

Abstract
The treatment of ischemic strokes is limited to the prevention of cerebrovascular risk factors and to the modulation of the coagulation cascade during the acute phase. A new therapeutic strategy could be to preventively protect the brain against noxious biological reactions induced by cerebral ischemia such as oxidative stress and inflammation to minimize their neurological consequences. Here, we show that a peroxisome proliferator-activated receptor (PPAR-α) activator, fenofibrate, protects against cerebral injury by anti-oxidant and anti-inflammatory mechanisms. A 14 d preventive treatment with fenofibrate reduces susceptibility to stroke in apolipoprotein E-deficient mice as well as decreases cerebral infarct volume in C57BL/6 wild-type mice. The neuroprotective effect of fenofibrate is completely absent in PPAR-α-deficient mice, suggesting that PPAR-α activation is involved as a mechanism of the protection against cerebral injury. Furthermore, this neuroprotective effect appears independently of any improvement in plasma lipids or glycemia and is associated with (1) an improvement in middle cerebral artery sensitivity to endothelium-dependent relaxation unrelated to an increase in nitric oxide synthase (NOS) type III expression, (2) a decrease in cerebral oxidative stress depending on the increase in numerous antioxidant enzyme activities, and (3) the prevention of ischemia-induced expression of vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 in cerebral vessels without any change in NOS II expression. These data demonstrate that PPAR-α could be a new pharmacological target to preventively reduce the deleterious neurological consequences of stroke in mice and suggest that PPAR-α activators could preventively decrease the severity of stroke in humans.
Keywords: stroke, PPAR-α, neuroprotection, prevention, oxidative stress, inflammation, adhesion proteins, mouse
Introduction
Ischemic stroke is the third leading cause of death and the first leading cause of disability in adults in industrialized countries (Hankey and Warlow, 1999). Currently, the therapeutic strategy available for the acute phase of stroke is limited to aspirin and thrombolysis (Hankey and Warlow, 1999). To date, clinical trials have not demonstrated any protective effects of drugs during the acute phase of cerebral ischemia (Lee et al., 2000), and therefore interest has turned to alternative therapeutic strategies such as preventive neuroprotection based on the induction of cerebral resistance to ischemia before its occurrence (Jonas, 1995). On the basis of experimental brain ischemic tolerance data (Chen and Simon, 1997), there is evidence that brain could be rendered resistant to the deleterious effects of ischemia by the induction of cytoprotective proteins such as anti-oxidant enzymes, endothelial nitric oxide synthase (NOS), or heat shock proteins as well as by the inhibition of deleterious inflammatory, oxidative, and apoptotic pathways (Chen and Simon, 1997; Bordet et al., 2000; Puisieux et al., 2000). Pharmacological agents able to mimic the biological effects observed in brain ischemic tolerance might then increase the resistance to ischemia of patients with high risk for stroke (Fisher et al., 1994). Recent evidence indicates that peroxisome proliferator-activated receptor (PPAR)-α activators (fibrates) could reduce the incidence of stroke by improving plasma lipid metabolism (Amarenco, 2001; Bloomfield Rubins et al., 2001). In addition, by their so-called pleiotropic lipid-independent effects (Fruchart et al., 1999) they could influence biological targets involved in experimental brain ischemic tolerance (Chen and Simon, 1997).
PPAR-α is one of the three subtypes of the nuclear receptor PPAR family (Fruchart et al., 1999). The activation of PPAR-α, by both natural ligands such as fatty acids and derivates or synthetic ligands such as hypolipidemic fibrates, stimulates target gene transcription via the formation of heterodimeric transcription factor complexes with the retinoid x receptor (RXR) (Fruchart et al., 1999). PPAR-α activation also induces a negative transcriptional regulation of NF-κB and of the activator protein-1 (AP-1) signaling pathways (Delerive et al., 1999), thereby modulating inflammation and oxidative stress (Poynter and Daynes, 1998; Fruchart et al., 1999). It has been demonstrated that PPAR-α inhibits inducible NOS (NOS II) in macrophages and prevents the expression of cyclooxygenase-2 (COX-2) (Staels et al., 1998) in smooth muscle cells. PPAR-α activation also inhibits vascular cell adhesion molecule-1 (VCAM-1) expression (Marx et al., 1999) and, to a lesser degree, intercellular adhesion molecule-1 (ICAM-1) (Wayman et al., 2002). Moreover, PPAR-α activation stimulates antioxidant enzyme expression, such as superoxide dismutase and catalase (Poynter and Daynes, 1998; Inoue et al., 2001), both of which are involved in brain ischemic tolerance (Jander et al., 1996; Chen and Simon, 1997; Bordet et al., 2000).
PPAR-α is expressed in cerebral cells such as neurons and astrocytes (Kainu et al., 1994; Cullingford et al., 1998) and in cerebral vascular endothelial and smooth muscle cells, as well as in monocytes/macrophages (Bishop-Bailey, 2000), which are all involved in cerebral ischemia pathophysiology (Bastide et al., 1999; Deplanque et al., 2000; Lee et al., 2000; Chan, 2001). Activation of PPAR-α in all of these biological sites could contribute to change the pathophysiological process during cerebral ischemia. Therefore, the purpose of this study was to determine whether chronic PPAR-α activation by fenofibrate reduces cerebral infarct size in mice, independently of any change in plasma lipid levels, by modulating endothelial function of cerebral arteries as well as by upregulating brain antioxidant enzyme activities and by inhibiting inflammation pathways through a decrease in ischemia-induced VCAM-1, ICAM-1, or NOS II expression.
Materials and Methods
Animals and treatment administration. All experiments were performed within the framework of the French legislation that controls animal experimentation. To assess the neuroprotective effect of prophylactic treatment with fenofibrate, male apolipoprotein (Apo) E-deficient mice (F6 homozygotes; C57BL/6 genetic background; Transgenic Alliance, L'Arbresle, France), C57BL/6 and SV129 wild-type mice (IFFA Credo, L'Arbresle, France), and PPAR-α-deficient mice (PPAR-α -/-) (F6 homozygotes; SV129 genetic background) (Lee et al., 1995; Delerive et al., 1999) were fed a diet containing 0.2% fenofibrate (UAR, Villemoissonsur-orge, France) or placebo for 14 d. To assess the acute neuroprotective effect of fenofibrate, three other groups of C57BL/6 wild-type mice received fenofibrate (50 or 250 mg/kg, i.p.) (Sigma-Aldrich, Saint Quentin Fallavier, France) or its vehicle (DMSO solution, 50 μl), 1 and 6 hr after the beginning of middle cerebral artery (MCA) occlusion. To assess the vascular effects of fenofibrate, C57BL/6 mice and Wistar Kyoto rats (IFFA Credo) were fed a diet containing 0.2% fenofibrate (UAR) or placebo for 14 d.
