

Abstract
Markers of inhibitory neurotransmission are altered in the prefrontal cortex (PFC) of subjects with schizophrenia, and several lines of evidence suggest that these alterations may be most prominent in the subset of GABA-containing neurons that express the calcium-binding protein, parvalbumin (PV). To test this hypothesis, we evaluated the expression of mRNAs for PV, another calcium-binding protein, calretinin (CR), and glutamic acid decarboxylase (GAD67) in postmortem brain specimens from 15 pairs of subjects with schizophrenia and matched control subjects using single- and dual-label in situ hybridization. Signal intensity for PV mRNA expression in PFC area 9 was significantly decreased in the subjects with schizophrenia, predominately in layers III and IV. Analysis at the cellular level revealed that this decrease was attributable principally to a reduction in PV mRNA expression per neuron rather than by a decreased density of PV mRNA-positive neurons. In contrast, the same measures of CR mRNA expression were not altered in schizophrenia. These findings were confirmed by findings from cDNA microarray studies using different probes. Across the subjects with schizophrenia, the decrease in neuronal PV mRNA expression was highly associated (r = 0.84) with the decrease in the density of neurons containing detectable levels of GAD67 mRNA. Furthermore, simultaneous detection of PV and GAD67 mRNAs revealed that in subjects with schizophrenia only 55% of PV mRNA-positive neurons had detectable levels of GAD67 mRNA. Given the critical role that PV-containing GABA neurons appear to play in regulating the cognitive functions mediated by the PFC, the selective alterations in gene expression in these neurons may contribute to the cognitive deficits characteristic of schizophrenia.
Keywords: calretinin, GABA neurons, GAD67, parvalbumin, prefrontal cortex, schizophrenia
Introduction
Alterations in the circuitry of the prefrontal cortex (PFC) may contribute to impairments of certain cognitive functions, such as working memory, that are commonly observed in individuals with schizophrenia (Weinberger et al., 1986; Goldman-Rakic, 1994; Lewis and Lieberman, 2000). Convergent lines of evidence indicate that local circuit neurons, which play a critical role in regulating PFC information processing during working memory tasks (Rao et al., 2000; Constantinidis et al., 2002), exhibit altered GABA neurotransmission in the PFC of subjects with schizophrenia (for review, see Blum and Mann, 2002). For example, reduced amounts of the mRNA and protein for the 67 kDa isoform of glutamate decarboxylase (GAD67), a synthesizing enzyme for GABA, in the PFC (Akbarian et al., 1995; Guidotti et al., 2000; Volk et al., 2000) are among the most robust and diagnostically specific findings in postmortem studies of schizophrenia (Knable et al., 2002).
These alterations appear to involve only a subset of GABA neurons, because the expression levels of GAD67 and GABA membrane transporter-1 (GAT-1) mRNAs were reported to be decreased below the limit of detection in 25–35% of GABA neurons in the PFC of subjects with schizophrenia, whereas the majority of GABA neurons exhibited normal levels of expression for these two genes (Volk et al., 2000, 2001). Given the heterogeneity in axonal arbors, synaptic targets, and electrophysiological properties of different subclasses of PFC GABA neurons (Lund and Lewis, 1993; Kawaguchi and Kubota, 1997; Krimer and Goldman-Rakic, 2001), identification of the affected subset is critical for understanding the neural circuitry basis of impaired working memory in schizophrenia.
Most cortical GABA neurons express one of three calcium-binding proteins [parvalbumin (PV), calretinin (CR), or calbindin D-28], and these markers have been used to identify specific morphological and functional subgroups of GABA neurons (Condé et al., 1994; Gabbott and Bacon, 1996; Kawaguchi and Kubota, 1997). For example, in monkey PFC, PV-containing neurons include chandelier and wide arbor (basket) neurons (Williams et al., 1992; Lund and Lewis, 1993), whereas CR is expressed in double-bouquet neurons (Lund and Lewis, 1993; Condé et al., 1994). Among PFC GABA neurons, altered neurotransmission may be more prominent in PV-than in CR-containing neurons in subjects with schizophrenia because the density of axon cartridges immunoreactive for GAT-1, the vertical arrays of axon terminals furnished by chandelier neurons, was observed to be selectively decreased in the PFC of subjects with schizophrenia, whereas the densities of the remaining GAT-1-positive terminals and of CR-positive axon terminals appeared to be unchanged (Woo et al., 1998; Pierri et al., 1999).
Consequently, we hypothesized (1) that the selective involvement of PV-containing neurons in the pathophysiology of schizophrenia results in altered levels of PV, but not CR, mRNA expression in the PFC of subjects with schizophrenia, (2) that a marker of GABA neurotransmission, namely GAD67 mRNA, is reduced in PV-containing neurons, and (3) that these abnormalities in PV-containing neurons reflect the disease process and not the treatment of schizophrenia. To test these hypotheses, we used single-label in situ hybridization and cDNA microarray approaches to quantify the regional, laminar, and cellular expression of PV and CR mRNAs in the PFC of matched pairs of schizophrenic and control subjects and of monkeys exposed to antipsychotic medication in a manner that mimics clinical use. In addition, a dual-label approach was used to assess GAD67 mRNA expression in PV mRNA-positive neurons. The findings of this study suggest that the pathophysiology of PFC-mediated cognitive dysfunction in schizophrenia involves selective alterations in a specific class of GABA neurons.
Materials and Methods
Subjects. With the approval of the University of Pittsburgh's Institutional Review Board for Biomedical Research and the consent of the surviving next-of-kin, brain specimens were obtained during autopsies performed at the Allegheny County Coroner's Office (Pittsburgh, PA). Fifteen pairs of schizophrenic and control subjects, matched for sex, age, and postmortem interval (PMI), were used in this study (Table 1). Individual pairs were completely matched for sex, and the mean ± SD differences within pairs were 4.4 ± 3.6 years for age and 4.8 ± 2.5 hr for PMI. Two-tailed paired t tests revealed that the schizophrenia and control groups did not differ in terms of age (t = 0.18; p = 0.86), PMI (t = 0.06; p = 0.95), brain pH (t = 0.32; p = 0.76), or tissue storage time (t = -1.4; p = 0.18).
Table 1.
Characteristics of subjects
|
|
Control subjects |
Schizophrenic subjects |
||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pair
|
Case
|
Sex/race
|
Age
|
PMI a
|
Brain pH
|
Storage time b
|
Cause of death c
|
Case
|
DSM IV diagnosis
|
Sex/race
|
Age
|
PMI a
|
Brain pH
|
Storage time b
|
Cause of death c
|
|||||||||||||
| 1 | 592 | M/B | 41 | 22.1 | 6.72 | 59 | ASCVD | 533 | Chronic undifferentiated schizophrenia | M/W | 40 | 29.1 | 6.82 | 69 | Accidental asphyxiation | |||||||||||||
| 2 | 567 | F/W | 46 | 15.0 | 6.72 | 63 | Mitral valve prolapse | 537 | Schizoaffective disorder k | F/W | 37 | 14.5 | 6.68 | 68 | Suicide by hanging | |||||||||||||
| 3 | 516 | M/B | 20 | 14.0 | 6.86 | 70 | Homicide by gun shot | 547 | Schizoaffective disorder | M/B | 27 | 16.5 | 6.95 | 66 | Heat stroke | |||||||||||||
| 4 | 630 | M/W | 65 | 21.2 | 6.95 | 53 | ASCVD | 566 | Chronic undifferentiated schizophrenia d | M/W | 63 | 18.3 | 6.80 | 63 | ASCVD | |||||||||||||
| 5 | 604 | M/W | 39 | 19.3 | 7.08 | 56 | Hypoplastic coronary artery | 581 | Chronic paranoid schizophrenia ef | M/W | 46 | 28.1 | 7.22 | 61 | Accidental combined drug overdose | |||||||||||||
| 6 | 546 | F/W | 37 | 23.5 | 6.74 | 67 | ASCVD | 587 | Chronic undifferentiated schizophrenia d | F/B | 38 | 17.8 | 7.02 | 60 | Myocardial hypertrophy | |||||||||||||
| 7 | 558 g | M/W | 47 | 6.6 | 6.99 | 64 | ASCVD | 317 | Chronic undifferentiated schizophrenia | M/W | 48 | 8.3 | 6.07 | 115 | Pneumonia | |||||||||||||
| 8 | 635 h | F/W | 54 | 17.8 | 6.47 | 52 | ASCVD | 597 | Schizoaffective disorder | F/W | 46 | 10.1 | 7.02 | 58 | Pneumonia | |||||||||||||
| 9 | 551 | M/W | 61 | 16.4 | 6.63 | 66 | Cardiac tamponade | 625 | Chronic disorganized schizophrenia g | M/B | 49 | 23.5 | 7.32 | 54 | ASCVD | |||||||||||||
| 10 | 685 | M/W | 56 | 14.5 | 6.57 | 46 | Hypoplastic coronary artery | 622 | Chronic undifferentiated schizophrenia k | M/W | 58 | 18.9 | 6.78 | 54 | Right MCA infarction | |||||||||||||
| 11 | 681 | M/W | 51 | 11.6 | 7.15 | 46 | Hypertrophic cardiomyopathy | 640 | Chronic paranoid schizophrenia | M/W | 49 | 5.2 | 6.86 | 52 | Pulmonary embolism | |||||||||||||
| 12 | 806 | M/W | 57 | 24.0 | 6.94 | 25 | Pulmonary thrombo-embolism | 665 | Chronic paranoid schizophrenia e | M/B | 59 | 28.1 | 6.92 | 49 | Intestinal hemorrhage | |||||||||||||
| 13 | 822 | M/B | 28 | 25.3 | 7.04 | 22 | ASCVD | 787 | Schizoaffective disorder i | M/B | 27 | 19.2 | 6.67 | 29 | Suicide by gun shot | |||||||||||||
| 14 | 727 | M/B | 19 | 7.0 | 7.15 | 39 | Trauma | 829 | Schizoaffective disorder ejk | M/W | 25 | 5.0 | 6.80 | 20 | Suicide by drug overdose | |||||||||||||
| 15
|
871
|
M/W
|
28
|
16.5
|
7.14
|
12
|
Trauma
|
878
|
Disorganized schizophrenia e
|
M/W
|
33
|
10.8
|
6.72
|
11
|
Myocardial fibrosis
|
|||||||||||||
| Mean | 43.3 | 17.0 | 6.88 | 49.3 | 43.0 | 16.9 | 6.84 | 52.3 | ||||||||||||||||||||
|
|
SD
|
|
14.6
|
5.8
|
0.22
|
17.8
|
|
|
|
|
12.0
|
8.0
|
0.28
|
24.1
|
|
|||||||||||||
PMI indicates postmortem interval in hours
storage time (months) at −80°C
ASCVD indicates atherosclerotic coronary vascular disease
alcohol abuse, in remission at time of death
alcohol dependence, current at time of death
other substance abuse, current at time of death
alcohol abuse, current at time of death
depressive disorder
other substance dependence, current at time of death
other substance abuse, in remission at time of death
schizophrenic subjects off medications at time of death.
