

Abstract
Adaptive responses mediated by the hypothalamus require sustained activation until homeostasis is achieved. Increases in excitatory drive to the magnocellular neuroendocrine cells that mediate these responses, however, result in the activation of a presynaptic metabotropic glutamate receptor (mGluR) that curtails synaptic excitability. Recent evidence that group III mGluRs can be inhibited by protein kinase C prompted us to test the hypothesis that activation of PKC by noradrenaline (NA) inhibits group III mGluRs and increases excitatory synaptic input to these cells. To examine the effects of NA on miniature EPSCs (mEPSCs), we obtained whole-cell recordings from magnocellular vasopressin and oxytocin neurons in the paraventricular nucleus of the hypothalamus. All of the neurons tested in the current study displayed an α1 adrenoceptor-mediated increase in mEPSC frequency in response to NA (1–200 μm). The excitatory effects of NA were mimicked by the phorbol ester PMA and blocked by the PKC inhibitor calphostin C. The activation of PKC inhibits the efficacy of group III mGluRs, resulting in an increase in mEPSC frequency in response to a subsequent exposure to NA. By removing feedback inhibition, this mechanism effectively primes the synapses such that subsequent activation is more efficacious. The novel form of synaptic rescaling afforded by this cross-talk between distinct metabotropic receptors provides a means by which ascending catecholamine inputs can facilitate the control of homeostasis by hypothalamic networks.
Keywords: magnocellular, hypothalamus, presynaptic inhibition, G-protein-coupled receptors, noradrenaline, paraventricular nucleus
Introduction
In the CNS, the release of neurotransmitter from the nerve terminal is regulated by a number of inhibitory feedback mechanisms (Zucker and Regehr, 2002). These mechanisms include, but are not limited to, the activation of high-affinity presynaptic receptors by neurotransmitters such as acetylcholine (MacDermott et al., 1999), adenosine (Oliet and Poulain, 1999; Dunwiddie and Masino, 2001), or GABA (Mouginot et al., 1998). At the majority of excitatory synapses, however, this feedback is initiated by the binding of glutamate to presynaptic, G-protein-coupled autoreceptors (for review, see Schoepp, 2001). Activation of these metabotropic glutamate receptors (mGluRs) results in a decrease in the release of neurotransmitter (Baskys and Malenka, 1991; Schoppa and Westbrook, 1997), thereby providing a local, short-term mechanism through which synaptic strength (Anwyl, 1991; Scanziani et al., 1997; Oliet et al., 2001) and hyperexcitability (Sansig et al., 2001) may be regulated.
In certain physiological situations, however, neuronal output must be sustained for longer durations. Under these conditions, a process that limits the continuous release of glutamate may be unfavorable. In particular, adaptive responses mediated by the hypothalamus generally require the sustained activation of neuronal populations until homeostasis is achieved. In the supraoptic nucleus (SON) and paraventricular nucleus (PVN), the magnocellular neuroendocrine cells exhibit prolonged discharges in response to specific physiological perturbations (Wakerley et al., 1978). This increase in neuronal activity may be achieved by changes in intrinsic conductances (Bourque and Renaud, 1984; Legendre and Poulain, 1992; Hatton and Li, 1998; Shibuya et al., 2000) or, alternatively, by a persistent increase in excitatory synaptic drive (Nissen et al., 1995; Moos et al., 1997; Jourdain et al., 1998; Shibuya et al., 2000) to these neurons. Because this glutamatergic input to magnocellular neuroendocrine cells is regulated by presynaptic mGluRs (Schrader and Tasker, 1997; Oliet et al., 2001), this raises the possibility that targeting mGluR autoreceptor activity may be an attractive means for potentiating this input and ultimately augmenting the output of these neurons.
