

Abstract
Increasing evidence has suggested an important role for environmental toxins such as pesticides in the pathogenesis of Parkinson's disease (PD). Chronic exposure to rotenone, a common herbicide, reproduces features of Parkinsonism in rats. Mechanistically, rotenone-induced dopaminergic neurodegeneration has been associated with both its inhibition of neuronal mitochondrial complex I and the enhancement of activated microglia. Our previous studies with NADPH oxidase inhibitors, diphenylene iodonium and apocynin, suggested that NADPH oxidase-derived superoxide might be a major factor in mediating the microglia-enhanced rotenone neurotoxicity. However, because of the relatively low specificity of these inhibitors, the exact source of superoxide induced by rotenone remains to be further determined. In this study, using primary mesencephalic cultures from NADPH oxidase—null (gp91phox-/-) or wild-type (gp91phox+/+) mice, we demonstrated a critical role for microglial NADPH oxidase in mediating microglia-enhanced rotenone neurotoxicity. In neuron—glia cultures, dopaminergic neurons from gp91phox-/- mice were more resistant to rotenone neurotoxicity than those from gp91phox+/+ mice. However, in neuron-enriched cultures, the neurotoxicity of rotenone was not different between the two types of mice. More importantly, the addition of microglia prepared from gp91phox+/+ mice but not from gp91phox-/- mice to neuron-enriched cultures markedly increased rotenone-induced degeneration of dopaminergic neurons. Furthermore, apocynin attenuated rotenone neurotoxicity only in the presence of microglia from gp91phox+/+ mice. These results indicated that the greatly enhanced neurotoxicity of rotenone was attributed to the release of NADPH oxidase-derived superoxide from activated microglia. This study also suggested that microglial NADPH oxidase may be a promising target for PD treatment.
Keywords: pesticides, microglia, superoxide, NADPH oxidase, Parkinson's disease, inflammation
Introduction
Parkinson's disease (PD) is a movement disorder characterized by a progressive and selective degeneration of the nigrostriatal dopaminergic pathway (Olanow and Tatton, 1999). Although its etiology remains unclear, many lines of evidence have indicated an environmental exposure basis for the disease. Living in a rural environment, farming, drinking well water, and occupational exposure to agricultural chemicals have been linked to nigrostriatal dopaminergic neurodegeneration (Rajput and Uitti, 1987; Semchuk et al., 1992; Liou et al., 1997; Priyadarshi et al., 2001). A number of epidemiological and case-controlled studies have suggested a positive correlation between early exposure to pesticides and increased risk for PD (Liou et al., 1997; Gorell et al., 1998; Ritz and Yu, 2000; Herishanu et al., 2001). Experimental evidence also indicates pesticides and/or related agrochemicals as risk factors for PD. Chronic systemic exposure to rotenone reproduces Parkinsonian features in rats (Betarbet et al., 2000; Alam and Schmidt, 2002). Mechanistically, rotenone-induced degeneration of dopaminergic neurons may not be solely attributed to an impairment of neuronal mitochondrial complex I activity (Greenamyre et al., 1999; Jenner, 2001) but may also involve the participation of the resident immune cells in the brain, microglia (Gao et al., 2002a; Sherer et al., 2003). Combined exposure to the herbicide paraquat and the fungicide maneb during both adult and postnatal periods results in lesions of the nigrostriatal dopaminergic system, and the developmental exposure enhances adult susceptibility to these pesticides (Thiruchelvam et al., 2000, 2002). In addition, there seems to be an interplay between environmental factors and genetic vulnerability, because several pesticides, including rotenone, dieldrin, and paraquat, have been reported to directly accelerate the rate of α-synuclein fibril formation that is associated with α-synuclein aggregation (Uversky et al., 2001, 2002). Moreover, various environmental factors may also work in concert to induce dopaminergic neurodegeneration. For example, nontoxic or minimally toxic concentrations of rotenone combined with the inflammogen lipopolysaccharide synergistically induced a selective degeneration of dopaminergic neurons in primary mesencephalic neuron—glia cultures (Gao et al., 2003).
Recently, a number of studies have demonstrated that microglial activation, the hallmark of neuroinflammation, plays an important role in the pathogenesis of PD (Vila et al., 2001; Liu and Hong, 2003). We reported that the presence of microglia markedly increased rotenone neurotoxicity, which was associated with the release of superoxide from activated microglia. Studies with NADPH oxidase inhibitors suggested that rotenone-induced extracellular superoxide production was catalyzed by NADPH oxidase (Gao et al., 2002a). In that study, two mechanistically dissimilar inhibitors, diphenylene iodonium (DPI) and apocynin, were used to examine the involvement of NADPH oxidase, also called phagocyte oxidase (PHOX), in rotenone-induced neuro-degeneration. However, the lack of specificity of these inhibitors, with the former inhibiting other flavoenzymes (Irani et al., 1997; Li and Trush, 1998) and the latter increasing γ-glutamylcysteine synthetase activity (Lapperre et al., 1999), calls for the determination of the exact source of the rotenone-induced superoxide production. In the current study, we used mesencephalic cultures from NADPH oxidase—null (gp91 phox-/-) or wild-type (gp91phox+/+) mice to directly test the critical role of microglial NADPH oxidase in mediating the elevated rotenone neurotoxicity.
Materials and Methods
Animals. Wild-type C57BL/6J (gp91 phox+/+) and NADPH oxidase—null (gp91 phox-/-) mice were obtained from The Jackson Laboratory (Bar Harbor, ME). Breeding of the mice was conducted to achieve timed pregnancy with an accuracy of ±0.5 d. Housing and breeding of the animals were performed in strict accordance with National Institutes of Health guidelines.