An independent committee of experienced research clinicians made consensus DSMIV (Diagnosis and Statistical Manual of Mental Disorders, 1994) diagnoses for each subject on the basis of medical records and the results of structured interviews conducted with family members of the deceased. This diagnostic procedure revealed a history of depressive disorder (not otherwise specified) in one control subject (635) and the presence of alcohol abuse (current at the time of death) in another control subject (558). Eight subjects with schizophrenia also had a history of substance (including alcohol) abuse or dependence disorder, or both (Table 1), although only four met criteria for dependence at time of death. Toxicology studies for all subjects revealed positive plasma alcohol levels (0.01–0.06%) in three control subjects; no other substances of abuse were detected in any subjects. Three subjects with schizophrenia (537, 622, and 829) were free of antipsychotic medications at the time of death (Table 1). The length of time without the medications before death was 9.6 and 1.2 months for the former two subjects, respectively, and unknown for the last subject. The mean ± SD age of schizophrenic subjects at the onset of illness was 25.1 ± 9.0 years, and the average duration of illness was 18.7 ± 9.4 years. Because the brain specimens used in our study were obtained from a community-based population, most subjects (12 with schizophrenia and 14 controls) died suddenly outside of a hospital setting.
Neuropathological examination of each brain revealed that subject 622 had an infarction limited to the distribution of the inferior branch of the right middle cerebral artery, but PFC area 9 appeared unaffected. Alzheimer's disease was excluded in each subject on the basis of clinical and neuropathological criteria (Mirra et al., 1991).
The first 10 pairs (Table 1) of subjects with schizophrenia and matched controls were used in our previous studies of GAD67 and GAT-1 gene expression in PFC area 9 (Volk et al., 2000, 2001).
Tissue preparation and in situ hybridization. For each subject, the right PFC was blocked coronally, immediately frozen, and stored at -80°C. Serial sections (20 μm) containing the superior frontal gyrus were cut, thaw mounted onto glass slides, and stored at -80°C until processed. The location of PFC area 9 was identified by cytoarchitectonic criteria in Nissl-stained sections as described previously (Glantz et al., 2000; Volk et al., 2000, 2001). For both PV and CR mRNAs, three sections evenly spaced at 400 μm were chosen with the rostrocaudal locations of the sections matched within each pair. Thirty sections (one section from each subject) were processed in a single run with sections from each pair processed side by side. Three runs were performed for each mRNA.
Templates for synthesis of riboprobes were obtained by PCR. Specific primer sets amplified a 345 base pair (bp) fragment for PV and a 748 bp fragment for CR, corresponding to bases 59–403 of the human PV gene (GenBank NM_002854) and bases 393–1140 of the human CR gene (GenBank X56667), respectively. Nucleotide sequencing revealed 100% homologies for both amplified fragments to the previously reported sequences. These fragments were subcloned into the plasmid pSTBlue-1 (Novagen, Madison, WI). Antisense and sense probes were transcribed in vitro in the presence of 35S-CTP (Amersham Biosciences, Piscataway, NJ), using T7 or SP6 RNA polymerase. The templates were then digested with DNase I, and riboprobes were purified by centrifugation through the RNeasy mini spin columns (Qiagen, Valencia, CA). Hybridization was performed as described previously (Mirnics et al., 2000; Middleton et al., 2002). After fixation with 4% paraformaldehyde in PBS, the sections were acetylated with 0.25% acetic anhydrate in 0.1 m triethanolamine/0.9% NaCl for 10 min, dehydrated through a graded ethanol series, and defatted in chloroform for 10 min. The sections were then hybridized with 35S-labeled riboprobes (2 × 107 dpm/ml) in hybridization buffer containing 50% formamide, 0.75 m NaCl, 20 mm 1,4-piperazine diethane sulfonic acid, pH 6.8, 10 mm EDTA, 10% dextran sulfate, 5× Denhardt's solution (0.2 mg/ml Ficoll, 0.2 mg/ml polyvinylpyrrolidone, 0.2 mg/ml BSA), 50 mm dithiothreitol, 0.2% SDS, and 100 μg/ml yeast tRNA at 56°C for 16 hr. The sections were washed in a solution containing 0.3 m NaCl, 20 mm Tris-HCl, pH 8.0, 1 mm EDTA, pH 8.0, and 50% formamide at 63°C, treated with RNase A (20 μg/ml) at 37°C, and washed in 0.1× SSC (150 mm NaCl, 15 mm sodium citrate) at 67°C. Sections were then dehydrated through a graded ethanol series, air dried, and exposed to BioMax MR film (Kodak, Rochester, NY) for 72 hr. After the exposure to film, sections were coated with NTB2 emulsion (Kodak) diluted 1:1 with water. To ensure the consistency of emulsion thickness across sections, slides were dipped by using a mechanical dipper, Auto-dip Emulsion Coater (Ted Pella, Redding, CA), at a constant withdrawal speed (64 mm/min) and temperature (43°C). Sections were exposed for 12 d at 4°C, developed with D-19 (Kodak), and counterstained with cresyl violet.
Quantification of mRNA expression levels. Quantification was performed without knowledge of diagnosis or subject number by random coding of the sections. Trans-illuminated autoradiographic film images were captured by a video camera under precisely controlled conditions, digitized, and analyzed using a Microcomputer Imaging Device (MCID) system (Imaging Research Inc, London, Ontario, Canada). Images of adjacent sections stained with cresyl violet were also captured and superimposed onto the autoradiographic images to draw contours of the full thickness of the portion of area 9 gray matter cut perpendicular to the pial surface. The area included in the contour was as large as possible within each section. For PV mRNA, the mean ± SD total sampled areas per subject were 364 ± 117 and 358 ± 132 mm 2 for control and schizophrenic subjects, respectively; for CR mRNA, these values were 291 ± 95 and 325 ± 135 mm 2 for control and schizophrenic subjects, respectively. Optical density was measured within the contours and expressed as nanocuries per gram of tissue by reference to radioactive standards (Carbon-14 standards; ARC Inc., St. Louis, MO) exposed on the same film. To assess the laminar specificity of differences in optical density, three cortical traverses, 1–2 mm in width, were sampled from each section (nine traverses per subject). Within these traverses, the ratios of the depth of each laminar border to the total cortical thickness, determined from adjacent Nissl-stained sections, were applied to the autoradiograms to obtain optical densities for each layer. All cortical optical density measures were corrected by subtracting background measures in the white matter (for PV mRNA) and in deep layer 6 (for CR mRNA).
For evaluation of mRNA expression at the cellular level, grain analysis was performed after the sections were coated with emulsion. Using the MCID system equipped with a motor-driven stage, sampling frames with a size of 220 × 220 μm were systematically and randomly placed within contours of the gray matter of area 9 for each section (Fig. 1 A), as outlined for the film autoradiography. Because RNase A treatment of sections, which destroys Nissl-stainable substances within the cytoplasm, made it difficult to draw contours of neuronal soma, we counted grains within circles with a fixed size of 22 μm diameter that covered the largest cross-sectional area of GABA neurons (∼400 μm 2) observed in previous studies (Volk et al., 2000). Grain counts using a circle with a fixed diameter may potentially be influenced by differences in somal size. However, our findings are unlikely to be confounded in this respect because somal size has been reported to be unchanged for both GAD67 mRNA-positive neurons (Akbarian et al., 1995; Volk et al., 2000) and PV-immunoreactive neurons (Woo et al., 1997) in PFC area 9 in subjects with schizophrenia. In a bright-field image of the sampling frame, the circles were centered over every Nissl-stained neuronal nucleus (Fig. 1 B). In a dark-field image of the same sampling frame, the number of grains within each circle was determined (Fig. 1C). Using the same sampling frames, background grain density was determined for each section by counting the number of grains within 22-μm-diameter circles centered on glial nuclei. Small glial nuclei, stained darkly with cresyl violet, were easily discriminated from large and faintly stained neuronal nuclei (Fig. 1 B). Totals of 92,206 and 86,211 neurons were sampled for PV and CR mRNAs, respectively.
Figure 1.
