

Abstract
Mutations in SCN1A, the gene encoding the brain voltage-gated sodium channel α1 subunit (NaV1.1), are associated with at least two forms of epilepsy, generalized epilepsy with febrile seizures plus (GEFS+) and severe myoclonic epilepsy of infancy (SMEI). We examined the functional properties of four GEFS+ alleles and one SMEI allele using whole-cell patch-clamp analysis of heterologously expressed recombinant human SCN1A. One previously reported GEFS+ mutation (I1656M) and an additional novel allele (R1657C), both affecting residues in a voltage-sensing S4 segment, exhibited a similar depolarizing shift in the voltage dependence of activation. Additionally, R1657C showed a 50% reduction in current density and accelerated recovery from slow inactivation. Unlike three other GEFS+ alleles that we recently characterized, neither R1657C nor I1656M gave rise to a persistent, noninactivating current. In contrast, two other GEFS+ mutations (A1685V and V1353L) and L986F, an SMEI-associated allele, exhibited complete loss of function. In conclusion, our data provide evidence for a wide spectrum of sodium channel dysfunction in familial epilepsy and demonstrate that both GEFS+ and SMEI can be associated with nonfunctional SCN1A alleles.
Keywords: epilepsy, sodium channel, GEFS+, SMEI, SCN1A, electrophysiology
Introduction
Voltage-gated sodium channels are responsible for the generation and propagation of action potentials in excitable tissues. Mutations in genes encoding sodium channel pore-forming α subunits and an accessory β1 subunit have been associated with disorders of membrane excitability, including inherited forms of epilepsy (Ptacek, 2002). Two distinct clinical syndromes with overlapping features have been linked recently to mutations in the neuronal sodium channel α1 subunit gene SCN1A, and a more complete understanding of the precise molecular and biophysical mechanisms underlying these disorders is needed.
In generalized epilepsy with febrile seizures plus (GEFS+), convulsions associated with fever continue to occur beyond the age when typical febrile seizures abate (Scheffer and Berkovic, 1997). Affected individuals with GEFS+ may also experience afebrile ictal episodes of varying types, including generalized tonic-clonic, absence, myoclonic, atonic, and myoclonic-astatic seizures. Missense mutations in SCN1A predominate among the genetic defects associated with GEFS+ (Escayg et al., 2000, 2001; Abou-Khalil et al., 2001; Sugawara et al., 2001a; Wallace et al., 2001b), but heritable alterations in two other sodium channel genes (SCN1B and SCN2A) (Wallace et al., 1998; Sugawara et al., 2001b) and a GABA receptor subunit (GABRG2) (Baulac et al., 2001; Wallace et al., 2001a) have been implicated as well. In most cases, GEFS+ is considered to be a benign disorder.
In contrast, severe myoclonic epilepsy of infancy (SMEI), also known as Dravet syndrome (Online Mendelian Inheritance in Man 607208), is a rare convulsive disorder characterized by febrile seizures with onset during the first year of life, followed by intractable epilepsy, impaired psychomotor development, and ataxia (Dravet and Bureau, 1981; Scheffer et al., 2001). Seizures in this disorder are usually unresponsive to anticonvulsant drugs. Recently, several de novo heterozygous SCN1A mutations have been reported in SMEI probands including missense, nonsense, and insertion/deletion alleles (Claes et al., 2001, 2003; Ohmori et al., 2002; Sugawara et al., 2002; Fujiwara et al., 2003; Nabbout et al., 2003). The observed clinical similarities between SMEI and GEFS+, including the frequent occurrence of febrile seizures and shared molecular genetic etiology, have prompted the idea that these disorders represent two extremes in clinical presentation of the same condition (Singh et al., 2001).
To improve our understanding of the molecular basis of GEFS+ and SMEI specifically, and to broaden our knowledge of epileptogenesis in general, we and others have begun characterizing the biophysical properties of mutant voltage-gated sodium channels associated with these disorders (Alekov et al., 2000, 2001; Spampanato et al., 2001, 2003; Lossin et al., 2002; Cossette et al., 2003). Recently, we reported that three SCN1A mutations associated with GEFS+ caused similar defects in fast inactivation gating characterized by a persistent, noninactivating current during membrane depolarizations when recombinant human SCN1A was coexpressed heterologously with its accessory subunits (Lossin et al., 2002). Our findings suggested that, in some cases of GEFS+, SCN1A mutations may promote a gain of function in sodium channels, leading to neuronal hyperexcitability. Whether all SCN1A mutations associated with GEFS+ exhibit the same pattern of channel dysfunction has not been determined.
We have now characterized four additional SCN1A missense mutations associated with GEFS+, including a novel allele and one missense mutation reported in an SMEI proband. Surprisingly, two of the four GEFS+ mutations, as well as the SMEI mutation, exhibited no function in heterologous cells. In contrast, the other two GEFS+ alleles were functional but had altered gating properties. Our findings suggest that a spectrum of functional sodium channel defects rather than a single biophysical mechanism contribute to the pathogenesis of inherited epilepsy.
Materials and Methods
Mutation screening. Family NY-005 is a small, three-generation pedigree with Ashkenazi Jewish heritage ascertained in the New York City area. The proband (male) developed generalized tonic-clonic seizures associated with high fever at 2 years of age. Over the subsequent 18 months, he experienced at least three additional generalized febrile convulsions generally lasting 2-5 min. In addition, he experienced episodic myoclonic jerks associated with momentary impairment of consciousness and conjugate upward eye deviation. The frequency of these episodes was variable, but fatigue, loud noise, or sudden bright light exposure were identified as possible triggering events. The proband's mother, maternal grandmother, and a maternal aunt all experienced febrile convulsions during childhood, although the age of termination of febrile seizures is unknown. This family was diagnosed with probable GEFS+.
