

Abstract
The systemic injection of γ-hydroxybutyric acid (GHB) elicits spike and wave discharges (SWDs), the EEG hallmark of absence seizures, and represents a well established, widely used pharmacological model of this nonconvulsive epilepsy. Despite this experimental use of GHB, as well as its therapeutic use in narcolepsy and its increasing abuse, however, the precise cellular mechanisms underlying the different pharmacological actions of this drug are still unclear.
Because sensory thalamic nuclei play a key role in the generation of SWDs and sleep rhythms, and because direct injection of GHB in the ventrobasal (VB) thalamus elicits SWDs, we investigated GHB effects on corticothalamic EPSCs and GABAergic IPSCs in VB thalamocortical (TC) neurons. GHB (250 μm-10 mm) reversibly decreased the amplitude of electrically evoked EPSCs and GABAA IPSCs via activation of GABAB receptors; however, ∼60% of the IPSCs were insensitive to low (250 μm-1.0 mm) GHB concentrations. The putative GHB receptor antagonist NSC 382 applied alone had a number of unspecific effects, whereas it either had no action on, or further increased, the GHB-elicited effects on synaptic currents. Low GHB concentrations (250 μm) were also effective in increasing absence-like intrathalamic oscillations evoked by cortical afferent stimulation.
These results indicate that low concentrations of GHB, similar to the brain concentrations that evoke SWDs in vivo, differentially affect excitatory and inhibitory synaptic currents in TC neurons and promote absence-like intrathalamic oscillations. Furthermore, the present data strengthen previous suggestions on the GHB mechanism of sleep promotion and will help focus future studies on the cellular mechanisms underlying its abuse.
Keywords: GABAB receptors, inhibition, excitation, sleep, intrathalamic oscillations, abuse
Introduction
Administration of γ-hydroxybutyric acid (GHB) elicits euphoric sensations, sedation, normalization of sleep parameters, and spike and wave discharges (SWDs), the EEG hallmark of absence seizures (Tunnicliff and Cash, 2002). Some GHB effects are clinically exploited (i.e., in anesthesia, alcohol withdrawal, and narcolepsy) (Lammers et al., 1993; Kleinschmidt et al., 1998; Poldrugo and Addolorato, 1999), whereas others underlie its illicit use as a recreational and so-called “rape” drug (Boyce et al., 2000; Nicholson and Balster, 2001). Experimentally, GHB-elicited SWDs are a well established model of absence seizures (Snead, 1995) that are used to assess the effect of pharmacological-transgenic manipulations of neuronal channels and receptors on the expression of this paroxysmal activity (Snead et al., 2000; Kim et al., 2001).
The mechanisms of GHB actions, however, are unclear. The existence of (1) binding sites with a specific brain distribution [different from those of GABAA receptors (GABAARs) and GABAB Rs] (Bernasconi et al., 1999), (2) high-affinity ligands (that do not displace GABA or baclofen from GABAA and GABAB sites) (Maitre et al., 1990), and (3) biochemical and behavioral responses that are blocked by NCS 382 (the only available GHB antagonist) (Maitre et al., 1990; Schmidt et al., 1991; Hechler et al., 1993; Snead, 1996; Ferraro et al., 2001; Mehta et al., 2001) has led to the proposition that there are physiologically relevant brain GHB receptors (Maitre, 1997). In contrast, other biochemical and behavioral data indicate that GHB only acts as a weak GABAB receptor agonist (Bernasconi et al., 1999), and isolation of GHB receptor(s) has been elusive (Cash and Tunnicliff, 2002).
Electrophysiological results are also contradictory (Crunelli and Leresche, 2002b), because in vivo studies show GHB responses that are mediated by putative GHB receptors, whereas most of the in vitro investigations highlight GABAB receptor-mediated responses, a result confirmed by data from recombinant GABAB receptors (Lingenhoehl et al., 1999). The only exception to this scenario is the GHB-mediated modulation of Ca2+ currents in NCB20 cells, which is NCS 382 sensitive (Kemmel et al., 2003).
Sensory thalamic nuclei play a key role in SWDs (Crunelli and Leresche, 2002a) and sleep rhythms (Steriade et al., 1997). Taking advantage of current knowledge of thalamic GABAB (Emri et al., 1996a; Ulrich and Huguenard, 1996; Kim et al., 1997; Le Feuvre et al., 1997) and putative GHB receptors (Hechler et al., 1992), and because GHB injection in the ventrobasal (VB) thalamus elicits SWDs (Snead, 1991), we investigated in VB thalamocortical (TC) neurons whether, and via which receptors, GHB affects corticothalamic EPSCs and GABAAergic IPSCs originating from the nucleus reticularis thalami (NRT). We report that GHB elicits a GABAB-mediated reduction of EPSCs and IPSCs, but only 41% of the IPSCs are sensitive to concentrations ≤1 mm. Low GHB concentrations also increase slow intrathalamic oscillations evoked by cortical stimulation. These results indicate that low GHB concentrations differentially affect excitatory and inhibitory synaptic currents in TC neurons and are discussed in the context of the ability of similar brain GHB concentrations to generate SWDs in vivo.
Preliminary data have been published in abstract form (Monnier et al., 2000).
Materials and Methods
Whole-cell recordings. A block containing the thalamus was dissected from the brain of male 10- to 22-d-old Wistar rats (mean 14 ± 2), and corticothalamic (400 μm thick) or horizontal (220 μm thick) slices containing the NRT and ventral posterolateral and ventral posteromedial nuclei were prepared in ice-cold saline, as described by Agmon and Connors (1991) and Le Feuvre et al. (1997), respectively, using a Vibroslice (Leica VT1000S). Slices were kept at 35°C in oxygenated saline (95% O2/5% CO2), before being transferred, one at the time, to the experimental setup, where they were maintained at room temperature and perfused at a rate of 1-2 ml/min with an oxygenated medium of composition (in mm): 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, and 25 glucose, pH 7.3.
Membrane currents were recorded from the soma of TC neurons visualized under Nomarski optics. Recording pipettes were pulled from borosilicate glass using a Narishige (Tokyo, Japan) micropipette puller and coated with wax. For recording synaptic currents, electrodes were filled with the following solution (in mm): 115 CsCl, 1 CaCl2, 5 MgCl2, 10 EGTA, 10 HEPES, 4 Na-ATP, 0.4 Na-GTP, 15 phosphocreatine, and 50 U/ml creatine phosphokinase, pH 7.3 (osmolarity, 305 mOsm) (electrode resistance, 1.5-2 MΩ). Once a high-resistance seal had been established, the holding potential was set to -60 mV, and the whole-cell, voltage-clamp configuration was obtained. Values of access resistance ranged from 4 to 6 MΩ at the beginning of the recording to 4-8 MΩ at the end. At least 70% of these values were compensated. Patch-clamp electrodes were connected to an Axopatch 200B amplifier (Axon Instruments, Foster City, CA). Voltage protocols and acquisition were controlled by Axograph 4.8. The membrane currents were filtered by a four-pole Bessel filter set at a corner frequency of 5 kHz, digitalized on-line at a sampling rate of 10 or 20 kHz, and later analyzed using Axograph 4.8 (Axon Instruments). Data were not collected until ≥10 min after patch rupture to allow the internal and external solutions to equilibrate.
Isolation of glutamatergic EPSCs was obtained by adding the GABAergic antagonist, gabazine (1-10 μm), to the perfusion medium. Isolation of GABAA IPSCs was obtained by adding to the perfusion medium the glutamate antagonists, dl-APV (50 μm) and CNQX (10 μm) (Eaton and Salt, 1996; Salt, 2002). Evoked postsynaptic currents were recorded from a holding potential of -60 mV. Evoked IPSCs were elicited by extracellular electrical stimulation, using an electrode consisting of a glass pipette (6-8 μm tip diameter) filled with the extracellular medium, and a remote 100-μm-thick platinum wire. The stimulating electrode was positioned 25-60 μm from the recorded neuron. Evoked corticothalamic EPSCs were elicited using a bipolar metallic electrode located in the internal capsula, close to the NRT. Square pulses of 20 μsec duration and 10-40 V amplitude were applied at a frequency of 0.05 Hz to evoke EPSCs or IPSCs.
