

Abstract
The p75 neurotrophin receptor (p75NTR), a member of the tumor necrosis factor receptor superfamily, facilitates apoptosis during development and after injury to the CNS. The signaling cascades activated by p75NTR that result in apoptosis remain poorly understood. In this study, we show that overexpression of p75NTR in primary cortical neurons, in pheochromocytoma cell line (PC12) cells, and in glioma cells results in activation of Jun kinase (JNK), accumulation of cytochrome c within the cytosol, and activation of caspases 9, 6, and 3. To link p75NTR-dependent JNK activation to mitochondrial cytochrome c release, regulation of BH3-domain-only family members was examined. Transcription of BH3-domain-only family members was not induced by p75NTR, but p75NTR-dependent JNK activation resulted in phosphorylation and oligomerization of the BH3-domain-only family member Bad. Loss of function experiments using Bad dominant negatives or RNA interference demonstrated a requirement for Bad in p75NTR-induced apoptosis. Together, these studies provide the first data linking apoptosis induced by p75NTR to the phosphorylation of BH3-domain-only family members.
Keywords: apoptosis, jun kinase, neurotrophin, receptor, trk, cell death
Introduction
The four mammalian neurotrophins comprise a family of related growth factors required for differentiation, survival, development, and death of specific populations of neurons and non-neuronal cells. The effects of the neurotrophins are mediated by binding to cell surface TrkA (tyrosine kinase A), TrkB, and TrkC tyrosine kinase receptors and to the p75 neurotrophin receptor (p75NTR). Roles for Trk receptors in neurotrophin action in neuronal survival, growth, and synaptic modulation are now well established (Kaplan and Miller, 2000; Patapoutian and Reichardt, 2001). The functions of the p75NTR receptor are complex and have been more difficult to ascertain (Dechant and Barde, 2002; Roux and Barker, 2002; Chao, 2003). It is clear that p75NTR functions as a Trk co-receptor that increases neurotrophin binding affinity (Barker and Shooter, 1994; Ryden et al., 1997; Esposito et al., 2001), and recent studies suggest that it may be a critical element in a receptor complex that responds to myelin-based growth inhibitory signals (Wang et al., 2002; Wong et al., 2002) and regulates myelination (Cosgaya et al., 2002). p75NTR also has autonomous signaling roles, particularly in facilitating apoptosis. In vitro analyses have shown that p75NTR induces cell death in primary trigeminal (Davies et al., 1993), hippocampal (Friedman, 2000; Brann et al., 2002), and sympathetic neurons (Lee et al., 1994; Bamji et al., 1998), as well as retinal precursor (Frade et al., 1996; Frade and Barde, 1998), Schwann (Soilu-Hanninen et al., 1999; Syroid et al., 2000; Petratos et al., 2003), oligodendrocyte (Casaccia-Bonnefil et al., 1996; Yoon et al., 1998) and neuroblastoma cells (Bunone et al., 1997). In vivo, p75NTR plays a prominent role in apoptosis that occurs in glia and neurons after traumatic injury to the spinal cord (Casha et al., 2001; Beattie et al., 2002) or brain (Roux et al., 1999; Troy et al., 2002) and has been implicated in developmental apoptosis in somites (Cotrina et al., 2000), retina, and spinal cord (Frade and Barde, 1999) and in the peripheral nervous system (Bamji et al., 1998).
The signaling events that link p75NTR activation to apoptosis are beginning to emerge and p75NTR-dependent apoptosis is associated with an increase in Rac and Jun kinase (JNK) activity and caspase activation (Gu et al., 1999; Tournier et al., 2000; Wang et al., 2001; Harrington et al., 2002). The precise ligand requirements for p75NTR apoptotic signaling are not clear, but recent studies have shown that unprocessed NGF (proNGF) is a more efficacious p75NTR ligand than mature NGF (Lee et al., 2001; Beattie et al., 2002). A plethora of p75NTR interacting proteins have been identified (Roux and Barker, 2002), and some of these, including neurotrophin receptor-interacting MAGE homolog (NRAGE) (Salehi et al., 2000), neurotrophin receptor-interacting factor (NRIF) (Casademunt et al., 1999), and p75NTR-associated cell death executor (NADE) (Mukai et al., 2000), facilitate p75NTR-dependent apoptosis. We have recently shown that NRAGE activates a mitochondrial death pathway involving JNK-dependent cytochrome c release and the activation of caspases (Salehi et al., 2002), but establishing the precise roles of each of the cytosolic interactors of p75NTR remains a significant challenge.
Despite this progress, several important questions remain unresolved. The proximal elements that connect p75NTR to apoptotic pathways remain uncertain, and it is not clear whether JNK activation is a prerequisite for p75NTR-induced apoptosis in all responsive cells. Furthermore, the mechanisms used by p75NTR to induce mitochondrial cytochrome c release and caspase activation are unknown. In this report, we addressed the mechanism of p75NTR-induced apoptosis in primary mouse cortical neurons and in pheochromocytoma, glioma, neuroblastoma, and medulloblastoma cells. Our findings reveal that activated p75NTR invariably causes JNK activation, mitochondrial cytochrome c release, and caspase 9, 6, and 3 activation. We show that JNK activation is necessary for p75NTR-dependent caspase cleavage in all responsive cell types. To link p75NTR-induced JNK activation to mitochondrial dysfunction, we examined the ability of p75NTR to increase expression of BH3-domain-only proteins but found that p75NTR did not activate transcription of BH3-domain-only genes. Instead, we demonstrate that p75NTR activation results in JNK-dependent phosphorylation of the BH3-domain-only protein Bad and show that Bad is required for p75NTR-induced apoptosis.
Materials and Methods
Materials. Cell culture reagents were purchased from BioWhittaker (Walkersville, MD), unless otherwise indicated. The p75NTR antibody αP1 is directed against the p75NTR intracellular domain (Roux et al., 1999). The antipeptide antibody to phosphorylated serine 128 BAD was directed against phosphopeptide C-EGMEEELpSPFRGRS conjugated to keyhole limpet cyanin and characterized as previously described (Konishi et al., 2002). The phospho-Thr83 p53 antibody was a kind gift of Ze'ev Ronai (Mount Sinai School of Medicine, New York, NY). JNK1 antibody (C-17, catalog #sc-474), the two Bad antibodies (C-20, catalog #sc-943 and N-19 catalog #sc-6542) and the actin antibody (C-2, catalog #sc-8432) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-flag antibody (M2, catalog #F-3165) was obtained from Sigma (St. Louis, MO), cytochrome c antibody was purchased from PharMingen (San Diego, CA) (catalog #556433), β-galactosidase (LacZ) antibody was purchased from Promega (Madison, WI) (catalog #23781), and anti-influenza hemagglutinin (anti-HA) antibody (12CA5, catalog #1583816) was purchased from Roche (Hertforshire, UK). Phospho-Thr183/Tyr185 JNK (G9, catalog #9255), SAPK/JNK (catalog #9252), phospho-Ser63 c-Jun (catalog #9261), phospho-Ser73 c-Jun (catalog #9164S), c-Jun (catalog #9162), caspase-9 (catalog #9502), cleaved caspase-3 (Asp175, catalog #9661), cleaved caspase-6 (Asp198, catalog #9761S), and cleaved Poly(ADP-ribose) polymerase (PARP) (Asp214, catalog #9541) -specific antibodies were obtained from Cell Signaling Technology (Beverly, MA). Cleaved caspase-9 antibody was generously provided by Merck-Frosst (Dorval, Quebec, Canada). Horseradish peroxidase-conjugated secondary antibodies were purchased from Jackson ImmunoResearch (West Grove, PA). Immunoreactive bands were detected using enhanced chemiluminescence purchased from PerkinElmer Life Sciences (Emeryville, CA). All other reagents were from Sigma, Calbiochem (La Jolla, CA), or ICN Biochemicals (Costa Mesa, CA), unless otherwise indicated.