Genomic DNA (25 ng) was screened for SCN1A mutations using PCR-single-strand conformational analysis, as described previously. Mutation R1657C was independently verified using allele-specific oligonucleotide hybridization coupled with chemiluminescent detection (Pierce, Rockford, IL). The probe sequence was 5′-ATCTGCACGCTGCT-biotin-3′ (mutant base italicized). Hybridizations and washes were performed at 47.5°C.
Mutagenesis and heterologous expression of human SCN1A. Full-length human SCN1A (NaV1.1) cDNA was prepared and mutated as described previously (Lossin et al., 2002). To reconstitute the native brain sodium channel complex, SCN1A was coexpressed heterologously with human accessory β1 and β2 subunits in human tsA201 cells, as described previously (Lossin et al., 2002). All mutant cDNAs were fully sequenced before their use in transfection experiments. At least two different recombinant clones of each mutant cDNA were evaluated. Concentration of each plasmid DNA was determined spectrophotometrically (UV) as well as fluorometrically (PicoGreen; Molecular Probes, Eugene, OR).
Electrophysiology and data analysis. Whole-cell voltage-clamp experiments addressing fast inactivation were described previously (Lossin et al., 2002). Whole-cell capacitance and access resistance were determined by integrating capacitive transients of voltage steps from -120 to -110 mV filtered at 10 kHz. Series resistance (2 ± 0.1 MΩ) was compensated 87-95% to assure that the command potential was reached within microseconds and with a voltage error <3 mV. All data were acquired at 10-50 kHz and low-pass Bessel filtered at 5 kHz. Persistent current was evaluated by 100 msec depolarizations to -10 mV in the presence and absence of 10 μm TTX, a potent sodium channel blocker (Sigma, St. Louis, MO). Representative traces for each condition were selected to have the same initial leak current, a composite TTX-sensitive trace was generated, and the mean current of the composite trace between 80 and 100 msec was calculated as percentage of the peak current. Data of recovery and steady-state inactivation experiments were digitally adjusted for leak current by subtracting the mean inward current apparent at 15-20 msec in a -10 mV step. Recording artifacts were further avoided by selecting traces with rapid capacitive transients and a holding current at -120 mV of <5% of the peak current. The range of peak current amplitude recorded at -10 mV in this study was 0.8-11.3 nA.
Slow inactivation was examined by paired-pulse protocols in which the response to a 50 msec test pulse to -10 mV was measured before (P1) and after (P2) variable conditioning protocols. Current measured during P2 was then normalized to the maximal current measured during P1. In detail, for onset of slow inactivation, the conditioning comprised voltage steps to -10 mV lasting from 100 msec to 100 sec, followed by a 50 msec recovery from fast inactivation at -120 mV. Conditioning for steady-state slow inactivation consisted of a 30 sec step to voltages between -140 and -10 mV and a 50 msec fast inactivation recovery at -120 mV. Conditioning for recovery from slow inactivation experiments used a 30 sec pulse to -10 mV, followed by recovery periods ranging from 100 msec to 100 sec. P1 pulses were repeated before each change of condition to minimize offset from run-down. In all cases, sufficient recovery time at -120 mV was allotted between different conditions to avoid accumulation of slow inactivation (determined in preliminary experiments; data not shown). Recordings with noticeable changes in P1 amplitude during the course of the experiment were discarded. Data from onset of slow inactivation experiments were plotted versus the length of the conditioning pulse and fitted to the two-exponential decay function:
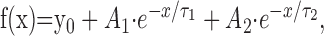 |
1 |
where Ai describes the fraction of the channels entering slow inactivation with time constant τi, and y0 describes fraction of the channels that remains active. Steady-state slow inactivation data were fitted to the Boltzmann function:
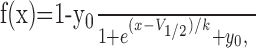 |
2 |
where V1/2 is the voltage at which half-maximal slow inactivation occurs, k is the slope factor of the fit, and y0 is the fraction of the channels that remains active. Recovery from slow inactivation was fitted to the two-exponential function:
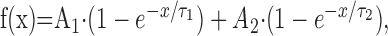 |
3 |
where Ai refers the fraction of the channels recovering with time constant τi. Data analysis was performed using Clampfit 8.2 (Axon Instruments, Union City, CA), Excel 2002 (Microsoft, Seattle, WA), and OriginPro 7.0 (OriginLab, Northampton, MA) software. All data were fit using a nonlinear least squares minimization method. Results are presented as means ± SE, and statistical comparisons were made between data from mutant sodium channels and wild-type (WT) SCN1A using the unpaired Student's t test. Statistical significance was assumed for p < 0.05 and deemed not significant (NS) for p ≥ 0.05. In some figures, the SE bars are smaller than the data symbols.
Single-channel current measurements were performed in the outside-out patch configuration at room temperature using the same solutions as the whole-cell experiments. Pipettes were pulled from thick-wall borosilicate glass (World Precision Instruments, Sarasota, FL) with a multistage P-97 Flaming-Brown micropipette puller (Sutter Instruments, San Rafael, CA). Fire-polished patch pipettes for single-channel studies were additionally coated with Sylgard 184 (Dow Corning, Midland, MI) and had resistance of ∼6 MΩ with standard pipette and bath solutions. Single-channel conductance was estimated by fitting the single-channel amplitude-voltage relationships in the range -30 to +10 mV by linear regression.