The perforated-patch technique was used to record outward K+ currents, because it provided more stable recordings. The patch electrodes were filled with 1 μl of the following solution (in mm): 130 KCl, 10 HEPES, 1 EGTA, 1 MgCl2, 0.1 CaCl2, pH 7.3 (osmolarity, 305 mOsm). A quartz capillary was loaded with 0.5 μl of pipette solution in which 100 μm amphotericin B from a fresh stock solution in DMSO and 10 μm fluorescein were added. The 10-μm-diameter tip of the capillary was placed close to the tip of the recording pipette, and the solution was ejected once the cell-attached configuration had been obtained, leading to a rapid decrease in the series resistance. The integrity of the membrane patch was monitored with the fluorescein fluorescence.
Extracellular recordings. Extracellular recordings were performed in adult rat horizontal slices (prepared as described previously) maintained at 35°C, using an interface recording chamber and glass pipettes filled with 0.5 m NaCl (resistance, 1-5 MΩ) connected to a Neurolog 104 amplifier (Digitimer Ltd., Welwyn Garden City, UK). The extracellular medium contained (in mm): 134 NaCl, 3.75 KCl, 1.25 KH2PO4, 1 MgSO4, 2 CaCl2, 16 NaHCO3, 10 glucose. Multiunit activity was evoked by delivering a brief electrical stimulus (100-200 μsec; 50-100 μV) to the internal capsule using a bipolar tungsten electrode. The oscillatory index was calculated as the ratio of the first satellite peak to the central peak of the autocorrelogram, expressed as a percentage (Huntsman et al., 1999).
Quantitative data obtained with both extracellular and patch electrode recordings are given as mean ± SD, and the statistical significance was tested using Student's t test.
Drugs. Solutions containing (±)baclofen, GHB, NCS 382, NCS 356, trans-4-hydroxycrotonic acid (4-HCN), CGP 56999A, and CGP 35348 were prepared immediately before the experiment. All drugs were bath applied. We thank Novartis (Basel, Switzerland) for their gift of CGP 56999A and CGP 35348, and Dr. J. J. Bourguignon (Unité Mixte de Recherche Centre National de la Recherche Scientifique 7081, Strasbourg, France) for NCS 356. All other chemicals were purchased from Sigma and Tocris.
Results
GABAB receptors on corticothalamic afferents
Before assessing the action of GHB on corticothalamic EPSCs, we investigated the effects of the GABAB receptor agonist baclofen at this synapse, because no study has so far addressed this issue. As shown in Figures 1 and 2, electrical stimulation of the corticothalamic afferents evoked in VB TC neurons two types of EPSCs that differ in their kinetics, with the longer-lasting type of EPSCs being recorded more frequently, as reported previously by Kao and Coulter (1997).
Figure 1.

Effect of (±)baclofen on the corticothalamic EPSC. Baclofen (0.1 and 10 μm) reversibly decreases the amplitude of the EPSC by 23% (A) and 76% (B), respectively. The GABAB antagonist CGP56999A (100 nm) antagonizes the effect of baclofen at both concentrations and increases the EPSCs by 12% compared with the control amplitude. Each trace in A and B is the average of 10-20 EPSCs. C, Dose-response curve of the effect of baclofen on the corticothalamic EPSCs.
Figure 2.
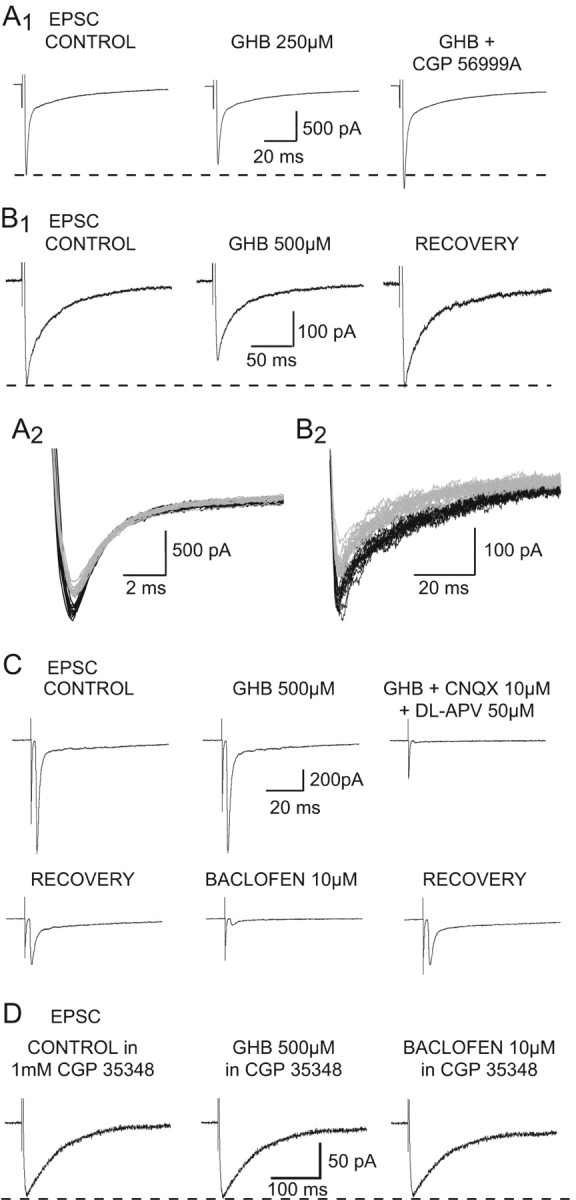
Effect of GHB on the corticothalamic EPSC. A1, GHB (250 μm) decreases the amplitude of the EPSC by 11%. This effect is antagonized by CGP56999A (100 nm), which also increases the EPSC size by 16% compared with control. A2, Superimposition of the individual synaptic currents used to build up the averages presented in A1 (control in black; GHB in gray). B1, GHB (500 μm) produces a reversible 22% decrease of the EPSC. B2, Superimposition of the individual synaptic currents used to build up the averages presented in B1 (control in black; GHB in gray). C, Recordings from one of the few TC neurons in which GHB fails to decrease the EPSC amplitude show that 10 μm baclofen decreases this synaptic current by 90%. Perfusion with the excitatory amino acid antagonists (CNQX and APV) blocks the electrically evoked corticothalamic EPSC. D, In the continuous presence of the GABAB antagonist CGP35348 (1 mm), GHB (500 μm) has no effect on the EPSC. The lack of effect of 10 μm baclofen on the EPSC after washout of GHB demonstrates that this concentration of CGP 35348 fully blocks presynaptic GABAB receptors. Each trace in A1, B1, C, and D is the average of 10-20 EPSCs.
Application of (±)baclofen dose-dependently and reversibly decreased the amplitude of corticothalamic EPSCs in all neurons tested (Fig. 1A,B). The effect of baclofen reached steady-state between 2 and 3 min from the beginning of the application, and the recovery was obtained between 10 and 12 min after the start of the washout. The reduction observed with 0.1, 0.5, 1, 10, and 20 μm baclofen was 15 ± 4% (n = 7), 47 ± 9 (n = 8), 55 ± 6% (n = 9), 78 ± 11% (n = 14), and 85 ± 6% (n = 4), respectively (IC50, 0.48 μm) (Fig. 1C). The decreases obtained with 10 and 20 μm baclofen were not statistically different (p = 0.05), suggesting that 10 μm baclofen already provides a maximal activation of the GABAB receptors present at this synapse. The GABAB receptor antagonists CGP 35348 (1 mm) (n = 3) or CGP 56999A (100 nm) (n = 15) not only fully antagonized the 10 μm baclofen-induced reduction of the EPSC, but also increased the amplitude of the EPSC above the control level (Fig. 1A,B), as has already been shown for both the sensory EPSP (Emri et al., 1996a) and the NRT-derived IPSPs in TC neurons (Le Feuvre et al., 1997). No difference in baclofen potency and the effect of GABAB antagonists was observed between EPSCs with different kinetics.