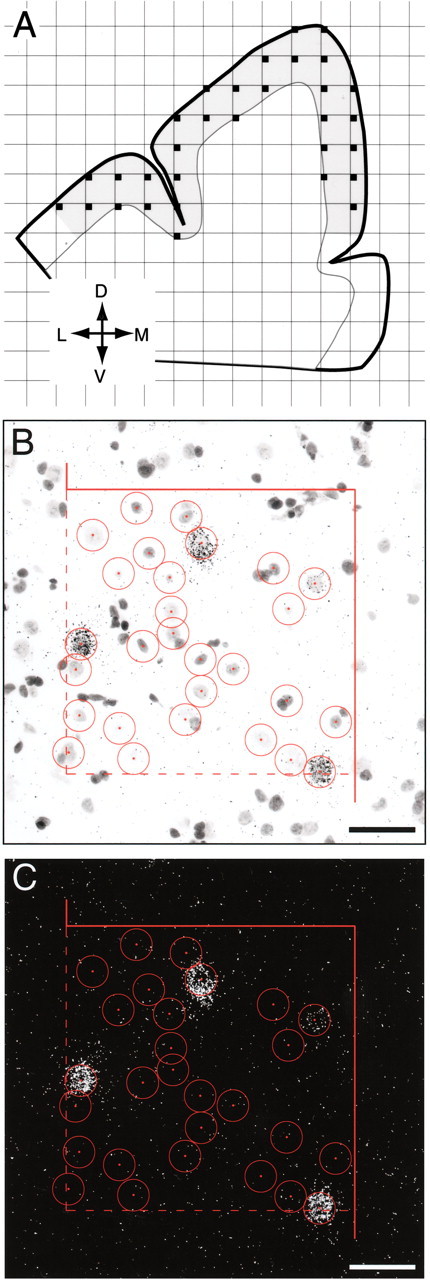
Schematic representation of the sampling strategy for grain analysis of mRNA expression. A, Camera lucida drawing of the dorsal PFC, with gray shading indicating the boundaries of a typical contour used for sampling. A sampling grid was randomly super imposed on this contour to designate sampling frames (small filled squares). Orientation is indicated in the bottom left: L, lateral; D, dorsal; M, medial; V, ventral. B, C, Representative bright-field and dark-field images, respectively, of a sampling site for PV mRNA grain analysis. In the bright-field image (B), Nissl-stained neuronal nuclei were identified and included for study according to unbiased inclusion and exclusion rules (broken and solids lines indicate inclusion and exclusion boundaries, respectively). Circles of 22 μm diameter were centered over every neuronal nucleus, and the number of grains within the circle was counted in dark-field image (C) of the same sampling frame. Scale bars, 50 μm.
Grain density per neuron (number of grains within the circle of 22 μm diameter) was determined for each sampled neuron. A threshold of grain density per neuron was then established to identify specifically labeled neurons (Volk et al., 2000, 2001). For PV and CR mRNAs, histograms of grain density per neuron (natural log transformed) for all sampled neurons revealed a distribution that appeared bimodal in both subject groups, representing the modes of unlabeled and specifically labeled neuron populations (Gerfen et al., 1991). Similar histograms including only neurons with a grain density >5× background showed a distribution that appeared normal and unimodal in both the schizophrenia and control groups for both PV and CR mRNAs. Therefore, a threshold of 5× background provided a cutoff at the point of rarity in the distribution of all cells that permitted the identification of specifically labeled neurons, referred to as PV or CR mRNA-positive neurons. Mean ± SD numbers of PV mRNA-positive neurons sampled per subject were 203 ± 43.5 and 159 ± 37.3 for control and schizophrenia subjects, respectively. Mean ± SD numbers of CR mRNA-positive neurons sampled per subject were 179 ± 35.5 and 183 ± 41.3 for control and schizophrenia subjects, respectively. The mean ± SD coefficients of error for positive neuron counts did not differ between the control (0.09 ± 0.01 for PV mRNA, 0.11 ± 0.01 for CR mRNA) and schizophrenia (0.10 ± 0.01 for PV mRNA, 0.11 ± 0.01 for CR mRNA) groups. The grain density per positive neuron, indicating the relative level of PV or CR mRNA expression per positive neuron, was corrected for nonspecific background labeling by subtracting the average background grain density for each section.
cDNA microarrays. To verify the results of the film level in situ hybridization analyses with different probes against the PV and CR mRNAs, we analyzed previously obtained (Mirnics et al., 2000) cDNA microarray data from a subset of these same subject pairs. Balanced signal intensity ratios for CR to PV expression were constructed for each subject, and the results were compared with a one-tailed t test.
Dual-label in situ hybridization. To directly assess the possibility of GAD67 mRNA expression deficits in PV mRNA-positive neurons, we performed dual-label in situ hybridization. In addition to the 35S-labeled riboprobe for PV mRNA, a digoxigenin (DIG)-labeled riboprobe for GAD67 mRNA was transcribed in vitro in the presence of DIG-11-UTP (Roche, Mannheim, Germany). For the in vitro transcription, a 360 bp template fragment, corresponding to bases 1757–2116 of the human GAD67 gene (GenBank NM_000817), was obtained by PCR and its sequence verified. Two sections from each subject were processed in two runs, with one section from each subject included in each run. Sections were treated as described above except for the combination of the DIG- and 35S-labeled probes at concentrations of 100 ng/ml and 2 × 107 dpm/ml, respectively, in hybridization buffer. After the post-hybridization washing, sections were preincubated with 3% BSA in 100 mm Tris-HCl, pH 7.5, 150 mm NaCl, 0.05% Triton X-100 for 30 min, incubated with anti-DIG antiserum conjugated with alkaline phosphatase (Roche) diluted 1:2000 in 1% BSA, 100 mm Tris-HCl, pH 7.5, 150 mm NaCl, 0.05% Triton X-100 for 12 hr at 4°C, washed in 100 mm Tris-HCl, pH 7.5, 150 mm NaCl, rinsed in 100 mm Tris-HCl, pH 9.5, 100 mm NaCl, 50 mm MgCl2, and air dried. To detect PV mRNA signal, sections were coated with nuclear emulsion and developed as described above. For detection of GAD67 mRNA signal, sections were incubated in 0.34 mg/ml nitroblue tetrazolium and 0.18 mg/ml bromo-chloroindolylphosphate (Roche) in 100 mm Tris-HCl, pH 9.5, 100 mm NaCl, 50 mm MgCl2 for 24 hr. Because the qualitative difference between control and schizophrenic subjects appeared similar across both runs, we used the sections in the second run for counting of positive profiles. For each coded slide, numbers of single- and double-labeled profiles were counted separately in a 500-μm-wide traverse from the pial surface through the entire thickness of cortex.
To assess the possible quenching of signal caused by interference between the DIG- and 35S-labeled probes, we performed control in situ hybridization experiments combining the DIG-labeled GAD67 riboprobe and another 35S-labeled GAD67 riboprobe, complementary to a 324 bp region corresponding to bases 711–1034 of the mRNA (GenBank NM_000817), that was separated by 700 bp from the region recognized by the DIG-labeled probe. Among 625 labeled neurons randomly sampled in sections from five control subjects, signals from the DIG- and 35S-labeled probes were co-detected in 98% of the neurons (Fig. 2), whereas only 1.4 and 0.5% of the neurons were single labeled with DIG- or 35S-labeled probes, respectively. These results indicate that the presence of one signal type did not interfere with the detection of the other signal in our dual-label in situ hybridization procedure.
Figure 2.

Representative photomicrograph of dual-label in situ hybridization verifying the simultaneous detection of GAD67 mRNA by DIG- and 35S-labeled riboprobes in the same neurons. The DIG- and 35S-labeled probes recognize different regions of the mRNA molecule (see Materials and Methods), and their specific hybridization was visualized as color reaction product and silver grain accumulation, respectively. Note the coexistence of both types of signals in all labeled profiles. Scale bar, 50 μm.
Haloperidol-treated monkeys. To mimic the clinical treatment of subjects with schizophrenia, four male cynomolgus (Macaca fascicularis) monkeys, matched individually to a control monkey for sex, age, and weight, were treated with haloperidol decanoate (mean ± SD trough serum level: 4.3 ± 1.1 ng/ml) and benztropine mesylate (to relieve extra-pyramidal side effects) for 9–12 months as described previously (Pierri et al., 1999). Three sections evenly spaced at 400 μm intervals and selected from coronal sections serially cut from tissue blocks containing the middle one-third of the principal sulcus, were processed for PV in situ hybridization as described above. The optical density of PV mRNA signals was measured in both areas 9 and 46.
Statistical analyses. Analysis of covariance (ANCOVA) models (Neter et al., 1996) were performed to examine the differences in PV and CR mRNA expression levels between the schizophrenia and control groups. Because three sections were measured for every subject, each dependent variable (film optical density, grain density per positive neuron, positive neuron density) was represented by three observations for each subject. Thus, the three observations for each subject can be considered to be exchangeably correlated and treated as repeated measures with compound symmetric covariance structure (Neter et al., 1996). Because no data were missing and the numbers of observations for each subject were identical, the ANCOVA models produce the same results as a multivariate ANCOVA in terms of testing the diagnosis effect. The corresponding ANCOVA models required averaging across the three sections for each dependent variable before the statistical analyses were conducted. The first ANCOVA model, used for each dependent variable, had diagnostic group as a main effect, pair as a blocking effect, and storage time and brain pH as covariates. The inclusion of pair effect reflects the matching of individual subject pairs for sex, age, and PMI. Postmortem brain pH was included as a covariate because it may reflect the integrity of some mRNA species (Harrison et al., 1995). Because long-term tissue storage may also affect mRNA preservation, tissue storage time was included in the model. Subject pairing may be considered an attempt to balance the two diagnostic groups with regard to the experimental factors instead of a true statistical paired design. Thus, to validate the first model, a second ANCOVA model was performed with main effect of diagnostic group and covariates of all pairing factors (sex, age, PMI), as well as brain pH and storage time. Because these two ANCOVAs produced similar results, only the results from the first analysis are reported. These analyses were implemented in SAS PROC GLM (SAS Inc., Cary, NC) and SAS PROC MIXED (Littell et al., 1996).
The influence of sex and substance abuse and the diagnosis of schizoaffective disorder on the differences within subject pairs in film optical density and grain density per positive neuron for PV mRNA were assessed using one-way ANOVAs.