Plasmids and recombinant adenovirus. Preparation of recombinant adenovirus expressing enhanced green fluorescent protein (AdGFP), β-galactosidase (AdLacZ), full-length p75NTR (Adp75NTR), the Flag-tagged JNK-binding domain of JNK interacting protein (JIP1) (AdJBD), and HA-epitope tagged MLK-3 (adMLK3) have been previously described (Roux et al., 2002). All adenoviruses were amplified in 293A cells and purified on sucrose gradients, as previously described (Roux et al., 2002). Viruses were titered by optical density and using the tissue culture infectious dose 50 (TCID) assay in 293A cells. Titers are expressed in term of plaque-forming units. The Bad dominant-negative plasmid consisting of GFP fused to a Bad nonapeptide in which Ser128 was substituted by Ala and the parental GFP vector have both been previously described (Konishi et al., 2002). The Bad RNAi construct was generated by targeting the sequence GGGAGCATCGTTCAGCAGCAGC in rat BAD, as previously described (Gaudilliere et al., 2002).
Cell culture, infection, and transfection. Human glioma (U343, U373, U87, and U251) and medulloblastoma (UW228-1, UW228-3, UW228-3, and Daoy) cell lines were provided by Dr. Roland Del Maestro (McGill University, Montreal, Quebec, Canada) and maintained in 5% CO2 at 37°C in either DMEM or RPMI medium supplemented with 10% fetal calf serum (FCS) (Clontech, Cambridge, UK), 2 mm l-glutamine, and 100 μg/ml penicillin-streptomycin. Neuroblastoma cell lines (SY5Y, SKNAS, 15N, and NGP) were provided by Dr. David Kaplan (University of Toronto, Toronto, Canada) and maintained as above. The rat pheochromocytoma cell line PC12 was maintained as previously described (Roux et al., 2001), and the PC12rtTA cell line (PC12) was purchased from Clontech and maintained in 10% CO2 at 37°C in DMEM supplemented with 10% FCS, 5% horse serum, 2 mm l-glutamine, 100 μg/ml penicillin-streptomycin, and 100 μg/ml G418. Cell lines were plated 18-24 hr before transfection and typically harvested 24-48 hr after infection. Primary cortical cultures were prepared from embryonic day 14 (E14)-E16 CD1 mouse telencephalon as described previously (Bhakar et al., 2002). Neuronal cultures were infected before plating and maintained in Neurobasal media (Invitrogen, Gaithersburg, MD) supplemented with 1× B27 supplement (Invitrogen), 2 mm l-glutamine, and 100 μg/ml penicillin-streptomycin. PC12 cells were plated onto poly-l-lysine-coated plates and transfected using Lipofectamine2000 as directed by the manufacturer (Invitrogen). Cell lines were infected with adenovirus 24 hr after plating.
Cytochrome c release assay. Cytosol-enriched subcellular fractions were prepared as described in Salehi et al. (2002). In brief, five million cells were harvested, washed once in Tris-buffered saline (10 mm Tris, pH 8.0, 150 mm NaCl), once in Buffer A (100 mm sucrose, 1 mm EGTA, and 20 mm 3-(N-morpholino)propanesulfonic acid, pH 7.4), and then resuspended in 500 μl Buffer B (Buffer A plus 5% Percoll, 0.01% digitonin, 1 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin, 1 mm sodium orthovanadate, and 1 mm phenylmethylsulfonyl fluoride). A sample of this suspension was retained as total cell lysate. The remainder was incubated on ice for 15 min and then centrifuged at 2500 × g for 10 min to remove intact cells and nuclei. The supernatant was then centrifuged at 15,000 × g for 15 min to pellet mitochondria. The final supernatant was designated cytosol.
Immunoblotting. Cells were lysed in radioimmunoprecipitation assay buffer (10 mm Tris, pH 8.0, 150 mm NaCl, 1% Nonidet P-40, 0.5% deoxycholate, 0.1% SDS, 1 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin, 1 mm sodium orthovanadate, and 1 mm phenylmethylsulfonyl fluoride) and analyzed for protein content using the BCA assay (Pierce, Rockford, IL). Samples were normalized for protein content, suspended in Laemmli sample buffer, separated by SDS-polyacrylamide gel electrophoresis, and electroblotted onto nitrocellulose. Blocking and secondary antibody incubations of immunoblots were performed in Tris-buffered saline-Tween (10 mm Tris, pH 8.0, 150 mm NaCl, and 0.2% Tween 20) supplemented with 5% (w/v) dried skim milk powder or 5% (w/v) bovine serum albumin (BSA) (Pierce). All primary antibody incubations were performed in the blocking solution, except for those involving phosphospecific antibodies that were performed in Tris-buffered saline-Tween supplemented with 5% BSA. Immunoreactive bands were detected by chemiluminescence (Perkin-Elmer Life Sciences), according to the manufacturer's instructions.
Survival assay. Analysis of cell survival was performed by MTT assay using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), which was added at a final concentration of 1 mg/ml for the last 4 hr of a 48 hr infection. The reaction was ended by the addition of one volume of solubilization buffer (20% SDS, 10% dimethylformamide, and 20% acetic acid). After overnight solubilization, specific and nonspecific absorbencies were read at 570 and 690 nm, respectively. Each data point was performed in triplicate or quadruplicate, and experimental results were analyzed by multiple ANOVAs with statistical probabilities assigned using the Tukey test for multiple comparisons. Each experiment was performed independently at least three times.
RT-PCR. 450,000 U373 cells or primary cortical neurons were infected with virus, and 24 hr later mRNA was isolated using the RNEasy Mini kit according to the manufacturer's instructions (Qiagen, Hilden, Germany). cDNA was generated using the Omniscript RT kit (Qiagen) and random hexamers (Roche) as primers. PCR was performed for 30 cycles using 300 nm of the following primers for U373 cells: actin sense, 5′ CACCACTTTCTACAATGAGC; antisense, 5′ CGGTCAGGATCTTCATGAGG; hBIMEL sense, 5′ TGGCAAAGCAACCTTCTGATG; antisense, 5′ AGTCGTAAGATAACCATTCGTGGG; hBMF sense, 5′ CTTGCTCTCTGCTGACCTGTTTG; antisense, 5′ AAGCCGATAGCCAGCATTGC; hHrk/Dp5 sense, 5′ TCGGCAGGCGGAACTTGTAG; antisense, 5′ GCTGTATGTAAATAGCATTGGGGTG; hBIK sense, 5′ AACCCCGAGATAGTGCTGGAAC; antisense, 5′ GCTGGAAACCAACATTTTATTGAGC; hPUMA sense, 5′ ACTGTGAATCCTGTGCTCTGCC; antisense, 5′ ACCCCCCAAATGAATGCCAG; hNOXA sense, 5′ CCAAACTCTTCTGCTCAGGAACC; antisense, 5′ CGGTAATCTTCGGCAAAAACAC.
For mouse cortical neurons, PCR was performed using the same conditions as above using the following primers: mBimEL sense, 5′ CCCCTACCTCCCTACAGACAGAA; antisense, 5′ CCAGACGGAAGATAAAGCGTAACAG; mBMF sense, 5′ CTTGCTCTCTGCTGACCTCTTTG; antisense, 5′ GTTGCGTATGAAGCCGATGG; mHrk/Dp5 sense, 5′ TGGAAACACAGACAGAGGAAGCC; antisense, 5′ AAAGGAAAGGGACCACCACG; mBIK sense, 5′ TCACCAACCTCAGGGAAAACATC; antisense, 5′ AGCAGGGGTCAAGAGAAGAAGG; mNOXA sense, 5′ TGATGTGATGAGAGAAACGCTCG; antisense, 5′ AAAGCAATCCCAAACGACTGCC; p75NTR sense, 5′ TGAATTCTGGAACAGCTGCAAAC; antisense, 5′ CCTTAAGTCACACTGGGGATGTG. Five percent of the cDNA prepared was used in a 25 μl PCR reaction, and the reaction product was separated on an 8% polyacrylamide gel, stained with ethidium bromide, and visualized under UV light.