Effect of GHB on the corticothalamic EPSC
Having established the action of baclofen, we then investigated the effect of GHB on electrically evoked corticothalamic EPSCs. Application of GHB reversibly decreased the amplitude of these EPSCs in 96% of TC neurons. The effect of GHB reached steady-state between 2 and 3 min from the beginning of the application, and the recovery was obtained between 10 and 12 min after the start of the washout. No effect was observed with 100 μm GHB (n = 3), whereas 250 μm evoked a reduction (17 ± 3%) in seven of eight neurons (Fig. 2A). Increasing the GHB concentration to 500 μm, 1 mm, 10 mm, and 20 mm elicited a decrease of 27 ± 10% (n = 34 of 36), 25 ± 10% (n = 6 of 6), 56 ± 11% (n = 5 of 5), and 61 ± 13% (n = 5 of 5), respectively (Figs. 2B, 4A,B). In the two neurons in which 500 μm GHB was ineffective, subsequent application of baclofen markedly decreased the amplitude of the EPSC (Fig. 2C).
Figure 4.
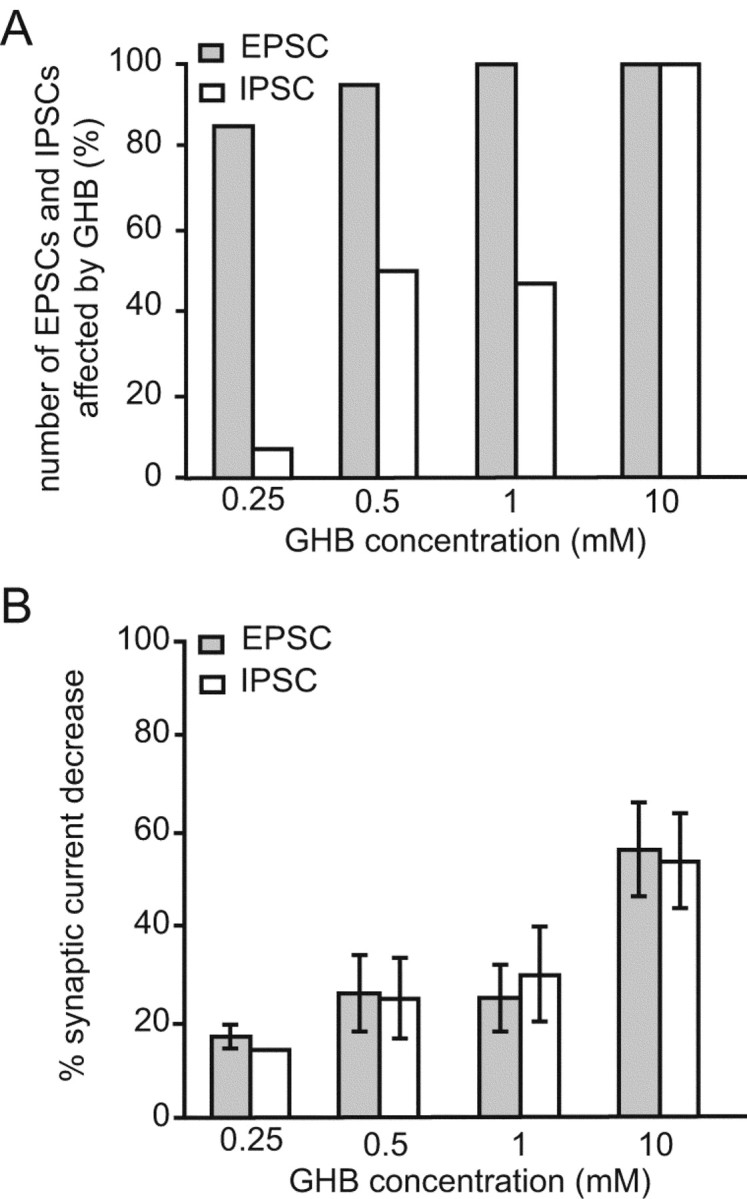
Summary of the effects of GHB on the corticothalamic EPSC and the NRT-derived GABAA IPSC. A, Percentage of EPSCs and IPSCs affected by different GHB concentrations. Note that although almost all EPSCs are affected by GHB (0.25-1.0 mm), a much smaller proportion of IPSCs is sensitive to these concentrations. At 10 mm, all EPSCs and IPSCs are decreased by GHB. B, Effect of different GHB concentrations on the amplitude of EPSCs and IPSCs. No difference in the decrease elicited by GHB is observed between EPSCs and those IPSCs that are sensitive to GHB.
The GHB-elicited reduction of the corticothalamic EPSC was antagonized by CGP 56999A (100 nm) in 3 of 3, 10 of 12, and 4 of 4 neurons tested with 250 μm, 500 μm, and 1 mm GHB, respectively (Fig. 2A). Similar to what had been observed with baclofen, CGP 56999A produced an increase over the control amplitude in the presence of GHB (Fig. 2A), and no differences in the effect of GHB were observed between EPSCs with different kinetics.
These results indicate that the totality of GHB effects on the corticothalamic EPSCs recorded in TC neurons occurs through activation of GABAB receptors. In the hippocampus, however, an effect of GHB on the EPSPs and IPSPs evoked in CA1 neurons after stimulation of Schaeffer collateral-commissural fibers could still be observed when GHB was applied in the continuous presence of the GABAB receptor antagonists CGP 35348 (500 μm) (Berton et al., 1999) or CGP 55845 (1 μm) (Cammalleri et al., 2002). To match this experimental paradigm, we applied GHB in the continuous presence of 1 mm CGP 35348, a concentration that fully antagonizes the response to a maximal concentration of baclofen (Fig. 2D). Also in this condition, however, GHB was never capable of inducing a reduction of the corticothalamic EPSC (n = 8) (Fig. 2D).
Effect of GHB on NRT-mediated IPSCs
GHB also decreased the amplitude of NRT-derived GABAA IPSCs with a similar potency as against the EPSCs (see below), but at low concentrations (250 μm-1 mm) it was effective on only 41% of the IPSCs. In particular, 250 μm GHB did not produce any effect on 13 of 14 neurons (Fig. 3A), whereas a 14% reduction was observed in 1 neuron (Fig. 4A,B). Increasing the GHB concentration to 500 μm induced a 25 ± 8% reduction of the IPSCs in 21 of 42 neurons (Fig. 3B), whereas 1 mm GHB produced a 29 ± 10% reduction in 9 of 19 neurons (Fig. 4A,B); however, application of 10 mm GHB induced a 54 ± 10% decrease in every TC neuron tested (n = 7) (Figs. 3C, 4A,B).
Figure 3.