Primary mesencephalic neuron—glia cultures. Neuron—glia cultures were prepared from the ventral mesencephalic tissues of embryonic day 12 (E12)—E13 mice, as described previously (Gao et al., 2002a, 2003). Briefly, dissociated cells were seeded to 24 well (6 × 105/well) culture plates precoated with poly-d-lysine (20 μg/ml) and maintained in 0.5 ml/well of minimum essential medium supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 10% heat-inactivated horse serum, 1 gm/l glucose, 2 mm l-glutamine, 1 mm sodium pyruvate, 100 μm nonessential amino acids, 50 U/ml penicillin, and 50 μg/ml streptomycin. Cultures were maintained at 37°C in a humidified atmosphere of 5% CO2 and 95% air. Cultures were replenished with fresh medium (0.5 ml/well) 3 d later. Seven-day-old cultures were used for treatment. Immunocytochemical analysis indicated that at the time of treatment, the cultures were made up of ∼11% F4/80-immunoreactive (IR) microglia, 49% glial fibrillary acidic protein (GFAP)-IR astrocytes, and 40% neuron-specific nuclear protein (NeuN)-IR neurons, of which 2.5—3.5% were tyrosine hydroxylase (TH)-IR neurons. There was no significant difference in the composition of the neuron—glia cultures between gp91 phox-/- and gp91 phox+/+ mice.
Mesencephalic neuron-enriched cultures. Neuron-enriched cultures were prepared from the ventral mesencephalic tissues of E12—E13 mice as described previously (Gao et al., 2002a). Dissociated cells were first seeded at 6 × 105 cells/well into poly-d-lysine-coated 24-well culture plates. Two days later, cytosine β-d-arabinofuranoside (8 —10 μm) was added to suppress the proliferation of glial cells. Seven-day-old cultures that contained <0.1% F4/80-IR microglia and 10% GFAP-IR astrocytes were used for treatment. Of the NeuN-IR neurons, 2.5—3.5% were TH-IR neurons. In neuron-enriched cultures, the cell composition was not different between gp91 phox-/- and gp91 phox+/+ mice.
Microglia-enriched cultures. Microglia were prepared from whole brains of 1-d-old mice as described previously (Liu et al., 2001). After reaching confluence (14 d), microglia were separated from astrocytes by shaking the flasks for 5 hr at 150 rpm. The enriched microglia were >95% pure, as determined by immunostaining with antibodies against F4/80 and GFAP.
Immunocytochemistry. Immunostaining was performed as described previously (Liu et al., 2000; Gao et al., 2003) with the following primary antibodies: anti-NeuN (1:2000; Chemicon, Temecula, CA), anti-F4/80 antigen (1:20; Serotec, Raleigh, NC), anti-GFAP (1:1000; Dako, Carpinteria, CA), and anti-TH (1:20,000; a gift from GlaxoSmithKline, Research Triangle Park, NC). Briefly, after blocking, formaldehyde-fixed cells were incubated overnight at 4°C with primary antibodies diluted in antibody diluent. The bound primary antibody was visualized by incubation with an appropriate biotinylated secondary antibody (Vector Laboratories, Burlingame, CA) followed by Vectastain ABC reagents (Vector Laboratories) and color development with 3,3′-diaminobenzidine. Images were recorded with a CCD camera and Metamorph software (Universal Imaging, West Chester, PA). For visual enumeration of the immunostained cells in cultures, 10 representative areas per well were counted. The overall dendrite length for individual TH-IR neurons was measured by following our previously published protocol (Gao et al., 2002a). Three wells of identical treatment from each experiment and 50 TH-IR neurons per well were measured. Results were obtained from two separate experiments and were expressed as a percentage of the control cultures.
[3H]Dopamine uptake assay. Uptake of [3H]dopamine (DA) was determined by incubation of cultures for 15 min at 37°C with 1 μm [3H]DA (30 Ci/mmol; NEN, Boston, MA) as described previously (Gao et al., 2002a). Nonspecific uptake for DA was determined in the presence of 10 μm mazindol.
Measurement of superoxide release. The release of superoxide was determined by measuring the superoxide dismutase (SOD)-inhibitable reduction of cytochrome c, as described previously (Liu et al., 2000; Gao et al., 2002a). Primary microglia (1 × 105/well), grown overnight in 96 well plates in DMEM containing 10% FBS, were switched to phenol red-free HBSS (100 μl/well). Fifty microliters of HBSS containing vehicle or rotenone were then added to each well, followed by 50 μl of ferricyto-chrome c (100 μm) in HBSS, with and without 600 U/ml SOD. The cultures were incubated for 30 min at 37°C, and the absorbance at 550 nm was read with a SpectraMax Plus microplate spectrophotometer (Molecular Devices, Sunnyvale, CA). To determine the effect of the NADPH oxidase inhibitor apocynin on superoxide release, microglia were preincubated for 30 min with vehicle or apocynin before the addition of rotenone.
Statistical analysis. Statistical significance was assessed by ANOVA followed by Bonferroni's t test using the StatView program (Abacus Concepts, Berkeley, CA). A value of p < 0.05 was considered statistically significant.