Rescue experiments. To test for rescue of trafficking-defective mutants, tsA201 cells were transfected with L986F plasmid DNA and both β subunits at 37°C, as described above. Immediately after the transfection, the medium was replaced with drug-supplemented media or the cells were transferred to a low temperature (26°C) incubator. The cells were washed three times with prewarmed DMEM and incubated in drug-free media for another 1.5 hr at 37°C before data acquisition. Low temperature rescue only was analogously attempted for V1353L and A1685V.
Results
A novel SCN1A mutation associated with epilepsy
We screened a family with Ashkenazi Jewish heritage segregating probable GEFS+ and discovered a novel C→T transition at nucleotide 4969 (nucleotide numbering based on the SCN1A open reading frame). The mutation was present in all affected members but not in 342 normal controls, including 164 samples from individuals of Ashkenazi descent. The nucleotide change predicts the replacement of arginine-1657 with cysteine in the S4 segment of domain IV (D4/S4), part of the sodium channel voltage-sensing apparatus. The replaced arginine is the innermost positively charged residue in D4/S4 and is highly conserved among vertebrate and invertebrate sodium channels. We designated this mutation R1657C.
Biophysical characterization of I1656M and R1657C
The novel mutation described above lies immediately adjacent to another previously reported GEFS+ allele (I1656M) (Wallace et al., 2001b) and in close proximity to R1648H, an SCN1A mutation originally described by Escayg et al. (2000) that we recently demonstrated to exhibit abnormal inactivation (Lossin et al., 2002). We determined the biophysical characteristics of both R1657C and I1656M using recombinant human SCN1A cDNA expressed heterologously with the recombinant human accessory sodium channel subunits β1 and β2. Figure 1, a-c, shows representative whole-cell voltage-clamp recordings from cells expressing either WT SCN1A, R1657C, or I1656M channels. Cells expressing either mutant exhibited voltage-dependent inward current that resembled cells transfected with WT channels. Inactivation proceeded in a rapid biexponential manner, with no apparent difference between WT and the mutants (Fig. 1d). However, cells expressing R1657C exhibited significantly lower sodium current density (p < 0.005) across a wide range of test potentials compared with cells transfected with identical amounts of WT or I1656M plasmids (Fig. 2a). Similarly, both I1656M and R1657C were less efficiently expressed, as measured by the percentage of patch-clamped cells that exhibited voltage-gated inward current greater than the -0.7 nA threshold we used to exclude endogenous channel activity (mean, less than -100 pA; untransfected cells, n = 6). Only 29.6% (n = 309; p < 0.0001) of cells transfected with R1657C and 54.6% (n = 97; p < 0.05) of cells transfected with I1656M exhibited voltage-gated current compared with 70.7% (n = 254) for WT. These findings were independent of the time period after transfection. The lower peak current amplitude observed for R1657C was not caused by a reduced single-channel conductance (16.8 ± 1.0 pS, n = 6 patches; compared with WT: 18.0 ± 0.5 pS, n = 15 patches; NS).
Figure 1.

WT and mutant whole-cell currents from tsA201 cells. a-c, Cells were stepped to various potentials between -80 and +60 mV in 10 mV intervals from a holding potential of -120 mV (see Fig. 2b). All experiments were performed in the whole-cell patch-clamp configuration at room temperature 24-72 hr after transfection of the indicated DNA. Calibration: 1 nA vs. 2 msec. d, Inactivation time constants of WT and mutant SCN1A currents. The decay phase of voltage-sensitive inward currents was fitted with a two-exponential function, as described in Materials and Methods, generating τfast (open symbols) and τslow (closed symbols). No significant difference between WT and mutant currents was found.
Figure 2.

Biophysical characterization of GEFS+-associated mutants. a, Current-voltage relationships of whole-cell currents from transiently transfected cells. Currents were elicited by test pulses to various potentials (b, inset) and normalized to cell capacitance. WT, n = 17; I1656M, n = 8; R1657C, n = 17. R1657C current density is significantly smaller than WT between -30 and +40 mV (p < 0.005). b, Voltage dependence of activation of whole-cell currents. The voltage dependence of channel activation was estimated by measuring peak sodium current during a variable test potential from a holding potential of -120 mV. The current at each membrane potential was divided by the electrochemical driving force for sodium ions and normalized to the maximum sodium conductance. c, Voltage dependence of inactivation. The two-pulse protocol outlined in the inset was used to examine channel availability after conditioning at various potentials. Currents were normalized to the peak current amplitude. d, Recovery from fast inactivation. Channels were inactivated by a 100 msec pulse and stepped back to -120 mV for increasingly long periods. Currents were normalized to the peak current amplitude measured during the inactivation pulse and fitted to a two-exponential function generating a fast and a slower recovery time constant. Fit parameters for all experiments shown are provided in Table 1.
Disturbances in activation and inactivation
Because both R1657C and I1656M affect conserved residues in a voltage-sensor domain (D4/S4 segment), we assessed the voltage dependence of activation and discovered that both mutants caused significant depolarizing shifts in this relationship. The voltage at which channels were half-maximally activated was -23.6 ± 1.2 mV (n = 17) for WT channels and -17.4 ± 1.5 mV (n = 8; p < 0.01) and -16.6 ± 1.3 mV (n = 17; p < 0.0005) for I1656M and R1657C, respectively (Fig. 2b). Additionally, both mutants had statistically decreased voltage sensitivity, as suggested by comparison of their slope factors (k) with that of WT (Table 1). These findings indicate that R1657C and I1656M sodium channels will require a greater degree of membrane depolarization to activate, and this may contribute to altered neuronal excitability.