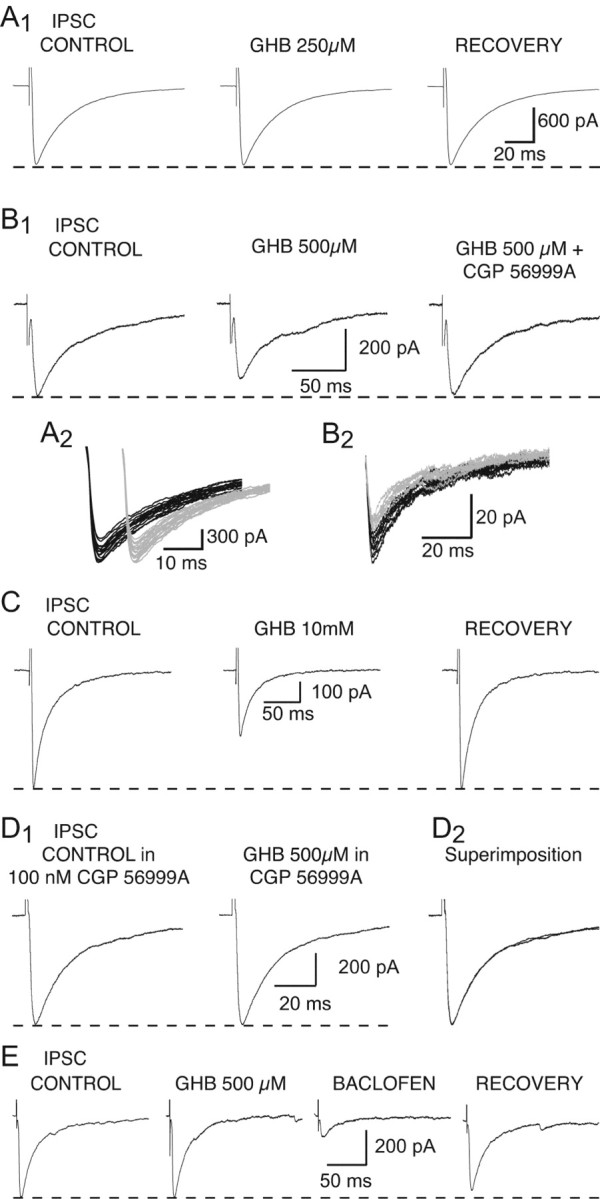
Effect of GHB on the NRT-derived IPSC. A1, GHB (250 μm) has no effect on the amplitude of the GABAA IPSC. A2, Superimposition of the individual synaptic currents used to build up the averages presented in A1 (control in black; GHB in gray). The IPSCs recorded in control condition and during GHB application have been offset for clarity. B1, GHB (500 μm) decreases the amplitude of the IPSC by 20%. This effect is antagonized by CGP 56999A (100 nm). B2, Superimposition of the individual synaptic currents used to build up the averages presented in B1 (control in black; GHB in gray). C, GHB at 10 mm reversibly decreases the IPSC amplitude by 48%. D1, In the continuous presence of CGP 56999A (100 nm), GHB (500 μm) has no effect on the IPSC. D2, Superimposition of the traces shown in D1. E, Recordings from one of the VB TC neurons in which 500 μm GHB fails to decrease the IPSC amplitude show that 10 μm baclofen reduces this synaptic current by 72%. Each trace in A1, B1, and C-E is the average of 10-20 IPSCs.
The GHB-induced reduction of the NRT-derived IPSCs was always fully antagonized by CGP 56999A (100 nm) (n = 1 and 4, for 250 and 500 μm GHB, respectively) (Fig. 3B). In addition, in the continuous presence of CGP 56999A (100 nm), application of GHB (500 μm) never decreased the IPSC amplitude (n = 10) (Fig. 3D).
In 10 of the 31 neurons that were insensitive to either 500 μm or 1 mm GHB, we were able to apply baclofen (5-10 μm) after washout of GHB. In each of these 10 neurons, baclofen decreased the IPSC amplitude by the same amount as in those neurons in which GHB was active (Fig. 3E).
It has been claimed that some of the actions of GHB observed in vitro when using high micromolar to low millimolar concentrations could be caused by its conversion to GABA via the GHB dehydrogenase (Hechler et al., 1997). If this were the case under the experimental conditions used to record IPSCs, one would have expected to see a change in holding current caused by the continuous activation of GABAA receptors. No change in holding current, however, was observed in any experiments on the IPSCs, nor has any change been reported during similar experiments in cortex and hippocampus (Jensen and Mody, 2001).
Effects of NCS 382
To completely exclude the involvement of putative GHB receptors in the action of GHB on thalamic synaptic currents, we then looked at the effect of the putative GHB receptor antagonist NCS 382. We started by investigating the action of this compound applied alone, because no study has thoroughly addressed this issue so far.
NCS 382 at a concentration of 50-100 μm, which has been shown to be effective against GHB action in NCB20 cells (Kemmel et al., 1998), had no effect on the EPSC amplitude (n = 5) (Fig. 5A). At concentrations ≥300 μm, however, NCS 382 produced a reversible increase in the EPSC amplitude. In particular, at 300 μm, 500 μm, and 1 mm, NCS 382 increased the EPSC by 10 and 20% (n = 2), 10 and 12% (n = 2), and 24 ± 4% (n = 6), respectively (Fig. 5B). Conversely, application of 1 mm NCS 382 decreased the amplitude of ISPCs by 21 ± 5% (n = 4) (Fig. 5C).
Figure 5.

Effects of NCS 382 applied alone. A1, NCS 382 (100 μm) has no effect on the corticothalamic EPSCs amplitude. A2, Superimposition of the traces shown in A1. B, NCS 382 (1 mm) produces a reversible 18% increase in the corticothalamic EPSC. C, NCS 382 (1 mm) produces a reversible 18% decrease in the NRT-derived IPSC. Each trace in A-C is the average of 10-20 EPSCs or IPSCs.
Despite the narrow range within which NCS 382 alone had no action on synaptic currents, we then tested the ability of NCS 382 to antagonize the effect of GHB. Application of NCS 382 at 100 μm-1 mm had no effect on the GHB-elicited reduction of the EPSC (Fig. 6A), or it prevented the full block of the GHB response by a GABAB receptor antagonist. In addition, at concentration ≥500 μm, NCS 382 could elicit an additional decrease to that produced by GHB (Fig. 6B) (i.e., 20% reduction in one of three neurons tested with 500 μm GHB and 18% in two of seven neurons tested with 1 mm GHB), as reported previously for the sensory EPSP in the same thalamic nucleus (Emri et al., 1996b).
Figure 6.

Effect of NCS 382 on the GHB-induced decrease of EPSCs and IPSCs. A, NCS 382 (1 mm) does not antagonize the GHB-induced decrease of the corticothalamic EPSC, whereas CGP 56999A (100 nm) does. B, Recordings from one of the neurons in which NCS 382 (1 mm) produces a decrease of the EPSC additional to that evoked by GHB on the EPSC. C, Lack of effect of NCS 382 (1 mm) on the GHB-induced decrease of the IPSC. Each trace in A-C is the average of 10-20 EPSCs or IPSCs.
Similarly, application of 500 μm or 1 mm NCS 382 did not antagonize the effect of GHB (250 μm, n = 1; 500 μm, n = 4) on the IPSC amplitude (Fig. 6C); however, in three of five neurons tested with 1 mm NCS 382, it produced an additional 18 ± 7% decrease in the IPSC amplitude. Overall, therefore, NCS 382 shows a number of nonspecific actions on synaptic currents that drastically limit its use.
Lack of effect of NCS 356 and trans-4-hydroxycrotonic acid
Although GHB actions on synaptic currents were fully blocked by GABAB receptor antagonists, it might be that GHB receptor may be disclosed using more potent agonists. We therefore tested the effect of NCS 356 and 4-HCN, two high-affinity ligands of putative GHB receptors that have been suggested to possess agonist-like actions (Hechler et al., 1990, 1993). Application of NCS 356 (500 μm) failed to produce any change in the amplitude of the corticothalamic EPSC (n = 18) (Fig. 7A) or the NRT-derived GABAA IPSC (n = 8) (Fig. 7B). In the same neurons, however, subsequent application of either 500 μm GHB or 1-10 μm baclofen produced the usual reduction in the synaptic current amplitude (Fig. 7A,B). 4-HCN (500 μm) was also unable to affect the IPSC (n = 3) (Fig. 7C).
Figure 7.

Lack of effect of NCS356 and 4-HCN on EPSCs and IPSCs. A, NCS356 (500 μm) has no effect on the EPSC, whereas the decrease induced by baclofen (1 μm) indicates that the synaptic current in this neuron is sensitive to GABAB receptor activation. B, NCS356 (500 μm) fails to affect the NRT-derived IPSC, whereas the decrease induced by baclofen (10 μm) indicates that the synaptic current in this neuron is sensitive to GABAB receptor activation. C1, 4-HCN (500 μm) has no effect on the GABAA IPSC amplitude. C2, Superimposition of the traces shown in C1. Each trace in A-C is the average of 10-20 EPSCs or IPSCs.
Postsynaptic action of GHB
GHB has already been shown to hyperpolarize TC neurons, an effect that is mediated by activation of postsynaptic GABAB receptors and insensitive to NCS 382 (Williams et al., 1995). Because in the continuous presence of GABAB antagonists the unmasking of a GHB receptor-mediated response on hippocampal synaptic currents has been reported recently (Berton et al., 1999; Cammalleri et al., 2002), we wonder whether under similar conditions a postsynaptic effect of GHB on TC neurons could also be highlighted.