Recent evidence demonstrates that an increase in protein kinase C activity functionally inhibits mGluRs (Macek et al., 1998, 1999; Nakajima et al., 1999). Thus, substrates that activate signaling pathways linked to PKC may exert long-lasting changes in synaptic efficacy by decreasing mGluR activity. In the hypothalamus, the physiological trigger for this override of mGluR feedback may be the activation of presynaptic, G-protein-coupled α1 adrenoceptors by noradrenaline (NA), which is released from ascending afferents in response to a number of physiological challenges (Crowley et al., 1987; Leibowitz et al., 1990). Because α1 adrenoceptors are positively coupled to PKC, we hypothesize that the functional inactivation of mGluRs by NA primes excitatory glutamatergic synapses terminating on magnocellular neuroendocrine cells.
Using whole-cell, voltage-clamp recordings from magnocellular neurons in the PVN of the hypothalamus, we demonstrate that α1 adrenoceptor activation increases glutamate release and inactivates presynaptic mGluRs. This PKC-mediated inhibition of negative feedback is long lasting, as evidenced by an amplification of transmitter release in response to subsequent application of NA. The rescaling of synaptic input that results from cross-talk between distinct metabotropic receptors provides a means by which ascending catecholamine inputs can facilitate the control of homeostasis by hypothalamic networks.
Materials and Methods
Hypothalamic slices containing the PVN were prepared from young adult, postnatal day (P) 21–27, male Sprague Dawley rats. Animals were anesthetized with sodium pentobarbitol (3 mg/kg) and decapitated, and the brains were rapidly removed into ice-cold high-sucrose slicing solution saturated with 95% O2 and 5% CO2 and allowed to cool for ∼3 min. The brain was then blocked and mounted on a vibrating slicer (Leica, Nussloch, Germany) and submerged in ice-cold slicing solution containing the following (in mm): 87 NaCl, 2.5 KCl, 25 NaHCO3, 0.5 CaCl2, 7 MgCl2, 1.25 NaH2PO4, 25 glucose, 75 sucrose, 3 pyruvic acid, and 1 ascorbic acid, saturated with 95% O2 and 5% CO2. The brain was cut in the coronal plane, and hemisected slices of 300 μm thickness containing the hypothalamus were incubated at 32.5°C in a submerged chamber of artificial CSF (ACSF) for a minimum of 60 min before recording. ACSF contained the following (in mm): 126 NaCl, 2.5 KCl, 26 NaHCO3, 2 CaCl2, 2 MgCl2, 1.25 NaH2PO4, 10 glucose, and 1 ascorbic acid, saturated with 95% O2 and 5% CO2. Whole-cell recordings were obtained from magnocellular neurons visually identified using an upright microscope (AxioskopII FS Plus; Zeiss, Oberkochen, Germany) fitted with infrared differential interference contrast. All recordings were obtained at 32.5°C using borosilicate glass microelectrodes (tip resistance, 3–7 MΩ). The intracellular solution contained the following (in mm): 123 potassium gluconate, 2 MgCl2, 8 NaCl, 1 potassium EGTA, 4 potassium ATP, and 0.3 sodium GTP, buffered with 16 mm KHCO3. The internal solution was filtered before use.
With the exception of the initial confirmation of cell type based on the electrical fingerprint in bridge mode, all experiments were performed in voltage-clamp mode, and recordings were accepted when access resistance changes were limited to <15%. All experiments were performed on PVN magnocellular neurons first identified visually and confirmed by their prominent delay to first spike in response to depolarizing current steps from hyperpolarized potentials (Tasker and Dudek, 1991). The perfusate always contained picrotoxin (100 μm) to block inhibitory GABAA synaptic currents and tetrodotoxin (1 μm) to block voltage-gated Na + channel and inhibit action potential-driven release of neurotransmitter.
Signals were amplified with the Multiclamp 700A amplifier (Axon Instruments, Foster City, CA), low-pass filtered at 1 kHz, and digitized at 5–10 kHz using the Digidata 1322 (Axon Instruments). Data were collected (pClamp; Axon Instruments) and stored on computer for off-line analysis using software designed to detect miniature synaptic events using a variable threshold (MiniAnalysis; Synaptosoft, Decatur, GA). Experimental values are presented as means ± SEM, and statistical analyses were performed using Student's t test when comparing two groups and ANOVA with a post hoc Newman–Keuls test for comparisons across multiple groups. p < 0.05 was accepted as statistically significant (*p < 0.05; **p < 0.01).