Middle cerebral artery occlusion method. Animals were anesthetized with chloral hydrate (300 mg/kg, i.p.) (Sigma-Aldrich). According to the mouse strain (Connolly et al., 1996), cerebral infarcts were produced by a 60 min (C57BL/6) or a 90 min (SV129) MCA occlusion followed by a 24 hr reperfusion period. The ostium of the right MCA was occluded intraluminally with an adapted method from that described previously in rat (Bastide et al., 1999). Under surgical microscope control, an aneurysm clip was place across the carotid bifurcation, and an arteriotomy was made in the common carotid artery stump to allow the introduction of a 20-mm-long 6-0 nylon monofilament suture with its tip rounded by flame heating. This was secured in place and the aneurysm clip was removed; then the suture was advanced gently into the internal carotid artery and passed into the intracranial circulation (12–13 mm distal to the carotid bifurcation), thereby occluding the origin of MCA. After 60 or 90 min, to allow reperfusion, the suture was removed carefully until its tip was blocked by ligature placed on the common carotid artery. A rectal probe was inserted, and core temperature was maintained at ∼37°C by the use of a heating pad and a heating lamp.
Histology and measurement of infarct size. After 24 hr, mice were killed, and the brains were removed and frozen in isopentane for histological analysis. Cryostat-cut coronal brain sections (20 μm) were stained with cresyl violet, and infarct volume was assessed by numerical integration of the infarcted areas calculated by an image analysis software (Color Image 1.32, NIH, Bethesda, MD) after digitization by scanner as described (Bordet at al., 2000). Corrected infarct volumes were calculated to compensate for the effect of brain edema as described (Bordet et al., 2000).
Analysis of metabolic parameters. Serum cholesterol [total cholesterol, very low-density lipoprotein (VLDL) plus intermediate-density lipoprotein (IDL) plus low-density lipoprotein (LDL) cholesterol, high-density lipoprotein (HDL) cholesterol], triglycerides, and glucose were determined from blood samples obtained by retro-orbital puncture under chloral hydrate-induced anesthesia 24 hr before surgical procedure as described (Peters et al., 1997). Because of the well documented liver peroxisome proliferation in rodents (Fruchart at al., 1999), liver weights were recorded after animals were killed to confirm that fenofibrate was pharmacologically active.
Blood–brain barrier model. To examine the ability of fenofibrate to cross the blood–brain barrier, an in vitro model of blood–brain barrier consisting of a coculture of bovine brain capillary endothelial cells and rat astrocytes was used as described (Dehouck et al., 1990). The major active metabolite of fenofibrate, fenofibric acid (Laboratoire Fournier, Dijon, France), was added to the cultured medium in contact with endothelial cells at a dose of 100 μm for 45 min at 37°C. Transports were measured by HPLC quantification of fenofibric acid in the basal compartment at each time point. To obtain a concentration-independent transport parameter, the clearance principle was used. The slopes of the clearance curves for the cultured inserts and the control filters were Pst and Psf, respectively, with Ps = permeability × surface area product. Permeability of endothelial cells monolayers (Pse) was calculated from 1/Pse = 1/Pst - 1/Psf. The permeability coefficient of endothelial cells (Pe) was obtained by dividing Pse values by the surface area of the insert as described (Dehouck et al., 1990).
Cerebral blood flow measurement. Regional cerebral blood flow (rCBF) was evaluated at baseline, during MCA occlusion and until 30 min after reperfusion by laser Doppler flow (Periflux PF3, Perimed, Järfälla, Sweden) in treated and untreated mice (five in each group) submitted to a 1 hr cerebral ischemia using a flexible 0.5 mm fiber-optic probe (Perimed) that was attached to the intact skull overlying the core region of the MCA territory (2 mm posterior, 6 mm lateral from bregma).
MCA vasoreactivity. Because of methodological limitations according to the size of cerebral vessels in mice, MCA vascular reactivity was evaluated in Wistar Kyoto rats (five in each groups) after a 14 d period of fenofibrate or placebo treatment using a technique described previously (Bastide et al., 2003). A segment of the dissected right MCA was mounted in a small vessel arteriograph (Living Systems Intrumentation, Burlington, VT), and then the lumen diameter was analyzed in the no-flow condition through a camera coupled to a video dimension analyzer (Bastide et al., 2003). Before testing the endothelium-dependent and endothelium-independent relaxation, a precontraction of the vessel was realized using serotonin (5-HT) at a concentration of 10-6m. When the diameter reached a steady state, the vessel was exposed to increasing concentrations of acetylcholine (ACh) (10-9 to 3 × 10-5m) or to the NO donor sodium nitroprusside (SNP) (3 × 10-5m). In these conditions, vasorelaxation was measured as the percentage of increase from the preconstricted artery diameter
Oxidative stress and antioxidant enzymes. Markers of oxidative stress, thiobarbituric acid-reactive substances (TBARS), were measured in brain homogenates of fenofibrate-treated and untreated animals as described (Ramassamy et al., 2001). This was completed by measuring antioxidant enzyme activities (copper/zinc superoxide dismutase, manganese superoxide dismutase, glutathione peroxidase, glutathione reductase, glutathione S-transferase, and catalase) in brain homogenates of fenofibrate-treated and untreated animals by spectrophotometric assay as described (Paglia and Valentine, 1967; Habig et al., 1974; Goldberg and Spooner, 1983; Bordet et al., 2000).
VCAM-1 and ICAM-1 immunohistochemistry. Ischemia-induced VCAM-1 and ICAM-1 expressions (Wang et al., 1994; Jander et al., 1996) were evaluated in fenofibrate-treated and untreated animals (five in each group). Twenty-four hours after the cerebral ischemia–reperfusion procedure, mice were anesthetized with a lethal dose of pentobarbital and fixed by transcardial perfusion with 4% paraformaldehyde in PBS, pH 7.4. Brains were removed and postfixed in the same fixative during 4 hr at 4°C and then processed for paraffin embedding and cut into 4-μm-thick sections. Endogenous peroxidases were inhibited with 0.3% H2O2 and 10% methanol in PBS, pH 7.4, and protein nonspecific binding was blocked with 5% normal rabbit serum in PBS, pH 7.4. Sections were then incubated overnight at 4°C with a goat polyclonal antibody (anti-VCAM-1, 1:50; anti-ICAM-1, 1:100; Santa Cruz Biotechnology, Santa Cruz, CA). Antibodies were visualized with an anti-goat Vectastain ABC Elite kit (Vector Laboratories, Burlingame, CA) and DAB/H2O2 exposure (Fast DAB tablets set; Sigma, St. Louis, MO) according to the manufacturers' procedures. The sections incubated with anti-VCAM-1 but not anti-ICAM-1 were counterstained with cresyl violet. As negative control, some sections were incubated without primary antibody. The intensity of staining for VCAM-1 and ICAM-1 was semiquantitatively evaluated in number of + signs (0, no staining; +++, important staining), with treatment and ischemic conditions concealed.
Western blot analysis. To investigate the role of fenofibrate treatment on the putative protective or deleterious proteins, we first analyzed the expression of NOS III and copper/zinc superoxide dismutase in brain homogenates of treated and untreated animals by Western blot analysis (anti-NOS III, 1:200; anti-superoxide dismutase (SOD)-1, 1:5000; Upstate Biotechnology). According to manufacturer's procedures, mouse brain extract and epidermal growth factor (EGF)-stimulated A431 cell lysate were used, respectively, as control. Using the same technique, we analyzed the expression of VCAM-1, ICAM-1, and NOS II 24 hr after brain–ischemia reperfusion in treated and untreated mice (anti-VCAM-1, 1:1000; anti-ICAM-1, 1:1000; anti-NOS II, 1:1000; Upstate Biotechnology) when non-ischemic mice injected with lipopolysaccharide (1.5 mg/kg, i.p.; 24 hr before) were used as positive control. Moreover, for all of these experiments, selected blots were probed with β-actin (1:250; Santa Cruz Biotechnology) as internal control. After digitization, densitometric values were evaluated using an image analysis software (Perfect-image V6.1, Claravision, Orsay, France).
