PubMed]
- Chen M, Won DJ, Krajewski S, Gottlieb RA (2002) Calpain and mitochondria in ischemia/reperfusion injury. J Biol Chem 277:29181-29186. [DOI] [PubMed]
- Cohen GM (1997) Caspases: the executioners of apoptosis. Biochem J 326:1-16. [DOI] [PMC free article] [PubMed]
- Cosi C, Suzuki H, Milani D, Facci L, Menegazzi M, Vantini G, Kanai Y, Skaper SD (1994) Poly(ADP-ribose) polymerase: early involvement in glutamate-induced neurotoxicity in cultured cerebellar granule cells. J Neurosci Res 39:38-46. [DOI] [PubMed]
- Currie RW, Ellison JA, White RF, Feuerstein GZ, Wang X, Barone FC (2000) Benign focal ischemic preconditioning induces neuronal Hsp70 and prolonged astrogliosis with expression of Hsp27. Brain Res 863:169-181. [DOI] [PubMed]
- D'Amours D, Germain M, Orth K, Dixit VM, Poirier GG (1998) Proteolysis of poly(ADP-ribose) polymerase by caspase 3: kinetics of cleavage of mono(ADP-ribosyl)ated and DNA-bound substrates. Radiat Res 150:3-10. [PubMed]
- D'Amours D, Desnoyers S, D'Silva I, Poirier GG (1999) Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J 342:249-268. [PMC free article] [PubMed]
- De Maria R, Zeuner A, Eramo A, Domenichelli C, Bonci D, Grignani F, Srinivasula SM, Alnemri ES, Testa U, Peschle C (1999) Negative regulation of erythropoiesis by caspase-mediated cleavage of GATA-1. Nature 401:489-493. [DOI] [PubMed]
- Dirnagl U, Simon RP, Hallenbeck JM (2003) Ischemic tolerance and endogenous neuroprotection. Trends Neurosci 26:248-254. [DOI] [PubMed]
- Eliasson MJ, Sampei K, Mandir AS, Hurn PD, Traystman RJ, Bao J, Pieper A, Wang ZQ, Dawson TM, Snyder SH, Dawson VL (1997) Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat Med 3:1089-1095. [DOI] [PubMed]
- Endres M, Wang ZQ, Namura S, Waeber C, Moskowitz MA (1997) Ischemic brain injury is mediated by the activation of poly(ADP-ribose)polymerase. J Cereb Blood Flow Metab 17:1143-1151. [DOI] [PubMed]
- Feldenberg LR, Thevananther S, del Rio M, de Leon M, Devarajan P (1999) Partial ATP depletion induces Fas- and caspase-mediated apoptosis in MDCK cells. Am J Physiol 276:F837-846. [DOI] [PubMed]
- Francois F, Godinho MJ, Dragunow M, Grimes ML (2001) A population of PC12 cells that is initiating apoptosis can be rescued by nerve growth factor. Mol Cell Neurosci 18:347-362. [DOI] [PubMed]
- Germain M, Affar EB, D'Amours D, Dixit VM, Salvesen GS, Poirier GG (1999) Cleavage of automodified poly(ADP-ribose) polymerase during apoptosis. Evidence for involvement of caspase-7. J Biol Chem 274:28379-28384. [DOI] [PubMed]
- Glazner GW, Chan SL, Lu C, Mattson MP (2000) Caspase-mediated degradation of AMPA receptor subunits: a mechanism for preventing excitotoxic necrosis and ensuring apoptosis. J Neurosci 20:3641-3649. [DOI] [PMC free article] [PubMed]
- Gonzalez-Zulueta M, Feldman AB, Klesse LJ, Kalb RG, Dillman JF, Parada LF, Dawson TM, Dawson VL (2000) Requirement for nitric oxide activation of p21(ras)/extracellular regulated kinase in neuronal ischemic preconditioning. Proc Natl Acad Sci USA 97:436-441. [DOI] [PMC free article] [PubMed]
- Grabb MC, Choi DW (1999) Ischemic tolerance in murine cortical cell culture: critical role for NMDA receptors. J Neurosci 19:1657-1662. [DOI] [PMC free article] [PubMed]
- Ha HC, Snyder SH (1999) Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc Natl Acad Sci USA 96:13978-13982. [DOI] [PMC free article] [PubMed]