To test the association (using Pearson's correlation analysis) between PV mRNA expression and our previous observations of GAD67 mRNA expression (Volk et al., 2000), percentage differences within subject pairs were calculated for GAD67 mRNA-positive neuron density, grain densities per positive neuron for PV and CR mRNAs, and the densities of PV or CR mRNA-positive neurons within each of the 10 subject pairs shared by both studies. Because the GAD67 data were collected for each layer, we averaged the values for each layer using the weighing ratio of 1:1:3:1:2:2 for layers I, II, III, IV, V, and VI, respectively (Akil et al., 1999), to obtain a single estimate of GAD67 mRNA-positive neuron density across all cortical layers.
For the dual-label in situ hybridization analysis, we calculated the percentage of PV mRNA-positive grain clusters that lacked GAD67 mRNA signal and the percentage of all labeled profiles that were single labeled for GAD67 mRNA for each subject. These percentages were compared between the subject groups using the same ANCOVA models described above. Because the dependent variables are binomial proportions, transformations were performed to stabilize the variance.
For the haloperidol-treated monkeys, two-tailed paired t tests were used to determine the effect of treatment group on PV mRNA expression in the PFC.
Results
Specificity of riboprobes for PV and CR mRNAs
Microscopic observation of emulsion-coated slides revealed silver grains clustered around Nissl-stained neuronal nuclei, indicating specific hybridization within the cytoplasm for both PV (Fig. 3D) and CR mRNAs (Fig. 3E). All labeled cells were recognized as neurons on the basis of the faint Nissl staining of their large nuclei and distinguished from unlabeled glial cells containing intensely stained small nuclei (Fig. 3D,E). The specificity of the antisense riboprobes was also confirmed by the distinctive laminar distribution patterns of the labeled neurons. PV mRNA-positive neurons were distributed across layers II–VI, with the highest densities in layers III and IV (Fig. 3B), whereas CR mRNA-positive neurons were present in highest density in layers I, II, and superficial III and were rarely found in layer VI (Fig. 3C). These distribution patterns match the reported laminar locations of PV- and CR-immunoreactive cell bodies in human and monkey PFC (Condé et al., 1994; Daviss and Lewis, 1995; Woo et al., 1997). Specificity was also confirmed by the absence of signal above background in sections treated with the sense riboprobes for these mRNAs (data not shown).
Figure 3.
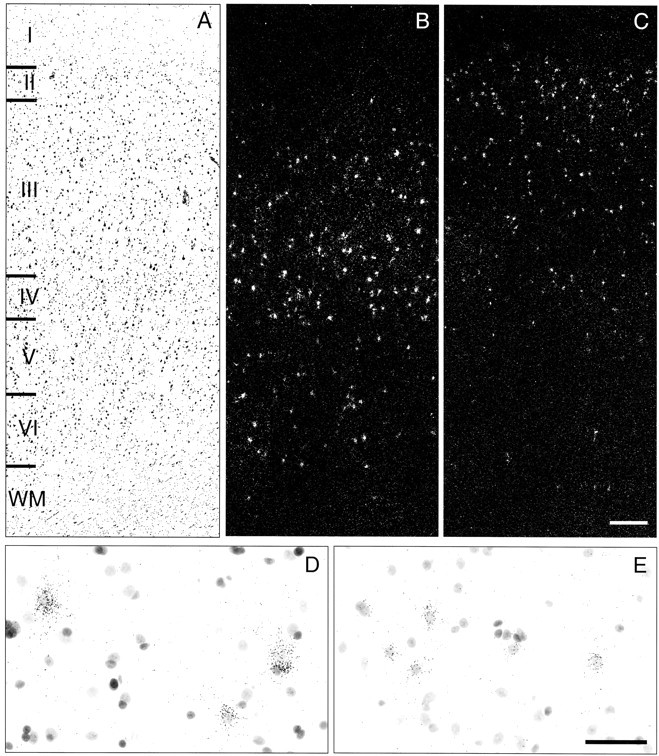
Distribution of silver grain clusters representing PV and CR mRNA-positive neurons. Three serial sections of PFC area 9 of a control subject (604) were stained for Nissl substance (A) or hybridized with antisense riboprobes for PV (B, D) or CR (C, E) mRNAs and then stained for Nissl substance. Note that the density of PV mRNA-positive neurons appears greatest in layers III and IV (B), whereas the density of CR mRNA-positive neurons is higher in layers II and superficial III (C). Representative high-magnification photomicrographs illustrate the expression of PV (D) and CR (E) mRNAs in deep layers III and layer II, respectively. For both probes, silver grains accumulated around neuronal nuclei. The size of grain clusters for PV mRNA appears larger than those for CR mRNA, reflecting the difference in somal sizes of PV- and CR-containing neurons (Gabbott and Bacon, 1996). Roman numerals indicate cortical layers, and WM indicates white matter. Scale bar: (in C), A–C, 300 μm; (in E), D, E, 50 μm.
Expression of PV and CR mRNAs in area 9 of schizophrenic and control subjects
In macroscopic observation of film autoradiograms, PV mRNA signal in PFC area 9 appeared to be decreased in subjects with schizophrenia (Fig. 4B,F) compared with the matched control subjects (Fig. 4A,E), whereas CR mRNA signal appeared unchanged (Fig. 4, compare C,D and G,H). Comparison of film optical density for PV mRNA in PFC area 9 between the subject groups revealed a significant effect of diagnostic group (F(1,12) = 10.85; p = 0.006). Mean ± SD optical density of PV mRNA was decreased by 30% [95% confidence interval (CI) = 9.7–48.4%] in the subjects with schizophrenia (21.5 ± 9.8 nCi/gm) compared with the matched controls (30.8 ± 11.0 nCi/gm) (Fig. 5A).
Figure 4.
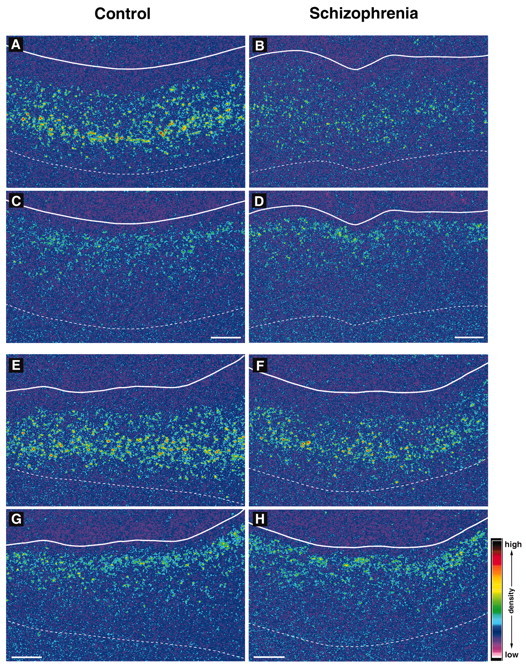
Representative film autoradiograms showing signals for PV and CR mRNAs in PFC area 9 of subject pairs 3 (A–D) and 5 (E–H). The densities of hybridization signals are presented in pseudocolor manner according to the scale in bottom right. In both pairs, the PV mRNA signals appear to be weaker in subjects with schizophrenia (B, F) than in matched controls (A, E), whereas in the same region of the adjacent sections, CR mRNA signals do not appear to differ between subjects with schizophrenia (D, H) and matched controls (C, G). Solid and broken white lines indicate the pial surface and the border between white matter and gray matter, respectively. Scale bars: (apply to both sections from a given subject), 1 mm.
Figure 5.
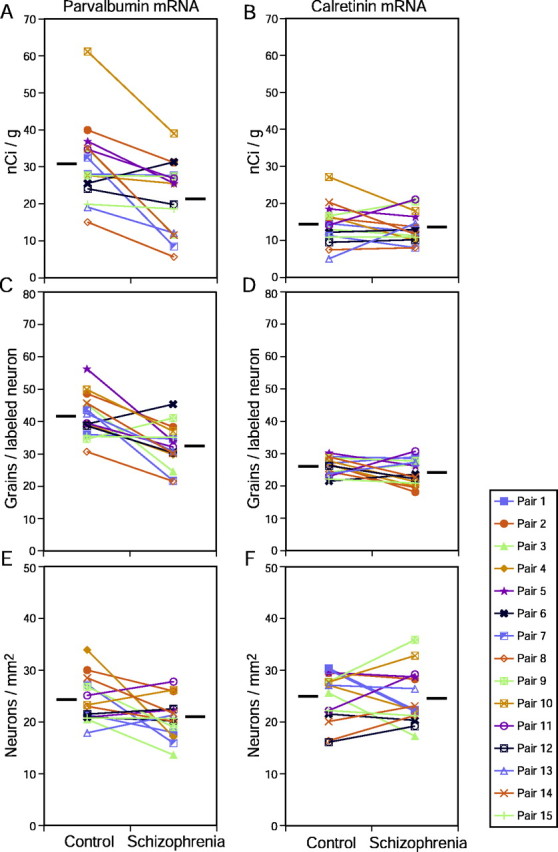
Pair-wise comparisons of film optical density (A, B), grain density per positive neuron (C, D), and positive neuron density (E, F) for parvalbumin (A, C, E) and calretinin (B, D, F) mRNA expression. Mean levels of expression for each subject group are indicated by the horizontal bars.
In the grain analysis, there was a significant effect of diagnostic group for grain density per PV mRNA-positive neuron (F(1,12) = 11.98; p = 0.005). Mean ± SD grain density per PV mRNA-positive neuron exhibited a 22% decrease (95% CI = 7.4–32%) in the subjects with schizophrenia (32.4 ± 6.7 per neuron) compared with controls (41.7 ± 6.7 per neuron) (Fig. 5C). In contrast, the mean ± SD density of PV mRNA-positive neurons did not differ (F(1,12) = 3.18; p = 0.10) between the subjects with schizophrenia (20.8 ± 3.9/mm2) and the matched controls (24.1 ± 4.4/mm2) (Fig. 5E).