Single-cell caspase-3 activation assay. PC12 cells were transfected with plasmids encoding either GFP alone, GFP fused to a dominant interfering Bad nonapeptide, or GFP plasmid and pU6/BS-Bad RNA interference plasmid at a 1:2 ratio. Cells were infected with either adLacZ or with adp75NTR 48 hr after transfection and fixed 24 hr later using 4% paraformaldehyde in PBS. Cells were blocked in TBS supplemented with 5% donkey serum and 0.3% Triton X-100 for 30 min and then incubated for 18 hr at 4°C with control rabbit sera or with antibodies directed against cleaved caspase 3. Secondary antibodies (donkey anti-rabbit conjugated Cy3) and Hoescht 33248 were applied for 2 hr at 4°C. GFP-positive cells were scored for the presence of activated caspase 3 by a blinded observer, with 300 cells counted per condition. This experiment was repeated three times, and the composite data were analyzed for statistical significance by ANOVA (Tukey's honestly significant difference (HSD) multiple comparison).
Results
The physiological conditions that result in activation of p75NTR apoptotic pathways are complex and likely regulated by multiple ligands and co-receptors. We have previously shown that recombinant adenovirus expressing full-length p75NTR or the p75NTR intracellular domain efficiently induces apoptosis in the absence of added ligand (Roux et al., 2001), and this approach was used to define apoptotic signaling pathways activated by p75NTR. We began by testing cell lines and primary cell types for susceptibility to p75NTR-induced death. Figure 1 shows that primary mouse cortical neurons, PC12 pheochromocytoma cells, and U343 and U373 glioma lines all showed reduced viability when infected with adenovirus-expressing p75NTR, whereas infection with control adenovirus expressing β-galactosidase (LacZ) had no significant effect. Other lines tested, including other glioma lines (U251 and U87), various medulloblastoma lines (Daoy, UW288-1, UW288-2, and UW288-3), and neuroblastoma lines (SY5Y, 15N, NGP, and SKNAS) were resistant to p75NTR-induced death in this assay (data not shown). For the remainder of this study, we focused our attention on p75NTR-dependent apoptosis in primary mouse cortical neurons, rat PC12 cells, and human U343 and U373 glioma lines.
Figure 1.

Overexpression of p75NTR reduces survival in a variety of cell types. A, Cortical neurons; B, PC12; C, U343 (wild-type p53); D, U373 (mutant p53) cells were infected with increasing multiplicities of infection (MOI) of LacZ or p75NTR recombinant adenovirus and then analyzed for survival by the MTT assay (see Materials and Methods). Error bars indicate SD. Results were analyzed for statistical significance by ANOVA (Tukey's HSD multiple comparison). Statistically significant differences of p < 0.001 are indicated by an asterisk.
Activation of the extrinsic apoptotic pathway by death receptors that are structurally related to p75NTR results in autocleavage and activation of caspase 8. Activation of the intrinsic apoptotic pathway results in release of mitochondrial contents and activation of caspase 9. We therefore determined the activation status of apical caspases 8 and 9 and effector caspases 3 and 6 during p75NTR-induced apoptosis. Expression of p75NTR resulted in a reduction in levels of full-length caspase 9, a corresponding increase in activated caspase 9, caspase 3, and caspase 6 and accumulation of the cleaved form of PARP, a caspase 3 substrate (Fig. 2A,B). In contrast, p75NTR-dependent caspase 8 cleavage was not observed in any of the cell types examined (data not shown). p75NTR-dependent caspase activation was not caused by adenoviral toxicity because cells infected with comparable quantities of LacZ adenovirus did not exhibit caspase activation. These data indicate that p75NTR-induced apoptosis occurs primarily through an intrinsic death pathway that involves release of mitochondrial contents and activation of caspase 9.
Figure 2.

p75NTR activates caspases and induces accumulation of cytosolic cytochrome c. A, Cortical neurons infected with 10, 50, or 100 MOI of LacZ or p75NTR recombinant adenovirus were lysed and analyzed by immunoblot for levels of LacZ, p75NTR, and full-length caspase 9 protein or, using cleavage-specific antibodies, for levels of cleaved caspases 3 and 6 and cleaved PARP. B, U373 cells were infected with 50 or 100 MOI of either LacZ or p75NTR adenovirus for 48 hr or treated with etoposide 50 μm (+), and then lysed and analyzed for increases in cleaved caspase 9. C, E15 cortical neurons, U373, and PC12 cells were left uninfected (0) or were infected with 100 MOI of LacZ (Lz), or p75NTR (p75) recombinant adenovirus. Thirty hours later, cells were fractionated for cytosolic components as described in Materials and Methods. Cytosolic fractions normalized for protein content were analyzed by immunoblotting with an antibody directed against cytochrome c.
Caspase 9 activation requires formation of an apoptosome complex consisting of caspase 9, Apaf-1, and cytosolic cytochrome c. Release of cytochrome c from mitochondria into the cytosol is a key regulatory step in this process. To determine if cytochrome c is released during p75NTR-induced apoptosis, cells were left uninfected or were infected with p75NTR or a control adenovirus, then lysed, subjected to subcellular fractionation, and cytosolic fractions were analyzed for cytochrome c levels by immunoblot. Figure 2C shows that cytochrome c was not detected in the cytosol of uninfected cells or in cells infected with control adenovirus, whereas cytosolic cytochrome c was readily detected in the cytosol of cells expressing p75NTR. Thus, p75NTR induces cytochrome c release from mitochondria of multiple cell types.
Activation of the JNK pathway is an important regulator of apoptotic events in several neuronal death paradigms, and JNK can be activated by p75NTR in several cell types. Consistent with this, we found that p75NTR expression in primary mouse cortical neurons and in PC12 and U373 cells consistently resulted in phosphorylation of JNK (Fig. 3A,B) and induced a dose-responsive increase in the phosphorylation of c-Jun, a JNK target (Fig. 3C). These results indicate that 75NTR-induced JNK activation is a consistent feature of p75NTR-responsive cell types.
Figure 3.

p75NTR activates the JNK pathway. A, U373 cells were infected with 0, 50, 100, or 200 MOI of control AdLacZ or with Adp75NTR. B, PC12 cells were injected with 0 or 50 MOI of AdLacZ or Adp75NTR. C, Cortical neurons were infected with 10, 50, or 150 MOI of AdLacZ or Adp75NTR. Lysates were prepared 30-48 hr after infection and examined by immunoblot for LacZ, p75NTR, phosphorylated JNK (pJNK), total JNK (SC-474 for A, CS-9252 for B), phosphorylated c-Jun (pJun), and total c-Jun as indicated.
To begin to address the role of the JNK pathway in p75NTR-induced apoptosis, we tested the effect of CEP1347, a MAP3K inhibitor that exhibits anti-apoptotic effects in several neuronal and non-neuronal systems (Saporito et al., 2002). We first tested the ability of CEP1347 to block c-Jun phosphorylation in PC12 cells overexpressing MLK3, an MAP3K identified as a target of CEP1347 (Maroney et al., 2001; Roux et al., 2002). Figure 4A shows that the compound almost completely blocked the robust c-Jun phosphorylation induced by this kinase. We next examined whether CEP1347 reduced c-Jun phosphorylation or caspase 3 activation, which was induced by p75NTR. CEP1347 did indeed reduce p75NTR-dependent c-Jun phosphorylation and caspase 3 activation but only at high concentrations (500-1000 nm) (Fig. 4B,C) (data not shown). These findings indicate that reductions in MAP3K and JNK signaling attenuates apoptosis induced by p75NTR yet suggest that blockade of a nonpreferred target of CEP1347 is required for this effect.
Figure 4.