Table 1.
Biophysical parameters for activation and fast inactivation
|
|
Voltage dependence of activation |
Voltage dependence of fast inactivation |
Recovery from fast inactivation |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
V½(mV)
|
k(mV)
|
n
|
V½ (mV)
|
k (mV)
|
n
|
τ1 (msec)
|
τ2 (msec)
|
n
|
||||||
| WT | −23.6 ± 1.2 | 7.4 ± 0.3 | 17 | −64.1 ± 1.1 | −5.7 ± 0.1 | 18 | 3.9 ± 0.5 | 96 ± 16 | 19 | ||||||
| (83 ± 2%) | (17 ± 2%) | ||||||||||||||
| l1656M | −17.4 ± 1.5† | 8.3 ± 0.2* | 8 | −68.2 ± 1.7* | −7.1 ± 0.4††† | 8 | 5.6 ± 0.6 | 60 ± 14 | 7 | ||||||
| (87 ± 3%) | (13 ± 3%) | ||||||||||||||
| R1657C | −16.6 ± 1.3*** | 8.3 ± 0.2† | 18 | −67.1 ± 1.4 | −5.2 ± 0.1** | 18 | 1.9 ± 0.2** | 29 ± 3†† | 15 | ||||||
|
|
|
|
|
|
|
|
(76 ± 2%**)
|
(24 ± 2%**)
|
|
||||||
Values significantly different from WT are marked with asterisks or daggers: *p < 0.05; **p < 0.005; ***p < 0.0005; †p < 0.01; ††p < 0.001; †††p < 0.0001.
The two mutants also displayed subtle, yet significant, defects in fast inactivation. I1656M exhibited a slight hyperpolarizing shift in half-maximal steady-state inactivation (V1/2) (Table 1; Fig. 2c), and both mutants had slope factors for inactivation that differed significantly from WT, albeit in opposite directions (Table 1). Recovery from inactivation of WT channels was distributed into two populations, with the majority (83 ± 2%) recovering within a few milliseconds (τ1 = 3.9 ± 0.5 msec) and a minor population of channels (17 ± 2%) recovering more slowly (τ2 = 96 ± 16 msec) (Fig. 2d). In cells expressing R1657C, recovery from fast inactivation was faster in both populations (p < 0.005 and 0.001 for τ1 and τ2, respectively), but there was a significant (p < 0.005) redistribution such that more channels partitioned to the more slowly recovering state (Table 1).
Alterations in slow inactivation
We also tested cells expressing either WT or mutant channels for differences in slow inactivation properties (Fig. 3). In these experiments, the effects of fast inactivation were eliminated by a recovery pulse to -120 mV lasting at least 50 msec (see Materials and Methods). Entry into the slow inactivated state was biexponential, with more than half (57 ± 5%) of WT channels entering with a time constant smaller than 1000 msec (τ1) (Table 2). For R1657C, we observed a significantly (p < 0.05) greater proportion of channels entering the slow inactivated state with the larger time constant (τ2) (Table 2). Steady-state availability after 30 sec depolarizations to various potentials was similar in WT and both mutants, but slow inactivation voltage sensitivity was significantly (p < 0.01) reduced in I1656M (Fig. 3b). When subjected to a 30 sec depolarization, WT channels recovered in a biexponential manner, with a large proportion of the channels (69 ± 2%) repriming quickly (τ1 = 281 ± 45 msec) and the remainder (31 ± 2%) requiring more sustained (τ2 = 4896 ± 1542) hyperpolarization. Under the same conditions, cells expressing R1657C recovered significantly (p < 0.01) faster (τ1 = 146 ± 17 msec), whereas cells expressing I1656M displayed a significantly (p < 0.05) larger proportion of rapidly recovering channels. These observations are in keeping with a subtle impairment in slow inactivation for both mutants.
Figure 3.

Slow inactivation properties of I1656M and R1657C. a, Onset of slow inactivation. Cells were stepped to -10 mV for 0.001-100 sec, allowed to recover from fast inactivation at -120 mV for 50 msec, and subjected to a -10 mV test pulse. b, Steady-state slow inactivation after a 30 sec depolarization to potentials between -140 and -10 mV. Cells were allowed to recover from fast inactivation at -120 mV for 50 msec before the actual test pulse to -10 mV. c, Recovery from slow inactivation. Cells were conditioned at -10 mV for 30 sec, allowed to recover at -120 mV for 0.1-100 sec, and immediately tested at -10 mV. Because the intermediate recovery period always exceeded 100 msec, effects of fast inactivation were deemed to be negligible. All data were fitted to a two-exponential or Boltzmann function, as described in Materials and Methods. Fit parameters for all experiments shown are provided in Table 2.
Table 2.