Reagents were obtained from the following sources. TTX, picrotoxin, calphostin C, PMA, 1-[3,4-dihydroxyphenyl]-2-aminoethanol (NA/arterenol), and 1-[4-amino-6,7-dimethoxy-2-quinazolinyl]-4-[2-furanyl-carbonyl]-piperazine (prazosin), were purchased from Sigma (St. Louis, MO). TTX was also purchased from Alomone Labs (Jerusalem, Israel). l-(+)-2-amino-4-phosphonobutyric acid (l-AP4) and (S)-2-amino-2-methyl-4-phosphonobutanoic acid (MAP4) were purchased from Tocris Cookson (Ballwin, MO). Picrotoxin and calphostin C were dissolved in dimethylsulfoxide in which the final bath concentration of DMSO was <0.1%. Prazosin was dissolved in methanol, in which the final bath concentration of methanol was <0.05%. All other reagents were dissolved in ACSF.
Results
In contrast to neurons in most cortical regions in which the spontaneous, action potential-independent release of neurotransmitter is relatively infrequent (Staley, 1999), the magnocellular neuroendocrine cells of the hypothalamus are subjected to relatively high rates of quantal input. In the cells tested in the current study, the rate of stochastic release under basal conditions ranged from 0.46 to 8 Hz (mean, 2.74 ± 0.35 Hz). This high frequency of events combined with the demonstration that in magnocellular neurons, increases in mEPSC frequency lead to increases in firing (Kombian et al., 2000) suggests that quantal glutamatergic release conveys important signaling information to these neurons.
NA exerts predominantly excitatory effects on magnocellular neurons (Day et al., 1984; Armstrong et al., 1986; Randle et al., 1986b). These are partially attributable to a direct α1-mediated depolarization of the postsynaptic membrane (Randle et al., 1986a) and an increase in TTX-sensitive glutamate release (Daftary et al., 1998). The effects of NA on quantal release at the presynaptic nerve terminal, however, have been less well defined. To clarify a role for NA at afferent excitatory terminals synapsing on magnocellular neurons, we examined the effects of this compound on TTX-insensitive mEPSCs.
NA increases the frequency and amplitude of mEPSCs
In response to a brief application of NA (2 min, 100 μm), all of the neurons tested exhibited an increase in mEPSC frequency (354.2 ± 34.9% of control; n = 24; p < 0.01) (Fig. 1A–C). Although in the majority of cells tested (63%, n = 15 of 24) NA had no effect on mEPSC amplitude (99.8 ± 2.8% of control; p > 0.05) (Fig. 1B,D), an increase in amplitude was observed in the remaining cells (123.3 ± 3.0% of control; p < 0.01) (Fig. 1D). We also noted a mean inward current of 22.1 ± 2.2 pA in cells tested at 100 μm (p < 0.05; data not shown), consistent with a direct postsynaptic effect of NA on magnocellular neurons (Randle et al., 1986a; Daftary et al., 1998). We next tested the effects of longer NA applications on mEPSC frequency and amplitude. In response to NA applications that ranged from 4 to 10 min, the mEPSC frequency increased to 612.2 ± 114.2% of control (p < 0.01; n = 28) (Fig. 1E). Interestingly, in these longer applications, the majority of cells (86%; n = 24 of 28) exhibited an increase in mEPSC amplitude (141.8 ± 4.2% of control; p < 0.01) (Fig. 1F). The remaining cells (14%; n = 4 of 28) displayed no change in mEPSC amplitude (99.7 ± 2.3% of control; p > 0.05) (Fig. 1F). This discrepancy in response profiles for short versus long applications of NA suggests a time-dependent component to the excitation induced by NA.
Figure 1.
