

Abstract
We discovered a novel alternatively spliced form of synaptotagmin I (Syt I). This splicing event is conserved over evolution and, in Aplysia, results in a two amino acid insert in the juxtamembrane domain of Syt I (Syt IVQ). Both Syt I and Syt IVQ are localized to synaptic vesicles; however, we also observed punctae that contained one or the other spliced products. Both Syt I and Syt IVQ are phosphorylated at the adjacent PKC site. Overexpression of Syt IVQ, but not of Syt I, in Aplysia neurons blocked the ability of serotonin to reverse synaptic depression. This effect is upstream of PKC activation, because neither Syt IVQ nor Syt I blocked the effects of phorbol esters on reversing synaptic depression or the effects of serotonin on facilitating nondepressed synapses. Our results demonstrate a physiological role for splicing in the juxtamembrane domain of Syt I.
Keywords: synaptotagmin, transmitter release, protein kinase C, PKC, Aplysia, depressed synapses, alternative splicing
Introduction
Synaptotagmins (Syts) are membrane proteins thought to act as calcium sensors during membrane fusion. In particular, Syt I is important for the release of neurotransmitters from synaptic vesicles because loss of Syt I function removes the fast Ca2+-dependent phase of neurotransmitter release (Nonet et al., 1993; DiAntonio and Schwarz, 1994; Geppert et al., 1994). Moreover, as would be expected from a Ca2+ sensor, mutations that affect the Ca2+ sensitivity of Syt I affect the Ca2+ sensitivity of transmitter release (Fernandez-Chacon et al., 2001; Mackler et al., 2002). So far, 13 isoforms of Syts have been identified in mammals (Syt I–Syt XIII), as well as six to eight homologues in Drosophila and Caenorhabditis elegans (for review, see Schiavo et al., 1998; Adolfsen and Littleton, 2001; Sudhof, 2002). Syts comprise a lumenal N terminal, a transmembrane domain, and a short juxtamembrane linker, followed by two C2 domains (C2A and C2B). The function of synaptotagmin in membrane fusion is mediated by protein–protein and protein–lipid interactions of these C2 domains (for review, see Schiavo et al., 1998; Adolfsen and Littleton, 2001).
The juxtamembrane domain of Syt I is well conserved within Syt I-like isoforms (Syt II, Syt IX, and invertebrate Syt Is) but is not conserved in other Syts. This suggests that the juxtamembrane region may play a role in specific functions of Syt I-like isoforms. Indeed, this segment has been proposed to play a role in the specific cellular localization of Syts (Fukuda et al., 2001). The juxtamembrane domain is also the location of all of the well characterized sites for Syt I phosphorylation (Bennett et al., 1993; Davletov et al., 1993; Hilfiker et al., 1999).
Aplysia sensorimotor (SM) neuron synapses show a remarkable synaptic depression to repeated stimulation that is thought to underlie behavioral depression (Byrne and Kandel, 1996). Both depression and the reversal of this depression by serotonin (5-HT) involve regulation of the release machinery (Byrne and Kandel, 1996). While recloning Aplysia Syt I to examine a possible role for Syt I phosphorylation in regulating the reversal of depression, we discovered a novel alternatively spliced form of this protein with two amino acids, V and Q, added in the juxtamembrane domain. This splicing event is conserved in Drosophila and mammalian Syt I. Overexpression of this synaptotagmin spliced product (Syt IVQ), but not of Syt I, blocked 5-HT-mediated reversal of depression. These results reveal the first evidence for the importance of splicing in the juxtamembrane domain of Syt I and suggest additional roles for Syt I in regulating membrane trafficking.
Materials and Methods
Aplysia californica (50–200 gm) were purchased from Marine Specimens Unlimited (Pacific Palisades, CA) or the Aplysia resource facility at the University of Miami (Miami, FL) and kept in an aquarium for at least 3 d before experimentation. Dissections and isolation of tissues and cultured neurons was as described previously (Manseau et al., 2001).
Plasmid construction. We designed exact primers to the cytoplasmic domain of Aplysia Syt I (5′, CGCGAATTCAAGAAGGAGGGCAAGAAAGG; 3′, GCGCCCGGGTTAGTTCTTCTCTGGCA) based on the published sequence including restriction sites, allowing us to insert the PCR product into pGEX-5X-1 vector (Amersham Biosciences, Oakville, Ontario, Canada). The full-length Syt I was amplified by PCR using distinct 5′ primers, again based on the published sequence (CGCGAATTCACCATGGACTCCCTTCTGGCG). These constructs were subsequently excised from pGEX-5X-1 and inserted into enhanced green fluorescent protein (EGFP)-C2 vectors (Clontech, Palo Alto, CA) using EcoRI and SmaI. The EGFP-C2 Syt I clones were then excised by NheI and SmaI and inserted into the Aplysia expression vector pNEX-3 (Manseau et al., 2001) cut with XbaI and SmaI. All pNEX-3 EGFP-Syt I clones were checked by sequencing over their entire length. EGFP was replaced by enhanced cyan fluorescent protein (ECFP) and enhanced yellow fluorescent protein (EYFP) (Clontech) using SphI and BsrGI. The S→ A mutant was generated using the Syt I cytoplasmic domain of Syt IVQ cloned into pGEX-5X-1 or the pNEX-3 EGFP-Syt IVQ in a two-step mutagenic procedure as described previously (Manseau et al., 2001). A new MluI site was formed in Syt IVQ; S-A mutant. All constructs were sequenced over the entire amplified region to confirm that no changes were made. The DsRed VAMP (vesicle-associated membrane protein) was made using primers based on the published Aplysia VAMP sequence (Yamasaki et al., 1994) for insertion into EGFP-C2 and then insertion into pNEX-3. The EGFP was then replaced with DsRed (Clontech) with appropriate enzymes.