In pair-wise comparisons of PV mRNA expression, the subjects with schizophrenia exhibited decreased film optical density measures in 14 of 15 pairs and decreased grain density per positive neuron in 13 of 15 pairs (Fig. 5A,C). Neither the optical density nor grain density per positive neuron measures of PV mRNA expression in the subjects with schizophrenia relative to their matched controls varied as a function of sex (F(1,13) < 1.06; p > 0.32), history of substance abuse (F(1,13) < 3.04; p > 0.11), or a diagnosis of schizoaffective disorder (F(1,3) < 2.22; p > 0.16) (Fig. 6). In addition, these measurements were not significantly correlated with age in either the control or schizophrenia groups (|r| < 0.22; p > 0.25).
Figure 6.
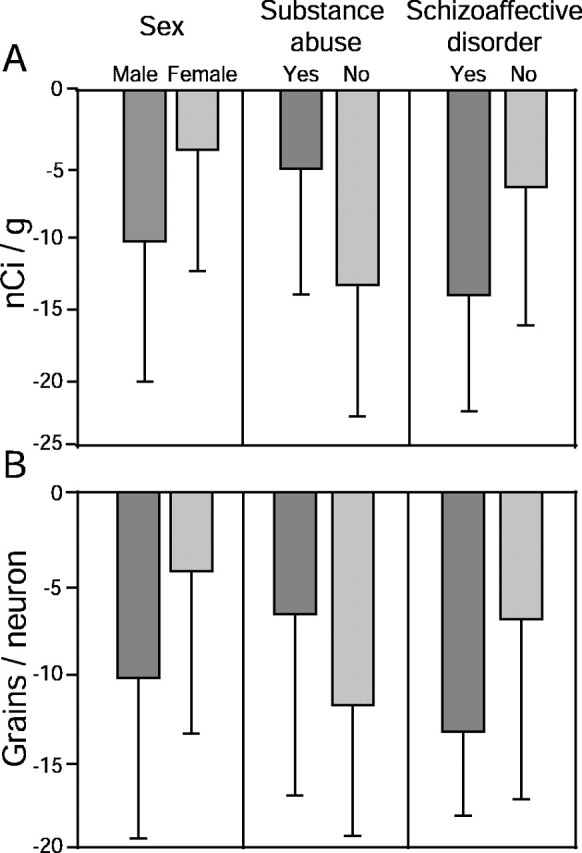
Mean ± SD differences from control subjects in film optical density (A) and grain density per positive neuron (B) for parvalbumin mRNA across subject pairs grouped by sex (male, n = 12; female, n = 3), substance abuse history (yes, n = 8; no, n = 7), and the diagnosis of schizoaffective disorder (yes, n = 5; no, n = 10).
In contrast to these alterations in PV mRNA expression, no significant differences in film optical density (F(1,12) = 0.36; p = 0.56), in grain density per positive neuron (F(1,12) = 1.76; p = 0.21), or in positive neuron density (F(1,12) = 0.07; p = 0.80) were observed for CR mRNA (Fig. 5B,D,F).
To verify the differential effect of schizophrenia on PV and CR mRNA expression using a complementary method, we analyzed previously obtained cDNA microarray data (Mirnics et al., 2000) from a subset of the subject pairs used in the present study. The sequences used to detect PV and CR mRNAs were different in the microarray and in situ hybridization analyses. Consistent with a selective decrease in PV mRNA expression level in subjects with schizophrenia, the balanced signal intensity ratio for CR to PV mRNA expression was significantly higher (t = -2.96; p < 0.03) in subjects with schizophrenia (4.05 ± 0.33) than in control subjects (2.91 ± 0.91).
Because PV and CR mRNAs were expressed in distinct laminar patterns, we further evaluated the film optical density for the mRNAs in each cortical layer (Fig. 7). There was a significant effect of diagnostic group on PV mRNA expression in layers III (F(1,12) = 8.67; p = 0.012) and IV (F(1,12) = 11.97; p = 0.005), but not in layer I (F(1,12) = 0.98; p = 0.34), layer II (F(1,12) = 0.87; p = 0.37), or layers V/VI (F(1,12) = 2.08; p = 0.12). PV mRNA expression was significantly decreased by 29% (95% CI = 0.2–52.9%) in layer III and by 35% (95% CI = 4.5–59.5%) in layer IV. In contrast, no significant differences between subject groups in CR mRNA expression were found in any layers (F(1,12) < 0.88; p > 0.37 for all layers).
Figure 7.
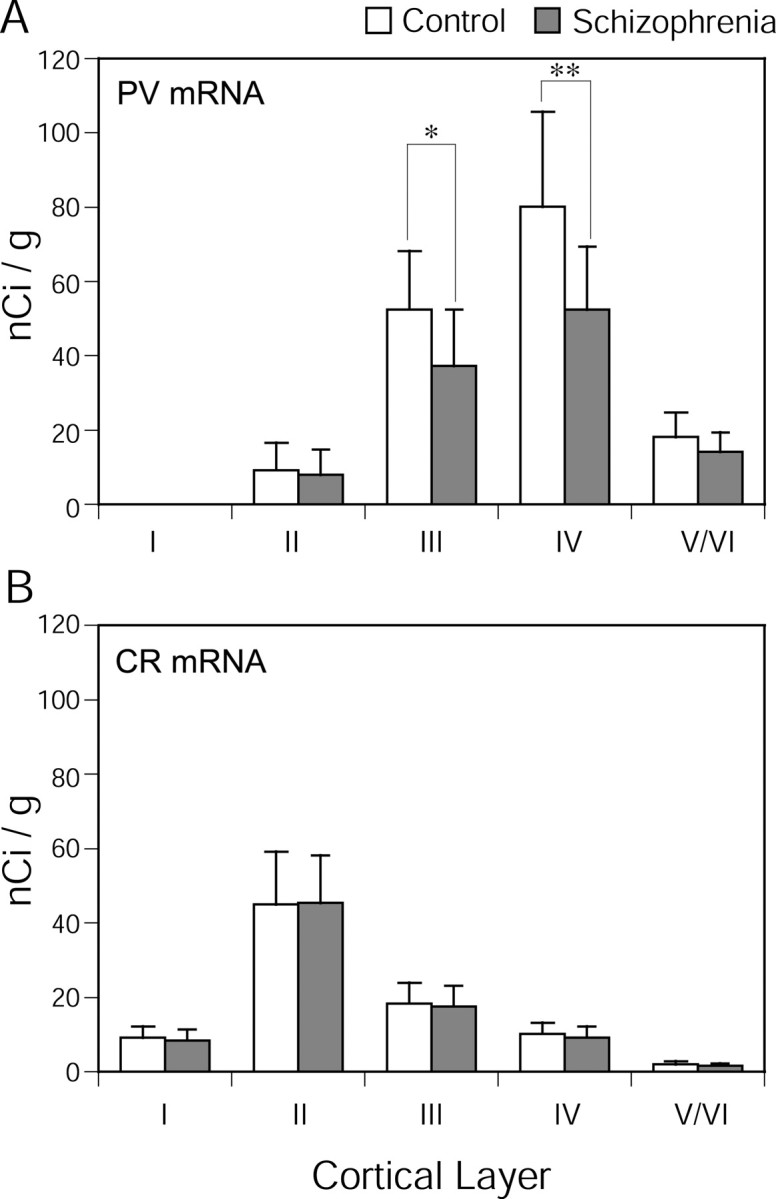
Comparison by cortical layer of mean ± SD film optical density for PV (A) and CR (B) mRNA expression between the subject groups. Asterisks indicate statistical significance (*p = 0.012; **p = 0.005).
Relationship to decreased GAD67 mRNA expression in the PFC of subjects with schizophrenia
To explore the relationship between the changes in PV mRNA expression and our previous observation of decreased GAD67 mRNA-positive neuron density (Volk et al., 2000) among the 10 subject pairs shared by both studies, we performed Pearson's correlation analyses. The within-pair differences in GAD67 mRNA-positive neuron density and in grain density per PV mRNA-positive neuron were strongly correlated (r = 0.84; p = 0.002), whereas the differences in GAD67 mRNA-positive neuron density and in PV mRNA-positive neuron density were not (r = 0.08; p = 0.83). The within-pair differences in the density of GAD67 mRNA-positive neurons were not significantly correlated with the differences in CR mRNA-positive neuron density (r = 0.28; p = 0.43) or grain density per CR mRNA-positive neuron (r = 0.19; p = 0.61).
On the basis of the strong association between the decreased GAD67 mRNA-positive neuron density and decreased PV mRNA expression levels per neuron, we hypothesized that GAD67 mRNA expression is decreased below the limit of detection in some PV mRNA-positive neurons in schizophrenia. To directly test this hypothesis, GAD67 and PV mRNAs were simultaneously detected by combining a DIG-labeled antisense riboprobe for GAD67 mRNA and the 35S-labeled probe for PV mRNA. The nonpyramidal morphology and laminar distribution of the labeled cells (data not shown) confirmed the specificity of the DIG-labeled probe for GAD67 mRNA. In addition, no signal above background was detected after the hybridization with the sense probe (data not shown). In control subjects, PV mRNA-positive grain clusters were generally positive for GAD67 mRNA signals, whereas in subjects with schizophrenia, GAD67 mRNA signals were undetectable in a number of PV mRNA-positive grain clusters (Fig. 8). Quantitative assessments (Table 2) revealed that GAD67 mRNA was not detectable in ∼10% of PV mRNA-positive grain clusters in control subjects, whereas in subjects with schizophrenia, an average of 45% of the PV mRNA-positive grain clusters lacked GAD67 mRNA signal (F(1,12) = 40.16; p < 0.001). In contrast, the mean ± SD percentage of all labeled profiles that were single-labeled for GAD67 mRNA did not differ significantly (F(1,12) = 0.02; p = 0.90) between control (62.8 ± 7.3%) and schizophrenia (61.8 ± 7.6%) subjects.
Figure 8.