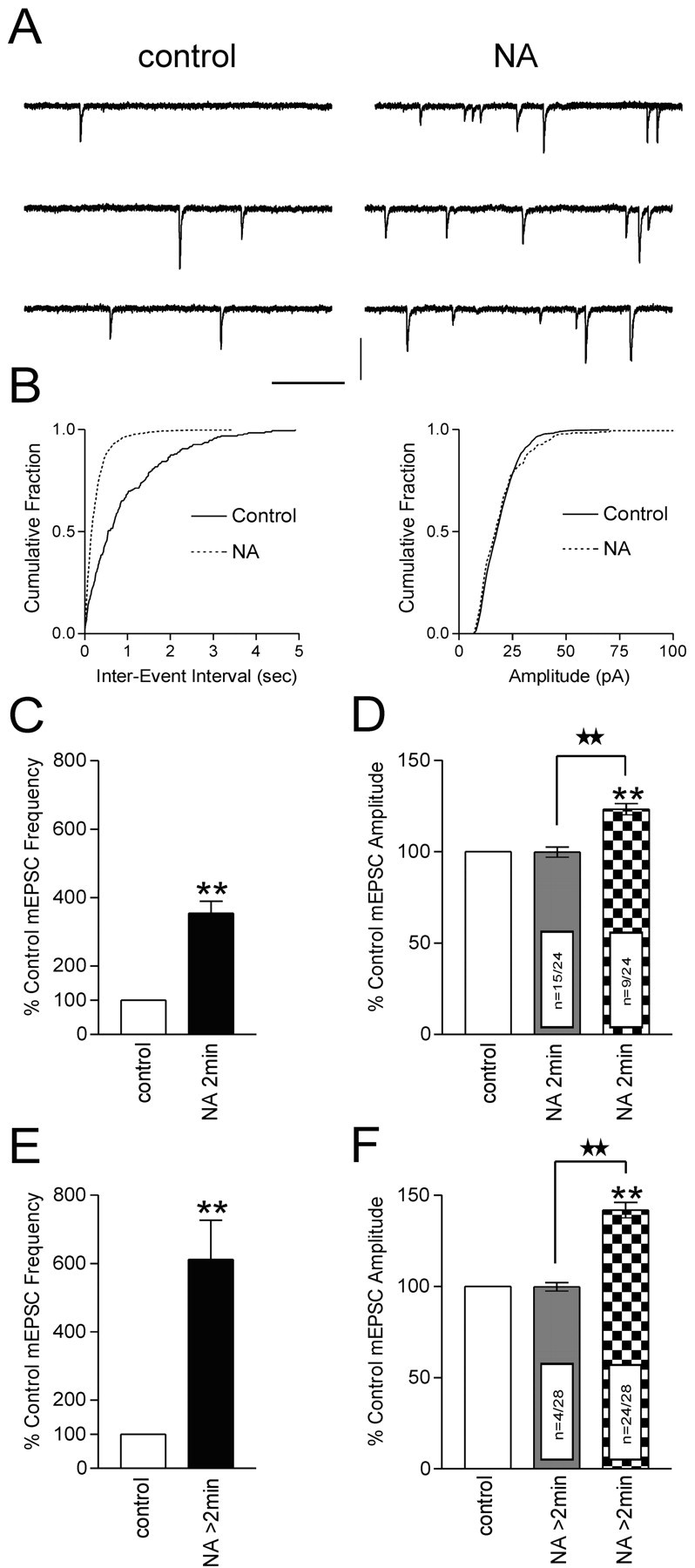
NA increases frequency and amplitude of mEPSCs. A, Representative voltage-clamp traces from a cell tested with a 2 min application of NA (100 μm). B, In the cumulative fraction plots, NA decreases the inter-event interval (left, p < 0.01) with no change in mEPSC amplitude (right, p > 0.05). C–F, Summary bar graphs of NA data. C, A 2 min application of NA increase smEPSC frequency to 354.2 ± 34.9% of control (p < 0.01; n = 24). D, In 63% of these cells (n = 15 of 24), there is no change in mEPSC amplitude (99.8 ± 2.8% of control1; p > 0.05). The remaining 37% of cells tested (n = 9 of 24) exhibit an increase in mEPSC amplitude (123.3 ± 3.0% of control; p < 0.01). E, When the NA application exceeds 2 min, the mEPSC frequency increases to 612.2 ± 114.2% of control (p < 0.01; n = 28). F, In these cells, 86% (n = 24 of 28) exhibited an increase in mEPSC amplitude (141.8 ± 4.2% of control; p < 0.01). No change in mEPSC amplitude (99.7 ± 2.3% of control; p > 0.05) was observed in the remaining 14% (n = 4 of 28). Calibration (in A): 15 pA, 250 msec. Stars indicate comparing treatment with treatment. Asterisks indicate comparing treatment with control.