From a holding potential of -60 mV, GHB (500 μm-10 mm; n = 32) produced an outward current (Fig. 8A,B), with an ED50 of 1.2 mm. In the continuous presence of CGP 56999A, however, at a concentration (300 nm) that fully blocked the maximal response to baclofen (10 μm) (n = 4) (Fig. 8C), GHB was unable to elicit any outward current (n = 12) (Fig. 8B).
Figure 8.

GHB-elicited outward current. A, Application of GHB dose-dependently activates an outward current in a VB TC neuron (A1). A2 shows the dose-response curve of the effect of GHB in one neuron. Numbers in the plot refer to the order of application, as indicated in A1. For comparison, the inset shows the outward current activated in the same neuron by 10 μm baclofen. B, The outward current elicited by 1 mm GHB is fully antagonized by CGP 56999A (300 nm). C, The outward current elicited by 10 μm baclofen is fully antagonized by CGP 56999A (300 nm).
Action of GHB on intrathalamic oscillations
Having established the action of GHB on corticothalamic EPSCs, NRT-evoked IPSCs, and TC neurons, we examined the ability of this drug to affect intrathalamic oscillations using extracellular recordings of multiunit activity in the VB. Stimulation of the cortical afferents, achieved by delivering a brief stimulus to the internal capsule, elicited slow intrathalamic oscillations of firing activity (frequency, 1.5 ± 0.4 Hz; n = 9 recordings in nine different slices) consisting of action potential bursts in few TC neurons (1.8 ± 0.4 U) with an oscillatory index of 51 ± 7% (Fig. 9A). After application of 250 μm GHB, the oscillatory response to internal capsule stimulation comprised a larger number of neurons (4.7 ± 0.5 U; p < 0.001) and occurred at a higher frequency (3.1 ± 1.0 Hz; p < 0.01), with an increased oscillatory index (72 ± 8%; p < 0.001) (Fig. 9B-D). Interestingly, in some experiments (n = 3), the oscillatory activity recorded in the presence of GHB became self-sustained, i.e., it no longer required electrical stimulation of the cortical afferents (data not shown). All of these effects of GHB were reversed after removal of the drug from the perfusion medium (frequency, 1.3 ± 0.4 Hz; number of units, 2.2 ± 0.3; oscillatory index, 62 ± 6%) (Fig. 9A-D). Application of 500 μm GHB, on the other hand, abolished evoked intrathalamic oscillations in four of five cases (Fig. 9E).
Figure 9.

Effect of GHB on population activity in the VB. A, In control conditions, the response to stimulation of the cortical afferents recorded in the VB shows a period of transient multiunit bursting with a dominant frequency of 1.8 Hz (see autocorellogram on the right; bin width 20 msec). After application of 250 μm GHB, the response comprises activity from an increased number of neurons, leading to an increase in the total spike count, and occurs at a higher frequency (2.2 Hz) (see autocorrelogram on the right). After washout of GHB, the response is again characterized by fewer bursting neurons and occurs at a lower frequency (1.7 Hz). All autocorrelograms were normalized to the peak value obtained in control conditions. The dotted lines in the autocorrelogram indicate the 99% confidence limit. B-D, Summary (from 9 multiunit recordings in 9 different slices) of the effect of 250 μm GHB on the oscillation frequency (B), the oscillatory index (see Materials and Methods) (C), and the number of units comprising the oscillatory response (D) (*p < 0.01; **p < 0.001). E, After a brief stimulus delivered to the internal capsule, an extracellular recording in the VB displays transient neuronal activity consisting of action potential bursts in a small number of TC neurons (see enlarged section). This response is reversibly abolished by application of 500 μm GHB.
Discussion
The main conclusion of this investigation is that in the concentration range (250-500 μm) that elicits SWDs, GHB instates an imbalance between the excitatory and inhibitory drives to TC neurons because it decreases 96% of corticothalamic glutamatergic EPSCs but only 43% of the NRT GABAAergic IPSCs. Low GHB concentrations (250 μm) also increase intrathalamic oscillations evoked by cortical stimulation.
In addition, our results demonstrate that these thalamic GHB effects are mediated by GABAB receptors and strongly question the use of NCS 382 and NCS 356 and 4-HCN as putative GHB receptor antagonist and agonists, respectively.
GABAB versus GHB receptors
That all GHB actions in VB are mediated by GABAB receptors is in agreement with previous in vitro electrophysiological experiments in another thalamic nucleus (Williams et al., 1995; Emri et al., 1996b) and any brain areas examined so far, i.e., substantia nigra (Harris et al., 1989), hippocampus (Xie and Smart, 1992b; King et al., 1997), ventral tegmental area (VTA) (Madden and Johnson, 1998), and cortex (Jensen and Mody, 2001). Berton et al. (1999) and Cammalleri et al. (2002), however, reported that in the presence of CGP 35348 (500 μm) or CGP 55845 (1 μm), application of GHB (600 μm) decreases EPSPs or GABAA IPSPs, respectively, in CA1 neurons in vitro, an effect abolished by NCS 382. Using a similar protocol, we did not detect any GHB receptor-mediated action on thalamic EPSCs and IPSCs. Although this discrepancy may reflect a real difference between thalamus and hippocampus, it is likely that GHB was still acting on presynaptic GABAB receptors in these two hippocampal studies, because 1 mm is necessary to fully block presynaptic GABAB receptors in this area (Davies et al., 1991). Indeed, Berton et al. (1999) and Cammalleri et al. (2002) did not test whether presynaptic GABAB receptors were fully blocked. Furthermore, although these studies reported no effect of 500 μm NCS 382 alone, an increase (11 and 65% at 100 μm and 2 mm, respectively) has been observed in hippocampal field potentials (King et al., 1997), and our analysis has highlighted nonspecific NCS 382 effects in thalamus.
Physiologically relevant GHB receptors?
Two critical issues still confound our understanding of brain GHB effects. It has been suggested that submicromolar to low micromolar GHB concentrations are needed to evoke GHB responses mediated by putative GHB receptors; however, the only in vitro studies that reported electrophysiological effects of GHB concentrations <100 μm (Crunelli and Leresche, 2002b) are in cell lines (Kemmel et al., 1998; Kemmel et al., 2003). In all other in vitro studies, including those in areas with high GHB binding sites (e.g., VTA, cortex, hippocampus, thalamus), a threshold GHB concentration of 100-300 μm was required to observe GHB effects (Harris et al., 1989; Xie and Smart, 1992a; Williams et al., 1995; Emri et al., 1996b; Madden and Johnson, 1998), and these responses were then invariably abolished by GABAB antagonists. With regard to in vivo investigations, most of the studies using systemic or iontophoretic applications of GHB (Diana et al., 1991; Engberg and Nissbrandt, 1993; Godbout et al., 1995; Aizawa et al., 1997) cannot help to clarify this issue because the brain GHB concentration was not measured. On the other hand, the expression of SWDs (i.e., the only behavioral effect of systemically injected GHB for which the brain GHB concentration-response curve is available) requires a threshold brain GHB concentration of 240 μm (Snead, 1991), and here we have indeed shown that 250 μm GHB fully acts on GABAB receptors (Williams et al., 1995; Emri et al., 1996b).
Furthermore, it has been claimed that the block by systemic NCS 382 of the behavioral and in vivo electrophysiological effects of GHB supports the existence of GHB receptors (Maitre, 1997). We would argue, however, that because the brain NCS 382 concentration after systemic administration has never been measured, any apparent block of GHB effects by NCS 382 in vivo should be treated with caution, because it may result from any of its nonspecific actions in thalamus (Emri et al., 1996b; this study) and hippocampus (King et al., 1997), as well as in the cerebellum, which has no GHB binding sites (Bernasconi et al., 2002). This negative view of the existence of physiologically relevant brain GHB receptors is also supported by the lack of action on thalamic EPSCs and IPSCs of NCS 356 and 4-HCN (this study), two high-affinity GHB receptor ligands-agonists in cell lines (Kemmel et al., 2003) and in biochemical tests (Hechler et al., 1990, 1993), and by the inability of 4-HCN to elicit SWDs in vivo (Depaulis et al., 1988).