Biophysical parameters for slow inactivation
|
|
Onset of slow inactivation |
Voltage dependence of slow inactivation |
Recovery from slow inactivation |
||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
τ1 (msec)
|
τ2 (msec)
|
Mean residual current (%)
|
n
|
V½ (mV)
|
k (mV)
|
Mean residual current (%)
|
n
|
τ1 (msec)
|
τ2 (msec)
|
n
|
||||||||
| WT | 881 ± 105 | 11479 ± 4446 | 16 ± 2 | 12 | −72.5 ± 1.8 | 6.4 ± 0.3 | 12 ± 2 | 10 | 281 ± 45 | 4896 ± 1542 | 12 | ||||||||
| (57 ± 5%) | (27 ± 5%) | (69 ± 2%) | (31 ± 2%) | ||||||||||||||||
| I1656M | 961 ± 217 | 4548 ± 1245 | 15 ± 2 | 8 | −71.3 ±1.5 | 7.5 ± 0.3† | 10 ± 2 | 9 | 244 ± 31 | 4586 ± 856 | 10 | ||||||||
| (46 ± 8%) | (39 ± 6%) | (76 ± 3%*) | (24 ± 3%) | ||||||||||||||||
| R1657C | 546 ± 126 | 6701 ± 3396 | 16 ± 4 | 9 | −72.9 ± 1.6 | 6.4 ± 0.4 | 17 ± 3 | 10 | 146 ± 17† | 2724 ± 515 | 13 | ||||||||
|
|
(30 ± 6%**)
|
(54 ± 10%*)
|
|
|
|
|
|
|
(73 ± 2%)
|
(27 ± 2%)
|
|
||||||||
Values significantly different from WT are marked with asterisks or daggers: *p < 0.05; **p < 0.005; †p < 0.01.
Absence of persistent current in I1656M and R1657C
We previously observed that three other SCN1A mutations associated with GEFS+ including R1648H, also in D4/S4, confer a biophysical defect characterized by persistent, noninactivating whole-cell current. However, neither I1656M nor R1657C generated persistent currents statistically larger than WT as determined by subtraction of TTX-insensitive current (magnitude of persistent current as percentage of peak current amplitude: WT: 0.60 ± 0.07%, n = 7; I1656M: 0.81 ± 0.15%, n = 8; R1657C: 0.84 ± 0.17%, n = 9). This finding was reinforced by the absence of observable channel activation during slow voltage ramps from -120 to +40 mV over 8 sec (data not shown). Therefore, it seems that this single biophysical defect is not shared by all SCN1A mutations associated with GEFS+.
Nonfunctional SCN1A mutations
During this study, we also examined three additional SCN1A mutants associated with either GEFS+ (V1353L and A1685V) (Sugawara et al., 2001a; Wallace et al., 2001b) or SMEI (L986F) (Claes et al., 2001). All three mutations were associated with complete absence of heterologously expressed SCN1A sodium current (peak current amplitude, less than -0.3 nA). We ruled out cloning artifacts as an explanation for nonfunctional sodium channels by verifying the complete coding sequencing of each mutant construct and by examining multiple independent recombinant clones.
The absence of sodium current could have several explanations with primary failure of sodium conductance or defects in protein trafficking representing the two most likely causes. In other ion channel disorders such as cystic fibrosis and congenital long QT syndrome, misfolded proteins can be functionally rescued with a variety of in vitro experimental maneuvers (French et al., 1996; Sato et al., 1996; Rubenstein et al., 1997; Egan et al., 2002; Rajamani et al., 2002; Valdivia et al., 2002). We tested several strategies to rescue the L986F mutation, including lowering the incubation temperature (26°C) of cultured cells after transfection, exposure of cells to sodium channel blockers (phenytoin, lidocaine, or mexiletine) that may stabilize protein conformation, and treatment with thapsigargin to inhibit Ca2+-dependent quality control mechanisms in the endoplasmic reticulum. However, none of these experimental maneuvers were successful in rescuing measurable sodium current. Similarly, low temperature incubation of cells transfected with V1353L or A1685V did not rescue sodium currents.
These findings suggest that nonfunctional alleles may explain either GEFS+ or SMEI and further emphasize that a single biophysical mechanism does not explain epilepsy in these disorders. Our results also suggest that the correlation between in vitro biophysical behavior and clinical syndrome is complex.
Discussion
The identification of human ion channel mutations in epilepsy has provided new opportunities to investigate the molecular basis of this complex and heterogeneous neurological condition (Meisler et al., 2001). We have focused our efforts on defining the biophysical defects associated with mutations in the human neuronal voltage-gated sodium channel gene SCN1A. Mutations in this gene are associated with GEFS+, often transmitted as an autosomal dominant trait, and SMEI (Dravet syndrome), a more severe disorder with an unclear inheritance pattern (Dravet and Bureau, 1981; Scheffer et al., 2001). In some families, both disorders may be present, suggesting that GEFS+ and SMEI represent a clinical spectrum of the same disease (Singh et al., 2001). More than one hundred SCN1A mutations have been defined to date, with missense mutations being most common in GEFS+ (Escayg et al., 2000, 2001; Abou-Khalil et al., 2001; Sugawara et al., 2001a; Wallace et al., 2001b; Ito et al., 2002) and more deleterious alleles (nonsense, frameshift) representing the majority of SMEI mutations (Claes et al., 2001; Sugawara et al., 2001a; Ohmori et al., 2002; Fujiwara et al., 2003; Nabbout et al., 2003). Given this mutational diversity, it is logical to assume that a range of functional defects account for the varied clinical presentation of these disorders.
Gain or loss of function in GEFS+?
We recently characterized three missense SCN1A mutations associated with GEFS+ (R1648H, T875M, and W1204R) and discovered a shared biophysical defect consistent with a gain of sodium conductance over time because of a persistent, noninactivating current component (Lossin et al., 2002). This functional defect is consistent with the notion of neuronal hyperexcitability attributable to the impact of sustained sodium current on action potential threshold. At least one other human brain sodium channel mutation associated with an inherited epilepsy (R187W in SCN2A) has been demonstrated to exhibit abnormal inactivation leading to increased sodium influx (Sugawara et al., 2001b). Similarly, transgenic mice with neuronal overexpression of an inactivation-impaired NaV1.2 sodium channel exhibit severe epilepsy (Kearney et al., 2001). These findings indicate that gain-of-function sodium channel mutations can lead to phenotypes attributed to neuronal hyperexcitability.