Quantitative reverse transcription-PCR. The relative amounts of Syt IVQ and Syt I mRNA was determined by quantitative reverse transcription (RT)-PCR. RNA isolation was performed using the RNAqueous-4PCR kit (Ambion, Austin, TX) according to the protocol provided by the manufacturer. Common forward and reverse primers were used, followed by an RsaI digest (addition of VQ introduces a new RsaI site into Syt I DNA sequence). Mixes of plasmids encoding Syt IVQ and Syt I were used in control reactions as part of each PCR set to generate a standard curve. The PCR products were subjected to RsaI digest and separated on agarose gels, illuminated under UV light, digitally scanned, and quantified using NIH Image.
In vitro phosphorylation assay. Phosphorylation was initiated by the addition of purified protein kinase C (PKC) Apl II (Sossin et al., 1996) to the phosphorylation mix [50 nm 12-O-tetradecanoylphorbol-13-acetate, 5 μg/ml phosphatidylserine, 500 μm CaCl2, 10 μm ATP, 1–3 μCi [γ-32P]ATP, 45 mm MgCl2, 180 mm Tris, pH 7.5, and various amounts of glutathione S-transferase (GST) fusion proteins]. Nonphosphorylated controls were incubated in a control mix (phosphorylation mix without ATP). These reactions were allowed to proceed at 25°C for 30 min and were stopped by the addition of 20 μl of Laemmli buffer and then loaded onto 9% SDS-polyacrylamide gels. After transfer to nitrocellulose, the blots were exposed to film to visualize the incorporation of radioactive phosphate.
Cell culture preparation. Injections of plasmid DNA and physiological paradigms were as described previously (Manseau et al., 2001). An arbitrary scale of fluorescence (from 0 to 5) was established to evaluate the labeling of each sensory neuron (Manseau et al., 2001). Sensory neurons that were positive for plasmid expression (3–5 on the scale) were individually paired with motor neurons (kept aside until then in the refrigerator at 4°C to prevent them from retracting their axons), in a 10% hemolymph-enriched medium. The day after the pairing, the Petri dish solution was exchanged with fresh 10% hemolymph–L-15. Electrophysiological recordings started on the fifth day, 2 d after pairing, to allow the formation of new synaptic contacts and a full maturation of the PKC transduction pathway involved in short-term plasticity (Sun and Schacher, 1996). All recordings were done in L-15 at room temperature (21–24°C) using Axoclamp-2A and Axoprobe-1A amplifiers (Axon Instruments, Foster City, CA) in the current-clamp mode. Membrane resistance of both sensory and motor neurons and the resting potential of motor neurons were measured at the start and at the end of each recording. The resting potential of sensory neurons was not measured until the end of experiment to prevent the generation of unwanted spikes. The major criterion for selection of healthy neurons was a stable resting potential. Throughout the experiments, the sensory neuron was held at -50 mV, and the siphon motor neuron of the LF cluster, which was impaled first, was hyperpolarized to -80 mV. In experiments on short-term facilitation of depressed synapses, a hyperpolarizing current was passed (glass pipette, 10–15 MΩ, filled with 2 m KAc) to prevent spike generation during the sensory neuron impalement. Short intracellular pulses were delivered, and, once the threshold for action potential was reached, the stimulation intensity and interval was kept constant through the experiment. We continued the experiment when the initial EPSP amplitude exceeded 2 mV. The series of EPSPs were evoked every 20 sec in the motor neuron. 5-HT (10 μm final concentration) was added directly to the bath near the cells and mixed gently after 40 EPSPs. Ten additional EPSPs were recorded. In another set of cocultures, 12,13-dibutyrate (PDBu) (100 nm final concentration) was added instead of 5-HT to determine whether the inhibition of facilitation was before or after PKC activation.
Changes in synaptic transmission. EPSPs were always normalized to the size of the initial EPSP. The amount of facilitation was calculated as the difference between EPSPs after treatment (averages of EPSPs 41–43) and EPSPs before treatment (averages of EPSPs 38–40). In experiments on short-term facilitation of rested synapses, a single depolarizing stimulus was applied to the sensory neuron, and the initial EPSP amplitude was recorded. At 2 min, 5-HT was applied to the bath (final concentration of 10 μm), and a second EPSP was recorded 3 min later in the presence of 5-HT. The amount of facilitation was calculated as the difference between EPSP 2 and EPSP 1 (EPSP 1 normalized to 100%). Data were acquired and analyzed digitally using CLAMPEX 7 and a modified version of pCLAMP (Axon Instruments) (Manseau et al., 2001). Experiments comparing the effects of the various constructs were always done in parallel.
Confocal laser microscopy on living cells. The cells were coinjected with constructs tagged with either ECFP or EYFP and were visualized with a Zeiss (Jena, Germany) LSM 510 confocal laser microscope. EYFP and ECFP were chosen because their emission spectra overlap minimally, so they can be distinguished when used simultaneously. For dual imaging of EYFP- and ECFP-injected cells, the cells were excited successively with multi-line argon lasers at 514 and 458 nm, respectively. Images were analyzed using Zeiss LSM 510 software. For EYFP, the cells were light collected through a 530 nm long-pass emission filter, passing by an infrared 480–520 nm bandpass dichroic mirror. For ECFP, the cells were light collected with a 480–520 nm bandpass emission filter. A DsRed-VAMP construct was used as an indicator for synapse localization. In these experiments, the cells were coinjected with DsRed and ECFP Syt I or ECFP Syt IVQ. For Ds-Red, the cells were excited with a helium–neon laser unit at 543 nm and light collected through a 558–583 bandpass filter.