The effects of NA were also dose dependent. Cells tested with brief applications of 1, 10, and 200 μm NA exhibited increases in mEPSC frequency of 104.6 ± 8.6% (n = 2), 188.4 ± 14.9% (n = 6), and 877.6 ± 37.8% (n = 2), respectively (data not shown). No inward current was observed at 1 μm NA, whereas 10 μm NA elicited significantly less inward current compared with 100 μm treatments (9.7 ± 2.4 pA; p < 0.05; n = 3; data not shown). Because of the robust increase in mEPSC frequency, we were unable to accurately assess changes in holding current for 200 μm NA.
NA primes glutamatergic synapses
Sustained increases in neuronal activity and hormonal output are reliably observed in magnocellular neuroendocrine cells in response to acute physiological stressors such as dehydration or hemorrhage. Although a single hemorrhagic stimulus elicits a sustained increase in hormone release from these cells, repeated hemorrhages result in an amplification of hormone release (Lilly et al., 1989). We examined whether repeated applications of NA would mimic these observations and provide an explanation at the synaptic level for the amplification of hormone release. We conducted experiments that used brief (2 min) repetitive applications of NA. The initial application of NA elicited an increase (Fig. 2A–C) in the frequency of mEPSCs (403.8 ± 77.2% of control; n = 6; p < 0.01) (Fig. 2E). After the recovery of mEPSC frequency to control levels, the synapses were rechallenged for 2 min with the same dose of NA. This application (15 min after the initial test) resulted in an additional amplification (Fig. 2A–C)in the mEPSC frequency (749.8 ± 126.4% of control; n = 6; p < 0.01) (Fig. 2E). We also observed an increase in mEPSC amplitude in response to the second application of NA only (Fig. 2B,D). We did not observe any changes in the kinetics of the mEPSCs after application of NA (Fig. 2D, inset scaled traces). The mEPSC amplitude increased from 113.4 ± 7.8% (p > 0.05) of control to 143.9 ± 7.8% (p < 0.05) (Fig. 2F) of control for NA1 and NA2, respectively. Additional applications of NA failed to increase the frequency of mEPSCs further (n = 3; data not shown). Our analysis failed to reveal any consistent changes in amplitude for subsequent applications. These results indicate that NA primes excitatory synapses for subsequent exposure to the agonist.
Figure 2.