In contrast to these previously characterized gain-of-function mutations, we now demonstrate that two mutant SCN1A alleles associated with GEFS+ (I1656M and R1657C) exhibited biophysical features consistent with either reduced function (reduced current density and expression efficiency, depolarizing shift in the voltage dependence of activation) or enhanced function (accelerated recovery from fast and slow inactivation in R1657C), whereas two other missense mutations associated with this disorder (V1353L and A1685V) completely abolished functional sodium channel activity in heterologous cells. Neither I1656M nor R1657C exhibited a noninactivating sodium current component. These data indicate that not all SCN1A mutations associated with GEFS+ cause the same biophysical defect and, thus, illustrate functional heterogeneity in this syndrome.
The functional heterogeneity observed for SCN1A mutations may help explain some of the clinical variability in phenotype severity between families segregating different alleles, but it does not explain intrafamilial variation in the clinical expression of the disorder. In the three-generation family in which we discovered R1657C and in many other GEFS+ families, considerable differences in epilepsy severity were evident among mutation carriers, suggesting possible involvement of other genetic or environmental modifiers.
The coexistence of biophysical characteristics with partially offsetting effects as observed for I1656M and R1657C makes simple classification of these alleles as either “gain” or “loss” of function seem inappropriate. Predicting the cellular consequences of these biophysical defects is very difficult, and additional experiments are clearly warranted to define mutation effects in native neurons. Although the subpopulation of neurons most likely affected by these sodium channel disturbances is uncertain, we can speculate that reduced excitability of inhibitory neurons contributes to the seizure susceptibility in this disorder. This notion is consistent with the fact that mutations in a GABA receptor subunit resulting in impaired receptor function cause a similar condition (Baulac et al., 2001; Wallace et al., 2001a; Bianchi et al., 2002). In contrast, subtle impairment of slow inactivation as observed for R1657C may enable more rapid recovery of channels from prolonged membrane depolarizations. This notion, however, disagrees with findings of other groups that have observed enhanced susceptibility to slow inactivation with different GEFS+-associated SCN1A mutants (Alekov et al., 2000, 2001; Spampanato et al., 2001). Experimental variations, such as the use of a mammalian expression system in our study or use of the human SCN1A versus SCN4A (Alekov et al., 2000, 2001) or rat Scn1A (Spamanato et al., 2001) may account, in part, for these observed differences. Alternatively, the observed differences may reflect the actual biophysical heterogeneity among SCN1A mutants. The expression pattern of SCN1A is broad, and specific localization in inhibitory neurons is not known (Whitaker et al., 2000). Careful analysis of the spatial and temporal distribution of SCN1A, its splice isoforms, and their regulation is expected to improve our understanding of these disorders as will development of animal models expressing GEFS+ mutations in defined brain regions.
Functional SCN1A haploinsufficiency in SMEI and GEFS+
The majority of SCN1A mutations found to date in SMEI have been de novo nonsense or frameshift alleles that are presumed to encode nonfunctional sodium channels (Claes et al., 2001; Sugawara et al., 2001a; Ohmori et al., 2002; Nabbout et al., 2003). We characterized one missense mutation (L986F) associated with SMEI and discovered that it is also nonfunctional, as were two alleles (V1353L and A1685V) associated with GEFS+. These findings suggest that haploinsufficiency for SCN1A explains SMEI and some cases of GEFS+. This was an unexpected finding for the two GEFS+ mutations because the severity of this disorder is much less than that of SMEI. The more subtle channel-gating disturbances we have defined for other GEFS+ alleles seem more consistent with a mild and paroxysmal clinical phenotype. Perhaps other factors, including genetic and environmental modifiers, alter the host response to SCN1A haploinsufficiency and determine the eventual severity of the disease. Additional studies are needed to examine SMEI and GEFS+ families carefully for phenotype variability.
Footnotes
This work was supported by National Institutes of Health Grant NS32387 (A.L.G.), by a predoctoral fellowship award from the Epilepsy Foundation (C.L.), by the Kathy Holden Genetic Research Fund of the Epilepsy Foundation, and by a grant from the Roland and Ruby Holden Foundation, on behalf of Ronald and Arlene Holden.