Results
Rotenone induced a progressive and selective dopaminergic neurodegeneration, which was enhanced by glia
Primary neuron—glia cultures were treated with 0.5—15 nm rotenone for 3 or 7 d, and the extent and selectivity of neurodegeneration were determined by multiple parameters. [3H]DA uptake assays demonstrated a concentration- and time-dependent reduction in DA uptake (Fig. 1A). Three days after treatment with rotenone, only the highest concentration of rotenone resulted in a significant reduction (37%) in DA uptake. As treatment time progressed, the reduction in DA uptake became more prominent. A 90% reduction was observed after treatment with 15 nm rotenone for 7 d, whereas 5 nm rotenone also caused a significant reduction in DA uptake. Immunocytochemical analysis illustrated that the progressive neurodegeneration induced by rotenone was selective to dopaminergic neurons (Fig. 1B). A significant decrease in the number of TH-IR neurons was seen in neuron—glia cultures treated for 7 d with 10 nm rotenone, whereas the decrease in NeuN-IR neurons was insignificant. The morphological analysis after immunocytochemical staining with antibodies against TH or NeuN indicated that rotenone neurotoxicity was selective to dopaminergic neurons (data not shown). Quantification of TH-IR and NeuN-IR neurons (Fig. 1B) and morphological analysis verified that rotenone-induced reduction in DA uptake was attributed not to an alteration in dopamine transporter (DAT) but rather to dopaminergic neuronal death. Consistent with our published results from rats (Gao et al., 2002a), the presence of microglia essentially increased rotenone neurotoxicity in mouse neuron—glia cultures (Fig. 1C). For example, treatment for 7 d with 5 nm rotenone, which was a nontoxic concentration in neuron-enriched cultures, induced a significant reduction (30%) in DA uptake in neuron—glia cultures.
Figure 1.

Glia enhanced dopaminergic neurodegeneration induced by rotenone. A, Primary neuron—glia cultures were treated for 3 or 7 d with vehicle or 0.5—15 nm rotenone, and the capability of cultures to uptake DA was evaluated. B, Neuron—glia cultures were treated for 7 d with vehicle or 10 nm rotenone, and the selectivity of rotenone-induced neurodegeneration was determined by quantification of TH-IR and NeuN-IR neurons after immunocytochemical staining. C, Neuron—glia (N/G; •) or neuron-enriched (N; ▪) cultures were treated for 7 d with vehicle or 2.5—10 nm rotenone. [3H]DA uptake was measured to assess neurotoxicity to dopaminergic neurons. The results for DA uptake (A, C) and cell counts (B) are expressed as a percentage of the control cultures and are means ± SEM of three experiments performed in triplicate. *p < 0.05, **p < 0.005 compared with the corresponding control (A, B). *p < 0.05 compared with the corresponding rotenone-treated neuron-enriched cultures (C).
Dopaminergic neurons from gp91phox-/- mice were more resistant to rotenone neurotoxicity than those from gp91phox+/+ mice in the neuron—glia culture system
Neuron—glia cultures, prepared from either gp91phox+/+ or gp91phox-/- mice, were treated with 2.5—10 nm rotenone for 7 d, and dopaminergic neurodegeneration was determined by the measurement of [3H]DA uptake, quantification of TH-IR neurons, or measurement of overall dendrite length of TH-IR neurons. As shown in Figure 2A, rotenone decreased [3H]DA uptake in a dose-dependent manner in both cell culture systems. However, the magnitude of decrease in the cultures from gp91phox+/+ mice was greater than that from gp91phox-/- mice. Consistent with the effect observed with the uptake assays, quantification of TH-IR neurons also demonstrated the resistance of dopaminergic neurons from gp91phox-/- mice to rotenone neurotoxicity (Fig. 2B). In addition, morphological analysis also illustrated that the number of TH-IR neurons in rotenone-treated cultures from gp91phox+/+ mice was significantly lower than that from gp91phox-/- mice, and that the dendritic network of rotenone-treated cultures from gp91phox+/+ mice was markedly less extensive compared with that from gp91phox-/- mice (Fig. 2C). Quantification of TH-IR dendrite length also indicated the resistance of dopaminergic neurons from gp91phox-/- mice to rotenone neurotoxicity. Treatment for 7 d with 10 nm rotenone reduced the average dendrite length of TH-IR neurons from gp91phox+/+ mice to 25.8 ± 10.5% of control cultures (p < 0.001 compared with control), whereas the dendrite length of TH-IR neurons from gp91phox-/- mice was 73.2 ± 8.7% of control cultures (p < 0.01 compared with corresponding control; p < 0.01 compared with the reduction in the dendrite length of TH-IR neurons from gp91phox+/+ mice).
Figure 2.