Statistical analysis. For statistical data comparison, a standard software package (SPSS 11.0 for windows) was used. Variables were compared between the different groups with a one-way variance analysis followed by least significant difference tests. All values are given as mean ± SEM. Values of p < 0.05 are considered significant.
Results
Chronic fenofibrate administration suppresses susceptibility to provoked stroke in apolipoprotein E-deficient mice
To investigate the putative neuroprotective effect of PPAR-α activation, we chose to study the effects of fenofibrate on Apo E-deficient mice, which are hypercholesterolemic and highly susceptible to the deleterious effects of a provoked stroke (Laskowitz et al., 1997). Apo E-deficient mice were pretreated with fenofibrate (0.2% w/w in standard diet) or with placebo for 14 d. In this first experimental step, wild-type mice carrying the same genetic background (C57BL/6) received placebo. There was no mortality in the three tested groups subjected to a 60 min middle cerebral artery occlusion followed by a recovery period of 24 hr. Placebo-treated Apo E-deficient mice had larger cerebral infarct volumes than placebo-treated wild-type mice (51 ± 6 versus 34 ± 6 mm3, respectively; p < 0.05), confirming the previously described higher susceptibility of Apo E-deficient mice to the deleterious effects of cerebral ischemia (Laskowitz et al., 1997). Fenofibrate pretreatment drastically reduced the susceptibility to stroke of Apo E-deficient mice, because fenofibrate-treated Apo E-deficient mice had lower cerebral infarct volumes than placebo-treated Apo E-deficient mice (32 ± 4 versus 51 ± 6 mm3, respectively; p < 0.05) and because infarct volumes in fenofibrate-treated Apo E-deficient mice were similar to those of placebo-treated wild-type mice. This protective effect against susceptibility to stroke was observed only in the cortical area and not in the striatal area (Fig. 1A). Although fenofibrate reduced cerebral infarct volumes in Apo E-deficient mice, it increased total cholesterol by increasing atherogenic cholesterol (VLDL + IDL + LDL cholesterol) and decreasing anti-atherogenic cholesterol (HDL cholesterol) plasma levels without affecting triglyceride concentrations (Table 1). It decreased fasting hyperglycemia as compared with the level measured in placebo-treated Apo E-deficient mice or in placebo-treated wild-type mice (Table 1). Liver weights were increased in fenofibrate-treated Apo E-deficient mice, because of the already well documented peroxisome proliferation in rodents (Fruchart et al., 1999), indicating that fenofibrate was pharmacologically active (Table 1).
Figure 1.
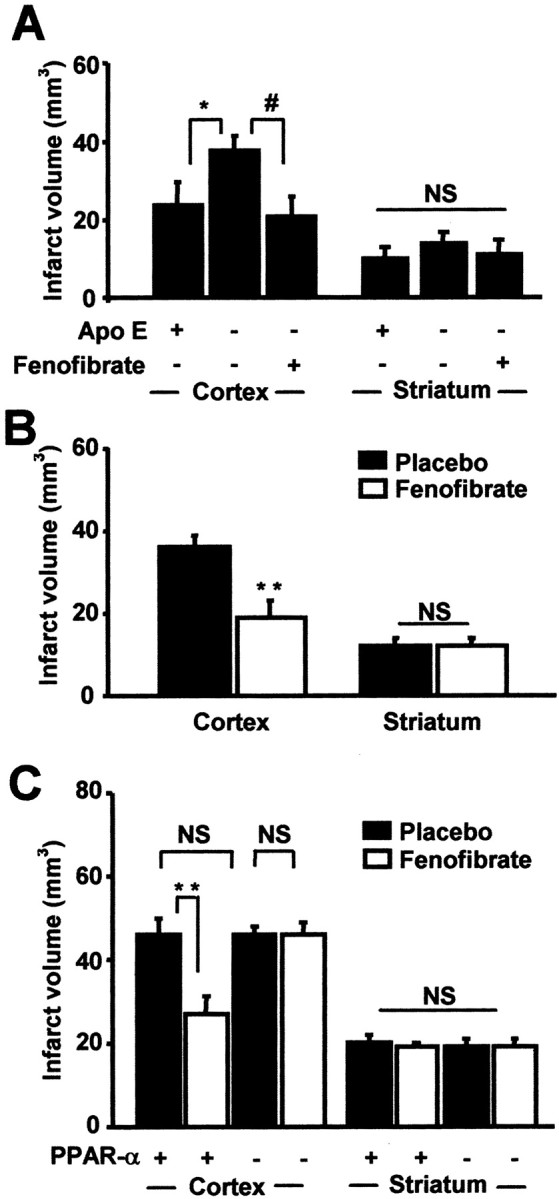
PPAR-α activation as a mechanism of fenofibrate-induced preventive neuroprotection. In A, Apo E-deficient mice were fed a diet containing 0.2% fenofibrate or placebo (n = 5 per group) for 14 d before a 60 min MCA occlusion was performed. In these conditions, the increased susceptibility to cerebral ischemia of Apo E-deficient mice and the neuroprotective effect of fenofibrate appear only in the cortical area. *, #p < 0.05. B, Using the same procedure, cortical infarct volumes in C57BL/6 wild-type mice (n = 6 per group) are reduced by 46% after fenofibrate treatment when striatal infarct volumes remain similar. **p < 0.01. C, Preventive neuroprotective effect of fenofibrate is confirmed in SV129 wild-type mice (n = 5 per group) submitted to a 90 min MCA occlusion. Such a neuroprotective effect is not reproduced in PPAR-α-deficient mice (n = 4 per group), just as PPAR-α deficiency does not induce any increase in cerebral infarct volumes. **p < 0.01. NS, Not significant.
Table 1.
Weight and metabolic variables in C57BL/6 and Apo E-deficient mice
|
Mice type |
Mice weight (g) |
Livers weight (g) |
Total cholesterol (mg/dl) |
VLDL+IDL+LDL (mg/dl) |
HDL (mg/dl) |
Triglycerides (mg/dl) |
Glycemia (mg/dl) |
|---|---|---|---|---|---|---|---|
| C57BL/6 wild-type mice | 33 ± 1 | 1.2 ± 0.1 | 96 ± 6 | 37 ± 4 | 58 ± 2 | 68 ± 7 | 173 ± 6 |
| Apo E-deficient mice | 33 ± 2 | 1.3 ± 0.1 | 545 ± 87 ** | 528 ± 84 ** | 16 ± 2 ** | 121 ± 11 * | 201 ± 5 * |
| Apo E-deficient and F
|
35 ± 3
|
2.9 ± 0.2 †
|
732 ± 74 **
|
728 ± 73 **#
|
3 ± 0.3 **##
|
111 ± 10 *
|
115 ± 5 *##
|
| C57BL/6 wild-type mice | 31 ± 1 | 1.2 ± 0.1 | 85 ± 9 | 32 ± 6 | 53 ± 3 | 61 ± 9 | 188 ± 29 |
| Wild-type mice and F
|
28 ± 1
|
2.2 ± 0.1 †
|
78 ± 10
|
30 ± 7
|
48 ± 2
|
53 ± 6
|
183 ± 37
|
p<0.0001
p<0.001
p<0.01
p<0.05 versus wild-type mice
p<0.001 versus untreated Apo E-deficient mice. F, Fenofibrate.