Correspondence should be addressed to Dr. Alfred L. George Jr, Division of Genetic Medicine, 529 Light Hall, Vanderbilt University, 2215 Garland Avenue, Nashville, TN 37232-0275. E-mail: al.george@vanderbilt.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/2311289-07$15.00/0
References
- Abou-Khalil B, Ge Q, Desai R, Ryther R, Bazyk A, Bailey R, Haines JL, Sutcliffe JS, George Jr AL ( 2001) Partial and generalized epilepsy with febrile seizures plus and a novel SCN1A mutation. Neurology 57: 2265-2272. [DOI] [PubMed] [Google Scholar]
- Alekov A, Rahman MM, Mitrovic N, Lehmann-Horn F, Lerche H ( 2000) A sodium channel mutation causing epilepsy in man exhibits subtle defects in fast inactivation and activation in vitro. J Physiol (Lond) 529: 533-539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alekov AK, Rahman MM, Mitrovic N, Lehmann-Horn F, Lerche H ( 2001) Enhanced inactivation and acceleration of activation of the sodium channel associated with epilepsy in man. Eur J Neurosci 13: 2171-2176. [DOI] [PubMed] [Google Scholar]
- Baulac S, Huberfeld G, Gourfinkel-An I, Mitropoulou G, Beranger A, Prud'homme JF, Baulac M, Brice A, Bruzzone R, LeGuern E ( 2001) First genetic evidence of GABA(A) receptor dysfunction in epilepsy: a mutation in the gamma2-subunit gene. Nat Genet 28: 46-48. [DOI] [PubMed] [Google Scholar]
- Bianchi MT, Song L, Zhang H, Macdonald RL ( 2002) Two different mechanisms of disinhibition produced by GABAA receptor mutations linked to epilepsy in humans. J Neurosci 22: 5321-5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claes L, Del Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, De Jonghe P ( 2001) De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet 68: 1327-1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claes L, Ceulemans B, Audenaert D, Smets K, Lofgren A, Del Favero J, Ala-Mello S, Basel-Vanagaite L, Plecko B, Raskin S, Thiry P, Wolf NI, Van Broeckhoven C, De Jonghe P ( 2003) De novo SCN1A mutations are a major cause of severe myoclonic epilepsy of infancy. Hum Mutat 21: 615-621. [DOI] [PubMed] [Google Scholar]
- Cossette P, Loukas A, Lafreniere RG, Rochefort D, Harvey-Girard E, Ragsdale DS, Dunn RJ, Rouleau GA ( 2003) Functional characterization of the D188V mutation in neuronal voltage-gated sodium channel causing generalized epilepsy with febrile seizures plus (GEFS). Epilepsy Res 53: 107-117. [DOI] [PubMed] [Google Scholar]
- Dravet C, Bureau M ( 1981) The benign myoclonic epilepsy of infancy (author's translation). Rev Electroencephalogr Neurophysiol Clin 11: 438-444. [DOI] [PubMed] [Google Scholar]
- Egan ME, Glockner-Pagel J, Ambrose C, Cahill PA, Pappoe L, Balamuth N, Cho E, Canny S, Wagner CA, Geibel J, Caplan MJ ( 2002) Calcium-pump inhibitors induce functional surface expression of delta F508-CFTR protein in cystic fibrosis epithelial cells. Nat Med 8: 485-492. [DOI] [PubMed] [Google Scholar]
- Escayg A, MacDonald BT, Meisler MH, Baulac S, Huberfeld G, An-Gourfinkel I, Brice A, LeGuern E, Moulard B, Chaigne D, Buresi C, Malafosse A ( 2000) Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat Genet 24: 343-345. [DOI] [PubMed] [Google Scholar]
- Escayg A, Heils A, MacDonald BT, Haug K, Sander T, Meisler MH ( 2001) A novel SCN1A mutation associated with generalized epilepsy with febrile seizures plus—and prevalence of variants in patients with epilepsy. Am J Hum Genet 68: 866-873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French PJ, van Doorninck JH, Peters RH, Verbeek E, Ameen NA, Marino CR, de Jonge HR, Bijman J, Scholte BJ ( 1996) A delta F508 mutation in mouse cystic fibrosis transmembrane conductance regulator results in a temperature-sensitive processing defect in vivo. J Clin Invest 98: 1304-1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara T, Sugawara T, Mazaki-Miyazaki E, Takahashi Y, Fukushima K, Watanabe M, Hara K, Morikawa T, Yagi K, Yamakawa K, Inoue Y ( 2003) Mutations of sodium channel alpha subunit type 1 (SCN1A) in intractable childhood epilepsies with frequent generalized tonic-clonic seizures. Brain 126: 531-546. [DOI] [PubMed] [Google Scholar]
- Ito M, Nagafuji H, Okazawa H, Yamakawa K, Sugawara T, Mazaki-Miyazaki E, Hirose S, Fukuma G, Mitsudome A, Wada K, Kaneko S ( 2002) Autosomal dominant epilepsy with febrile seizures plus with missense mutations of the (Na+)-channel alpha 1 subunit gene, SCN1A. Epilepsy Res 48: 15-23. [DOI] [PubMed] [Google Scholar]
- Kearney JA, Plummer NW, Smith MR, Kapur J, Cummins TR, Waxman SG, Goldin AL, Meisler MH ( 2001) A gain-of-function mutation in the sodium channel gene Scn2a results in seizures and behavioral abnormalities. Neuroscience 102: 307-317. [DOI] [PubMed] [Google Scholar]
- Lossin C, Wang DW, Rhodes TH, Vanoye CG, George Jr AL ( 2002) Molecular basis of an inherited epilepsy. Neuron 34: 877-884. [DOI] [PubMed] [Google Scholar]