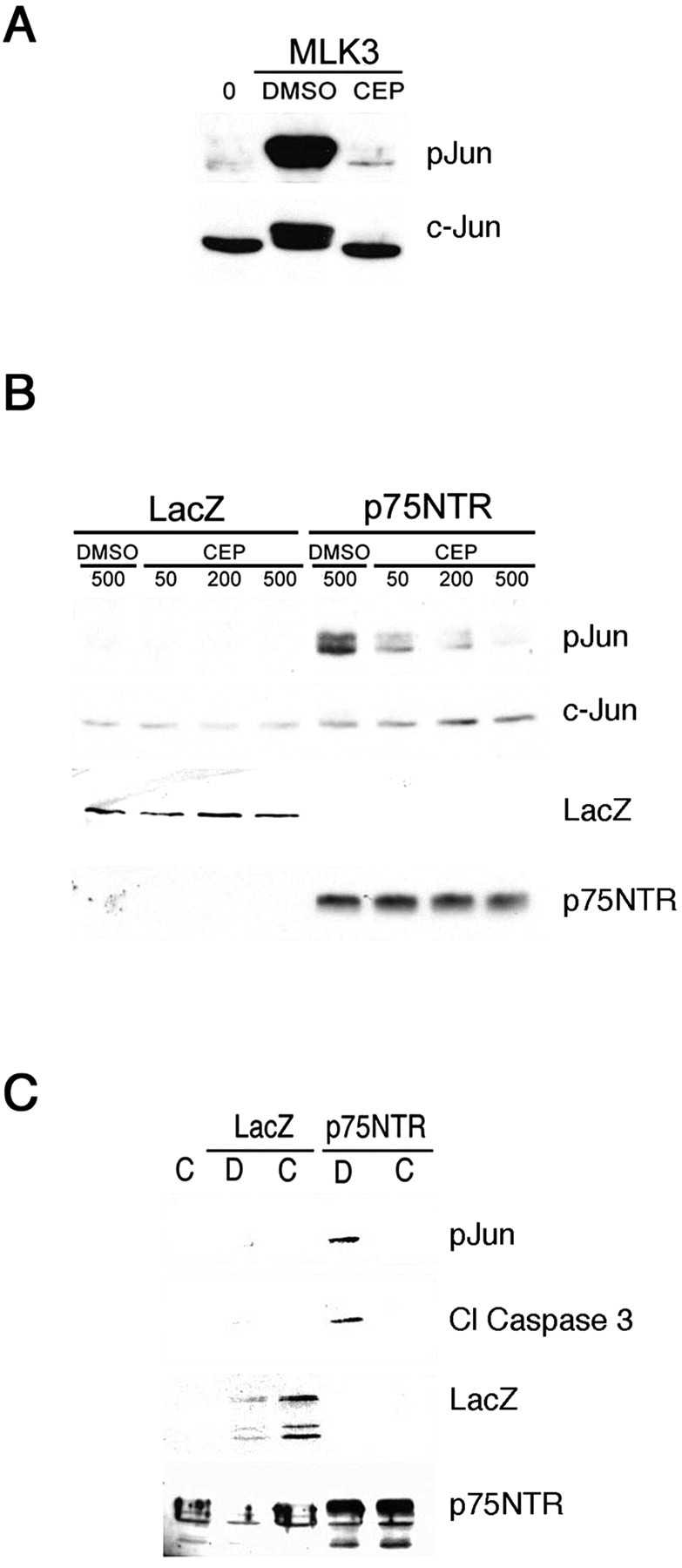
Inhibition of MAP3K signaling attenuates apoptosis induced by p75NTR. A, U373 cells infected with 100 MOI of MLK3 adenovirus or left uninfected (0) were treated 47 hr later with DMSO or CEP1347 at 200 nm for 1 hr. Cells were harvested, and lysates were subjected to immunoblot analysis for phospho-Ser63 c-Jun (pJun) and total c-Jun protein. B, Cortical neurons infected with 50 MOI of LacZ or p75NTR adenovirus were treated with DMSO or 50, 200, or 500 nm CEP1347 for 1 hr as in A. Lysates were analyzed by immunoblot as indicated (pJun, c-Jun, LacZ, p75NTR). C, AdLacZ or Adp75NTR-infected cortical neurons were treated with 500 nm CEP1347 (C) or DMSO (D) at the time of infection, and lysates were prepared 48 hr later and analyzed by immunoblot for levels of p75NTR, LacZ, phospho-Ser63 c-Jun (pJun), and cleaved caspase 3 (cl. caspase 3).
To directly assess the role of JNK activity in p75NTR-induced death, an adenovirus expressing the JNK binding domain of the JIP1 scaffolding molecule (AdJBD) was used to inhibit JNK activity in vivo. This JIP1 fragment is believed to sequester JNK and thus acts as an effective dominant inhibitor of JNK signaling (Harding et al., 2001). We first confirmed that AdJBD is capable of blocking JNK-dependent target phosphorylation by demonstrating that it blocked c-Jun phosphorylation induced by tumor necrosis factor (TNF)α, a well characterized JNK pathway inducer (Fig. 5A). Subsequent studies established that AdJBD was equally effective in blocking c-Jun phosphorylation induced by p75NTR expression (Fig. 5B). To determine if JNK inhibition blocked apoptotic signaling induced by p75NTR, cells were infected with p75NTR in the absence or presence of AdJBD and assessed for caspase 3 activation. Expression of AdJBD effectively blocked caspase 3 activation in all responsive cell types, indicating a crucial role for JNK activation in the p75NTR-induced apoptotic cascade (Fig. 5C) (data not shown).
Figure 5.

Activation of the JNK pathway is required for p75NTR-mediated caspase activation. Immunoblots for phospho-Ser63 c-Jun (pJun), c-Jun, Flag-JIP, LacZ, p75NTR, phospho-Thr183/Tyr185-JNK (pJNK), JNK, and cleaved caspase 3 were performed as indicated on lysates from U373 cells treated with TNF 20 ng/ml that were either left uninfected (0) or infected with JBD-JIP adenovirus (JBD) at 10 MOI (A), cortical neurons infected with 50 MOI of LacZ or p75NTR adenovirus together with increasing amounts (0, 0.05, 0.5, 2.5 MOI) of JBD-JIP adenovirus (B), and PC12 cells infected with 50 MOI of LacZ or p75NTR adenovirus supplemented with LacZ or JBD-JIP (JBD) adenovirus (both at 5 MOI) (C).
These data demonstrate that JNK activation is a prerequisite for p75NTR-induced apoptosis, but substrates of JNK that play a role in p75NTR-induced apoptosis are unknown. To begin to characterize targets of JNK involved in p75NTR-induced death, we first compared c-Jun phosphorylation induced by p75NTR or MLK3, a potent inducer of JNK activity (see above). Figure 6A shows that p75NTR and MLK3 induced robust phosphorylation of JNK. However, there was considerable discordance between the JNK activation, c-Jun phosphorylation, and caspase-3 activation induced by p75NTR versus MLK3. p75NTR and MLK3 induced comparable JNK phosphorylation, but only MLK3 produced a substantial increase in c-Jun phosphorylation, whereas only p75NTR induced substantial cleavage of caspase 3. To determine if our experimental design may have missed an early peak in p75NTR-induced c-Jun phosphorylation, JNK activation and c-Jun phosphorylation were examined at 12, 18, 24, and 30 hr after adenovirus infection. Figure 6B shows that phosphorylated JNK was first detected 18 hr after p75NTR infection and increased further by 24 and 30 hr. Cleaved caspase 3 was detectable 24 hr after infection, but c-Jun phosphorylation showed a significant lag, and an elevation in phospho-Jun levels were detected only after 30 hr infection. These data show that JNK activation correlates with p75NTR-induced death and suggests that c-Jun is not a preferred substrate of the JNK complex that is activated by p75NTR.
Figure 6.

p75NTR-induced caspase 3 cleavage does not correlate with phosphorylation of c-Jun. PC12 cells were infected with 50 MOI of AdLacZ (Lz), Adp75NTR (p75), or AdMLK3 (MLK) recombinant adenovirus, and lysates were prepared at 30 hr after infection (A, C), or at 12, 18, 24, and 30 hr after infection (B). Lysates normalized for protein content were analyzed for LacZ, p75NTR, cleaved caspase 3, phospho-Thr183/Tyr185-JNK (pJNK), total JNK, phospho-Ser63 c-Jun (pJun), and total c-Jun protein levels by immunoblot as indicated.