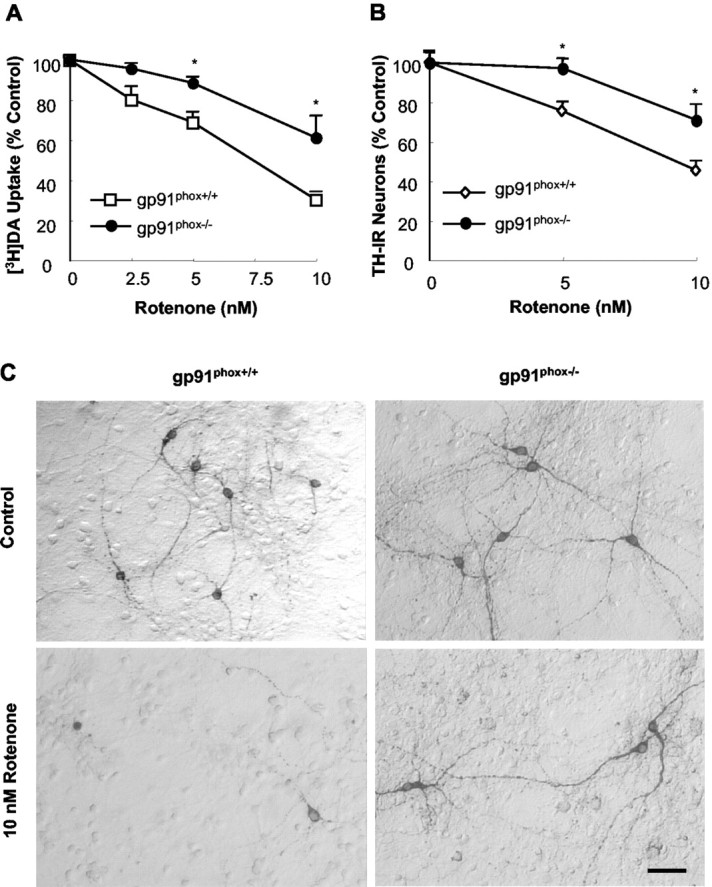
Resistance of dopaminergic neurons from gp91 phox-/- mice to rotenone neurotoxicity. Neuron—glia cultures were treated for 7 d with vehicle or desired concentrations of rotenone. Afterward, cultures were assayed for uptake of [3H]DA (A), quantification of the TH-IR neurons (B), or immunocytochemical analysis of TH-IR neurons (C). Results are expressed as a percentage of the control cultures and are means ± SEM of three experiments performed in triplicate. *p < 0.05 compared with the corresponding rotenone-treated cultures from gp91 phox+/+ mice. Scale bar, 50 μm.
Microglia from gp91phox+/+ but not gp91phox-/-mice enhanced rotenone-induced dopaminergic neurodegeneration
In neuron—glia cultures, the resistance of dopaminergic neurons from gp91phox-/- mice to rotenone neurotoxicity supported the notion that the rotenone neurotoxicity was partially mediated by the activation of microglia and the NADPH oxidase-mediated release of superoxide free radical (Gao et al., 2002a). To directly test the critical role of microglial NADPH oxidase in rotenone neurotoxicity, microglia prepared either from gp91phox+/+ mice or from gp91phox-/- mice were added to neuron-enriched cultures (prepared from gp91phox+/+ or gp91phox-/- mice), and [3H]DA uptake of the reconstituted cultures and neuron-enriched cultures was compared after treatment with 5 nm rotenone for 7 d. As shown in Figure 3, although the DA uptake of the neuron-enriched cultures was not affected by the addition of microglia from gp91phox-/- mice, the addition of microglia from gp91phox+/+ mice significantly increased the sensitivity of dopaminergic neurons (either from gp91phox+/+ mice or from gp91phox-/- mice) to rotenone neurotoxicity. Furthermore, in the neuron-enriched culture system, the neurotoxicity of rotenone was not different between gp91phox-/- mice and gp91phox+/+ mice. Moreover, the addition of microglia from gp91phox+/+ mice equally decreased the capability of neuron-enriched cultures from both types of mice to uptake DA. These data additionally strengthened the notion that microglial NADPH oxidase, but not neuronal NADPH oxidase, was responsible for the NADPH oxidase-mediated neurotoxicity of rotenone.
Figure 3.

Effect of the addition of microglia to neuron-enriched (N) cultures on rotenone-induced degeneration of dopaminergic neurons. Mesencephalic neuron-enriched cultures were supplemented with 5 × 104 microglia per well prepared from either gp91 phox+/+ or gp91 phox-/- mice. After 24 hr, the cultures were treated with vehicle or 5 nm rotenone, and [3H]DA uptake was determined 7 d after the treatment. Results are expressed as a percentage of the control cultures and are means ± SEM of three experiments performed in triplicate. p < 0.05 was considered statistically significant. All other comparisons were not significant.
Rotenone-induced release of superoxide from activated microglia was mediated by NADPH oxidase
We reported that activated microglia elevated rotenone neurotoxicity in rat cell cultures through the release of superoxide free radical (Gao et al., 2002a). The results described above indicated that microglial NADPH oxidase plays a pivotal role in microglia-enhanced rotenone neurotoxicity. Indeed, as shown in Figure 4, rotenone stimulated microglia from gp91phox+/+ mice to release superoxide free radical, which was attenuated by an inhibitor of NADPH oxidase, apocynin. In sharp contrast, microglia from gp91phox-/- mice failed to release superoxide free radical after the stimulation with rotenone. We observed similar profiles for the inhibition of superoxide production when using another inhibitor of NADPH oxidase, DPI (data not shown). In addition, in neuron-enriched cultures, superoxide production was undetectable (data not shown). These results indicated that the resistance of dopaminergic neurons of gp91phox-/- mice was attributable to the lack of superoxide release by microglia from gp91phox-/- mice.
Figure 4.

Rotenone stimulated release of superoxide in microglia from gp91 phox+/+ mice but not gp 91 phox-/- mice. Mouse primary microglia were seeded to 96 well plates. After 24 hr, microglia were pretreated with vehicle or 0.5 mm apocynin for 30 min. Afterward, microglia were stimulated with rotenone (5 or 10 nm) or corresponding vehicle. Superoxide production, measured as SOD-inhibitable cytochrome c reduction, was determined as described in Materials and Methods. There was no significant difference in the basal levels of superoxide production (control) between the cultures from gp91 phox-/- mice and those of their wild-type litter-mates. Results are a percentage of the control cultures and are expressed as means ± SEM of three experiments performed in triplicate. **p < 0.005 compared with the control. ++p < 0.005 compared with corresponding rotenone-treated cultures.