Antibody production and immunoblotting. Antibodies were raised against a GST fusion protein consisting of the cytoplasmic domain of Syt IVQ (nucleotides 279–1284). The antibodies were affinity purified using MBP-Syt IVQ fusion proteins encoding the cytoplasmic domain of Syt I. The MBP Syt I fusion proteins were immobilized on polyvinylidene difluoride membrane, and Syt I antibody was purified in a two-step purification procedure as described previously (Ramjaun et al., 1997).
We also generated a phospho-specific antibody against a peptide sequence [CQLLGNS(p)YKEK] from Aplysia Syt I, with serine 123 converted to a phosphoserine as described previously (Nakhost et al., 1999). Immunoblotting was performed as described previously (Nakhost et al., 1999).
Results
Identification of a novel alternatively spliced form of Aplysia synaptotagmin I
Aplysia Syt I was cloned previously and shares the putative domain structure of all other Syt isoforms (Martin et al., 1995). In the process of recloning the cytoplasmic domain of Aplysia Syt I, we identified an alternatively spliced form. Figure 1A shows the nucleotide alignment of the published Syt I sequence and our initial Syt I clone Syt IVQ in the region of the alternative splice. Our sequence has six additional nucleotides resulting in a VQ insert in the amino acid sequence. Interestingly, the VQ is also seen in the published sequences of squid and Drosophila Syt I (Fig. 1B). Searching the Drosophila Expressed Sequence Tag (EST) database, we found a number of cDNAs that are identical to the published Syt I but specifically lack the VQ residue (Fig. 1B). Although we have not cloned the genomic sequence of Aplysia synaptotagmin, examination of the Drosophila genome sequence reveals that the VQ sequence begins an exon of Syt I and thus suggests that alternative exon entry can explain the alternatively spliced forms. Interestingly, the VQ sequence is also located at the exon start in the Syt I-related Syt IX (also called Syt V) (Craxton et al., 1997). Alternative exon entry has also been described in this exact region of the linker region of rat Syt I in which the amino acids ALK are inserted instead of VQ (Perin et al., 1990) [Fig. 1B, Syt I(ALK)]. Syt I, II, and IX are more closely related to each other than to invertebrate Syt I isoforms, suggesting that they have diverged after the vertebrate–invertebrate separation (Marqueze et al., 2000). The juxtamembrane domain is well conserved in all Syt-I like isoforms (vertebrate and invertebrate) but not in other Syts, such as Syt IV (Fig. 1B). Although Syt I and Syt II are highly conserved in the juxtamembrane domain, it is also striking that, in the region of the VQ insertion, Syt II has a sizable deletion (Fig. 1B). Thus, there is an alternative exon entry site in the linker domain of Syt I that is conserved over evolution. However, the role for this splicing is unknown.
Figure 1.

Cloning of a novel spliced isoform of Syt I. A, Nucleotide sequence of two clones amplified from a nervous system library showing the insertion of six amino acids. Nucleotide numbering is from residues 303–449. B, Alignment of juxtamembrane domain from a number of species highlighting the conservation of this region in Syt Is and the conservation of the spliced forms. Drosophila Ib (Dros Ib) (from EST clones; accession numbers 15484159, 15504610, and 15505802) and Syt IALK (Perin et al., 1990). Syt IX has also been called Syt V in other publications. The black bar represents the site of alternative splicing, and the star represents the site of PKC phosphorylation. C, RT-PCR of Syt I and Syt IVQ demonstrates approximately equal amounts of both splice forms. The insertion of the VQ generates an RsaI site. We used PCR primers flanking the insert for RT-PCR from the Aplysia nervous system. The amplified product was then cut with RsaI to determine the proportion of RNAs with the insert. Different amounts of nervous system template were used to ensure that PCR amplification was in the linear range. Results are shown for two different animals (NS1 and NS2). To generate a standard curve, mixes of plasmids containing different proportions of Syt IVQ and Syt I were used as the template for PCR. D, The proportion of the two RNAs was calculated based on the standard curves. Values are mean ± SEM for four independent RT-PCRs from four individual animals.
Quantitative reverse transcription (RT-PCR) studies from the Aplysia nervous system indicate that mRNAs encoding Syt I and Syt IVQ are present in the nervous system of Aplysia at approximately a 1:1 ratio (Fig. 1C; quantitated in D). Similar results were obtained when RT-PCR was done with RNA isolated from sensory neuron clusters (data not shown). Treatment of sensory clusters with a paradigm that induces long-term facilitation [5 min pulses for five times each of 20 μm 5-HT (Montarolo et al., 1986)] did not alter the ratio of Syt I and Syt IVQ (percentage change in Syt I/Syt IVQ ratio, -4 ± 6%; n = 4).