Fenofibrate induces a preventive neuroprotection in wild-type C57BL/6 mice
To determine whether the neuroprotective effect of fenofibrate in mice was independent of any genetic susceptibility to cerebral ischemia such as Apo E deficiency, we tested whether preventive treatment with fenofibrate reduced cerebral infarct volume after a 60 min middle cerebral artery occlusion followed by a 24 hr reperfusion period in normolipidemic wild-type mice carrying the same genetic background (C57BL/6) as the previously tested Apo E-deficient mice. A 14 d prophylactic treatment with fenofibrate drastically (p < 0.05) reduced infarct volume in fenofibrate-treated wild-type mice (31 ± 5 mm3) as compared with placebo-treated wild-type mice (48 ± 4 mm3). As previously, infarct volumes were reduced only in the cortical area and not in the subcortical area (Fig. 1B). Fenofibrate was pharmacologically active as shown by increases in liver weights in treated mice but did not have any significant effect on glycemia and plasma lipid levels (Table 1) or on hemodynamic parameters during ischemia (data not shown).
Because we had shown previously that fenofibrate pretreatment decreases cerebral infarct size in mice, it was logical to test whether acute (and not preventive) treatment of mice with fenofibrate also reduces infarct size in this cerebral ischemia–reperfusion model. There was no mortality in the three tested groups. In the absence of pretreatment, two successive acute intraperitoneal administrations of fenofibrate (50 or 250 mg/kg) to C57BL/6 wild-type mice 1 and 6 hr after starting a 60 min middle cerebral artery occlusion did not induce any decrease in cerebral infarct volume (wild-type: 48 ± 4 mm3; fenofibrate 100 mg/kg: 45 ± 5 mm3; fenofibrate 500 mg/kg: 52 ± 2 mm3; NS).
Fenofibrate crosses the blood–brain barrier slowly
It could be suggested that the absence of any neuroprotective effect of acute administration of fenofibrate could be dependent on a poor capacity of its active metabolite (fenofibric acid) to cross the blood–brain barrier. In a cell culture model of blood–brain barrier consisting of a coculture of bovine brain capillary endothelial cells and rat astrocytes (Dehouck et al., 1990), the permeability coefficient of fenofibric acid (0.68 × 10-3 cm/min) was in the range of the permeability coefficient of sucrose (0.70 × 10-3 cm/min), a weak permeant molecule, indicating that fenofibric acid crosses the blood–brain barrier at a very slow rate (data not shown).
PPAR-α activation as a mechanism of fenofibrate-induced preventive neuroprotection
To evaluate the role of PPAR-α activation in preventive neuroprotection induced by fenofibrate, we compared its effect on brain infarct size in PPAR-α-deficient mice with its effect on wild-type mice positive for PPAR-α and carrying a genetic background similar to that of PPAR-α-deficient mice. PPAR-α -/- mice were created in animals carrying the SV129 genetic background (Lee et al., 1995; Delerive et al., 1999) but not in those carrying the C57BL/6 genetic background, as in our previous experiments. Because differences in susceptibility to middle cerebral artery occlusion have been reported between different types of mouse strains (Connolly et al., 1996), we adapted the middle cerebral artery occlusion model to wild-type SV129 mice. We chose a 90 min MCA occlusion because a 60 min duration did not induce reproducible cerebral infarct in SV129 mice.
Such a 90 min MCA occlusion did not induce mortality in either PPAR-α +/+ mice or PPAR-α -/- mice. Total cerebral infarct volumes were similar between untreated PPAR-α +/+ (66 ± 5 mm3) and PPAR-α -/- mice (65 ± 1 mm3) as were infarct volumes in cortical and striatal areas (Fig. 1C). Chronic fenofibrate pretreatment reduced total cerebral infarct volumes in PPAR-α +/+ mice (fenofibrate, 45 ± 5 mm3 versus placebo, 66 ± 5 mm3; p < 0.01). As previously, this beneficial effect of fenofibrate in PPAR-α +/+ mice was observed in cortical areas but not in subcortical areas (Fig. 1C). In contrast, in PPAR-α -/- mice, fenofibrate pretreatment had no effect on total infarct volume (fenofibrate, 65 ± 5 mm3 versus placebo, 65 ± 1 mm3; NS) or on cortical and subcortical infarct volume (Fig. 1C). Fenofibrate increased liver weights only in PPAR-α +/+ mice (Table 2).
Table 2.
Weight and metabolic variables in PPAR-α+/+ and −/− mice
|
Mice type |
Mice weight (g) |
Livers weight (g) |
Total cholesterol (mg/dl) |
Triglycerides (mg/dl) |
|---|---|---|---|---|
| PPAR-α +/+ | 25 ± 1 | 1.0 ± 0.1 | 97 ± 12 | 77 ± 20 |
| PPAR-α+/+ and F | 26 ± 1 | 2.7 ± 0.2 # | 92 ± 17 | 83 ± 18 |
| PPAR-α −/− | 27 ± 2 | 1.2 ± 0.1 | 129 ± 8 | 98 ± 31 |
| PPAR-α −/− and F
|
30 ± 1 *
|
1.3 ± 0.1
|
123 ± 4
|
122 ± 27
|
p<0.05 versus PPAR-α−/− untreated mice.
p<0.0001 versus PPAR-α +/+ untreated mice. F, Fenofibrate.
Chronic fenofibrate treatment improves sensitivity to endothelial relaxation but does not upregulate NOS III expression
rCBF measured by laser Doppler flow was not different during the ischemia and reperfusion period between mice treated with fenofibrate or placebo (Fig. 2A). Western blot analysis also showed that NOS III protein expression was not modified in brain homogenates from fenofibrate-treated animals (Fig. 2B). Chronic administration of fenofibrate induces in rat an increase in the sensitivity to acetylcholine-induced endothelium-dependent relaxation, without a change in maximal response as compared with untreated rats (Fig. 2C). Conversely, endothelium-independent relaxation to the NO donor SNP (3 × 10-5 M) was not affected, as shown by the lack of difference in the increase in diameter from the preconstricted level between placebo (40 ± 13%) and fenofibrate (38 ± 17%). Moreover, the contractile response of MCA to 5-HT (10-6 M) was not different in treated and untreated animals (32 ± 8 vs 35 ± 3%).
Figure 2.

Fenofibrate induces a slight improvement of MCA endothelial vasodilatation without any change in regional cerebral blood flow or NOS III expression. A, In mice treated with fenofibrate, there is no modification of regional cerebral blood flow (rCBF) as evaluated by laser Doppler (n = 5 in each group). NS, Not significant. B, NOS III is not overexpressed in brain of fenofibrate-treated mice. NS, Not significant. C, Application of low concentrations (3 × 10-9 to 3 × 10-8m) but not high concentrations (10-7 to 3 × 10-5m) of ACh on isolated MCA from rats treated with fenofibrate induces a slight improved sensitivity of endothelium-dependent relaxation as measured by the increase in artery diameter from 5-HT preconstricted level (n = 5 in each group). *p < 0.05.