Representative photomicrographs of dual-label in situ hybridization showing simultaneous detection of PV and GAD67 mRNAs in PFC area 9 of subject pair 14. Silver grain clusters represent PV mRNA, which was detected by a 35S-labeled probe, whereas GAD67 mRNA was visualized as color reaction product by a digoxigenin-labeled probe. We detected single-labeled GAD67 mRNA-positive profiles (solid arrowheads), single-labeled PV mRNA-positive silver grain clusters (open arrowheads), and double-labeled profiles (double arrowheads). In the control subject, GAD67 mRNA signals were detected in all PV mRNA-positive grain clusters (A), whereas in the schizophrenia subject, GAD67 mRNA signals were undetectable in some of the PV mRNA-positive grain clusters (B). Scale bar: (in A), A, B, 50 μm.
Table 2.
Mean ± SD numbers of profiles per subject with PV mRNA and/or GAD67 mRNA signals
|
|
|
PV mRNA(−) |
PV mRNA(+) |
||
|---|---|---|---|---|---|
|
|
Total profiles/traverse of cortex
|
GAD67 mRNA(+)
|
GAD67 mRNA(+)
|
GAD67 mRNA(m=-)
|
|
| Control | 106.7 ± 46.3 | 68.5 ± 33.9 | 34.3 ± 13.8 | 3.9 ± 1.0 | |
| Schizophrenia
|
107.3 ± 51.2
|
68.1 ± 37.4
|
23.4 ± 15.1
|
15.9 ± 5.1
|
|
PV mRNA expression in the PFC of haloperidol-treated monkeys
In both the haloperidol-treated and control monkeys, the laminar distribution of PV mRNA signal was similar to that observed in humans (Fig. 9). Silver grains were clustered around neuronal nuclei, and the laminar distribution of specifically labeled neurons (data not shown) was similar to the previously reported distribution of PV-immunoreactive neurons in monkey PFC (Condé et al., 1994). PV mRNA expression in the monkey PFC appeared to be unchanged after long-term treatment with haloperidol (Fig. 9). The mean optical density of PV mRNA signal in PFC area 9 did not differ (t = 0.64; p = 0.56) between control (28.7 ± 17.7 nCi/gm) and haloperidol-treated monkeys (26.4 ± 11.0 nCi/gm). Similarly, in area 46, the density of PV mRNA signal did not differ between control and haloperidol-treated animals (t = 1.0; p = 0.38).
Figure 9.
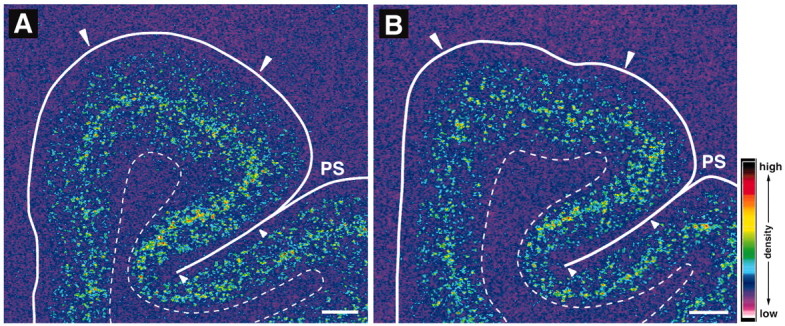
Representative autoradiograms showing signals for PV mRNA in the PFC of control (A) and haloperidol-treated (B) monkeys. The densities of hybridization signals are presented in pseudocolor manner according to the scale in bottom right. Note that the signal distribution and intensity appear to be unchanged in the PFC of the haloperidol-treated monkey. Solid and broken white lines indicate the pial surface and the border between gray and white matter, respectively. PS, Principal sulcus. Pairs of large and small white arrowheads indicate the quantified regions in areas 9 and 46, respectively. Scale bars, 1 mm.
Discussion
Abnormalities in PFC inhibitory neurotransmission in schizophrenia were hypothesized to be restricted to a subpopulation of GABA neurons on the basis of several observations (Blum and Mann, 2002), including the findings that the expression levels of GAD67 and GAT-1 mRNAs were decreased below the limit of detection in 25–35% of GABA neurons, whereas most of the GABA neurons exhibited normal levels of expression for these two genes (Volk et al., 2000, 2001). Both in situ hybridization and cDNA microarray findings in the present study support this hypothesis by demonstrating a selective decrease in PV, but not CR, mRNA expression in PFC area 9 in subjects with schizophrenia. This decrease appeared to be caused principally by a reduction in PV mRNA expression per neuron rather than by a reduced number of neurons with detectable levels of PV mRNA. This absence of a difference in the density of PV mRNA-positive neurons is consistent with two previous immunocytochemical studies that reported no significant changes in the densities of PV-immunoreactive neurons in PFC areas 9 or 46 of subjects with schizophrenia (Woo et al., 1997; Beasley et al., 2002). In addition, the reduced neuronal PV mRNA expression was significantly correlated with the decrease in density of GAD67 mRNA-positive neurons in the same subjects. Together, these findings suggested that the GAD67 mRNA expression deficit in schizophrenia is selective for the PV-containing subclass of PFC GABA neurons. Consistent with this prediction, simultaneous detection of GAD67 and PV mRNAs revealed that the proportion of PV mRNA-positive neurons without GAD67 mRNA signal was 4.5 times greater in the schizophrenia group than in the control group.
Although the typical long-term exposure of subjects with schizophrenia to antipsychotic medications represents a potential confound of this study, the available information suggests that PV mRNA expression is not likely to be downregulated by these medications. In rats, chronic treatment with haloperidol or clozapine was reported to increase expression of PV in the PFC (Scruggs and Deutch, 1999), and in our study, long-term treatment with haloperidol and benztropine did not affect PV mRNA expression in monkey PFC. Consistent with these observations, the three subjects with schizophrenia (pairs 2, 10, and 14) who were not receiving antipsychotic medications at the time of death still showed reduced PV mRNA expression levels compared with their matched controls (Fig. 5A,C).
Premortem agonal state events may affect postmortem levels of some mRNA species. Brain pH, reportedly an inverse correlate of the length/severity of the agonal state (Harrison et al., 1995), did not differ between the subject groups. Additionally, PV mRNA reduction was not attributable to general deterioration of mRNA integrity in subjects with schizophrenia because the expression of CR mRNA, and of a large number of other transcripts (at least in subject pairs 1–10) (Glantz et al., 2000; Mirnics et al., 2000; Middleton et al., 2002), did not differ between the subject groups.
The available data also indicate that comorbid substance abuse did not contribute to the decreased PV mRNA expression in the subjects with schizophrenia. First, there was no significant effect of substance abuse history on differences in the PV mRNA measures within the matched pairs of subjects with schizophrenia and controls (Fig. 6). Second, the only control subject (pair 7) with a history of alcohol abuse had greater PV mRNA expression levels than the matched subject with schizophrenia (Fig. 5A,C). Finally, the three control subjects (pairs 3, 7, 10) with positive plasma alcohol levels at the time of death still had higher PV mRNA expression levels than their matched subjects with schizophrenia (Fig. 5A,C).
The reduction in PV mRNA levels in schizophrenia could reflect alterations in transcriptional regulation secondary to the genetic liability for the illness. Interestingly, the human PV gene is localized to chromosome 22q12-q13.1 (Ritzler et al., 1992), a region that has been frequently suggested to contain susceptibility loci for schizophrenia (Riley and McGuffin, 2000). Within this region, the PV gene lies in the vicinity (∼0.8 mega bp telomeric) of the marker D22S278 (GenBank NT 011520), which has been repeatedly implicated in schizophrenia as well as in bipolar disorder (Schwab and Wildenauer, 2000). Alternatively, some common factor may induce both the decrease in PV mRNA expression and the GAD67 mRNA expression deficit in PV mRNA-positive neurons. For example, gene expression for both PV and GAD67 appears to change in an activity-dependent manner (Hendry and Jones, 1988; Benson et al., 1994; Carder et al., 1996; Nie and Wong-Riley, 1996). In layer III of monkey PFC, the local axon collaterals of neighboring pyramidal neurons are a major source of excitatory input to PV-containing but not to CR-containing neurons (Melchitzky et al., 2001; Melchitzky and Lewis, 2003). Interestingly, in the PFC of subjects with schizophrenia, layer III pyramidal neurons exhibit reductions in dendritic arborization and spine density (Garey et al., 1998; Glantz and Lewis, 2000), suggesting that they receive fewer excitatory inputs and thus may provide reduced excitatory output to PV-containing neurons. Finally, both PV and GAD67 mRNA expression may be regulated by brain-derived neurotrophic factor (BDNF) signaling (Marty et al., 1997), and altered mRNA and protein levels of BDNF and its receptor, tropomyosin-related kinase B, have been reported in the PFC in schizophrenia (Takahashi et al., 2000; Durany et al., 2001; Hashimoto et al., 2002).
The colocalization of decreased PV and GAD67 mRNA expression suggests that GABA neurotransmission mediated by the PV-containing subclass of GABA neurons is dysfunctional in the PFC of subjects with schizophrenia. In cortical GABA neurons, GAD67 is coexpressed with GAD65 (Soghomonian and Martin, 1998). On the basis of differences in subcellular localization and cofactor association (Soghomonian and Martin, 1998), GAD67 is thought to be responsible for basal levels of GABA, whereas GAD65 is specialized to respond to rapid changes in synaptic demand during intense neuronal activity. Indeed, cortical GABA content was significantly decreased in adult heterozygotes of GAD67 knockout mice (GAD65+/+, GAD67+/-) (Asada et al., 1997), whereas it was normal in GAD65 knock-out mice (GAD65-/-, GAD67+/+) (Asada et al., 1996). Thus, although GAD65 levels may be normal in the PFC in schizophrenia (Guidotti et al., 2000), the deficit in GAD67 expression in PV neurons is likely to cause alterations in GABA neurotransmission. Furthermore, a decrease in PV protein itself could contribute to functional alterations of these neurons by affecting a range of Ca2+-regulated neuronal processes such as neuronal excitation and synaptic transmission (Pauls et al., 1996), because PV appears to buffer transient increases in cytosolic Ca2+ released from extracellular and intracellular sources (Chard et al., 1993). For example, decreased PV levels could reduce the firing frequency of PV-containing neurons, because insufficient buffering of transiently increased Ca2+ after neuronal firing could cause larger afterhyperpolarization generated by calcium-dependent potassium channels (Savic et al., 2001). In addition, because presynaptic PV has been suggested to contribute to the maintenance of synaptic transmission during high-frequency firing (Caillard et al., 2000), decreased PV levels may cause deterioration in synaptic transmission in this subclass of GABA neurons. However, it is unclear whether the cellular decrease in PV mRNA expression levels is actually associated with a reduction in PV protein levels. For example, most, if not all, PV neurons in the PFC appear to retain detectable levels of PV protein (just as they do of PV mRNA) in schizophrenia, as evidenced by reports that the density of PV-immunoreactive neurons is not decreased in schizophrenia (Woo et al., 1997; Beasley et al., 2002). Interestingly, the densities of PV-immunoreactive puncta, putative axon terminals, have been reported to be selectively decreased in layers III and IV of subjects with schizophrenia (Lewis et al., 2001), the same layers in which PV mRNA expression was observed to be reduced. However, whether the apparent reduction in PV-labeled terminals is of thalamic or intrinsic origin cannot be determined at present.