- Heller B, Wang ZQ, Wagner EF, Radons J, Burkle A, Fehsel K, Burkart V, Kolb H (1995) Inactivation of the poly(ADP-ribose) polymerase gene affects oxygen radical and nitric oxide toxicity in islet cells. J Biol Chem 270:11176-11180. [DOI] [PubMed]
- Hoshida S, Yamashita N, Otsu K, Hori M (2002) The importance of manganese superoxide dismutase in delayed preconditioning. Involvement of reactive oxygen species and cytokines. Cardiovasc Res 55:495-505. [DOI] [PubMed]
- Ishida T, Yarimizu K, Gute DC, Korthuis RJ (1997) Mechanisms of ischemic preconditioning. Shock 8:86-94. [DOI] [PubMed]
- Kirino T (2002) Ischemic tolerance. J Cereb Blood Flow Metab 22:1283-1296. [DOI] [PubMed]
- Kroemer G, Reed JC (2000) Mitochondrial control of cell death. Nat Med 6:513-519. [DOI] [PubMed]
- Lazebnik YA, Kaufmann SH, Desnoyers S, Poirier GG, Earnshaw WC (1994) Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature 371:346-347. [DOI] [PubMed]
- Le DA, Wu Y, Huang Z, Matsushita K, Plesnila N, Augustinack JC, Hyman BT, Yuan J, Kuida K, Flavell RA, Moskowitz MA (2002) Caspase activation and neuroprotection in caspase-3-deficient mice after in vivo cerebral ischemia and in vitro oxygen glucose deprivation. Proc Natl Acad Sci USA 99:15188-15193. [DOI] [PMC free article] [PubMed]
- Leist M, Raab B, Maurer S, Rosick U, Brigelius-Flohe R (1996) Conventional cell culture media do not adequately supply cells with antioxidants and thus facilitate peroxide-induced genotoxicity. Free Radic Biol Med 21:297-306. [DOI] [PubMed]
- Lemasters JJ, Qian T, Bradham CA, Brenner DA, Cascio WE, Trost LC, Nishimura Y, Nieminen AL, Herman B (1999) Mitochondrial dysfunction in the pathogenesis of necrotic and apoptotic cell death. J Bioenerg Biomembr 31:305-319. [DOI] [PubMed]
- Lipton P (1999) Ischemic cell death in brain neurons. Physiol Rev 79:1431-1568. [DOI] [PubMed]
- Lobner D, Choi DW (1996) Preincubation with protein synthesis inhibitors protects cortical neurons against oxygen-glucose deprivation-induced death. Neuroscience 72:335-341. [DOI] [PubMed]
- Margolin N, Raybuck SA, Wilson KP, Chen W, Fox T, Gu Y, Livingston DJ (1997) Substrate and inhibitor specificity of interleukin-1 beta-converting enzyme and related caspases. J Biol Chem 272:7223-7228. [DOI] [PubMed]
- McLaughlin B, Hartnett KA, Erhardt JA, Legos JJ, White RF, Barone FC, Aizenman E (2003) Caspase 3 activation is essential for neuroprotection in preconditioning. Proc Natl Acad Sci USA 100:715-720. [DOI] [PMC free article] [PubMed]
- Nandagopal K, Dawson TM, Dawson VL (2001) Critical role for nitric oxide signaling in cardiac and neuronal ischemic preconditioning and tolerance. J Pharmacol Exp Ther 297:474-478. [PubMed]
- Oliver FJ, de la Rubia G, Rolli V, Ruiz-Ruiz MC, de Murcia G, Murcia JM (1998) Importance of poly(ADP-ribose) polymerase and its cleavage in apoptosis. Lesson from an uncleavable mutant. J Biol Chem 273:33533-33539. [DOI] [PubMed]
- Pieper AA, Verma A, Zhang J, Snyder SH (1999) Poly (ADP-ribose) polymerase, nitric oxide and cell death. Trends Pharmacol Sci 20:171-181. [DOI] [PubMed]
- Rajdev S, Reynolds IJ (1994) Glutamate-induced intracellular calcium changes and neurotoxicity in cortical neurons in vitro: effect of chemical ischemia. Neuroscience 62:667-679. [DOI] [PubMed]
- Rao RV, Peel A, Logvinova A, del Rio G, Hermel E, Yokota T, Goldsmith PC, Ellerby LM, Ellerby HM, Bredesen DE (2002) Coupling endoplasmic reticulum stress to the cell death program: role of the ER chaperone GRP78. FEBS Lett 514:122-128. [DOI] [PMC free article] [PubMed]