Fenofibrate regulates brain oxidative stress in parallel to preventive neuroprotection
To study one of the different molecular mechanisms that may explain how preventive PPAR-α activation induces a neuroprotective effect, we decided to study whether PPAR-α activation could decrease the chronic oxidative stress occurring in Apo E-deficient mice that forms the basis of the susceptibility to provoked stroke in this strain of mice (Laskowitz et al., 1997; Ramassamy et al., 2001). We confirmed that the TBARS level, a marker of oxidative stress, was significantly higher in the cortex of Apo E-deficient mice treated with placebo than in wild-type mice in similar conditions (Fig. 3A). In contrast, the striatal level of TBARS was unchanged in Apo E-deficient mice as compared with wild-type mice (Fig. 3B). A 14 d treatment with fenofibrate decreased TBARS concentration in the cortex of Apo E-deficient mice to levels similar to those measured in placebo-treated wild-type mice (Fig. 3A). There was no effect on TBARS concentration in the striatum (Fig. 3B), which agrees with the lack of any neuroprotective effect of fenofibrate in this brain area.
Figure 3.

Chronic administration of fenofibrate modulates oxidative stress and antioxidant enzyme activities in the brain. A, B, Thiobarbituric acid-reactive substances (TBARS) were measured in the cortex (A) and striatum (B) of fenofibrate-treated Apo E-deficient mice and untreated C57BL/6 wild-type mice and then compared with the TBARS level in untreated Apo E-deficient mice (n = 5 per group). It appears that in Apo E-deficient mice, the basal level of TBARS is increased in the cortical area, whereas this oxidative stress marker is normalized by 14 d fenofibrate pretreatment. Moreover, there is no TBARS level change in the striatum. **p < 0.001; #p < 0.05. C, D, Antioxidant enzyme activities were measured in brain homogenates of C57BL/6 wild-type mice after a 14 d period under fenofibrate (n = 5) or placebo (n = 7). Four of the main antioxidant enzymes, copper/zinc superoxide dismutase (SOD), glutathione peroxidase (GPX), glutathione reductase (GR), and glutathione S-transferase (GST), are significantly increased in fenofibrate-treated mice, whereas manganese SOD and catalase are not. *p < 0.05; **p < 0.01. E, Conversely to the measure of SOD activity, Western blot analysis does not show any difference in the expression of this enzyme in placebo and fenofibrate-treated animals as compared with control (EGF-stimulated A431 cells). Densitometric values from placebo and fenofibrate-treated animals were not different. NS, Not significant.
To further explore the molecular mechanism involved in the prophylactic activity of fenofibrate, the activity of antioxidant enzymes and the expression of copper/zinc superoxide dismutase were studied. Copper/zinc superoxide dismutase, glutathione reductase, glutathione peroxidase, and glutathione S-transferase activities were significantly increased in the brain of C57BL/6 wild-type mice treated with fenofibrate for 14 d as compared with placebo-treated mice (Fig. 3C,D). In contrast, manganese superoxide dismutase and catalase remained unchanged after 14 d fenofibrate treatment (Fig. 3C); there was no increase in copper/zinc superoxide dismutase expression on Western blot analysis (Fig. 3E) as well.
Fenofibrate decreases VCAM-1 and ICAM-1 expression in ischemic brain
To study the impact of PPAR-α activation on inflammatory pathways during cerebral ischemia, we decided to evaluate whether a 14 d treatment with fenofibrate could modulate VCAM-1 and ICAM-1 expression occurring in ischemic brain (Wang et al., 1994; Jander et al., 1996). We did not find any VCAM-1 or ICAM-1 expression in brain homogenates from sham animals (Fig. 4A,B), whereas these proteins were induced in the brain of animals submitted to a 1 hr ischemia period (Fig. 4C,D). In these conditions, fenofibrate significantly decreased ischemia-induced VCAM-1 and ICAM-1 expression (Fig. 4E,F). Such a result was confirmed by Western blot analysis, in which the expression of VCAM-1 and ICAM-1 was significantly reduced in fenofibrate-treated mice (Fig. 5A,B).
Figure 4.
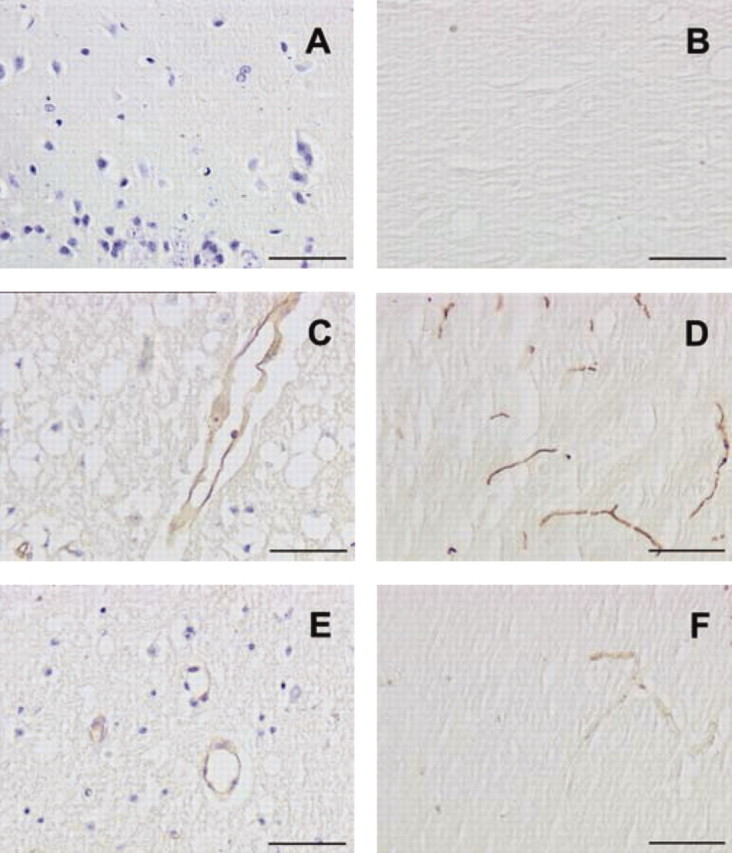
Chronic administration of fenofibrate prevents ischemia-induced vascular expression of VCAM-1 and ICAM-1. VCAM-1 and ICAM-1 expression were evaluated in sham or mice subjected to a cerebral ischemia after a 14 d treatment period with or without fenofibrate. The intensity of staining was measured as a mean of five animals in each group. A, B, No expression is shown of VCAM-1 (A; cresyl violet counterstaining) or ICAM-1 (B; without counterstaining) in the brain of sham animals. C, D, VCAM-1 (C) and ICAM-1 (D) are induced after a 1 hr ischemia after a 24 hr reperfusion period in untreated animals in which such expression involves mainly vessels in the ischemic area. E, F, Adhesion molecule expression is prevented in ischemic mice receiving the fenofibrate treatment. Magnification: 20×. Scale bars, 50 μm.
Figure 5.

A 14 d fenofibrate treatment prevents ischemia-induced VCAM-1 and ICAM-1 but not NOS II overexpression. Western blot analysis was performed in brain homogenates of placebo and fenofibrate-treated mice and then compared with control animals injected with lipopolysaccharide (1.5 mg/kg, 24 hr before). A, B, Representative blots as well as densitometric values show a decrease in ischemia-induced VCAM-1 (A) and ICAM-1 (B). *p < 0.05; **p < 0.01. C, Conversely, ischemia-induced NOS II overexpression was not modified by fenofibrate pretreatement. NS, Not significant.