Thalamic GABAB receptor subtypes
The existence of GABAB receptor subtypes (Bonanno and Raiteri, 1993; Guyon and Leresche, 1995) and of a presynaptic GHB-GABAB receptor complex (Banerjee and Snead, 1995) has been proposed, although no support from cloning studies is available yet. Interestingly, GABABR1a/b and GABABR2 immunoreactivity was detected only on postsynaptic elements in the VB (i.e., on TC neurons) (Kulik et al., 2002). The lack of GABAB immunoreactivity in VB presynaptic elements is surprising when compared with this and previous electrophysiological studies and has been explained by the existence of yet unidentified GABAB clones (Kulik et al., 2002). In this respect, our findings that ∼60% of GABAA IPSCs in VB are unaffected by low GHB concentrations but show a normal sensitivity to baclofen would suggest the existence of thalamic GABAB autoreceptor subtype(s) with a low sensitivity to GHB. Interestingly, the threshold concentration for GABAB-mediated effects of GHB in cortex and hippocampus (Jensen and Mody, 2001) is 10-fold higher than in thalamus.
Mechanism of GHB-elicited SWDs and decreased states of vigilance
A preferential effect of GHB on basal glutamate versus GABA release in vivo was shown in VB (Banerjee and Snead, 1995) but not in cortex (Hu et al., 2000), although the significance of changes in basal thalamic release for the EEG expression of a paroxysmal activity is unclear. Electrophysiologically, low (250 μm-1 mm) GHB concentrations hyperpolarize TC neurons (Williams et al., 1995) and decrease with a similar potency sensory EPSPs (Emri et al., 1996b), corticothalamic EPSCs, and 40% of GABAA IPSCs in VB, whereas the remaining IPSCs are unaffected (this study). On the basis of these results, and because SWDs can be elicited by direct injection of GHB into the VB (Snead, 1991), we suggest that a substantial component of the mechanism underlying the generation of SWDs by systemic GHB is caused by an imbalance of the physiological excitatory and inhibitory drives to TC neurons in favor of an increased inhibition. This would not only tend to slightly hyperpolarize TC neurons and decrease their afferent corticothalamic and sensory excitatory volleys, but would also leave the TC neuron population with a substantial proportion of unaltered NRT phasic inhibition, thus helping to impose that periodic inhibition that sculptures the electrical behavior of TC neuron during this paroxysmal activity (Steriade and Contreras, 1995; Pinault et al., 1998; Huntsman et al., 1999; Blumenfeld and McCormick, 2000; Tancredi et al., 2000; D'Arcangelo et al., 2002; Slaght et al., 2002). The ability of low (250 μm) GHB concentrations to increase absence-like intrathalamic oscillations clearly supports this view. It should be pointed out, however, that in addition to directly activating corticothalamic excitatory synapses, internal capsule stimulation will also strongly excite NRT neurons by activating both corticothalamic and thalamocortical fibers. Furthermore, because the NRT plays a key role in the maintenance and initiation of intrathalamic oscillations, the involvement of specific GHB-mediated effects in this nucleus should not be discounted when attempting to understand the pro-rhythmic actions of this drug. Whether an imbalance of excitatory and inhibitory drives might account for other GHB actions also remains to be elucidated, although it is worth noting that in VTA a differential control of EPSCs and IPSCs was recently suggested to underlie the rewarding properties of nicotine (Mansvelder et al., 2002).
Another critical feature of the GHB model of absence seizures that can also be explained by our results is the finding that SWDs are evoked within a narrow range of GHB concentrations (240-500 μm in the rat), because higher concentrations lead to a progressive slowing of the EEG associated with increasingly strong sedation and sleep promotion (Snead et al., 1976; Snead, 1991). Our results show that at a GHB concentration of 250 μm, the imbalance between the excitatory and inhibitory drives to TC neurons is already present and that slow intrathalamic oscillations are increased. At 500 μm, although the preferential effect on EPSCs versus IPSCs remains, the oscillations are partly abolished, probably because the (postsynaptic) activation of G-protein gated inward rectifying K+ current (GIRK) is stronger. At even higher concentrations, the dampening of both excitatory and inhibitory inputs, together with the increasing GIRK activation, would result in a stronger hyperpolarization of TC neurons, necessary to express those intrinsic-network activities associated with states of progressively decreasing vigilance (Steriade, 2000; Hughes et al., 2002). This thalamic action, together with similar effects on cortical neurons (Godbout et al., 1995; Jensen and Mody, 2001), might well account for the sedation and normalization of sleep patterns, which are exploited in the illicit use of GHB and its clinical use in narcolepsy, respectively.
Footnotes
The work was supported by the Wellcome Trust (Grant 71436) and partly by the National Institute on Drug Abuse (DA14830-01). We thank Dr. H. R. Parri for critical reading of this manuscript.
Correspondence should be addressed to N. Leresche, Neurobiologie des Processus Adaptatifs, Unité Mixte de Recherche, Centre National de la Recherche Scientifique 7102-Université Pierre et Marie Curie, 9 quai Saint-Bernard, F-75005 Paris, France. E-mail: nathalie.leresche@snv.jussieu.fr.
Z. Monnier's present address: Laboratoire de Neuroscience EA 481, Université de Franche-Comté, 25000 Besançon, France.
Copyright © 2003 Society for Neuroscience 0270-6474/03/2311469-10$15.00/0
N.G. and Z.M. contributed equally to this work.
References
- Agmon A, Connors BW ( 1991) Thalamocortical responses of mouse somatosensory (barrel) cortex in vitro. Neuroscience 41: 365-379. [DOI] [PubMed] [Google Scholar]
- Aizawa M, Ito Y, Fukuda H ( 1997) Roles of gamma-aminobutyric acid (GABA B) and gamma-hydroxybutyric acid receptors in hippocampal long-term potentiation and pathogenesis of absence seizures. Biol Pharmacol Bull 20: 1066-1070. [DOI] [PubMed] [Google Scholar]
- Banerjee PK, Snead III OC ( 1995) Presynaptic gamma-hydroxybutyric acid (GHB) and gamma-aminobutyric acid (GABAB) receptor-mediated release of GABA and glutamate (GLU) in rat thalamic ventrobasal nucleus (VB): a possible mechanism for the generation of absence-like seizures induced by GHB. J Pharmacol Exp Ther 273: 1534-1543. [PubMed] [Google Scholar]
- Bernasconi R, Mathivet P, Bischoff S, Marescaux C ( 1999) Gamma-hydroxybutyric acid: an endogenous neuromodulator with abuse potential? Trends Pharmacol Sci 20: 135-141. [DOI] [PubMed] [Google Scholar]
- Bernasconi R, Mathiver P, Otten U, Bettler B, Bischoff S, Marescaux C ( 2002) Part of the pharmacological actions of γ-hydroxybutyrate are mediated by GABAB receptors. In: Gamma-hydroxybutyrate: molecular, functional and clinical aspects (Tunnicliff G, Cash CD, eds), pp 29-63. New York: Taylor and Francis.
- Berton F, Brancucci A, Beghe F, Cammalleri M, Demuro A, Francesconi W, Gessa GL ( 1999) Gamma-hydroxybutyrate inhibits excitatory postsynaptic potentials in rat hippocampal slices. Eur J Pharmacol 380: 109-116. [DOI] [PubMed] [Google Scholar]
- Blumenfeld H, McCormick DA ( 2000) Corticothalamic inputs control the pattern of activity generated in thalamocortical networks. J Neurosci 20: 5153-5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonanno G, Raiteri M ( 1993) Multiple GABAB receptors. Trends Pharmacol Sci 14: 259-261. [DOI] [PubMed] [Google Scholar]
- Boyce SH, Padgham K, Miller LD, Stevenson J ( 2000) Gamma hydroxybutyric acid (GHB): an increasing trend in drug abuse. Eur J Emerg Med 7: 177-181. [DOI] [PubMed] [Google Scholar]
- Cammalleri M, Brancucci A, Berton F, Loche A, Gessa GL, Francesconi W ( 2002) Gamma-hydroxybutyrate reduces GABA(A)-mediated inhibitory postsynaptic potentials in the CA1 region of hippocampus. Neuropsychopharmacology 27: 960-969. [DOI] [PubMed] [Google Scholar]
- Cash C, Tunnicliff G ( 2002) The γ-hydroxybutyrate receptor in the brain. In: Gamma-hydroxybutyrate: molecular, functional and clinical aspects (Tunnicliff G, Cash D, eds), pp 17-27. New York: Taylor and Francis.