Apocynin protected dopaminergic neurons against rotenone neurotoxicity only in the presence of microglia from gp91phox+/+ mice
The relevance of rotenone-stimulated superoxide release in mediating microglia-enhanced rotenone neurotoxicity was established by the observation that inhibition of NADPH oxidase afforded neuroprotection (Fig. 5). Pretreatment of neuron—glia cultures from gp91phox+/+ mice with 0.5 mm apocynin significantly attenuated the rotenone-induced degeneration of dopaminergic neurons measured either by [3H]DA uptake (Fig. 5A) or by counts of TH-IR neurons (Fig. 5B). However, apocynin failed to protect dopaminergic neurons of gp91phox-/- mice against rotenone neurotoxicity (Fig. 5A). Additionally, the neuroprotective effect of apocynin was dependent on the presence of microglia from gp91phox+/+ mice: in neuron-enriched cultures from gp91phox+/+ mice, apocynin did not protect dopaminergic neurons against rotenone neurotoxicity, but did so quite effectively in neuron—glia cultures (Fig. 5C). We did not use DPI to determine the neuroprotective effect, because of its neurotoxicity in our long-term cell cultures (Gao et al., 2002a,b). These results gave strong support to the notion that the microglia-enhanced degeneration of dopaminergic neurons induced by rotenone was mediated by microglial NADPH oxidase.
Figure 5.

Effect of apocynin on rotenone-induced degeneration of dopaminergic neurons. A, B, Neuron—glia cultures were pretreated for 30 min with vehicle or 0.5 mm apocynin before treatment with 10 nm rotenone. Seven days later, degeneration of dopaminergic neurons was determined by [3H]DA uptake (A) and counts of TH-IR neurons (B). C, Neuron—glia (N/G) or neuron-enriched (N) cultures were pretreated for 30 min with the vehicle or 0.5 mm apocynin before treatment with 10 nm rotenone. [3H]DA uptake was measured 7 d later. Results are means ± SEM of three to four experiments performed in triplicate. *p < 0.05 compared with the corresponding control. +p < 0.05 compared with rotenone-treated cultures.
Discussion
Our previous studies with NADPH oxidase inhibitors (DPI and apocynin) have suggested that microglia, via the release of NADPH oxidase-derived superoxide, markedly enhanced rotenone-induced degeneration of dopaminergic neurons. However, because of the relatively low specificity of these two inhibitors, the exact source of rotenone-induced extracellular superoxide production remained to be determined. In this study, using primary neuron-enriched and neuron—glia cultures from the mesencephalon of gp91phox-/- mice or gp91phox+/+ mice, we demonstrated that the microglia-enhanced rotenone neurotoxicity was attributed to microglial NADPH oxidase-mediated release of superoxide free radical. The presence of microglia from gp91phox+/+ mice rendered dopaminergic neurons more sensitive to rotenone neurotoxicity. Rotenone stimulated microglia from gp91phox+/+ mice to release superoxide, but it failed to do so in microglia from gp91phox-/- mice. Furthermore, apocynin significantly attenuated rotenone neurotoxicity only in the presence of microglia from gp91phox+/+ mice. These results indicated that rotenone-induced activation of microglia and the release of NADPH oxidase-derived superoxide from activated microglia mediated the greatly enhanced dopaminergic neurodegeneration induced by rotenone.
Neuroinflammation, as an integral component of the progressive neurodegenerative process, has been increasingly implicated in the pathogenesis of PD. Microglial activation, a hallmark of neuroinflammation, has been detected frequently in PD patients and experimental animal models. Our recent work demonstrated that the presence of microglia markedly enhanced rotenone neurotoxicity, and that rotenone-stimulated microglial activation preceded dopaminergic neurodegeneration (Gao et al., 2002a). Similar to our in vitro observations, Sherer et al. (2003) reported recently that chronic rotenone exposure causes extensive microglial activation in the striatum and substantia nigra of the rat, with less activation of microglia in the cortex. They have also demonstrated that microglial activation occurs even in the absence of detectable nigrostriatal dopaminergic neurodegeneration and becomes more evident with the development of dopaminergic neurodegeneration.
Microglia play a pivotal role in the detection of invading pathogens, host defense, and tissue repair (Kreutzberg, 1996; Aloisi, 1999). In response to immunological challenges or injury, microglia become readily activated and produce a variety of proinflammatory and neurotoxic factors, including tumor necrosis factor-α, interleukin-1β, eicosanoids, nitric oxide, and superoxide (Cassarino et al., 1997; Liu et al., 2002; Liu and Hong, 2003). Although a few of these factors are thought to contribute to tissue repair (Streit et al., 1988), the majority are believed to induce or exacerbate neurodegeneration through mechanisms not yet fully understood (McGeer et al., 1988; Minghetti and Levi, 1998; Liu and Hong, 2003). Among the various cytotoxic factors released by activated microglia, reactive oxygen species (ROS) such as superoxide free radical appear to play a key role in the inflammation-mediated oxidative damage to neurons. Specifically, microglia-enhanced rotenone neurotoxicity was primarily mediated by NADPH oxidase-mediated release of superoxide, as evidenced by the significant release of superoxide by activated microglia, which was inhibitable by NADPH oxidase inhibitors and the essential neuroprotective effect of pharmacological inhibition and genetic inactivation of NADPH oxidase.