Syt I and Syt IVQ are both localized to synaptic vesicles
Splicing may effect localization of Syt Is as the juxtamembrane domain has been implicated in the localization of Syts (Fukuda et al., 2001). To test this, we examined colocalization of the expressed Syt Is with the synaptic vesicle protein VAMP/synaptobrevin. We coexpressed DsRed-tagged VAMP and either ECFPSyt IVQ or ECFP-Syt I in sensory neurons and examined their localization after 5 din coculture with motor neurons. FP-tagged VAMP has been used to mark synaptic vesicle pools in many systems (Nonet, 1999; Ahmari et al., 2000), and, in Aplysia sensory neurons, tagged VAMP colocalizes with antibodies to clustered glutamate at sensory-to-motor neuron synapses (data not shown). Both Syt I isoforms are colocalized with VAMP at concentrations of VAMP likely to mark pools of synaptic vesicles (Fig. 2A,B). This is not surprising because both Syt I and Syt IVQ contain the putative AP2 (adaptor protein 2) binding site required for endocytosis into synaptic vesicles (Zhang et al., 1994). However, because the juxtamembrane domain has been postulated to determine sorting from the trans-Golgi network (Fukuda et al., 2001), we examined whether Syt I and Syt IVQ were always localized together. In these experiments, we used colocalization of EYFP-Syt I and ECFP-Syt IVQ or ECFP-Syt I and EYFP-Syt IVQ to control for variations in the intensities of the different fluorescent proteins. Approximately 5% of punctae contained one or the other isoform (Fig. 2C), regardless of the combination of tags, whereas most punctae colocalized (Fig. 2D). Although the number of non-colocalized punctae is small, we did not observe any non-colocalized punctae in control experiments when we examined colocalization of ECFP-Syt I and EYFP-Syt I or ECFP-Syt IVQ and EYFP-Syt IVQ (data not shown). The punctae containing only Syt I or only Syt IVQ may be transport vesicles. The number of these vesicles in processes is probably small compared with the number of synaptic vesicles. The localization of ECFP- and EYFP-tagged proteins in the cell bodies was not possible because of the pigment granules in the cell body that could be seen even in the absence of injections.
Figure 2.

Localization of Syt I and Syt IVQ in sensory neurons. Plasmids encoding DsRed-labeled Aplysia VAMP and ECFP-labeled Syt I (A) or DsRed-labeld Aplysia VAMP and ECFP-labeled Syt IVQ (B) were injected into sensory neurons. Expressing neurons were then paired with motor neurons and visualized 3–5 d later. For five cells expressing Syt I and four cells expressing Syt IVQ, all DsRed VAMP clusters were completely overlapped with ECFP-Syt I (43 clusters) or ECFP-Syt IVQ (14 clusters). C, Plasmids encoding ECFP-labeled Syt I and EYFP-labeled Syt IVQ or ECFP-labeled Syt IVQ and EYFP-labeled Syt I were injected into sensory neurons. Expressing neurons were then paired with motor neurons and visualized 3–5 d later. D, In the majority of images, EYFP Syt IVQ and ECFP Syt I did completely overlap. Scale bars, 20 μm.
Syt I and Syt I VQ are both phosphorylated in vitro by PKC at serine 123
Serine 123 in Aplysia Syt I corresponds to the site phosphorylated by PKC in vertebrate Syt I (Hilfiker et al., 1999) and is located very close to the insertion of VQ (Fig. 1B). To examine whether the insertion of VQ affected PKC phosphorylation, we incubated PKC and PKC activators with GST fusion proteins with the cytoplasmic domain of Syt I, Syt IVQ, or Syt IVQ with serine 123 converted to alanine (Syt IVQ; S-A). Although these results confirm that serine 123 is a conserved PKC site because PKC phosphorylates both Syt I and Syt IVQ, but not Syt IVQ; S-A (Fig. 3A), there were no differences in the in vitro phosphorylation of Syt I and Syt IVQ (Fig. 3A). Further confirming serine 123 as an in vitro PKC site, a phospho-specific antibody raised to the serine 123 site in Aplysia Syt I recognized bacterially expressed Syt I only after phosphorylation by PKC (Fig. 3B).
Figure 3.

Syt I and Syt IVQ are phosphorylated at serine 123 by PKC in vitro. A, GST fusion proteins (0.15, 0.3, and 0.6 μg)encoding the intracellular region of Syt IVQ, Syt IVQ; S-A, or Syt I were incubated with purified PKC Apl II, PKC activators, and radioactive ATP for 30 min at room temperature. The proteins were separated on 9% SDS-PAGE, blotted to nitrocellulose, and then stained with Ponceau to visualize the fusion proteins. The blots were then exposed to radiography to visualize incorporation of ATP. This experiment was repeated three times with similar results. B, GST-Syt IVQ (0.6 μg) was incubated with purified PKCApl II and PKC activators in the presence or absence of radioactive ATP. The proteins were separated on 9% SDS-PAGE, blotted to nitrocellulose, and then probed first with a phospho-peptide-specific antibody raised to the serine 123 site in Syt I. The blot was then stripped and probed with an antibody raised to a peptide from Syt I. The blot was then exposed to radiography to visualize incorporation of ATP (Autorad).