- Meisler MH, Kearney J, Ottman R, Escayg A ( 2001) Identification of epilepsy genes in human and mouse. Annu Rev Genet 35: 567-588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabbout R, Gennaro E, Dalla BB, Dulac O, Madia F, Bertini E, Capovilla G, Chiron, C, Cristofori G, Elia M, Fontana E, Gaggero R, Granata T, Guerrini R, Loi M, La Selva L, Lispi ML, Matricardi A, Romeo A, Tzolas V, Valseriati D, Veggiotti P, Vigevano F, Vallee L, Dagna BF, Bianchi A, Zara F ( 2003) Spectrum of SCN1A mutations in severe myoclonic epilepsy of infancy. Neurology 60: 1961-1967. [DOI] [PubMed] [Google Scholar]
- Ohmori I, Ouchida M, Ohtsuka Y, Oka E, Shimizu K ( 2002) Significant correlation of the SCN1A mutations and severe myoclonic epilepsy in infancy. Biochem Biophys Res Commun 295: 17-23. [DOI] [PubMed] [Google Scholar]
- Ptacek L ( 2002) Channelopathies: episodic disorders of the nervous system. Novartis Found Symp 241: 87-104. [PubMed] [Google Scholar]
- Rajamani S, Anderson CL, Anson BD, January CT ( 2002) Pharmacological rescue of human K(+) channel long-QT2 mutations: human ether-a-go-go-related gene rescue without block. Circulation 105: 2830-2835. [DOI] [PubMed] [Google Scholar]
- Rubenstein RC, Egan ME, Zeitlin PL ( 1997) In vitro pharmacologic restoration of CFTR-mediated chloride transport with sodium 4-phenylbutyrate in cystic fibrosis epithelial cells containing delta F508-CFTR. J Clin Invest 100: 2457-2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S, Ward CL, Krouse ME, Wine JJ, Kopito RR ( 1996) Glycerol reverses the misfolding phenotype of the most common cystic fibrosis mutation. J Biol Chem 271: 635-638. [DOI] [PubMed] [Google Scholar]
- Scheffer IE, Berkovic SF ( 1997) Generalized epilepsy with febrile seizures plus. A genetic disorder with heterogeneous clinical phenotypes. Brain 120: 479-490. [DOI] [PubMed] [Google Scholar]
- Scheffer IE, Wallace R, Mulley JC, Berkovic SF ( 2001) Clinical and molecular genetics of myoclonic-astatic epilepsy and severe myoclonic epilepsy in infancy (Dravet syndrome). Brain Dev 23: 732-735. [DOI] [PubMed] [Google Scholar]
- Singh R, Andermann E, Whitehouse WP, Harvey AS, Keene DL, Seni MH, Crossland KM, Andermann F, Berkovic SF, Scheffer IE ( 2001) Severe myoclonic epilepsy of infancy: extended spectrum of GEFS+? Epilepsia 42: 837-844. [DOI] [PubMed] [Google Scholar]
- Spampanato J, Escayg A, Meisler MH, Goldin AL ( 2001) Functional effects of two voltage-gated sodium channel mutations that cause generalized epilepsy with febrile seizures plus type 2. J Neurosci 21: 7481-7490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spampanato J, Escayg A, Meisler MH, Goldin AL ( 2003) Generalized epilepsy with febrile seizures plus type 2 mutation W1204R alters voltage-dependent gating of Na(v)1.1 sodium channels. Neuroscience 116: 37-48. [DOI] [PubMed] [Google Scholar]
- Sugawara T, Mazaki-Miyazaki E, Ito M, Nagafuji H, Fukuma G, Mitsudome A, Wada K, Kaneko S, Hirose S, Yamakawa K ( 2001a) Nav1.1 mutations cause febrile seizures associated with afebrile partial seizures. Neurology 57: 703-705. [DOI] [PubMed] [Google Scholar]
- Sugawara T, Tsurubuchi Y, Agarwala KL, Ito M, Fukuma G, Mazaki-Miyazaki E, Nagafuji H, Noda M, Imoto K, Wada K, Mitsudome A, Kaneko S, Montal M, Nagata K, Hirose S, Yamakawa K ( 2001b) A missense mutation of the Na+ channel alpha II subunit gene Na(v)1.2 in a patient with febrile and afebrile seizures causes channel dysfunction. Proc Natl Acad Sci USA 98: 6384-6389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara T, Mazaki-Miyazaki E, Fukushima K, Shimomura J, Fujiwara T, Hamano S, Inoue Y, Yamakawa K ( 2002) Frequent mutations of SCN1A in severe myoclonic epilepsy in infancy. Neurology 58: 1122-1124. [DOI] [PubMed] [Google Scholar]
- Valdivia CR, Ackerman MJ, Tester DJ, Wada T, McCormack J, Ye B, Makielski JC ( 2002) A novel SCN5A arrhythmia mutation, M1766L, with expression defect rescued by mexiletine. Cardiovasc Res 55: 279-289. [DOI] [PubMed] [Google Scholar]
- Wallace RH, Wang DW, Singh R, Scheffer IE, George Jr AL, Phillips HA, Saar K, Reis A, Johnson EW, Sutherland GR, Berkovic SF, Mulley JC ( 1998) Febrile seizures and generalized epilepsy associated with a mutation in the Na+-channel beta1 subunit gene SCN1B. Nat Genet 19: 366-370. [DOI] [PubMed] [Google Scholar]
- Wallace RH, Marini C, Petrou S, Harkin LA, Bowser DN, Panchal RG, Williams DA, Sutherland GR, Mulley JC, Scheffer IE, Berkovic SF ( 2001a) Mutant GABA(A) receptor gamma2-subunit in childhood absence epilepsy and febrile seizures. Nat Genet 28: 49-52. [DOI] [PubMed] [Google Scholar]
- Wallace RH, Scheffer IE, Barnett S, Richards M, Dibbens L, Desai RR, Lerman-Sagie T, Lev D, Mazarib A, Brand N, Ben Zeev B, Goikhman I, Singh R, Kremmidiotis G, Gardner A, Sutherland GR, George Jr AL, Mulley JC, Berkovic SF ( 2001b) Neuronal sodium-channel alpha 1-subunit mutations in generalized epilepsy with febrile seizures plus. Am J Hum Genet 68: 859-865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitaker WR, Clare JJ, Powell AJ, Chen YH, Faull RL, Emson PC ( 2000) Distribution of voltage-gated sodium channel alpha-subunit and beta-subunit mRNAs in human hippocampal formation, cortex, and cerebellum. J Comp Neurol 422: 123-139. [DOI] [PubMed] [Google Scholar]