NADPH oxidase is an enzyme that is a major source of extracellular superoxide in immune cells, including microglia. This enzyme is a multisubunit enzymatic complex. Currently, at least seven subunits have been reported to be involved in NADPH oxidase assembly. In resting immune cells, these NADPH oxidase subunits are segregated into cytoplasmic and plasma membrane compartments. In response to stimuli, the cytosolic subunits (p47PHOX, p67PHOX, and p40PHOX) translocate to the membrane and associate with membrane components (gp91PHOX and p22PHOX) to assemble the functional oxidase and subsequently release superoxide free radical (DeLeo and Quinn, 1996; Babior, 1999). Two small G-proteins, Rac-1 and Rap1A, are also associated with the activation process of NADPH oxidase. The ability of immune cells, especially phagocytes, to generate prodigious quantities of superoxide anions is one of the main methods by which phagocytes kill invading pathogens. Although NADPH oxidase is primarily expressed in phagocytic cells, increasing evidence suggests that various subunits of NADPH oxidase are also expressed in nonphagocytic cells such as sympathetic ganglion neurons and cortical neurons (Fukui et al., 1995; Noh and Koh, 2000; Tammariello et al., 2000), which suggests the possibility that neurons in general may express NADPH oxidase. However, we illustrated that neuronal NADPH oxidase was not directly involved in the production of superoxide or the induction of dopaminergic neurodegeneration induced by rotenone, based on several observations. First, in neuron-enriched cultures, rotenone failed to induce superoxide release. Second, in the neuron-enriched culture system, the neurotoxicity of rotenone was not different between gp91phox-/- mice and gp91phox+/+ mice (Fig. 3). Third, the addition of microglia, either from gp91phox-/- mice or from gp91phox+/+ mice, to neuron-enriched cultures did not affect this trend (Fig. 3). Fourth, in the cultures from gp91phox+/+ mice, apocynin could not attenuate rotenone neurotoxicity in the absence of glia, but did so quite effectively in the presence of glia (Fig. 5). Together, these data suggest that microglial NADPH oxidase, but not neuronal NADPH oxidase, renders dopaminergic neurons more sensitive to rotenone neurotoxicity. Additionally, two mechanistically dissimilar inhibitors of NADPH oxidase were used to confirm that the extracellular superoxide production we measured was catalyzed by NADPH oxidase: DPI directly inhibits the catalytic activity of the enzyme (Irani et al., 1997), and apocynin prevents the assembly of the multisubunit enzyme complex (Stolk et al., 1994).
Superoxide and its downstream products (hydroxyl radicals, singlet oxygen species, and hydrogen peroxide) are quite reactive and can interact with proteins, DNA, or RNA to alter their functions or induce lipid peroxidation, leading to eventual cell death (Facchinetti et al., 1998). However, superoxide cannot directly cross cellular membranes (Halliwell, 2001), making it unlikely that microglia-derived extracellular superoxide would enter neurons and directly trigger intraneuron toxic events. Alternatively, superoxide may convert to membrane-permeable hydrogen peroxide or perhaps other downstream products in the extracellular space, which, in turn, can cross the cell membrane and cause direct injury to neurons. Of the various populations of neurons in the brain, dopaminergic neurons are uniquely vulnerable to oxidative stress because of their innate characteristics, including reduced antioxidant capacity, increased accumulation of iron, and the high concentration of neurochemicals such as dopamine that are prone to oxidative modification (Jenner, 1998; Greenamyre et al., 1999). Furthermore, it has been proposed that extracellular ROS appear to promote the mitochondria-mediated generation of intracellular ROS (Turrens and Boveris, 1980; Hasegawa et al., 1990). Thus, microglia-originated superoxide induced by rotenone, via formation of membrane-permeable downstream products, might damage dopaminergic neurons directly or promote neuronal mitochondria-originated free radical production, which was a result of the inhibition of mitochondria complex I by rotenone. Indeed, one of the downstream products of superoxide, hydrogen peroxide, has been reported to result in cytochrome c release from mitochondria, caspase-3 activation, and apoptosis of human neuroblastoma cells. Moreover, chronic exposure to a low concentration of rotenone potentiates hydrogen peroxide-induced caspase-dependent cell death (Sherer et al., 2002). Therefore, the combination of the intrinsic characteristics of dopaminergic neurons and their physical location in a microglia-rich microenvironment (Lawson et al., 1990; Kim et al., 2000) renders them more sensitive to attacks by free radicals generated both extracellularly and intracellularly.
The present study demonstrated that microglia-enhanced degeneration of dopaminergic neurons induced by rotenone was mediated by the activation of microglia and by the consequent release of microglial NADPH oxidase-mediated superoxide. This study also suggests that NADPH oxidase may be a promising target for PD treatment, especially in halting the progression of PD.
Footnotes
We thank Dr. Michelle Block for reading this manuscript.
Correspondence should be addressed to Hui-Ming Gao, F1-01, National Institute of Environmental Health Sciences, P.O. Box 12233, Research Triangle Park, NC 27709. E-mail: gao@niehs.nih.gov.
B. Liu's present address: Department of Pharmacodynamics, College of Pharmacy, P.O. Box 100487 HSC, University of Florida, Gaineville, FL 32610.