Overexpression of Syt IVQ specifically blocks the facilitation of depressed synapses upstream of PKC activation
We investigated the physiological role(s) of different Syt I isoforms in vivo by overexpressing them in Aplysia sensory neurons. Various plasmids encoding EGFP or EGFP-tagged Syt I constructs (EGFP-Syt I, EGFP-Syt IVQ, EGFP-Syt IS-A, or EGFP-Syt IVQ; S-A) were injected into Aplysia sensory neurons, which were subsequently used to make SM cell cultures. First, we examined the ability of 5-HT to reverse synaptic depression. Synaptic depression was produced by 40 repeated stimulations of the sensory cell. 5-HT (10 μm final concentration) was added to induce PKC-dependent facilitation (Ghirardi et al., 1992). Our electrophysiological results show that, in cells expressing EGFP-Syt I, EGFP-Syt IS-A, or EGFP alone, 5-HT-induced facilitation of depressed SM synapses was normal (Fig. 4 A, B). Injection of plasmids encoding EGFP alone had no effect on transmitter release or the reversal of synaptic depression in Aplysia (Manseau et al., 2001). Conversely, overexpression of EGFP-Syt IVQ blocked the facilitating effect of 5-HT (Fig. 4 A, B). Overexpression of EGFP-Syt IVQ; S-A also blocked the facilitating effect of 5-HT but appeared somewhat less effective than EGFP-Syt IVQ (Fig. 4 A, B). The difference between EGFP-Syt IVQ and EGFP-Syt IVQ; S-A became significant when examined a few stimuli after the 5-HT pulse [amount of facilitation measured using EPSPs (42–44 or 43–45) EGFP-Syt IVQ vs EGFP-Syt IVQ; S-A; p < 0.05; Tukey's post hoc test]. This result is not consistent with Syt IVQ being the PKC substrate that is important for the reversal of synaptic depression, because, in this case, converting the serine to ala-nine should reduce the ability to reverse synaptic depression. Moreover, mutating the serine to alanine in Syt I had no effect on the ability of 5-HT to reverse depression.
Figure 4.
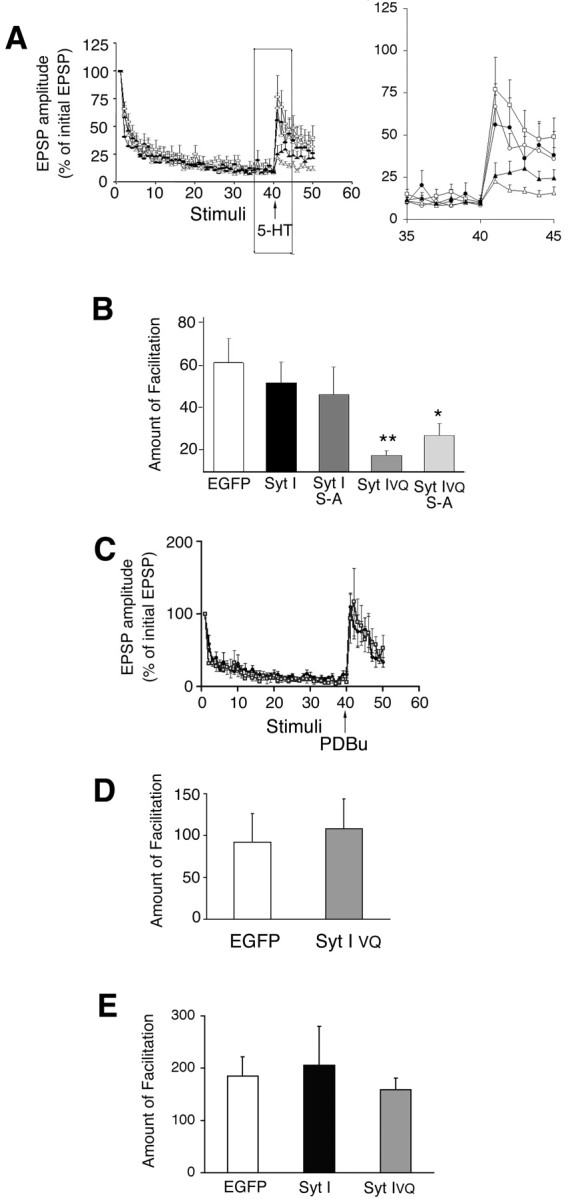
Syt IVQblocks the reversal of synaptic depression at a step upstream of PKC activation. A, Short-term facilitation of depressed synapses is blocked by overexpression of Syt IVQ but not Syt I. Sensory to motor transmission was depressed by a series of 40 repeated intracellular stimuli (inter-stimulus interval, 20sec), and 5-HT (10 μm) was applied to induce synaptic facilitation, followed by an additional 10 stimuli. Averaged normalized EPSPs (mean ± SEM) for cells expressing EGFP (open squares;n = 23), Syt I (open circles;n = 8),Syt IS-A (filled circles;n = 5), Syt IVQ (open triangles;n = 19), or Syt I VQ; S-A (filled triangles; n = 12) are shown. The region around 5-HT addition has been expanded for clarity below. B, The amount of facilitation was calculated as the difference between the average of the three normalized EPSPs after 5-HT and the three normalized EPSPs before 5-HT [ANOVA; p < 0.005; **post hoc Tukey's test showed that Syt IVQ (p < 0.01) and Syt IVQ; S-A (p < 0.05) were significantly different from EGFP]. C, Same as in A, but PDBu (100 nm) was added after the 40th stimuli in cells expressing EGFP (open squares; n = 4) or Syt IVQ (filled triangles; n = 6). D, The amount of facilitation was calculated as the difference between the average of the three normalized EPSPs after PDBu and the three normalized EPSPs before PDBu. There was no significant difference between EGFP- and Syt IVQ-expressing cells (ANOVA;p > 0.5). E, Facilitation of rested SM synapses is unaffected by overexpression of either Syt I or Syt IVQ. An initial EPSP was induced by single extracellular stimulation to the sensory neuron. After 5-HT (10 μm), a second EPSP was recorded. The interstimulus interval between the two EPSPs was 5 min. The EPSP amplitude was normalized to the initial control value. Facilitation was determined by comparing the difference between the two normalized EPSPs [EPSP 2 (after 5-HT) - EPSP 1 (before 5-HT); EGFP, n = 5; Syt I, n = 8; Syt IVQ, n = 6]. No significant differences were observed (ANOVA; p > 0.5).