- Ratan RR, Murphy TH, Baraban JM (1994) Macromolecular synthesis inhibitors prevent oxidative stress-induced apoptosis in embryonic cortical neurons by shunting cysteine from protein synthesis to glutathione. J Neurosci 14:4385-4392. [DOI] [PMC free article] [PubMed]
- Shamloo M, Wieloch T (1999) Changes in protein tyrosine phosphorylation in the rat brain after cerebral ischemia in a model of ischemic tolerance. J Cereb Blood Flow Metab 19:173-183. [DOI] [PubMed]
- Shamloo M, Rytter A, Wieloch T (1999) Activation of the extracellular signal-regulated protein kinase cascade in the hippocampal CA1 region in a rat model of global cerebral ischemic preconditioning. Neuroscience 93:81-88. [DOI] [PubMed]
- Shimazaki K, Ishida A, Kawai N (1994) Increase in bcl-2 oncoprotein and the tolerance to ischemia-induced neuronal death in the gerbil hippocampus. Neurosci Res 20:95-99. [DOI] [PubMed]
- Shimizu S, Nagayama T, Jin KL, Zhu L, Loeffert JE, Watkins SC, Graham SH, Simon RP (2001) bcl-2 Antisense treatment prevents induction of tolerance to focal ischemia in the rat brain. J Cereb Blood Flow Metab 21:233-243. [DOI] [PubMed]
- Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC (1985) Measurement of protein using bicinchoninic acid. Anal Biochem 150:76-85. [DOI] [PubMed]
- Sordet O, Rebe C, Plenchette S, Zermati Y, Hermine O, Vainchenker W, Garrido C, Solary E, Dubrez-Daloz L (2002) Specific involvement of caspases in the differentiation of monocytes into macrophages. Blood 100:4446-4453. [DOI] [PubMed]
- Swanson RA, Benington JH (1996) Astrocyte glucose metabolism under normal and pathological conditions in vitro. Dev Neurosci 18:515-521. [DOI] [PubMed]
- Szabo C, Dawson VL (1998) Role of poly(ADP-ribose) synthetase in inflammation and ischaemia-reperfusion. Trends Pharmacol Sci 19:287-298. [DOI] [PubMed]
- Takahashi K, Pieper AA, Croul SE, Zhang J, Snyder SH, Greenberg JH (1999) Post-treatment with an inhibitor of poly(ADP-ribose) polymerase attenuates cerebral damage in focal ischemia. Brain Res 829:46-54. [DOI] [PubMed]
- Tokime T, Nozaki K, Sugino T, Kikuchi H, Hashimoto N, Ueda K (1998) Enhanced poly(ADP-ribosyl)ation after focal ischemia in rat brain. J Cereb Blood Flow Metab 18:991-997. [DOI] [PubMed]
- Virag L, Szabo C (2002) The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev 54:375-429. [DOI] [PubMed]
- Wang KK (2000) Calpain and caspase: can you tell the difference? Trends Neurosci 23:20-26. [DOI] [PubMed]
- Wang ZQ, Auer B, Stingl L, Berghammer H, Haidacher D, Schweiger M, Wagner EF (1995) Mice lacking ADPRT and poly(ADP-ribosyl)ation develop normally but are susceptible to skin disease. Genes Dev 9:509-520. [DOI] [PubMed]
- Ying W, Han SK, Miller JW, Swanson RA (1999) Acidosis potentiates oxidative neuronal death by multiple mechanisms. J Neurochem 73:1549-1556. [DOI] [PubMed]
- Ying W, Sevigny MB, Chen Y, Swanson RA (2001) Poly(ADP-ribose) glycohydrolase mediates oxidative and excitotoxic neuronal death. Proc Natl Acad Sci USA 98:12227-12232. [DOI] [PMC free article] [PubMed]
- Ying W, Chen Y, Alano CC, Swanson RA (2002) Tricarboxylic acid cycle substrates prevent PARP-mediated death of neurons and astrocytes. J Cereb Blood Flow Metab 22:774-779. [DOI] [PubMed]
- Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, Poirier GG, Dawson TM, Dawson VL (2002) Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 297:259-263. [DOI] [PubMed]