Fenofibrate does not modify the expression of NOS II in the ischemic brain
To complete the study of inflammatory pathways and putative proteins involved in fenofibrate preventive neuroprotection, we analyzed NOS II expression in the ischemic brain of fenofibrate-treated and untreated mice. Western blot analysis realized on brain homogenates 24 hr after the brain ischemia–reperfusion procedure did not show any modification of NOS II expression in fenofibrate-treated animals (Fig. 5C).
Discussion
The major finding of this study is that preventive treatment with a PPAR-α activator protects the brain against the deleterious consequences of focal cerebral ischemia. The neuroprotective effect of fenofibrate is predominant in the cortical brain area, as is usual with potentially protective agents administered during stroke (Lee et al., 2000). This neuroprotective effect is independent of the well documented lipid-lowering effects of PPAR-α activators reported in various mammals (Fruchart et al., 1999), because in hypercholesterolemic Apo E-deficient mice, fenofibrate suppresses susceptibility to stroke, although it increases total cholesterol plasma level by increasing atherogenic cholesterol (VLDL + IDL + LDL cholesterol) and decreasing anti-atherogenic cholesterol (HDL cholesterol) as previously reported in such a model (Duez et al., 2002). Apo E-deficient mice had higher glycemia than wild-type mice, which might increase their susceptibility to stroke because hyperglycemia is an aggravating factor of ischemic injury (Kawai et al., 1997). The normalization of this hyperglycemia by fenofibrate might contribute to its neuroprotective effect observed in Apo E-deficient mice. However, the other two tested strains (C57BL/6 and SV129) are normolipidemic and normoglycemic, and fenofibrate also exerted a neuroprotective effect, without affecting plasma lipids or glycemia, suggesting that the neuroprotective effect of fenofibrate is independent of any effects on glucose and lipid metabolism.
In addition, we provide some arguments about the molecular target activated by fenofibrate to protect the brain against the consequences of ischemia. Previous data had already suggested that bezafibrate could decrease mortality after experimental cerebral ischemia in gerbils, in part through a decrease in anaerobic metabolism in ischemic tissue (Aspey et al., 1989). However, PPARs had not been discovered when this experiment was performed, but it has now been demonstrated that bezafibrate activates both PPAR-α and PPAR-γ (Willson et al., 2000; Inoue et al., 2001). Because fibrates have previously been reported to exert their pleiotropic pharmacological effects by activating PPAR-α, we hypothesized that the fenofibrate neuroprotective effect was the result of this nuclear factor activation (Fruchart et al., 1999). Despite the ability of fenofibrate to activate both PPAR-α and PPAR-γ and recent evidence that PPAR-γ activation could induce a neuroprotection (Uryu et al., 2002), our study strongly argues in favor of PPAR-α activation as a mechanism of the neuroprotective effect of fenofibrate because fenofibrate did not absolutely reduce infarct size in PPAR-α-deficient mice. Moreover, PPAR-α deficiency did not induce any increase in susceptibility to cerebral ischemia when animals were treated with placebo, suggesting that the basal activity of PPAR-α is not involved in endogenous neuroprotection against cerebral ischemia. Therefore, chronic PPAR-α activation might be a new pharmacological procedure to induce preventive neuroprotection. We show here that PPAR-α-induced neuroprotection is associated with the modulation of vascular function as well as oxidative stress and inflammatory pathways.
In our study, the occlusion-induced decrease in rCBF was comparable between the two groups, suggesting that the neuroprotective effect is not explained by differences in rCBF in treated and untreated mice. Nevertheless, we have shown that chronic treatment with fenofibrate induces a slight increase in endothelial vasodilatation of MCA from treated rats. Such a beneficial effect has been discussed recently during myocardial ischemia (Malik et al., 2001; Tabernero et al., 2002), in which fenofibrate treatment was able to enhance endothelial vasodilatation without modifying coronary blood flow (Tabernero et al., 2002). Moreover, the absence of NOS III overexpression after fenofibrate treatment suggests that the enhanced endothelium-dependent relaxation cannot be attributed to an increased activity of endothelial NOS. The lack of a significant difference in the vascular effect of the NO donor SNP suggests that sensibility to the transduction system of the NO pathway is not increased in fenofibrate-treated rats. An increase in sensitivity of MCA to ACh could be the result of a better bioavailability of NO related to an increase in antioxidant capacity (Tabernero et al., 2002).
Indeed, we have demonstrated that chronic treatment with a PPAR-α activator induces an increase in activity of the major antioxidant enzymes in the brain, in particular copper/zinc superoxide dismutase and enzymes involved in glutathione metabolism, without affecting the activity of other antioxidant enzymes such as catalase. Despite the presence of a peroxisome proliferator-responsive element in the promoter of the copper/zinc superoxide dismutase gene (Yoo et al., 1999), we did not demonstrate any increase in superoxide dismutase expression, suggesting a non-transcriptional underlying mechanism. Increases in antioxidant enzyme activities in the brains of fenofibrate-treated mice might contribute to the capacity of this molecule to protect against the deleterious consequences of cerebral ischemia. Moreover, fenofibrate decreases susceptibility to stroke in Apo E-deficient mice partly by inhibiting chronic high oxidative stress levels in the brains of these animals, because fenofibrate normalizes the TBARS level in the brains of Apo E-deficient mice.
Another hypothesis is that chronic PPAR-α activation prevents cerebral ischemia-induced expression of the adhesion proteins (Delerive et al., 1999; Marx et al., 1999). Indeed, as a consequence of oxidative stress and cytokine release (Chan, 2001), adhesion proteins such as VCAM-1 or ICAM-1 are overexpressed during cerebral ischemia and may play an important role in the inflammatory response in this condition (Wang et al., 1994; Jander et al., 1996). We have shown that chronic treatment with fenofibrate decreases ischemia-induced VCAM-1 expression in brain vessels in accordance with previous data showing that PPAR-α activation inhibits VCAM-1 expression in endothelial cells through the inhibition of NF-κB (Marx et al., 1999). We also show a decrease in ischemia-induced ICAM-1 expression in brain. Although the modulation of ICAM-1 by fenofibrate remains controversial (Marx et al., 1999), recent data indicate that the expression of such an adhesion molecule could be modulated through PPAR-α activation in a myocardial ischemia model (Wayman et al., 2002). In the brain, the decreased expression of these adhesion molecules in turn should inhibit the infiltration of the brain ischemic area by polynuclear neutrophils and the induction of NOS II and COX-2, which contribute to the extent of the ischemic lesions (Lee et al., 2000; Chan, 2001). Nevertheless, our data suggest that the NOS II pathway is not involved in the neuroprotective effect of fenofibrate. This result suggests that fenofibrate exerts its main effect on cerebral vessels in accordance with the poor blood–brain barrier crossing.