Copyright © 2003 Society for Neuroscience 0270-6474/03/236181-07$15.00/0
References
- Alam M, Schmidt WJ ( 2002) Rotenone destroys dopaminergic neurons and induces parkinsonian symptoms in rats. Behav Brain Res 136: 317—324. [DOI] [PubMed] [Google Scholar]
- Aloisi F ( 1999) The role of microglia and astrocytes in CNS immune surveillance and immunopathology. Adv Exp Med Biol 468: 123—133. [DOI] [PubMed] [Google Scholar]
- Babior BM ( 1999) NADPH oxidase: an update. Blood 93: 1464 —1476. [PubMed] [Google Scholar]
- Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT ( 2000) Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci 3: 1301—1306. [DOI] [PubMed] [Google Scholar]
- Cassarino DS, Fall CP, Swerdlow RH, Smith TS, Halvorsen EM, Miller SW, Parks JP, Parker Jr WD, Bennett Jr JP ( 1997) Elevated reactive oxygen species and antioxidant enzyme activities in animal and cellular models of Parkinson's disease. Biochim Biophys Acta 1362: 77—86. [DOI] [PubMed] [Google Scholar]
- DeLeo FR, Quinn MT ( 1996) Assembly of the phagocyte NADPH oxidase: molecular interaction of oxidase proteins. J Leukoc Biol 60: 677—691. [DOI] [PubMed] [Google Scholar]
- Facchinetti F, Dawson VL, Dawson TM ( 1998) Free radicals as mediators of neuronal injury. Cell Mol Neurobiol 18: 667—682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukui T, Lassegue B, Kai H, Alexander RW, Griendling KK ( 1995) Cytochrome b-558 alpha-subunit cloning and expression in rat aortic smooth muscle cells. Biochim Biophys Acta 1231: 215—219. [DOI] [PubMed] [Google Scholar]
- Gao HM, Hong JS, Zhang W, Liu B ( 2002a) Distinct role for microglia in rotenone-induced degeneration of dopaminergic neurons. J Neurosci 22: 782—790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao HM, Jiang J, Wilson B, Zhang W, Hong JS, Liu B ( 2002b) Microglial activation-mediated delayed and progressive degeneration of rat nigral dopaminergic neurons: relevance to Parkinson's disease. J Neurochem 81: 1285—1297. [DOI] [PubMed] [Google Scholar]
- Gao HM, Hong JS, Zhang W, Liu B ( 2003) Synergistic dopaminergic neurotoxicity of the pesticide rotenone and inflammogen lipopolysaccharide: relevance to the etiology of Parkinson's disease. J Neurosci 23: 1228 —1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorell JM, Johnson CC, Rybicki BA, Peterson EL, Richardson RJ ( 1998) The risk of Parkinson's disease with exposure to pesticides, farming, well water, and rural living. Neurology 50: 1346 —1350. [DOI] [PubMed] [Google Scholar]
- Greenamyre JT, MacKenzie G, Peng TI, Stephans SE ( 1999) Mitochondrial dysfunction in Parkinson's disease. Biochem Soc Symp 66: 85—97. [DOI] [PubMed] [Google Scholar]
- Halliwell B ( 2001) Role of free radicals in the neurodegenerative diseases: therapeutic implications for antioxidant treatment. Drugs Aging 18: 685—716. [DOI] [PubMed] [Google Scholar]
- Hasegawa E, Takeshige K, Oishi T, Murai Y, Minakami S ( 1990) 1-Methyl-4-phenylpyridinium (MPP+) induces NADH-dependent superoxide formation and enhances NADH-dependent lipid peroxidation in bovine heart submitochondrial particles. Biochem Biophys Res Commun 170: 1049 —1055. [DOI] [PubMed] [Google Scholar]
- Herishanu YO, Medvedovski M, Goldsmith JR, Kordysh E ( 2001) A case-control study of Parkinson's disease in urban population of southern Israel. Can J Neurol Sci 28: 144 —147. [DOI] [PubMed] [Google Scholar]
- Irani K, Xia Y, Zweier JL, Sollott SJ, Der CJ, Fearon ER, Sundaresan M, Finkel T, Goldschmidt-Clermont PJ ( 1997) Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science 275: 1649 —1652. [DOI] [PubMed] [Google Scholar]
- Jenner P ( 1998) Oxidative mechanisms in nigral cell death in Parkinson's disease. Mov Disord 13: 24 —34. [PubMed] [Google Scholar]
- Jenner P ( 2001) Parkinson's disease, pesticides and mitochondrial dysfunction. Trends Neurosci 24: 245—247. [DOI] [PubMed] [Google Scholar]
- Kim WG, Mohney RP, Wilson B, Jeohn GH, Liu B, Hong JS ( 2000) Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: role of microglia. J Neurosci 20: 6309 —6316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreutzberg GW ( 1996) Microglia: a sensor for pathological events in the CNS. Trends Neurosci 19: 312—318. [DOI] [PubMed] [Google Scholar]
- Lapperre TS, Jimenez LA, Antonicelli F, Drost EM, Hiemstra PS, Stolk J, MacNee W, Rahman I ( 1999) Apocynin increases glutathione synthesis and activates AP-1 in alveolar epithelial cells. FEBS Lett 443: 235—239. [DOI] [PubMed] [Google Scholar]
- Lawson LJ, Perry VH, Dri P, Gordon S ( 1990) Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 39: 151—170. [DOI] [PubMed] [Google Scholar]
- Li Y, Trush MA ( 1998) Diphenyleneiodonium, an NAD(P)H oxidase inhibitor, also potently inhibits mitochondrial reactive oxygen species production. Biochem Biophys Res Commun 253: 295—299. [DOI] [PubMed] [Google Scholar]