Thus, the significance of the present findings is the demonstration that schizophrenia is associated with selective gene expression abnormalities in a specific class of PFC GABA neurons. Interestingly, in contrast to the consistent findings across research groups of a deficit in GAD67 expression in the PFC (Akbarian et al., 1995; Guidotti et al., 2000; Volk et al., 2000; Knable et al., 2002), Heckers et al. (2002) reported no significant alteration in the density of GAD67 mRNA-positive cells or in the cellular expression of GAD67 mRNA in the hippocampus of subjects with schizophrenia. Thus, the GABA-related gene expression abnormalities in the PFC might be rather specific to working memory function, for which the PFC plays a central role, as opposed to other forms of memory, such as those mediated by the hippocampus (Squire and Zola-Morgan, 1991).
In the primate PFC, GABA-mediated inhibition has been shown to shape the firing pattern of pyramidal neurons that appears to encode the spatiotemporal specificity of information needed to guide the correct response in working memory tasks (Rao et al., 2000; Constantinidis et al., 2002). Among GABA neurons, the PV-containing chandelier and wide arbor (basket) neurons furnish inhibitory synapses on pyramidal neurons at the axon initial segment and soma, respectively (Williams et al., 1992), and thus provide more potent inhibitory regulation of pyramidal neuron firing than do inputs to distal dendrites (Cobb et al., 1995; Pouille and Scanziani, 2001) provided by other classes of GABA neurons such as CR-containing double-bouquet neurons. In slice electrophysiological experiments, chandelier and wide arbor (basket) neurons may synchronize the firing of multiple pyramidal neurons (Cobb et al., 1995); in addition, inhibitory inputs to the soma, but not to the dendrites, of pyramidal neurons regulate the temporal summation of excitatory inputs needed to evoke pyramidal neuron firing (Pouille and Scanziani, 2001). Because PV-containing neurons in the primate PFC receive direct excitatory inputs from thalamic afferents and from the local axon collaterals of cortical pyramidal neurons (Melchitzky et al., 1999; Melchitzky and Lewis, 2003), the inhibitory output of PV-containing neurons is thought to be important both in coupling the specific excitatory inputs conveying relevant information to synchronized patterns of pyramidal neuron firing and in suppressing pyramidal neuron activity driven by inputs conveying irrelevant information (Lewis and Gonzalez-Burgos, 2000). Consequently, selective disturbances in GABA neurotransmission by PV-containing GABA neurons are likely to disrupt the functional coordination of PFC pyramidal neuron activity and thus contribute to the working memory deficits frequently present in subjects with schizophrenia.
Footnotes
This work was supported by Grants MH45156 and MH43784 from the National Institutes of Health. We thank Frank A. Middleton and Deborah Hollingshead for helpful advice; Lansha Peng, Kathie Douglass, and Dianne A. Cruz for technical assistance; and Leonid Krimer, Karl-Anton Dorph-Petersen, Nadya Povysheva, Darlene S. Melchitzky, and Susan L. Erickson for thoughtful comments and discussion.
Correspondence should be sent to Dr. David A. Lewis, University of Pittsburgh, 3811 O'Hara Street, W1650 Biomedical Science Tower, Pittsburgh, PA 15213. E-mail: lewisda@msx.upmc.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/236315-12$15.00/0
References
- Akbarian S, Kim JJ, Potkin SG, Hagman JO, Tafazzoli A, Bunney Jr WE, Jones EG ( 1995) Gene expression for glutamic acid decarboxylase is reduced without loss of neurons in prefrontal cortex of schizophrenics. Arch Gen Psychiatry 52: 258–266. [DOI] [PubMed] [Google Scholar]
- Akil M, Pierri JN, Whitehead RE, Edgar CL, Mohila C, Lewis DA ( 1999) Lamina-specific alteration in the dopamine innervation of the prefrontal cortex in schizophrenic subjects. Am J Psychiatry 156: 1580–1589. [DOI] [PubMed] [Google Scholar]
- Asada H, Kawamura Y, Maruyama K, Kume H, Ding R, Ji FY, Kanbara N, Kuzume H, Sanbo M, Yagi T, Obata K ( 1996) Mice lacking the 65 kDa isoform of glutamic acid decarboxylase (GAD65) maintain normal levels of GAD67 and GABA in their brains but are susceptible to seizures. Biochem Biophys Res Commun 229: 891–895. [DOI] [PubMed] [Google Scholar]
- Asada H, Kawamura Y, Maruyama K, Kume H, Ding R, Kanbara N, Kuzume H, Sanbo M, Yagi T, Obata K ( 1997) Cleft palate and decreased brain γ-aminobutyric acid in mice lacking the 67-kDa isoform of glutamic acid decarboxylase. Proc Natl Acad Sci USA 94: 6496–6499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beasley CL, Zhang ZJ, Patten I, Reynolds GP ( 2002) Selective deficits in prefrontal cortical GABAergic neurons in schizophrenia defined by the presence of calcium-binding proteins. Biol Psychiatry 52: 708–715. [DOI] [PubMed] [Google Scholar]
- Benson DL, Huntsman MM, Jones EG ( 1994) Activity-dependent changes in GAD and preprotachykinin mRNAs in visual cortex of adult monkeys. Cereb Cortex 4: 40–51. [DOI] [PubMed] [Google Scholar]
- Blum BP, Mann JJ ( 2002) The GABAergic system in schizophrenia. Int J Neuropsychopharmacol 5: 159–179. [DOI] [PubMed] [Google Scholar]
- Caillard O, Moreno H, Schwaller B, Llano I, Celio MR, Marty A ( 2000) Role of the calcium-binding protein parvalbumin in short-term synaptic plasticity. Proc Natl Acad Sci USA 97: 13372–13377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carder RK, Leclerc SS, Hendry SHC ( 1996) Regulation of calcium-binding protein immunoreactivity in GABA neurons of macaque primary visual cortex. Cereb Cortex 6: 271–287. [DOI] [PubMed] [Google Scholar]
- Chard PS, Bleakman D, Christakos S, Fullmer CS, Miller RJ ( 1993) Calcium buffering properties of calbindin D28k and parvalbumin in rat sensory neurones. J Physiol (Lond) 472: 341–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb SR, Buhl EH, Halasy K, Paulsen O, Somogyi P ( 1995) Synchronization of neuronal activity in hippocampus by individual GABAergic interneurons. Nature 378: 75–78. [DOI] [PubMed] [Google Scholar]
- Condé F, Lund JS, Jacobowitz DM, Baimbridge KG, Lewis DA ( 1994) Local circuit neurons immunoreactive for calretinin, calbindin D-28k, or parvalbumin in monkey prefrontal cortex: distribution and morphology. J Comp Neurol 341: 95–116. [DOI] [PubMed] [Google Scholar]
- Constantinidis C, Williams GV, Goldman-Rakic PS ( 2002) A role for inhibition in shaping the temporal flow of information in prefrontal cortex. Nat Neurosci 5: 175–180. [DOI] [PubMed] [Google Scholar]
- Daviss SR, Lewis DA ( 1995) Local circuit neurons of the prefrontal cortex in schizophrenia: selective increase in the density of calbindin-immunoreactive neurons. Psychiatry Res 59: 81–96. [DOI] [PubMed] [Google Scholar]
- Durany N, Michel T, Zöchling R, Boissl KW, Cruz-Sánchez FF, Riederer P, Thome J ( 2001) Brain-derived neurotrophic factor and neurotrophin 3 in schizophrenic psychoses. Schizophr Res 52: 79–86. [DOI] [PubMed] [Google Scholar]
- Gabbott PLA, Bacon SJ ( 1996) Local circuit neurons in the medial prefrontal cortex (areas 24a, b, c, 25 and 32) in the monkey: I. Cell morphology and morphometrics. J Comp Neurol 364: 567–608. [DOI] [PubMed] [Google Scholar]
- Garey LJ, Ong WY, Patel TS, Kanani M, Davis A, Mortimer AM, Barnes TRE, Hirsch SR ( 1998) Reduced dendritic spine density on cerebral cortical pyramidal neurons in schizophrenia. J Neurol Neurosurg Psychiatry 65: 446–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR, McGinty JF, Young WS ( 1991) Dopamine differentially regulates dynorphin, substance P, and enkaphalin expression in striatal neurons: in situ hybridization histochemical analysis. J Neurosci 11: 1016–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glantz LA, Lewis DA ( 2000) Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry 57: 65–73. [DOI] [PubMed] [Google Scholar]
- Glantz LA, Austin MC, Lewis DA ( 2000) Normal cellular levels of synaptophysin mRNA expression in the prefrontal cortex of subjects with schizophrenia. Biol Psychiatry 48: 389–397. [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS ( 1994) Working memory dysfunction in schizophrenia. J Neuropsychiatry 6: 348–357. [DOI] [PubMed] [Google Scholar]
- Guidotti A, Auta J, Davis JM, Gerevini VD, Dwivedi Y, Grayson DR, Impagnatiello F, Pandey G, Pesold C, Sharma R, Uzunov D, Costa E ( 2000) Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder. Arch Gen Psychiatry 57: 1061–1069. [DOI] [PubMed] [Google Scholar]
- Harrison PJ, Heath PR, Eastwood SL, Burnet PWJ, McDonald B, Pearson RCA ( 1995) The relative importance of premortem acidosis and postmortem interval for human brain gene expression studies: selective mRNA vulnerability and comparison with their encoded proteins. Neurosci Lett 200: 151–154. [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Volk DW, Buchheit SE, Lewis DA ( 2002) Expression of BDNF and trkB mRNAs in prefrontal cortex of subjects with schizophrenia. Soc Neurosci Abstr 28: 703.7. [Google Scholar]
- Heckers S, Stone D, Walsh J, Schick J, Koul P, Benes FM ( 2002) Differential hippocampal expression of glutamic acid decarboxylase 65 and 67 messenger RNA in bipolar disorder and schizophrenia. Arch Gen Psychiatry 59: 521–529. [DOI] [PubMed] [Google Scholar]
- Hendry SHC, Jones EG ( 1988) Activity-dependent regulation of GABA expression in the visual cortex of adult monkeys. Neuron 1: 701–712. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Kubota Y ( 1997) GABAergic cell subtypes and their synaptic connections in rat frontal cortex. Cereb Cortex 7: 476–486. [DOI] [PubMed] [Google Scholar]
- Knable MB, Barci BM, Bartko JJ, Webster MJ, Torrey EF ( 2002) Molecular abnormalities in the major psychiatric illnesses: classification and regression tree (CRT) analysis of post-mortem prefrontal markers. Mol Psychiatry 7: 392–404. [DOI] [PubMed] [Google Scholar]
- Krimer LS, Goldman-Rakic PS ( 2001) Prefrontal microcircuits: membrane properties and excitatory input of local, medium, and wide arbor neurons. J Neurosci 21: 3788–3796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA, Gonzalez-Burgos G ( 2000) Intrinsic excitatory connections in the prefrontal cortex and the pathophysiology of schizophrenia. Brain Res Bull 52: 309–317. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Lieberman JA ( 2000) Catching up on schizophrenia: natural history and neurobiology. Neuron 28: 325–334. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Cruz DA, Melchitzky DS, Pierri JN ( 2001) Lamina-specific reductions in parvalbumin-immunoreactive axon terminals in the prefrontal cortex of subjects with schizophrenia: evidence for decreased projections from the thalamus. Am J Psychiatry 158: 1411–1422. [DOI] [PubMed] [Google Scholar]
- Littell RC, Milliken GA, Stroup WW, Wolfinger RD ( 1996) SAS system for mixed models. Cary, NC: SAS Institute.