Overexpression of EGFP-Syt I, EGFP-Syt IVQ, EGFP-Syt I S-A, or EGFP-Syt IVQ; S-A did not affect the resting membrane potential or the rate of synaptic depression (Table 1). Overexpression of EGFP-Syt I showed a trend to lower initial EPSPs, although, because of the large variability in initial EPSPs, this was not significant in an ANOVA (Table 1). The reduction in EPSP size is similar to that seen in a previous study in which Syt I was overexpressed in this system (Martin et al., 1995).
Table 1.
Comparison of intrinsic and synaptic properties of sensory neurons expressing EGFP or various EGFP-Syt I constructs
|
|
Syt IVQ |
Syt I |
Syt IS-A |
Syt IVQ:S-A |
EGFP |
ANOVA |
|---|---|---|---|---|---|---|
| Ihold (pA) | -0.19 ± 0.16 | -0.16 ± 0.07 | -0.19 ± 0.10 | -0.19 ± 0.13 | -0.16 ± 0.15 | NS |
| Rin (MΩ) | 79 ± 35 | 71 ± 30 | 71 ± 29 | 96 ± 45 | 79 ± 31 | NS |
| Vr (mV) | -32 ± 9.0 | -30 ± 7.5 | -40 ± 10 | -32 ± 5.3 | -32 ± 4.8 | NS |
| EGFP levels | 3.2 ± 0.4 | 3.1 ± 0.4 | 3.2 ± 0.3 | 3.3 ± 0.5 | 4.2 ± 0.8 | NS |
| Initial EPSP (mV) | 11.4 ± 9.8 | 6.2 ± 4.0 | 17.1 ± 10.5 | 14.3 ± 10.5 | 10.3 ± 6.3 | NS |
| EPSPs 6-10 (% of initial) | 25.1 ± 10.6 | 30.8 ± 14.9 | 22.2 ± 7.3 | 28.0 ± 12.4 | 26.8 ± 17.0 | NS |
| EPSPs 36-40 (% of initial)
|
14.5 ± 7.9
|
11.6 ± 10.9
|
13.1 ± 9.0
|
10.9 ± 6.8
|
11.1 ± 11
|
NS
|
The holding current (Ihold) is the current needed to hold the sensory neuron (SN) at -50 mV before the first stimulus. Resting potential (Vr) and input resistance (Rin) were taken at the end of each experiment. The values for Vt are therefore underestimated because, at this point, 5-HT or PDBu was present in the bathing solution and the SNs were often spontaneously active when hyperpolarization was removed. EGFP levels were scored from 1 to 5; only cells expressing levels over 3 were used. EPSPs 6-10, Early depression; EPSPs 36-40, late depression; ANOVA, One-way ANOVA; NS, no significance; p>0.05. All values are mean ± SD. Rows 1-5, Syt IVQ, n = 27; Syt I, n = 21; Syt IS-A, n = 5; Syt IVQ: S-A, n = 12; EGFP, n = 30. Rows 6 and 7, Syt IVQ, n = 23; Syt I, n = 13; Syt IS-A, n = 5; Syt IVQ: S-A, n = 12; EGFP, n = 24.
Whereas overexpression of EGFP-Syt IVQ significantly decreased the magnitude of the response to 5-HT in depressed synapses, the kinetics of the remaining effect of 5-HT was normal (Fig. 4A). This is in contrast to what is observed with overexpression of a dominant-negative form of PKC Apl II, in which the onset of facilitation was considerably delayed as might be expected for a true dominant-negative effect (Manseau et al., 2001). This suggested that the blockade was not attributable to a competition between wild-type and overexpressed EGFP-Syt IVQ for PKC phosphorylation, but instead, that 5-HT does not activate PKC as well in the cells expressing EGFP-Syt IVQ. To test whether facilitation downstream of PKC was intact, we examined whether EGFP-Syt IVQ could block the effect of PDBu, which activates PKC independently of 5-HT. Indeed, EGFP-Syt IVQ did not block phorbol ester-mediated reversal of synaptic depression (Fig. 4C,D), suggesting that the deficit lay in the ability of 5-HT to activate PKC. It should be noted that, although PDBu can increase transmitter release independently of PKC in some systems, the effect of PDBu on transmitter release at these synapses is blocked by inhibitors of PKC (Braha et al., 1990).
To determine whether there was a general deficit in 5-HT-mediated signal transduction, we looked at the facilitation of nondepressed synapses. This effect is meditated by 5-HT activation of PKA and not PKC (Ghirardi et al., 1992). In this case, facilitation was not affected by the overexpression of either EGFP-Syt I or EGFP-Syt IVQ (Fig. 4E). This demonstrates that overexpression of EGFP-Syt IVQ specifically interferes with the ability of 5-HT to reverse synaptic depression without generally affecting synaptic transmission or the ability of 5-HT to activate PKA.
Discussion
We found a novel alternative splice form of Syt I with a two amino acid VQ insert in the juxtamembrane region that joins the transmembrane region to C2A. Both Syt I and Syt IVQ are expressed at equal levels. This splicing is evolutionary well conserved and physiologically significant because expression of Syt IVQ, but not Syt I, blocked the reversal of synaptic depression.