BH3-domain-only proteins directly and indirectly induce the association of Bax and Bak, which in turn facilitate release of mitochondrial proteins such as cytochrome c into the cytosol. Transcriptional activation of BH3-domain-only genes through c-Jun- or p53-dependent pathways is important in apoptosis in several neuronal and non-neuronal settings. We therefore examined whether p75NTR-induced apoptotic signaling correlated with accumulation of BH3-domain-only gene products. PC12 and U373 cells and cortical neurons were infected with LacZ or p75NTR adenovirus and alterations in mRNA levels of the BH3-domain-only family members Bim, Bmf, Hrk, Bik, Puma, and Noxa were determined by RT-PCR. mRNA corresponding to each of these family members were readily detected in both cell types examined, but p75NTR-dependent increases in their levels were not detected (Fig. 7) (data not shown). This indicates that JNK activation induced by p75NTR does not induce transcription of BH3-domain-only genes and suggests that alternate pathways are responsible for p75NTR-induced cytochrome c release and caspase 3 activation.
Figure 7.

p75NTR does not transcriptionally regulate BH3-domain-only proteins. Cortical neurons were infected with 0, 50, or 200 MOI of LacZ or p75NTR (p75) adenovirus, and 24 hr later mRNA was isolated as described in Materials and Methods. RT-PCR was performed using primers directed against Bim, Bmf, Hrk, Bik, Puma, Noxa, p75NTR, and Actin as indicated.
BH3-domain-only proteins can, in some instances, be regulated by post-translational mechanisms. Akt-dependent phosphorylation of Bad on Ser112 and Ser136 allows it to associate with 14-3-3 proteins and thereby suppresses its pro-apoptotic activity. Apoptotic kinases including JNK directly activate the cell death machinery by phosphorylating Bad at Serine 128 (Donovan et al., 2002). The phosphorylation of Bad at this residue disrupts the interaction of Bad with 14-3-3 proteins, thus allowing Bad to induce apoptosis (Konishi et al., 2002). We therefore determined if p75NTR activation resulted in phosphorylation of Bad on Ser128. PC12 and U373 cells were infected with LacZ or p75NTR adenovirus and alterations in Bad phosphostatus were examined by immunoblot. Figures 8, A and B, shows that p75NTR expression had little effect on the levels or phosphostatus of monomeric Bad (∼25 kDa) but rather induced the accumulation of a higher molecular weight species (∼75 kDa). This product was detected by two antibodies directed against distinct epitopes in Bad (N19, C20) as well as by a phosphospecific antibody directed against the JNK phosphorylation site within Bad. The 75 kDa product therefore appears to represent a stable oligomeric complex containing Bad phosphorylated on Serine 128. To determine if JNK activity contributes to p75NTR-dependent Bad phosphorylation and oligomerization, PC12 cells were infected with adenovirus expressing p75NTR in the absence or presence of AdJBD, lysed and examined by Bad immunoblot. Figure 8C shows that inhibiting JNK activity with AdJBD prevented formation of the Bad complex, indicating that JNK activity is required for p75NTR-dependent Bad phosphorylation and oligomerization.
Figure 8.

p75NTR activates JNK-dependent phosphorylation and oligomerization of Bad. A, U373 cells were infected with 0, 50, 100, or 200 MOI of LacZ or p75NTR adenovirus, and lysates were analyzed by immunoblot for LacZ, p75NTR, phospho-Ser128 Bad, and Bad (C-20, shown; N19, data not shown). B, PC12 cells were left uninfected (0) or were infected with LacZ (Lz) or p75NTR (p75) adenovirus aqt 100 MOI, and lysates were analyzed by immunoblot for LacZ, p75NTR, phospho-Ser128 Bad, and Bad (C-20). C, PC12 cells were infected with nothing (0), LacZ (Lz), or p75NTR (p75) adenovirus together with either 5 MOI of LacZ or JBD-JIP (JBD) adenovirus. Lysates were compared for expression of Bad, cleaved caspase 3, LacZ, p75NTR, and Flag-JIP (Flag) by immunoblot as indicated.
To determine if phosphorylation of Serine 128 within Bad is necessary for p75NTR-induced caspase activation, PC12 cells were transfected with a dominant-negative Bad serine 128 mutant allele (Konishi et al., 2002) and then infected with p75NTR or control virus. The ability of the dominant-negative Bad construct to inhibit p75NTR-dependent caspase 3 activation was assessed after 24 hr of virus infection by scoring transfected cells for the presence of cleaved caspase 3. Figure 9 shows that expression of the dominant-negative Bad serine 128 mutant allele confers significant protection from p75NTR-induced caspase 3 cleavage, suggesting that Bad phosphorylation is necessary for p75NTR-induced apoptosis. To confirm that caspase 3 cleavage induced by p75NTR requires Bad, p75NTR-induced apoptosis was assessed in cells in which the endogenous level of Bad were reduced using RNA interference. The ability of the RNAi construct to reduce Bad levels was first validated in 293 and PC12 cells (supplementary Fig. 2, available at www.jneurosci.org) and then used to reduce Bad levels in PC12 cells before adenovirus infection. PC12 cells were transfected with GFP alone or with GFP together with the Bad-RNAi plasmid and, 48 hr later, were infected with either p75NTR or LacZ adenovirus for 24 hr. PC12 cells transfected with Bad-RNAi do not generate pyknotic nuclei (data not shown) and are highly resistant to p75NTR-induced caspase-3 cleavage (Fig. 9), indicating a crucial role for Bad in the p75NTR apoptotic pathway.
Figure 9.

Bad is required for p75NTR-induced apoptosis. PC12 cells were transfected with GFP plasmid alone or with GFP plasmid together with plasmids encoding DN-Bad (S128A) or expressing Bad-RNAi. Cells were infected 48 hr later with LacZ or p75NTR adenovirus and, at 24 hr after infection, were fixed and immunostained for cleaved caspase 3 as described in Materials and Methods. Control experiments established that cleaved caspase 3 immunoreactivity correlates with TUNEL staining and is thus a valid surrogate for direct measurement of apoptosis (see supplementary Fig. 1, available at www.jneurosci.org). Transfected cells were scored for caspase 3 cleavage by a blind observer (n = 300 cells/condition). **Indicates a difference of p < 0.001 between GFP/Mock (Bar 1) and GFP/p75NTR (Bar 5), and *indicates a difference of p < 0.001 between GFP/p75NTR (Bar 5) and both DN-Bad/p75NTR (Bar 6) and with Bad RNAi/p75NTR (Bar 7), indicated by ANOVA.
Discussion
The mechanisms used by p75NTR to induce apoptosis are unique and bear little similarity to cell death signaling pathways used by other pro-apoptotic members of the TNF receptor superfamily. In this report, we show that p75NTR-induced death correlates with cytosolic accumulation of cytochrome c and activation of caspase 9 and caspase 3. Using the JNK binding domain of JIP as a dominant suppressor of JNK activity, we show that JNK is required for p75NTR-induced caspase 3 activation. Under conditions in which p75NTR induces JNK phosphorylation and death, p75NTR does not increase mRNA levels of BH3-domain-only family members that are transcriptionally regulated by c-Jun or p53. Instead, we demonstrate that p75NTR specifically increases phosphorylation and oligomerization of Bad and show that Bad plays a crucial role in p75NTR-induced death.
Ligand binding to cell surface apoptotic receptors such as Fas and DR3 induces cell death by initiating formation of a death-inducing signaling complex that facilitates FAS-associated death domain-dependent caspase 8 aggregation and activation. Other death stimuli induce apoptosis primarily via cytochrome c-dependent activation of caspase 9 (Shi, 2002). Activation of caspase 8 versus caspase 9 is therefore a distinguishing regulatory event that provides insight into the precise apoptotic pathways invoked by an extracellular stimulus. We have found that in glioma cells, PC12 cells, and primary cortical neurons, p75NTR-induced apoptosis is invariably accompanied by the activation of caspase 9, caspase 6, and caspase 3, whereas p75NTR-dependent caspase 8 activation was never observed. This suggests that activation of the intrinsic death pathway is crucial for p75NTR-induced apoptosis and indicates that cytosolic mitochondrial cytochrome c accumulation is an important regulatory step in p75NTR-induced death. These findings are in substantial agreement with other studies examining p75NTR-dependent caspase activation and are consistent with a recent study showing that blockade of caspase 9 activity significantly attenuates p75NTR-induced apoptosis (Gu et al., 1999; Wang et al., 2001; Troy et al., 2002). Together, these results show that p75NTR induces apoptosis through an intrinsic death pathway that results in mitochondrial cytochrome c release and caspase 9 activation.