Experimental data concerning fenofibrate-induced PPAR-α activation support the hypothesis that the brain can be rendered pharmacologically and preventively resistant to cerebral ischemia. The other class of lipid-lowering drugs, the hydroxymethyl-glutaryl-CoA reductase inhibitors (statins) can also preventively reduce cerebral infarct size and improve neurological functioning in normocholesterolemic mice, via upregulation of NOS III contrasting with the mechanism of fenofibrate-induced neuroprotection (Endres et al., 1998; Amin-Hanjani et al., 2001). Nevertheless, differences in mechanisms involved in the neuroprotective effect of the two classes of lipid-lowering drugs suggest that brain ischemic tolerance could be better mimicked pharmacologically by their association (Chen and Simon, 1997; Vaughan and Delanty, 1999).
In conclusion, we have demonstrated that a PPAR-α activator decreases the deleterious consequences of ischemic stroke, independently of any effect on lipid metabolism, and that this neuroprotective effect could result from PPAR-α activation. Enhanced brain antioxidant enzyme activities and decreased adhesion protein expression are probably two of the mechanisms by which this agent protects against cerebral injury. This preventive neuroprotective property of PPAR-α activators could be useful in addition to primary prevention for patients with a high risk for stroke and to secondary prevention in patients with a previous history of stroke or transient ischemic attack as well as for patients undergoing elective cardiovascular or radiological procedures with high risk for stroke (Fisher et al., 1994). Moreover, evaluation of a potential synergistic protective effect of statins and fibrates could be interesting in regard to their probable complementary properties.
Footnotes
P.G. was supported by a grant from “Conseil Régional Nord Pas de Calais et Centre Hospitalier Régional et Universitaire de Lille. ” This work was supported by grants from “ Génopole Lille Nord-Pas de Calais ”(#01360124) and Institut National de la Santé et de la Recherche Médicale (CReS4CR02F). We thank Alexandra Tavernier-Sommerville for her assistance with the English version of this paper.
Correspondence should be addressed to R. Bordet, Laboratoire de Pharmacologie, Université de Lille 2, Faculté de Médecine, 1 Place de Verdun, Lille, 59045 France. E-mail: bordet@univ-lille2.fr.
Copyright © 2003 Society for Neuroscience 0270-6474/03/236264-08$15.00/0
D.D. and P.G. participated equally in this study.
References
- Amarenco P ( 2001) Hypercholesterolemia, lipid-lowering agents, and the risk for brain infarction. Neurology 57[Suppl 2]: S35–44. [DOI] [PubMed] [Google Scholar]
- Amin-Hanjani S, Stagliano NE, Yamada M, Huang PL, Liao JK, Moskowitz MA ( 2001) Mevastatin, an HMG-CoA reductase inhibitor, reduces stroke damage and upregulates endothelial nitric oxide synthase in mice. Stroke 32: 980–986. [DOI] [PubMed] [Google Scholar]
- Aspey BS, Ehteshami S, Hurst CM, Pereira S, Harrison MJ ( 1989) Effect of bezafibrate on the sequelae of acute experimental focal cerebral ischaemia. J Neurol Neurosurg Psychiatry 52: 1432–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastide M, Bordet R, Pu Q, Robin E, Puisieux F, Dupuis B ( 1999) Relationship between inward rectifier potassium current impairment and brain injury after cerebral ischemia/reperfusion. J Cereb Blood Flow Metab 19: 1309–1315. [DOI] [PubMed] [Google Scholar]
- Bastide M, Gelé P,Pétrault O, Pu Q, Caliez A, Robin E, Deplanque D, Duriez P, Bordet R. ( 2003) Delayed cerebrovascular protective effect of lipopolysaccharide in parallel to brain ischemic tolerance. J Cereb Blood Flow Metab 23: 399–405. [DOI] [PubMed] [Google Scholar]
- Bishop-Bailey D ( 2000) Peroxisome proliferator-activated receptors in the cardiovascular system. Br J Pharmacol 129: 823–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloomfield Rubins H, Davenport J, Babikian V, Brass LM, Collins D, Wexler L, Wagner S, Papademetriou V, Rutan G, Robins SJ ( 2001) Reduction in stroke with gemfibrozil in men with coronary heart disease and low HDL cholesterol: the Veterans Affairs HDL Intervention Trial. VA-HIT Study Group. Circulation 103: 2828–2833. [DOI] [PubMed] [Google Scholar]
- Bordet R, Deplanque D, Maboudou P, Puisieux F, Pu Q, Robin E, Martin A, Bastide M, Leys D, Lhermitte M, Dupuis B ( 2000) Increase in endogenous brain superoxide dismutase as a potential mechanism of lipopolysaccharide-induced brain ischemic tolerance. J Cereb Blood Flow Metab 20: 1190–1196. [DOI] [PubMed] [Google Scholar]
- Chan PH ( 2001) Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab 21: 2–14. [DOI] [PubMed] [Google Scholar]
- Chen J, Simon R ( 1997) Ischemic tolerance in the rat brain. Neurology 48: 306–311. [DOI] [PubMed] [Google Scholar]
- Connolly ES, Winfree CJ, Stern DM, Solomon RA, Pinsky DJ ( 1996) Procedural and strain-related variables significantly affect outcome in a murine model of focal cerebral ischemia. Neurosurgery 38: 523–532. [DOI] [PubMed] [Google Scholar]
- Cullingford TE, Bhakoo K, Peuchen S, Dolphin CT, Patel R, Clark JB ( 1998) Distribution of mRNAs encoding the peroxisome proliferator-activated receptor alpha, beta, and gamma and the retinoid X receptor alpha, beta, and gamma in rat central nervous system. J Neurochem 70: 1366–1375. [DOI] [PubMed] [Google Scholar]
- Dehouck MP, Meresse S, Delorme P, Fruchart JC, Cecchelli R ( 1990) An easier, reproducible and mass-production method to study the blood-brain barrier in vitro. J Neurochem 54: 1798–1801. [DOI] [PubMed] [Google Scholar]
- Delerive P, De Bosscher K, Besnard S, Berghe WV, Peters JM, Gonzalez FJ, Fruchart JC, Tedgui A, Haegeman G, Staels B ( 1999) PPARalpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-κB and AP-1. J Biol Chem 274: 32048–32054. [DOI] [PubMed] [Google Scholar]
- Deplanque D, Bastide M, Bordet R ( 2000) Ischaemic preconditioning of the endothelium and smooth muscle of cerebral arteries. Trends Pharmacol Sci 21: 332–333. [DOI] [PubMed] [Google Scholar]
- Duez H, Chao YS, Hernandez M, Torpier G, Poulain P, Mundt S, Mallat Z, Teissier E, Burton CA, Tedgui A, Fruchart JC, Fievet C, Wright SD, Staels B ( 2002) Reduction of atherosclerosis by the peroxisome proliferator-activated receptor alpha agonist fenofibrate in mice. J Biol Chem 277: 48051–48057. [DOI] [PubMed] [Google Scholar]
- Endres M, Laufs U, Huang Z, Nakamura T, Huang P, Moskowitz MA, Liao JK ( 1998) Stroke protection by 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase inhibitors mediated by endothelial nitric oxide synthase. Proc Natl Acad Sci USA 95: 8880–8885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher M, Jones S, Sacco RL ( 1994) Prophylactic neuroprotection for cerebral ischemia. Stroke 25: 1075–1080. [DOI] [PubMed] [Google Scholar]
- Fruchart JC, Duriez P, Staels B ( 1999) Peroxisome proliferator-activated receptor alpha activators regulate genes governing lipoprotein metabolism, vascular inflammation and atherosclerosis. Curr Opin Lipidol 10: 245–257. [DOI] [PubMed] [Google Scholar]
- Goldberg DM, Spooner RJ ( 1983) Glutathione reductase in methods in enzymatic analysis (Bermeyer HU, ed) pp 258–265. New York: Academic.