A novel but conserved splice form in the juxtamembrane domain of Syt I
The juxtamembrane region between the transmembrane domain and C2A has not been studied extensively. Using antibodies for Syt I and Syt IV, Fukuda et al. (2001) found that these two isoforms were localized to distinct subcellular fractions in PC12 cells. Using chimeras, they showed that the localization signal was in the juxtamembrane region (Fukuda et al., 2001). Whereas invertebrates appear to have only one Syt I-like form, vertebrates have three isoforms, Syt I, II, and IX. Remarkably, although the juxtamembrane region of all three vertebrate Syt I-like isoforms is generally conserved, they are remarkably distinct in the segment in which the VQ splice exists, suggesting that this may be an important distinction between the different Syt I-like vertebrate isoforms. Moreover, the conservation of this splicing event throughout evolution points to an important role for this domain.
Overexpression of Syt IVQ blocks the reversal of depression mediated by 5-HT, but Syt I is not the PKC substrate important for the reversal of depression
The evidence that 5-HT mediates the reversal of synaptic depression through activation of PKC is strongly supported by both pharmacological inhibitors and activators of PKC and dominant-negative experiments (Braha et al., 1990; Ghirardi et al., 1992; Manseau et al., 2001). Syt I is a conserved PKC substrate involved in transmitter release, and the insertion is located quite near the PKC phosphorylation site. Thus, regulation of PKC phosphorylation represented an attractive step at which insertion of VQ could regulate Syt I. However, PKC phosphorylation in vitro does not differentiate between Syt I and Syt IVQ, and Syt IVQ does not block the reversal of synaptic depression mediated by phorbol esters. Moreover, the reversal of synaptic depression is not blocked by Syt IS-A. Thus, the block by Syt IVQ likely acts before PKC activation. For example, this could be attributable to less available 5-HT receptors that could activate PKC. Indeed, it has been suggested that these receptors were variably active early in synapse formation, suggesting that the receptors may be rate limiting in the ability of 5-HT to reverse synaptic depression (Sun and Schacher, 1996).
Interestingly, whereas PKC activity is required for the reversal of synaptic depression, overexpression of active PKC actually inhibited the ability of 5-HT to reverse synaptic depression (Manseau et al., 2001). This inhibition appeared kinetically similar to the block by Syt IVQ and may be attributable to an effect of PKC on regulating the availability of 5-HT receptors (Manseau et al., 2001). Our results would be consistent with Syt IVQ being the PKC substrate involved in this phenomenon because Syt IVQ; S-A showed reduced inhibition compared with Syt IVQ.
In summary, we discovered a well conserved splice in the juxtamembrane region of Syt I. We demonstrated different physiological effects attributable to overexpression of the two distinct products of this splicing. Our results demonstrate an important undiscovered role for the juxtamembrane domain of Syt I.
Footnotes
This work was supported by Natural Sciences and Engineering Research Council of Canada (NSERC) Grant 187018 (W.S.S.) and Canadian Institutes of Health Research (CIHR) Grants MOP-12046 (W.S.S.) and MOP-14142 (V.F.C.). A.N. is supported by an NSERC graduate studentship, and W.S.S. is a Killiam Scholar and supported by a CIHR Scientist award. We thank Peter McPherson, Ted Fon, Luc Desgroseillers, and Louis-Eric Trudeau for helpful comments on this manuscript.
Correspondence should be addressed to Dr. Wayne S. Sossin, Department of Neurology and Neurosurgery, McGill University, Montreal Neurological Institute, BT110, 3801 rue University, Montreal, Quebec, Canada H3A2B4. E-mail: wayne.sossin@mcgill.ca.
Copyright © 2003 Society for Neuroscience 0270-6474/03/236238-07$15.00/0
A.N. and G.H. contributed equally to this work.
References
- Adolfsen B, Littleton JT ( 2001) Genetic and molecular analysis of the synaptotagmin family. Cell Mol Life Sci 58: 393–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmari SE, Buchanan J, Smith SJ ( 2000) Assembly of presynaptic active zones from cytoplasmic transport packets. Nat Neurosci 3: 445–451. [DOI] [PubMed] [Google Scholar]
- Bennett MK, Miller KG, Scheller RH ( 1993) Casein kinase II phosphorylates the synaptic vesicle protein p65. J Neurosci 13: 1701–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braha O, Dale N, Hochner B, Klein M, Abrams TW, Kandel ER ( 1990) Second messengers involved in the two processes of presynaptic facilitation that contribute to sensitization and dishabituation in Aplysia sensory neurons. Proc Natl Acad Sci USA 87: 2040–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne JH, Kandel ER ( 1996) Presynaptic facilitation revisited: state and time dependence. J Neurosci 16: 425–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craxton M, Olsen A, Goedert M ( 1997) Human synaptotagmin V (SYT5): sequence, genomic structure, and chromosomal location. Genomics 42: 165–169. [DOI] [PubMed] [Google Scholar]