The JNK signaling cascade plays a crucial role in apoptosis induced by a variety of stimuli (Kuranaga and Miura, 2002). We examined the role of JNK in p75NTR-induced apoptotic signaling by expressing a fragment of the JIP scaffolding molecule that directly binds to JNK and thus acts as a dominant JNK suppressor. This approach revealed that JNK signaling is a critical prerequisite for p75NTR-dependent caspase activation in all cell types examined. We also report that CEP1347 reduces p75NTR-dependent death but only at high concentrations, suggesting that inhibition of p75NTR-induced death by CEP1347 likely results from blockade of a nonpreferred target distinct from MLK3. Together with other recent studies (Friedman, 2000; Harrington et al., 2002), these data therefore indicate a crucial role for JNK activation in p75NTR-induced apoptosis in all cell types examined to date and raise the possibility that enzymes in the JNK pathway may provide feasible targets for inhibiting p75NTR-induced apoptosis after traumatic CNS injury.
BH3-domain-only family members inhibit the action of anti-apoptotic Bcl-2 family members such as Bcl-2 and Bcl-xL and facilitate the action of Bax and Bak at the mitochondria (Letai et al., 2002). The regulation of BH3-domain-only proteins is a key step linking proximal signaling events to the induction of cell death (Huang and Strasser, 2000). In sympathetic neurons, JNK activation results in phosphorylation of c-Jun, which in turn results in transcription of the BH3-domain-only family members Bim and Hrk (Harris and Johnson, 2001; Putcha et al., 2001; Whitfield et al., 2001). In other systems, p53 activation results in transcription of distinct pro-apoptotic BH3-domain-only family members, such as Noxa and Puma (Wu and Deng, 2002). We therefore hypothesized that p75NTR-dependent apoptosis was associated with transcription of known BH3-domain-only family members. However, p75NTR does not appear to enhance transcription of BH3-domain-only family members, suggesting that alternative pathways are required for p75NTR-dependent caspase activation.
BH3-domain-only family members are present in normal cells in the absence of apoptotic stimuli and must be rendered inactive to prevent apoptosis. One mechanism that accomplishes this is sequestration through protein-protein interactions. For example, the BH3-domain-only protein Bad is bound to 14-3-3 (Zha et al., 1996; Datta et al., 2000), and Bim and Bmf are sequestered in the cytosol by binding to dynein light chain or myosin V (Puthalakath et al., 1999, 2001). Significantly, recent findings have revealed that the sequestration of these three BH3-domain-only proteins can be negatively regulated by JNK. UV irradiation of HEK293 cells results in JNK-dependent phosphorylation of Bmf and Bim, releasing these proteins from their cytoskeletal association and allowing them to contribute to the apoptotic cascade (Lei and Davis, 2003). The Serine 128 phosphorylation of BAD activates BAD specifically by inhibiting the interaction of Serine 136-phosphorylated BAD with 14-3-3 proteins (Konishi et al., 2002). Serine 136 is a target of survival factor-induced kinases, including Akt, and the observation that p75NTR induces phosphorylation of BAD at Serine 128 suggests that p75NTR promotes apoptosis by opposing survival factor signals that suppress the cell death machinery. p75NTR activation also results in the oligomerization of Bad through a JNK-dependent pathway. Aside from Bad itself, the components of this stable oligomeric complex remain unknown but may include anti-apoptotic proteins such as Bcl-2 and Bcl-Xl (Letai et al., 2002). These findings provide the first data linking p75NTR activation to the phosphorylation of BH3-domain-only family members and indicate that p75NTR regulates apoptosis through a JNK pathway that is independent of transcription.
Palmada et al. (2002) have recently found that c-Jun is not required for p75NTR-induced cell death. Consistent with this, our data show that levels of c-Jun phosphorylation induced by p75NTR are modest and do not induce transcription of c-Jun targets that include Bim and Hrk. Thus, although c-Jun phosphorylation is a useful surrogate to assess JNK activation, it does not appear to play a significant role in p75NTR-induced apoptosis. However, alternative JNK-dependent pathways may contribute to p75NTR-dependent apoptosis. One candidate pathway involves p53, which can be activated by direct JNK phosphorylation and has been implicated in p75NTR-induced apoptosis in one study (Aloyz et al., 1998). However, p75NTR readily induces apoptosis in U373 cells that lack functional p53 (Fig. 1) and phosphospecific antibodies directed against Thr 81, a JNK target residue in p53 (Buschmann et al., 2001), or against Ser15 or Ser20 (Dumaz et al., 2001) did not reveal significant p75NTR-dependent phosphorylation of p53 (data not shown). Nonetheless, we cannot rule out the possibility that p53 or related family members may play a role in p75NTR-induced apoptosis in specific circumstances.
In this and previous studies, we have used p75NTR overexpression as an experimental paradigm to study p75NTR-induced apoptosis. p75NTR levels often rise dramatically after neuronal injury, and the injury-induced accumulation of p75NTR is tightly associated with neuronal death in vivo (Roux et al., 1999; Troy et al., 2002). However, although p75NTR overexpression is a convenient paradigm for analyzing downstream, it is important to emphasize that p75NTR binds multiple ligands, and these are certain to modulate its activity. In particular, recent studies indicate that the pro-form of NGF is a potent activator of p75NTR-induced apoptosis in vitro and in vivo (Lee et al., 2001; Beattie et al., 2002) and a crucial next step will be to directly assess the effects of proneurotrophins on p75NTR signaling events.
p75NTR plays a prominent role in nervous system apoptosis, particularly after trauma, and identification of the pro-apoptotic signal transduction mechanisms activated by the receptor are beginning to emerge. In this study, we show that p75NTR-dependent JNK activation is required for caspase activation. We demonstrate that p75NTR-dependent JNK activation induces phosphorylation and activation of Bad, a BH3-domain-only protein, and we show that Bad is required for apoptosis induced by p75NTR.
Footnotes
This work was supported by Grant MOP37850 from the Canadian Institute of Health Research (CIHR) (P.A.B.) and by National Institutes of Health Grant RO1-NS41021-01 (A.B.). A.L.B. was supported by a studentship from the CIHR, J.L.H. is supported by a Jean Timmons Costello Foundation studentship, C.E.P. is supported by a Natural Science and Engineering Research Centre studentship, A.H.S. is supported by a National Cancer Institute of Canada studentship, and E.B. is supported by the Boehringer Ingelheim Fonds. P.A.B. is a Scientist of the CIHR. We are grateful to David Kaplan and Mathieu Boudreau for providing the MLK3 adenovirus, to Aviva Tolkovsky for providing the truncated-JIP1 adenovirus, to Ze'ev Ronai for providing phosphospecific antibodies to Threonine 81 in p53, to Mohanish Deshmukh for advice on cleaved caspase-3 immunostaining, and to Genevieve Dorval for technical assistance in titrating adenovirus.
Correspondence should be addressed to Philip A. Barker, Montreal Neurological Institute, McGill University, 3801 University Avenue, Montreal, Quebec, Canada, H3A 2B4. E-mail: phil.barker@mcgill.ca.