- Lund JS, Lewis DA ( 1993) Local circuit neurons of developing and mature macaque prefrontal cortex: Golgi and immunocytochemical characteristics. J Comp Neurol 328: 282–312. [DOI] [PubMed] [Google Scholar]
- Marty S, de Penha Berzaghi M, Berninger B ( 1997) Neurotrophins and activity-dependent plasticity of cortical interneurons. Trends Neurosci 20: 198–202. [DOI] [PubMed] [Google Scholar]
- Melchitzky DS, Lewis DA ( 2003) Pyramidal neuron local axon terminals in monkey prefrontal cortex: differential targeting of subclasses of GABA neurons. Cereb Cortex 13: 452–460. [DOI] [PubMed] [Google Scholar]
- Melchitzky DS, Sesack SR, Lewis DA ( 1999) Parvalbumin-immunoreactive axon terminals in monkey and human prefrontal cortex: laminar, regional and target specificity of Type I and Type II synapses. J Comp Neurol 408: 11–22. [PubMed] [Google Scholar]
- Melchitzky DS, Gonzalez-Burgos G, Barrionuevo G, Lewis DA ( 2001) Synaptic targets of the intrinsic axon collaterals of supragranular pyramidal neurons in monkey prefrontal cortex. J Comp Neurol 430: 209–221. [DOI] [PubMed] [Google Scholar]
- Middleton FA, Mirnics K, Pierri JN, Lewis DA, Levitt P ( 2002) Gene expression profiling reveals alterations of specific metabolic pathways in schizophrenia. J Neurosci 22: 2718–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirnics K, Middleton FA, Marquez A, Lewis DA, Levitt P ( 2000) Molecular characterization of schizophrenia viewed by microarray analysis of gene expression in prefrontal cortex. Neuron 28: 53–67. [DOI] [PubMed] [Google Scholar]
- Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Bell G ( 1991) The consortium to establish a registry for Alzheimer's disease (CERAD). Part II. Standardization of the neuropathological assessment of Alzheimer's disease. Neurology 41: 479–486. [DOI] [PubMed] [Google Scholar]
- Neter J, Kutner MH, Nachtsheim CJ, Wasserman W ( 1996) Applied linear statistical models. Chicago: Irwin.
- Nie F, Wong-Riley MT ( 1996) Metabolic and neurochemical plasticity of gamma-aminobutyric acid-immunoreactive neurons in the adult macaque striate cortex following monocular impulse blockade: quantitative electron microscopic analysis. J Comp Neurol 370: 350–366. [DOI] [PubMed] [Google Scholar]
- Pauls TL, Cox JA, Berchtold MW ( 1996) The Ca(2+)-binding proteins parvalbumin and oncomodulin and their genes: new structural and functional findings. Biochim Biophys Acta 1306: 39–54. [DOI] [PubMed] [Google Scholar]
- Pierri JN, Chaudry AS, Woo T-U, Lewis DA ( 1999) Alterations in chandelier neuron axon terminals in the prefrontal cortex of schizophrenic subjects. Am J Psychiatry 156: 1709–1719. [DOI] [PubMed] [Google Scholar]
- Pouille F, Scanziani M ( 2001) Enforcement of temporal fidelity in pyramidal cells by somatic feed-forward inhibition. Science 293: 1159–1163. [DOI] [PubMed] [Google Scholar]
- Rao SG, Williams GV, Goldman-Rakic PS ( 2000) Destruction and creation of spatial tuning by disinhibition: GABAA blockade of prefrontal cortical neurons engaged by working memory. J Neurosci 20: 485–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley BP, McGuffin P ( 2000) Linkage and associated studies of schizophrenia. Am J Med Genet 97: 23–44. [DOI] [PubMed] [Google Scholar]
- Ritzler JM, Sawhney R, Guerts van Kessel AH, Grzeschik KH, Schinzel A, Berchtold MW ( 1992) The genes for highly homologous Ca(2+)-binding proteins oncomodulin and parvalbumin are not linked in the human genome. Genomics 12: 567–572. [DOI] [PubMed] [Google Scholar]
- Savic N, Pedarzani P, Sciancalepore M ( 2001) Medium afterhyperpolarization and firing pattern modulation in interneurons of striatum radiatum in the CA3 hippocampal region. J Neurophysiol 85: 1986–1997. [DOI] [PubMed] [Google Scholar]
- Schwab SG, Wildenauer DB ( 2000) Chromosome 22 workshop report. Am J Med Genet 88: 276–278. [PubMed] [Google Scholar]
- Scruggs JL, Deutch AY ( 1999) Chronic antipsychotic drug administration alters calcium-binding protein levels but not GAD levels in the prefrontal cortex of rat. Schizophr Res 36: 119–120. [Google Scholar]
- Soghomonian J-J, Martin DL ( 1998) Two isoforms of glutamate decarboxylase: why? Trends Pharmacol Sci 19: 500–505. [DOI] [PubMed] [Google Scholar]
- Squire LR, Zola-Morgan S ( 1991) The medial temporal lobe memory system. Science 20: 1380–1386. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Shirakawa O, Toyooka K, Kitamura N, Hashimoto T, Maeda K, Koizumi S, Wakabayashi K, Takahashi H, Someya T, Nawa H ( 2000) Abnormal expression of brain-derived neurotrophic factor and its receptor in the corticolimbic system of schizophrenic patients. Mol Psychiatry 5: 293–300. [DOI] [PubMed] [Google Scholar]
- Volk DW, Austin MC, Pierri JN, Sampson AR, Lewis DA ( 2000) Decreased GAD67 mRNA expression in a subset of prefrontal cortical GABA neurons in subjects with schizophrenia. Arch Gen Psychiatry 57: 237–245. [DOI] [PubMed] [Google Scholar]
- Volk DW, Austin MC, Pierri JN, Sampson AR, Lewis DA ( 2001) GABA transporter-1 mRNA in the prefrontal cortex in schizophrenia: decreased expression in a subset of neurons. Am J Psychiatry 158: 256–265. [DOI] [PubMed] [Google Scholar]
- Weinberger DR, Berman KF, Zec RF ( 1986) Physiologic dysfunction of dorsolateral prefrontal cortex in schizophrenia. I. Regional cerebral blood flow evidence. Arch Gen Psychiatry 43: 114–124. [DOI] [PubMed] [Google Scholar]
- Williams SM, Goldman-Rakic PS, Leranth C ( 1992) The synaptology of parvalbumin-immunoreactive neurons in primate prefrontal cortex. J Comp Neurol 320: 353–369. [DOI] [PubMed] [Google Scholar]
- Woo T-U, Miller JL, Lewis DA ( 1997) Parvalbumin-containing cortical neurons in schizophrenia. Am J Psychiatry 154: 1013–1015. [DOI] [PubMed] [Google Scholar]
- Woo T-U, Whitehead RE, Melchitzky DS, Lewis DA ( 1998) A subclass of prefrontal gamma-aminobutyric acid axon terminals are selectively altered in schizophrenia. Proc Natl Acad Sci USA 95: 5341–5346. [DOI] [PMC free article] [PubMed] [Google Scholar]