- Crunelli V, Leresche N ( 2002a) Childhood absence epilepsy: genes, channels, neurons and networks. Nat Rev Neurosci 3: 371-382. [DOI] [PubMed] [Google Scholar]
- Crunelli V, Leresche N ( 2002b) Action of γ-hydroxybutyrate on neuronal excitability and underlying membrane conductances. In: Gamma-hydroxybutyrate: molecular, functional and clinical aspects (Tunnicliff G, Cash CD, eds), pp 75-110. New York: Taylor and Francis.
- D'Arcangelo G, D'Antuono M, Biagini G, Warren R, Tancredi V, Avoli M ( 2002) Thalamocortical oscillations in a genetic model of absence seizures. Eur J Neurosci 16: 2383-2393. [DOI] [PubMed] [Google Scholar]
- Davies CH, Starkey SJ, Pozza MF, Collingridge GL ( 1991) GABA autoreceptors regulate the induction of LTP. Nature 349: 609-611. [DOI] [PubMed] [Google Scholar]
- Depaulis A, Bourguignon JJ, Marescaux C, Vergnes M, Schmitt M, Micheletti G, Warter JM ( 1988) Effects of gamma-hydroxybutyrate and gamma-butyrolactone derivates on spontaneous generalized non-convulsive seizures in the rat. Neuropharmacology 27: 683-689. [DOI] [PubMed] [Google Scholar]
- Diana M, Mereu G, Mura A, Fadda F, Passino N, Gessa G ( 1991) Low doses of gamma-hydroxybutyric acid stimulate the firing rate of dopaminergic neurons in unanesthetized rats. Brain Res 566: 208-211. [DOI] [PubMed] [Google Scholar]
- Eaton SA, Salt TE ( 1996) Role of N-methyl-d-aspartate and metabotropic glutamate receptors in corticothalamic excitatory postsynaptic potentials in vivo. Neuroscience 73: 1-5. [DOI] [PubMed] [Google Scholar]
- Emri Z, Turner JP, Crunelli V ( 1996a) Tonic activation of presynaptic GABA(B) receptors on thalamic sensory afferents. Neuroscience 72: 689-698. [DOI] [PubMed] [Google Scholar]
- Emri Z, Antal K, Crunelli V ( 1996b) Gamma-hydroxybutyric acid decreases thalamic sensory excitatory postsynaptic potentials by an action on presynaptic GABAB receptors. Neurosci Lett 216: 121-124. [DOI] [PubMed] [Google Scholar]
- Engberg G, Nissbrandt H ( 1993) Gamma-hydroxybutyric acid (GHBA) induces pacemaker activity and inhibition of substantia nigra dopamine neurons by activating GABAB-receptors. Naunyn Schmiedebergs Arch Pharmacol 348: 491-497. [DOI] [PubMed] [Google Scholar]
- Ferraro L, Tanganelli S, O'Connor WT, Francesconi W, Loche A, Gessa GL, Antonelli T ( 2001) Gamma-hydroxybutyrate modulation of glutamate levels in the hippocampus: an in vivo and in vitro study. J Neurochem 78: 929-939. [DOI] [PubMed] [Google Scholar]
- Godbout R, Jelenic P, Labrie C, Schmitt M, Bourguignon JJ ( 1995) Effect of gamma-hydroxybutyrate and its antagonist NCS-382 on spontaneous cell firing in the prefrontal cortex of the rat. Brain Res 673: 157-160. [DOI] [PubMed] [Google Scholar]
- Guyon A, Leresche N ( 1995) Modulation by different GABAB receptor types of voltage-activated calcium currents in rat thalamocortical neurones. J Physiol (Lond) 485: 29-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris NC, Webb C, Greenfield SA ( 1989) The effects of gamma-hydroxybutyrate on the membrane properties of guinea-pig pars compacta neurons in the substantia nigra in vitro. Neuroscience 31: 363-370. [DOI] [PubMed] [Google Scholar]
- Hechler V, Schmitt M, Bourguignon JJ, Maitre M ( 1990) Trans-gamma-hydroxycrotonic acid binding sites in brain: evidence for a subpopulation of gamma-hydroxybutyrate sites. Neurosci Lett 110: 204-209. [DOI] [PubMed] [Google Scholar]
- Hechler V, Gobaille S, Maitre M ( 1992) Selective distribution pattern of gamma-hydroxybutyrate receptors in the rat forebrain and midbrain as revealed by quantitative autoradiography. Brain Res 572: 345-348. [DOI] [PubMed] [Google Scholar]
- Hechler V, Peter P, Gobaille S, Bourguignon JJ, Schmitt M, Ehrhardt JD, Mark J, Maitre M ( 1993) Gamma-hydroxybutyrate ligands possess antidopaminergic and neuroleptic-like activities. J Pharmacol Exp Ther 264: 1406-1414. [PubMed] [Google Scholar]
- Hechler V, Ratomponirina C, Maitre M ( 1997) Gamma-hydroxybutyrate conversion into GABA induces displacement of GABAB binding that is blocked by valproate and ethosuximide. J Pharmacol Exp Ther 281: 753-760. [PubMed] [Google Scholar]
- Hu RQ, Banerjee PK, Snead III OC ( 2000) Regulation of gamma-aminobutyric acid (GABA) release in cerebral cortex in the gamma-hydroxybutyric acid (GHB) model of absence seizures in rat. Neuropharmacology 39: 427-439. [DOI] [PubMed] [Google Scholar]
- Hughes SW, Cope DW, Blethyn KL, Crunelli V ( 2002) Cellular mechanisms of the slow (1 Hz) oscillation in thalamocortical neurons in vitro. Neuron 33: 947-958. [DOI] [PubMed] [Google Scholar]
- Huntsman MM, Porcello DM, Homanics GE, DeLorey TM, Huguenard JR ( 1999) Reciprocal inhibitory connections and network synchrony in the mammalian thalamus. Science 283: 541-543. [DOI] [PubMed] [Google Scholar]
- Jensen K, Mody I ( 2001) GHB depresses fast excitatory and inhibitory synaptic transmission via GABA(B) receptors in mouse neocortical neurons. Cereb Cortex 11: 424-429. [DOI] [PubMed] [Google Scholar]
- Kao CQ, Coulter DA ( 1997) Physiology and pharmacology of corticothalamic stimulation-evoked responses in rat somatosensory thalamic neurons in vitro. J Neurophysiol 77: 2661-2676. [DOI] [PubMed] [Google Scholar]
- Kemmel V, Taleb O, Perard A, Andriamampandry C, Siffert JC, Mark J, Maitre M ( 1998) Neurochemical and electrophysiological evidence for the existence of a functional gamma-hydroxybutyrate system in NCB-20 neurons. Neuroscience 86: 989-1000. [DOI] [PubMed] [Google Scholar]
- Kemmel V, Taleb O, Andriamampandry C, Aunis D, Maitre M ( 2003) Gamma-hydroxybutyrate receptor function determined by stimulation of rubidium and calcium movements from NCB-20 neurons. Neuroscience 116: 1021-1031. [DOI] [PubMed] [Google Scholar]
- Kim D, Song I, Keum S, Lee T, Jeong MJ, Kim SS, McEnery MW, Shin HS ( 2001) Lack of the burst firing of thalamocortical relay neurons and resistance to absence seizures in mice lacking alpha(1G) T-type Ca(2+) channels. Neuron 31: 35-45. [DOI] [PubMed] [Google Scholar]
- Kim U, Sanchez-Vives MV, McCormick DA ( 1997) Functional dynamics of GABAergic inhibition in the thalamus. Science 278: 130-134. [DOI] [PubMed] [Google Scholar]
- King MA, Thinschmidt JS, Walker DW ( 1997) Gammahydroxybutyrate (GHB) receptor ligand effects on evoked synaptic field potentials in CA1 of the rat hippocampal slice. J Neural Transm 104: 1177-1193. [DOI] [PubMed] [Google Scholar]