Copyright © 2003 Society for Neuroscience 0270-6474/03/2311373-09$15.00/0
References
- Aloyz RS, Bamji SX, Pozniak CD, Toma JG, Atwal J, Kaplan DR, Miller FD ( 1998) p53 is essential for developmental neuron death as regulated by the TrkA and p75 neurotrophin receptors. J Cell Biol 143: 1691-1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamji SX, Majdan M, Pozniak CD, Belliveau DJ, Aloyz R, Kohn J, Causing CG, Miller FD ( 1998) The p75 neurotrophin receptor mediates neuronal apoptosis and is essential for naturally occurring sympathetic neuron death. J Cell Biol 140: 911-923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker PA, Shooter EM ( 1994) Disruption of NGF binding to the low affinity neurotrophin receptor p75LNTR reduces NGF binding to trkA on PC12 cells. Neuron 13: 203-215. [DOI] [PubMed] [Google Scholar]
- Beattie MS, Harrington AW, Lee R, Kim JY, Boyce SL, Longo FM, Bresnahan JC, Hempstead BL, Yoon SO ( 2002) ProNGF induces p75-mediated death of oligodendrocytes following spinal cord injury. Neuron 36: 375-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhakar AL, Tannis LL, Zeindler C, Russo MP, Jobin C, Park DS, MacPherson S, Barker PA ( 2002) Constitutive nuclear factor-kappa B activity is required for central neuron survival. J Neurosci 22: 8466-8475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brann AB, Tcherpakov M, Williams IM, Futerman AH, Fainzilber M ( 2002) Nerve growth factor-induced p75-mediated death of cultured hippocampal neurons is age-dependent and transduced through ceramide generated by neutral sphingomyelinase. J Biol Chem 277: 9812-9818. [DOI] [PubMed] [Google Scholar]
- Bunone G, Mariotti A, Compagni A, Morandi E, Della VG ( 1997) Induction of apoptosis by p75 neurotrophin receptor in human neuroblastoma cells. Oncogene 14: 1463-1470. [DOI] [PubMed] [Google Scholar]
- Buschmann T, Potapova O, Bar-Shira A, Ivanov VN, Fuchs SY, Henderson S, Fried VA, Minamoto T, Alarcon-Vargas D, Pincus MR, Gaarde WA, Holbrook NJ, Shiloh Y, Ronai Z ( 2001) Jun NH2-terminal kinase phosphorylation of p53 on Thr-81 is important for p53 stabilization and transcriptional activities in response to stress. Mol Cell Biol 21: 2743-2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casaccia-Bonnefil P, Carter BD, Dobrowsky RT, Chao MV ( 1996) Death of oligodendrocytes mediated by the interaction of nerve growth factor with its receptor p75. Nature 383: 716-719. [DOI] [PubMed] [Google Scholar]
- Casademunt E, Carter BD, Benzel I, Frade JM, Dechant G, Barde YA ( 1999) The zinc finger protein NRIF interacts with the neurotrophin receptor p75(NTR) and participates in programmed cell death. EMBO J 18: 6050-6061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casha S, Yu WR, Fehlings MG ( 2001) Oligodendroglial apoptosis occurs along degenerating axons and is associated with FAS and p75 expression following spinal cord injury in the rat. Neuroscience 103: 203-218. [DOI] [PubMed] [Google Scholar]
- Chao MV ( 2003) Neurotrophins and their receptors: a convergence point for many signaling pathways. Nat Rev Neurosci 4: 299-309. [DOI] [PubMed] [Google Scholar]
- Cosgaya JM, Chan JR, Shooter EM ( 2002) The neurotrophin receptor p75NTR as a positive modulator of myelination. Science 298: 1245-1248. [DOI] [PubMed] [Google Scholar]
- Cotrina ML, Gonzalez-Hoyuela M, Barbas JA, Rodriguez-Tebar A ( 2000) Programmed cell death in the developing somites is promoted by nerve growth factor via its p75(NTR) receptor. Dev Biol 228: 326-336. [DOI] [PubMed] [Google Scholar]
- Datta SR, Katsov A, Hu L, Petros A, Fesik SW, Yaffe MB, Greenberg ME ( 2000) 14-3-3 proteins and survival kinases cooperate to inactivate BAD by BH3 domain phosphorylation. Mol Cell 6: 41-51. [PubMed] [Google Scholar]
- Davies AM, Lee KF, Jaenisch R ( 1993) p75-deficient trigeminal sensory neurons have an altered response to NGF but not to other neurotrophins. Neuron 11: 565-574. [DOI] [PubMed] [Google Scholar]
- Dechant G, Barde YA ( 2002) The neurotrophin receptor p75(NTR): novel functions and implications for diseases of the nervous system. Nat Neurosci 5: 1131-1136. [DOI] [PubMed] [Google Scholar]
- Donovan N, Becker EB, Konishi Y, Bonni A ( 2002) JNK phosphorylation and activation of BAD couples the stress-activated signaling pathway to the cell death machinery. J Biol Chem 277: 40944-40949. [DOI] [PubMed] [Google Scholar]
- Dumaz N, Milne DM, Jardine LJ, Meek DW ( 2001) Critical roles for the serine 20, but not the serine 15, phosphorylation site and for the polyproline domain in regulating p53 turnover. Biochem J 359: 459-464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito D, Patel P, Stephens RM, Perez P, Chao MV, Kaplan DR, Hempstead BL ( 2001) The cytoplasmic and transmembrane domains of the p75 and Trk A receptors regulate high affinity binding to nerve growth factor. J Biol Chem 276: 32687-32695. [DOI] [PubMed] [Google Scholar]
- Frade JM, Barde YA ( 1998) Microglia-derived nerve growth factor causes cell death in the developing retina. Neuron 20: 35-41. [DOI] [PubMed] [Google Scholar]
- Frade JM, Barde YA ( 1999) Genetic evidence for cell death mediated by nerve growth factor and the neurotrophin receptor p75 in the developing mouse retina and spinal cord. Development 126: 683-690. [DOI] [PubMed] [Google Scholar]
- Frade JM, Rodriguez-Tebar A, Barde YA ( 1996) Induction of cell death by endogenous nerve growth factor through its p75 receptor. Nature 383: 166-168. [DOI] [PubMed] [Google Scholar]
- Friedman WJ ( 2000) Neurotrophins induce death of hippocampal neurons via the p75 receptor. J Neurosci 20: 6340-6346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudilliere B, Shi Y, Bonni A ( 2002) RNA interference reveals a requirement for myocyte enhancer factor 2A in activity-dependent neuronal survival. J Biol Chem 277: 46442-46446. [DOI] [PubMed] [Google Scholar]
- Gu C, Casaccia-Bonnefil P, Srinivasan A, Chao MV ( 1999) Oligodendrocyte apoptosis mediated by caspase activation. J Neurosci 19: 3043-3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding TC, Xue L, Bienemann A, Haywood D, Dickens M, Tolkovsky AM, Uney JB ( 2001) Inhibition of JNK by overexpression of the JNL binding domain of JIP-1 prevents apoptosis in sympathetic neurons. J Biol Chem 276: 4531-4534. [DOI] [PubMed] [Google Scholar]
- Harrington AW, Kim JY, Yoon SO ( 2002) Activation of Rac GTPase by p75 is necessary for c-jun N-terminal kinase-mediated apoptosis. J Neurosci 22: 156-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris CA, Johnson EM Jr ( 2001) BH3-only Bcl-2 family members are coordinately regulated by the JNK pathway and require Bax to induce apoptosis in neurons. J Biol Chem 276: 37754-37760. [DOI] [PubMed] [Google Scholar]
- Huang DC, Strasser A ( 2000) BH3-Only proteins-essential initiators of apoptotic cell death. Cell 103: 839-842. [DOI] [PubMed] [Google Scholar]
- Kaplan DR, Miller FD ( 2000) Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol 10: 381-391. [DOI] [PubMed] [Google Scholar]
- Konishi Y, Lehtinen M, Donovan N, Bonni A ( 2002) Cdc2 phosphorylation of BAD links the cell cycle to the cell death machinery. Mol Cell 9: 1005-1016. [DOI] [PubMed] [Google Scholar]