- Habig WH, Pabst MJ, Jakoby WB ( 1974) Glutathione S-transferase, the first enzymatic step in mercapturic acid formation. J Biol Chem 249: 7130–7139. [PubMed] [Google Scholar]
- Hankey GJ, Warlow CP ( 1999) Treatment and secondary prevention of stroke: evidence, costs, and effects on individuals and populations. Lancet 354: 1457–1463. [DOI] [PubMed] [Google Scholar]
- Inoue I, Goto S, Matsunaga T, Nakajima T, Awata T, Hokari S, Komoda T, Katayama S ( 2001) The ligands/activators for peroxisome proliferator-activated receptor alpha (PPARalpha) and PPARgamma increase Cu 2+, Zn 2+-superoxide dismutase and decrease p22phox message expressions in primary endothelial cells. Metabolism 50: 3–11. [DOI] [PubMed] [Google Scholar]
- Jander S, Pohl J, Gillen C, Schroeter M, Stoll G ( 1996) Vascular cell adhesion molecule-1 mRNA is expressed in immune-mediated and ischemic injury of the rat nervous system. J Neuroimmunol 70: 75–80. [DOI] [PubMed] [Google Scholar]
- Jonas S ( 1995) Prophylactic pharmacologic neuroprotection against focal cerebral ischemia. Ann NY Acad Sci 765: 21–25. [DOI] [PubMed] [Google Scholar]
- Kainu T, Wikström AC, Gustafsson JA, Pelto-Huikko M ( 1994) Localization of the peroxisome proliferator-activated receptor in the brain. NeuroReport 5: 2481–2485. [DOI] [PubMed] [Google Scholar]
- Kawai N, Keep RF, Betz AL ( 1997) Hyperglycemia and the vascular effects of cerebral ischemia. Stroke 28: 149–154. [DOI] [PubMed] [Google Scholar]
- Laskowitz DT, Sheng H, Bart RD, Joyner KA, Roses AD, Warner DS ( 1997) Apolipoprotein E-deficient mice have increased susceptibility to focal cerebral ischemia. J Cereb Blood Flow Metab 17: 753–758. [DOI] [PubMed] [Google Scholar]
- Lee JM, Grabb MC, Zipfel GJ, Choi DW ( 2000) Brain tissue responses to ischemia. J Clin Invest 106: 723–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SS, Pineau T, Drago J, Lee EJ, Owens JW, Kroetz DL, Fernandez-Salguero PM, Westphal H, Gonzalez FZ ( 1995) Targeted disruption of the alpha isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol Cell Biol 15: 3012–3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik J, Melenovsky V, Wichterle D, Haas T, Simek J, Ceska R, Hradec J ( 2001) Both fenofibrate and atorvastatin improve vascular reactivity in combined hyperlipidaemia (fenofibrate versus atorvastatin trial–FAT). Cardiovasc Res 52: 290–298. [DOI] [PubMed] [Google Scholar]
- Marx N, Sukhova GK, Collins T, Libby P, Plutzky J ( 1999) PPARalpha activators inhibit cytokine-induced vascular cell adhesion molecule-1 expression in human endothelial cells. Circulation 99: 3125–3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paglia DE, Valentine WN ( 1967) Studies on the quantitative characterization of erythrocyte glutathione peroxidase. J Lab Clin Med 70: 158–169. [PubMed] [Google Scholar]
- Peters JM, Hennuyer N, Staels B, Fruchart JC, Fievet C, Gonzalez FZ, Auwerx J ( 1997) Alterations in lipoprotein metabolism in peroxisome proliferator-activated receptor alpha-deficient mice. J Biol Chem 272: 27307–27312. [DOI] [PubMed] [Google Scholar]
- Poynter ME, Daynes RA ( 1998) Peroxisome proliferator-activated receptor α activation modulates cellular redox status, represses nuclear factor-κB signaling and reduces inflammatory cytokine production in aging. J Biol Chem 273: 32833–32841. [DOI] [PubMed] [Google Scholar]
- Puisieux F, Deplanque D, Pu Q, Souil E, Bastide M, Bordet R ( 2000) Differential role of nitric oxide pathway and heat shock protein in preconditioning and lipopolysaccharide-induced brain ischemic tolerance. Eur J Pharmacol 389: 71–78. [DOI] [PubMed] [Google Scholar]
- Ramassamy C, Krzywkowski P, Averill D, Lussier-Cacan S, Theroux L, Christen Y, Davignon J, Poirier J ( 2001) Impact of Apo E deficiency on oxidative insults and antioxidant levels in the brain. Mol Brain Res 86: 76–83. [DOI] [PubMed] [Google Scholar]
- Staels B, Koenig W, Habib A, Merval R, Lebret M, Torra IP, Delerive P, Fadel A, Chinetti G, Fruchart JC, Najib J, Maclouf J, Tedgui A ( 1998) Activation of human aortic smooth-muscle cells is inhibited by PPARalpha but not by PPARgamma activators. Nature 393: 790–793. [DOI] [PubMed] [Google Scholar]
- Tabernero A, Schoonjans K, Jesel L, Carpusca I, Auwerx J, Andriantsitohaina R ( 2002) Activation of the peroxisome proliferator-activated receptor α protects against myocardial ischaemic injury and improves endothelial vasodilatation. BMC Pharmacol 2: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uryu S, Harada J, Hisamoto M, Oda T ( 2002) Troglitazone inhibits both post-glutamate neurotoxicity and low-potassium-induced apoptosis in cerebellar granule neurons. Brain Res 924: 229–236. [DOI] [PubMed] [Google Scholar]
- Vaughan CJ, Delanty N ( 1999) Neuroprotective properties of statins in cerebral ischemia and stroke. Stroke 30: 1969–1973. [DOI] [PubMed] [Google Scholar]
- Wang X, Siren AL, Liu Y, Yue TL, Barone FC, Feueurstein GZ ( 1994) Upregulation of intercellular adhesion molecule-1 (ICAM-1) on brain microvascular endothelial cells in rat ischemic cortex. Mol Brain Res 26: 61–68. [DOI] [PubMed] [Google Scholar]
- Wayman NS, Hattori Y, McDonald MC, Mota-Filipe H, Cuzzocrea S, Pisano B, Chatterjee PK, Thiemermann C ( 2002) Ligands of the peroxisome proliferator-activated receptors (PPAR-gamma and PPAR-alpha) reduce myocardial infarct size. FASEB J 16: 1027–1040. [DOI] [PubMed] [Google Scholar]
- Willson TM, Brown PJ, Sternbach DD, Henke BR ( 2000) The PPARs: from orphan receptors to drug discovery. J Med Chem 43: 527–550. [DOI] [PubMed] [Google Scholar]
- Yoo HY, Chang MS, Rho HM ( 1999) Induction of the rat Cu/Zn superoxide dismutase gene through the peroxisome proliferator-responsive element by arachidonic acid. Gene 234: 87–91. [DOI] [PubMed] [Google Scholar]