- Kleinschmidt S, Grundmann U, Knocke T, Silomon M, Bach F, Larsen R ( 1998) Total intravenous anaesthesia with gamma-hydroxybutyrate (GHB) and sufentanil in patients undergoing coronary artery bypass graft surgery: a comparison in patients with unimpaired and impaired left ventricular function. Eur J Anaesthesiol 15: 559-564. [DOI] [PubMed] [Google Scholar]
- Kulik A, Nakadate K, Nyiri G, Notomi T, Malitschek B, Bettler B, Shigemoto R ( 2002) Distinct localization of GABA(B) receptors relative to synaptic sites in the rat cerebellum and ventrobasal thalamus. Eur J Neurosci 15: 291-307. [DOI] [PubMed] [Google Scholar]
- Lammers GJ, Arends J, Declerck AC, Ferrari MD, Schouwink G, Troost J ( 1993) Gammahydroxybutyrate and narcolepsy: a double-blind placebo-controlled study. Sleep 16: 216-220. [DOI] [PubMed] [Google Scholar]
- Le Feuvre Y, Fricker D, Leresche N ( 1997) GABAA receptor-mediated IPSCs in rat thalamic sensory nuclei: patterns of discharge and tonic modulation by GABAB autoreceptors. J Physiol (Lond) 502: 91-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingenhoehl K, Brom R, Heid J, Beck P, Froestl W, Kaupmann K, Bettler B, Mosbacher J ( 1999) Gamma-hydroxybutyrate is a weak agonist at recombinant GABA(B) receptors. Neuropharmacology 38: 1667-1673. [DOI] [PubMed] [Google Scholar]
- Madden TE, Johnson SW ( 1998) Gamma-hydroxybutyrate is a GABAB receptor agonist that increases a potassium conductance in rat ventral tegmental dopamine neurons. J Pharmacol Exp Ther 287: 261-265. [PubMed] [Google Scholar]
- Maitre M ( 1997) The gamma-hydroxybutyrate signalling system in brain: organization and functional implications. Prog Neurobiol 51: 337-361. [DOI] [PubMed] [Google Scholar]
- Maitre M, Hechler V, Vayer P, Gobaille S, Cash CD, Schmitt M, Bourguignon JJ ( 1990) A specific gamma-hydroxybutyrate receptor ligand possesses both antagonistic and anticonvulsant properties. J Pharmacol Exp Ther 255: 657-663. [PubMed] [Google Scholar]
- Mansvelder HD, Keath JR, McGehee DS ( 2002) Synaptic mechanisms underlie nicotine-induced excitability of brain reward areas. Neuron 33: 905-919. [DOI] [PubMed] [Google Scholar]
- Mehta AK, Muschaweck NM, Maeda DY, Coop A, Ticku MK ( 2001) Binding characteristics of the gamma-hydroxybutyric acid receptor antagonist [(3)H](2E)-(5-hydroxy-5, 7, 8, 9-tetrahydro-6H-benzo[a][7]annulen-6-ylidene) ethanoic acid in the rat brain. J Pharmacol Exp Ther 299: 1148-1153. [PubMed] [Google Scholar]
- Monnier Z, Paupardin-Tritsch D, Leresche N ( 2000) Effect of γ-hydroxybutyric acid on cortical EPSCs and NRT-mediated IPSCs in the rat sensory thalamus. Soc Neurosci Abstr 26: 274.3. [Google Scholar]
- Nicholson KL, Balster RL ( 2001) GHB: a new and novel drug of abuse. Drug Alcohol Depend 63: 1-22. [DOI] [PubMed] [Google Scholar]
- Pinault D, Leresche N, Charpier S, Deniau JM, Marescaux C, Vergnes M, Crunelli V ( 1998) Intracellular recordings in thalamic neurones during spontaneous spike and wave discharges in rats with absence epilepsy. J Physiol (Lond) 509: 449-456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poldrugo F, Addolorato G ( 1999) The role of gamma-hydroxybutyric acid in the treatment of alcoholism: from animal to clinical studies. Alcohol Alcohol 34: 15-24. [DOI] [PubMed] [Google Scholar]
- Salt TE ( 2002) Glutamate receptor functions in sensory relay in the thalamus. Philos Trans R Soc Lond B Biol Sci 357: 1759-1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt C, Gobaille S, Hechler V, Schmitt M, Bourguignon JJ, Maitre M ( 1991) Anti-sedative and anti-cataleptic properties of NCS-382, a gamma-hydroxybutyrate receptor antagonist. Eur J Pharmacol 203: 393-397. [DOI] [PubMed] [Google Scholar]
- Slaght SJ, Leresche N, Deniau JM, Crunelli V, Charpier S ( 2002) Activity of thalamic reticular neurons during spontaneous genetically determined spike and wave discharges. J Neurosci 22: 2323-2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snead III OC ( 1991) The gamma-hydroxybutyrate model of absence seizures: correlation of regional brain levels of gamma-hydroxybutyric acid and gamma-butyrolactone with spike wave discharges. Neuropharmacology 30: 161-167. [DOI] [PubMed] [Google Scholar]
- Snead III OC ( 1995) Basic mechanisms of generalized absence seizures. Ann Neurol 37: 146-157. [DOI] [PubMed] [Google Scholar]
- Snead III OC ( 1996) Presynaptic GABAB- and gamma-hydroxybutyric acid-mediated mechanisms in generalized absence seizures. Neuropharmacology 35: 359-367. [DOI] [PubMed] [Google Scholar]
- Snead III OC, Yu RK, Huttenlocher PR ( 1976) Gamma hydroxybutyrate. Correlation of serum and cerebrospinal fluid levels with electroencephalographic and behavioral effects. Neurology 26: 51-56. [DOI] [PubMed] [Google Scholar]
- Snead III OC, Banerjee PK, Burnham M, Hampson D ( 2000) Modulation of absence seizures by the GABA(A) receptor: a critical role for metabotropic glutamate receptor 4 (mGluR4). J Neurosci 20: 6218-6224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M ( 2000) Corticothalamic resonance, states of vigilance and mentation. Neuroscience 101: 243-276. [DOI] [PubMed] [Google Scholar]
- Steriade M, Contreras D ( 1995) Relations between cortical and thalamic cellular events during transition from sleep patterns to paroxysmal activity. J Neurosci 15: 623-642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M, Jones EG, McCormick DA ( 1997) Thalamus. Oxford: Elsevier Science.
- Tancredi V, Biagini G, D'Antuono M, Louvel J, Pumain R, Avoli M ( 2000) Spindle-like thalamocortical synchronization in a rat brain slice preparation. J Neurophysiol 84: 1093-1097. [DOI] [PubMed] [Google Scholar]
- Tunnicliff G, Cash C ( 2002) Gamma-hydroxybutyrate: molecular, functional and clinical aspects. New York: Taylor and Francis.
- Ulrich D, Huguenard JR ( 1996) GABAB receptor-mediated responses in GABAergic projection neurones of rat nucleus reticularis thalami in vitro. J Physiol (Lond) 493: 845-854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SR, Turner JP, Crunelli V ( 1995) Gamma-hydroxybutyrate promotes oscillatory activity of rat and cat thalamocortical neurons by a tonic GABAB, receptor-mediated hyperpolarization. Neuroscience 66: 133-141. [DOI] [PubMed] [Google Scholar]
- Xie X, Smart TG ( 1992a) Gamma-hydroxybutyrate hyperpolarizes hippocampal neurones by activating GABAB receptors. Eur J Pharmacol 212: 291-294. [DOI] [PubMed] [Google Scholar]
- Xie X, Smart TG ( 1992b) Gamma-hydroxybutyrate depresses monosynaptic excitatory and inhibitory postsynaptic potentials in rat hippocampal slices. Eur J Pharmacol 223: 193-196. [DOI] [PubMed] [Google Scholar]