- Kuranaga E, Miura M ( 2002) Molecular genetic control of caspases and JNK-mediated neural cell death. Arch Histol Cytol 65: 291-300. [DOI] [PubMed] [Google Scholar]
- Lee K-F, Davies AM, Jaenisch R ( 1994) p75-deficient embryonic dorsal root sensory and neonatal sympathetic neurons display a decreased sensitivity to NGF. Development 120: 1027-1033. [DOI] [PubMed] [Google Scholar]
- Lee R, Kermani P, Teng KK, Hempstead BL ( 2001) Regulation of cell survival by secreted proneurotrophins. Science 294: 1945-1948. [DOI] [PubMed] [Google Scholar]
- Lei K, Davis RJ ( 2003) JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc Natl Acad Sci USA 100: 2432-2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ ( 2002) Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2: 183-192. [DOI] [PubMed] [Google Scholar]
- Maroney AC, Finn JP, Connors TJ, Durkin JT, Angeles T, Gessner G, Xu Z, Meyer SL, Savage MJ, Greene LA, Scott RW, Vaught JL ( 2001) Cep-1347 (KT7515), a semisynthetic inhibitor of the mixed lineage kinase family. J Biol Chem 276: 25302-25308. [DOI] [PubMed] [Google Scholar]
- Mukai J, Hachiya T, Shoji-Hoshino S, Kimura MT, Nadano D, Suvanto P, Hanaoka T, Li Y, Irie S, Greene LA, Sato TA ( 2000) NADE, a p75NTR-associated cell death executor, is involved in signal transduction mediated by the common neurotrophin receptor p75NTR. J Biol Chem 275: 17566-17570. [DOI] [PubMed] [Google Scholar]
- Palmada M, Kanwal S, Rutkoski NJ, Gustafson-Brown C, Johnson RS, Wisdom R, Carter BD ( 2002) c-jun is essential for sympathetic neuronal death induced by NGF withdrawal but not by p75 activation. J Cell Biol 158: 453-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patapoutian A, Reichardt LF ( 2001) Trk receptors: mediators of neurotrophin action. Curr Opin Neurobiol 11: 272-280. [DOI] [PubMed] [Google Scholar]
- Petratos S, Butzkueven H, Shipham K, Cooper H, Bucci T, Reid K, Lopes E, Emery B, Cheema SS, Kilpatrick TJ ( 2003) Schwann cell apoptosis in the postnatal axotomized sciatic nerve is mediated via NGF through the low-affinity neurotrophin receptor. J Neuropathol Exp Neurol 62: 398-411. [DOI] [PubMed] [Google Scholar]
- Putcha GV, Moulder KL, Golden JP, Bouillet P, Adams JA, Strasser A, Johnson EM ( 2001) Induction of BIM, a proapoptotic BH3-only BCL-2 family member, is critical for neuronal apoptosis. Neuron 29: 615-628. [DOI] [PubMed] [Google Scholar]
- Puthalakath H, Huang DC, O'Reilly LA, King SM, Strasser A ( 1999) The proapoptotic activity of the Bcl-2 family member Bim is regulated by interaction with the dynein motor complex. Mol Cell 3: 287-296. [DOI] [PubMed] [Google Scholar]
- Puthalakath H, Villunger A, O'Reilly LA, Beaumont JG, Coultas L, Cheney RE, Huang DC, Strasser A ( 2001) Bmf: a proapoptotic BH3-only protein regulated by interaction with the myosin V actin motor complex, activated by anoikis. Science 293: 1829-1832. [DOI] [PubMed] [Google Scholar]
- Roux PP, Barker PA ( 2002) Neurotrophin signaling through the p75 neurotrophin receptor. Prog Neurobiol 67: 203-233. [DOI] [PubMed] [Google Scholar]
- Roux PP, Colicos MA, Barker PA, Kennedy TE ( 1999) p75 neurotrophin receptor expression is induced in apoptotic neurons after seizure. J Neurosci 19: 6887-6896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux PP, Bhakar AL, Kennedy TE, Barker PA ( 2001) The p75 neurotrophin receptor activates Akt (protein kinase B) through a phosphatidylinositol 3-kinase-dependent pathway. J Biol Chem 276: 23097-23104. [DOI] [PubMed] [Google Scholar]
- Roux PP, Dorval G, Boudreau M, Angers-Loustau A, Morris SJ, Makkerh J, Barker PA ( 2002) K252a and CEP1347 are neuroprotective compounds that inhibit mixed-lineage kinase-3 and induce activation of Akt and ERK. J Biol Chem 277: 49473-49480. [DOI] [PubMed] [Google Scholar]
- Ryden M, Hempstead B, Ibanez CF ( 1997) Differential modulation of neuron survival during development by nerve growth factor binding to the p75 neurotrophin receptor. J Biol Chem 272: 16322-16328. [DOI] [PubMed] [Google Scholar]
- Salehi AH, Xanthoudakis S, Barker PA ( 2002) NRAGE, a p75 neurotrophin receptor-interacting protein, induces caspase activation and cell death through a JNK-dependent mitochondrial pathway. J Biol Chem 277: 48043-48050. [DOI] [PubMed] [Google Scholar]
- Salehi AH, Roux PP, Kubu CJ, Zeindler C, Bhakar A, Tannis LL, Verdi JM, Barker PA ( 2000) NRAGE, a novel MAGE protein, interacts with the p75 neurotrophin receptor and facilitates nerve growth factor-dependent apoptosis. Neuron: 279-288. [DOI] [PubMed]
- Saporito MS, Hudkins RL, Maroney AC ( 2002) Discovery of CEP-1347/KT-7515, an inhibitor of the JNK/SAPK pathway for the treatment of neurodegenerative diseases. Prog Med Chem 40: 23-62. [DOI] [PubMed] [Google Scholar]
- Shi Y ( 2002) Mechanisms of caspase activation and inhibition during apoptosis. Mol Cell 9: 459-470. [DOI] [PubMed] [Google Scholar]
- Soilu-Hanninen M, Ekert P, Bucci T, Syroid D, Bartlett PF, Kilpatrick TJ ( 1999) Nerve growth factor signaling through p75 induces apoptosis in Schwann cells via a Bcl-2-independent pathway. J Neurosci 19: 4828-4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syroid DE, Maycox PJ, Soilu-Hanninen M, Petratos S, Bucci T, Burrola P, Murray S, Cheema S, Lee KF, Lemke G, Kilpatrick TJ ( 2000) Induction of postnatal Schwann cell death by the low-affinity neurotrophin receptor in vitro and after axotomy. J Neurosci 20: 5741-5747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, Bar-Sagi D, Jones SN, Flavell RA, Davis RJ ( 2000) Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 288: 870-874. [DOI] [PubMed] [Google Scholar]
- Troy CM, Friedman JE, Friedman WJ ( 2002) Mechanisms of p75-mediated death of hippocampal neurons: role of caspases. J Biol Chem 277: 34295-34302. [DOI] [PubMed] [Google Scholar]
- Wang KC, Kim JA, Sivasankaran R, Segal R, He Z ( 2002) P75 interacts with the Nogo receptor as a co-receptor for Nogo, MAG and OMgp. Nature 420: 74-78. [DOI] [PubMed] [Google Scholar]
- Wang X, Bauer JH, Li Y, Shao Z, Zetoune FS, Cattaneo E, Vincenz C ( 2001) Characterization of a p75NTR apoptotic signaling pathway using a novel cellular model. J Biol Chem 276: 33812-33820. [DOI] [PubMed] [Google Scholar]
- Whitfield J, Neame SJ, Paquet L, Bernard O, Ham J ( 2001) Dominant-negative c-Jun promotes neuronal survival by reducing BIM expression and inhibiting mitochondrial cytochrome c release. Neuron 29: 629-643. [DOI] [PubMed] [Google Scholar]
- Wong ST, Henley JR, Kanning KC, Huang KH, Bothwell M, Poo MM ( 2002) A p75(NTR) and Nogo receptor complex mediates repulsive signaling by myelin-associated glycoprotein. Nat Neurosci 5: 1302-1308. [DOI] [PubMed] [Google Scholar]
- Wu X, Deng Y ( 2002) Bax and BH3-domain-only proteins in p53-mediated apoptosis. Front Biosci 7: d151-156. [DOI] [PubMed] [Google Scholar]
- Yoon SO, Casaccia-Bonnefil P, Carter B, Chao MV ( 1998) Competitive signaling between TrkA and p75 nerve growth factor receptors determines cell survival. J Neurosci 18: 3273-3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ ( 1996) Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L). Cell 87: 619-628. [DOI] [PubMed] [Google Scholar]