- Davletov B, Sontag JM, Hata Y, Petrenko AG, Fykse EM, Jahn R, Sudhof TC ( 1993) Phosphorylation of synaptotagmin I by casein kinase II. J Biol Chem 268: 6816–6822. [PubMed] [Google Scholar]
- DiAntonio A, Schwarz TL ( 1994) The effect on synaptic physiology of synaptotagmin mutations in Drosophila Neuron 12: 909–920. [DOI] [PubMed] [Google Scholar]
- Fernandez-Chacon R, Konigstorfer A, Gerber SH, Garcia J, Matos MF, Stevens CF, Brose N, Rizo J, Rosenmund C, Sudhof TC ( 2001) Synaptotagmin I functions as a calcium regulator of release probability. Nature 410: 41–49. [DOI] [PubMed] [Google Scholar]
- Fukuda M, Ibata K, Mikoshiba K ( 2001) A unique spacer domain of synaptotagmin IV is essential for Golgi localization. J Neurochem 77: 730–740. [DOI] [PubMed] [Google Scholar]
- Geppert M, Goda Y, Hammer RE, Li C, Rosahl TW, Stevens CF, Sudhof TC ( 1994) Synaptotagmin I: a major Ca 2+ sensor for transmitter release at a central synapse. Cell 79: 717–727. [DOI] [PubMed] [Google Scholar]
- Ghirardi M, Braha O, Hochner B, Montarolo PG, Kandel ER, Dale N ( 1992) Roles of PKA and PKC in facilitation of evoked and spontaneous transmitter release at depressed and nondepressed synapses in Aplysia sensory neurons. Neuron 9: 479–489. [DOI] [PubMed] [Google Scholar]
- Hilfiker S, Pieribone VA, Nordstedt C, Greengard P, Czernik AJ ( 1999) Regulation of synaptotagmin I phosphorylation by multiple protein kinases. J Neurochem 73: 921–932. [DOI] [PubMed] [Google Scholar]
- Mackler JM, Drummond JA, Loewen CA, Robinson IM, Reist NE ( 2002) The C2BCa 2+-binding motif of synaptotagmin is required for synaptic transmission in vivo. Nature 418: 340–344. [DOI] [PubMed] [Google Scholar]
- Manseau F, Fan X, Hueftlein T, Sossin WS, Castellucci VF ( 2001) Ca 2+-independent PKC Apl II mediates the serotonin induced facilitation at depressed synapses in Aplysia J Neurosci 21: 1247–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marqueze B, Berton F, Seagar M ( 2000) Synaptotagmins in membrane traffic: which vesicles do the tagmins tag? Biochimie 82: 409–420. [DOI] [PubMed] [Google Scholar]
- Martin KC, Hu Y, Armitage BA, Siegelbaum SA, Kandel ER, Kaang BK ( 1995) Evidence for synaptotagmin as an inhibitory clamp on synaptic vesicle release in Aplysia neurons. Proc Natl Acad Sci USA 92: 11307–11311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montarolo PG, Goelet P, Castellucci VF, Morgan J, Kandel ER, Schacher S ( 1986) A critical period for macromolecular synthesis in long-term heterosynaptic facilitation in Aplysia Science 234: 1249–1254. [DOI] [PubMed] [Google Scholar]
- Nakhost A, Dyer JR, Pepio AM, Fan X, Sossin WS ( 1999) Protein kinase C phosphorylated at a conserved threonine is retained in the cytoplasm. J Biol Chem 274: 28944–28949. [DOI] [PubMed] [Google Scholar]
- Nonet ML ( 1999) Visualization of synaptic specializations in live C. elegans with synaptic vesicle protein-GFP fusions. J Neurosci Methods 89: 33–40. [DOI] [PubMed] [Google Scholar]
- Nonet ML, Grundahl K, Meyer BJ, Rand JB ( 1993) Synaptic function is impaired but not eliminated in C. elegans mutants lacking synaptotagmin. Cell 73: 1291–1305. [DOI] [PubMed] [Google Scholar]
- Perin MS, Fried VA, Mignery GA, Jahn R, Sudhof TC ( 1990) Phospholipid binding by a synaptic vesicle protein homologous to the regulatory region of protein kinase C. Nature 345: 260–263. [DOI] [PubMed] [Google Scholar]
- Ramjaun AR, Micheva KD, Bouchelet I, McPherson PS ( 1997) Identification and characterization of a nerve terminal-enriched amphiphysin isoform. J Biol Chem 272: 16700–16706. [DOI] [PubMed] [Google Scholar]
- Schiavo G, Osborne SL, Sgouros JG ( 1998) Synaptotagmins: more isoforms than functions? Biochem Biophys Res Commum 248: 1–8. [DOI] [PubMed] [Google Scholar]
- Sossin WS, Fan XT, Saberi F ( 1996) Expression and characterization of Aplysia protein kinase C: a negative regulatory role for the E region. J Neurosci 16: 10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC ( 2002) Synaptotagmins: why so many? J Biol Chem 277: 7629–7632. [DOI] [PubMed] [Google Scholar]
- Sun ZY, Schacher S ( 1996) Development of short-term heterosynaptic facilitation at Aplysia sensorimotor synapses in vitro is accompanied by changes in the functional expression of presynaptic serotonin receptors. J Neurophysiol 76: 2250–2261. [DOI] [PubMed] [Google Scholar]
- Yamasaki S, Hu Y, Binz T, Kalkuhl A, Kurazono H, Tamura T, Jahn R, Kandel E, Niemann H ( 1994) Synaptobrevin/vesicle-associated membrane protein (VAMP) of Aplysia californica: structure and proteolysis by tetanus toxin and botulinal neurotoxins type D and F. Proc Natl Acad Sci USA 91: 4688–4692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JZ, Davletov BA, Sudhof TC, Anderson RG ( 1994) Synaptotagmin I is a high affinity receptor for clathrin AP-2: implications for membrane recycling. Cell 78: 751–760. [DOI] [PubMed] [Google Scholar]