- Liou HH, Tsai MC, Chen CJ, Jeng JS, Chang YC, Chen SY, Chen RC ( 1997) Environmental risk factors and Parkinson's disease: a case-control study in Taiwan. Neurology 48: 1583—1588. [DOI] [PubMed] [Google Scholar]
- Liu B, Hong JS ( 2003) Role of microglia in inflammation-mediated neurodegenerative diseases: mechanisms and strategies for therapeutic intervention. J Pharmacol Exp Ther 304: 1—7. [DOI] [PubMed] [Google Scholar]
- Liu B, Du L, Hong JS ( 2000) Naloxone protects rat dopaminergic neurons against inflammatory damage through inhibition of microglia activation and superoxide generation. J Pharmacol Exp Ther 293: 607—617. [PubMed] [Google Scholar]
- Liu B, Wang K, Gao HM, Mandavilli B, Wang JY, Hong JS ( 2001) Molecular consequences of activated microglia in the brain: overactivation induces apoptosis. J Neurochem 77: 182—189. [DOI] [PubMed] [Google Scholar]
- Liu B, Gao HM, Wang JY, Jeohn GH, Cooper CL, Hong JS ( 2002) Role of nitric oxide in inflammation-mediated neurodegeneration. Ann NY Acad Sci 962: 318 —331. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Itagaki S, Boyes BE, McGeer EG ( 1988) Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology 38: 1285—1291. [DOI] [PubMed] [Google Scholar]
- Minghetti L, Levi G ( 1998) Microglia as effector cells in brain damage and repair: focus on prostanoids and nitric oxide. Prog Neurobiol 54: 99 —125. [DOI] [PubMed] [Google Scholar]
- Noh KM, Koh JY ( 2000) Induction and activation by zinc of NADPH oxidase in cultured cortical neurons and astrocytes. J Neurosci 20: RC111(1—5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olanow CW, Tatton WG ( 1999) Etiology and pathogenesis of Parkinson's disease. Annu Rev Neurosci 22: 123—144. [DOI] [PubMed] [Google Scholar]
- Priyadarshi A, Khuder SA, Schaub EA, Priyadarshi SS ( 2001) Environmental risk factors and Parkinson's disease: a metaanalysis. Environ Res 86: 122—127. [DOI] [PubMed] [Google Scholar]
- Rajput AH, Uitti RJ ( 1987) Paraquat and Parkinson's disease. Neurology 37: 1820 —1821. [DOI] [PubMed] [Google Scholar]
- Ritz B, Yu F ( 2000) Parkinson's disease mortality and pesticide exposure in California 1984 —1994. Int J Epidemiol 29: 323—329. [DOI] [PubMed] [Google Scholar]
- Semchuk KM, Love EJ, Lee RG ( 1992) Parkinson's disease and exposure to agricultural work and pesticide chemicals. Neurology 42: 1328 —1335. [DOI] [PubMed] [Google Scholar]
- Sherer TB, Betarbet R, Stout AK, Lund S, Baptista M, Panov AV, Cookson MR, Greenamyre JT ( 2002) An in vitro model of Parkinson's disease: linking mitochondrial impairment to altered α-synuclein metabolism and oxidative damage. J Neurosci 22: 7006 —7015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherer TB, Betarbet R, Kim JH, Greenamyre JT ( 2003) Selective microglial activation in the rat rotenone model of Parkinson's disease. Neurosci Lett 341: 87—90. [DOI] [PubMed] [Google Scholar]
- Stolk J, Hiltermann TJ, Dijkman JH, Verhoeven AJ ( 1994) Characteristics of the inhibition of NADPH oxidase activation in neutrophils by apocynin, a methoxy-substituted catechol. Am J Respir Cell Mol Biol 11: 95—102. [DOI] [PubMed] [Google Scholar]
- Streit WJ, Graeber MB, Kreutzberg GW ( 1988) Functional plasticity of microglia: a review. Glia 1: 301—307. [DOI] [PubMed] [Google Scholar]
- Tammariello SP, Quinn MT, Estus S ( 2000) NADPH oxidase contributes directly to oxidative stress and apoptosis in nerve growth factor-deprived sympathetic neurons. J Neurosci 20: RC53(1—5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiruchelvam M, Richfield EK, Baggs RB, Tank AW, Cory-Slechta DA ( 2000) The nigrostriatal dopaminergic system as a preferential target of repeated exposures to combined paraquat and maneb: implications for Parkinson's disease. J Neurosci 20: 9207—9214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiruchelvam M, Richfield EK, Goodman BM, Baggs RB, Cory-Slechta DA ( 2002) Developmental exposure to the pesticides paraquat and maneb and the Parkinson's disease phenotype. Neurotoxicology 23: 621—633. [DOI] [PubMed] [Google Scholar]
- Turrens JF, Boveris A ( 1980) Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem J 191: 421—427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky VN, Li J, Fink AL ( 2001) Pesticides directly accelerate the rate of alpha-synuclein fibril formation: a possible factor in Parkinson's disease. FEBS Lett 500: 105—108. [DOI] [PubMed] [Google Scholar]
- Uversky VN, Li J, Bower K, Fink AL ( 2002) Synergistic effects of pesticides and metals on the fibrillation of alpha-synuclein: implications for Parkinson's disease. Neurotoxicology 23: 527—536. [DOI] [PubMed] [Google Scholar]
- Vila M, Jackson-Lewis V, Guegan C, Wu DC, Teismann P, Choi DK, Tieu K, Przedborski S ( 2001) The role of glial cells in Parkinson's disease. Curr Opin Neurol 14: 483—489. [DOI] [PubMed] [Google Scholar]