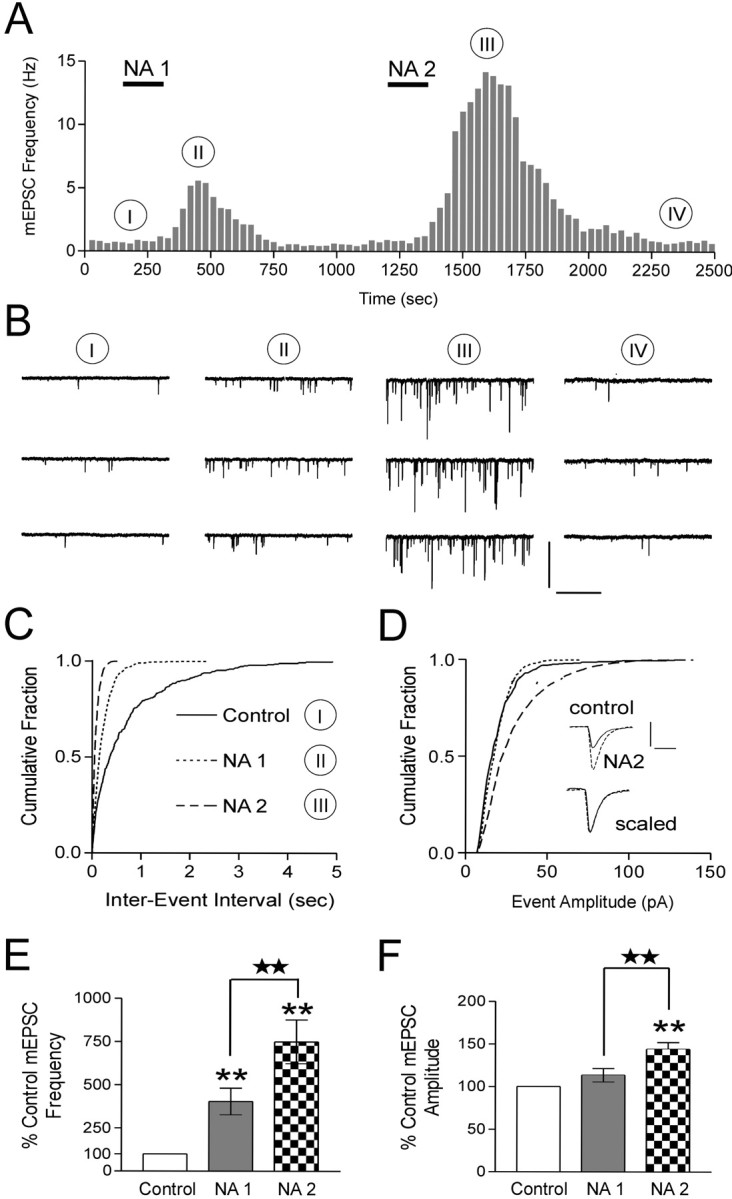
NA primes excitatory synapses. A, Two 2 min applications of NA (100 μm) separated by 15 min demonstrate an enhancement of the mEPSC frequency from NA1 to NA2. B, Representative voltage-clamp traces from the same neuron as A. The roman numerals in B correspond to the same time point in A. C, Cumulative fraction of mEPSC inter-event intervals before and during each NA application in this neuron. NA2 elicits an additional decrease in mEPSC inter-event interval (p < 0.01). D, Cumulative fraction of the amplitude of mEPSCs before and during each NA application. Here, only NA2 results in a significant increase in mEPSC amplitude (p < 0.05). Inset traces are averages of 40 spontaneous events in control and NA2. Control and NA2 traces are overlaid below to demonstrate that no change in the kinetics of the mEPSCs is observed. E, Summary bar graph showing the increase in mEPSC frequency after each successive NA application (NA1, 403.8 ± 77.2% of control; p < 0.01; NA2, 749.8 ± 126.4% of control; p < 0.01; n = 6). The mEPSC frequencies from NA1 and NA2 are significantly different (p < 0.05). F, Summary bar graph of increases in mEPSC amplitude (NA1, 113.4 ± 7.8% of control; p > 0.05; NA2, 143.9 ± 7.8% of control; p < 0.01, n = 6). The mEPSC amplitudes from NA1 and NA2 are significantly different (p < 0.05). Calibration: B, 50 pA, 1 sec; D, 20 pA, 5 msec. Stars indicate comparing treatment with treatment. Asterisks indicate comparing treatment with control.
Increase in mEPSC frequency is
α1 adrenoceptor mediated and PKC dependent The excitatory effects of NA on magnocellular neurons involve the activation of the α1 adrenoceptor (Day et al., 1984; Armstrong et al., 1986; Randle et al., 1986b). These effects have been characterized as a direct α1-mediated depolarization of the postsynaptic membrane and an α1-mediated increase in TTX-sensitive glutamate release (Randle et al., 1986a; Daftary et al., 1998). On the basis of these findings, we tested whether activation of the α1 adrenoceptor was necessary for the NA-induced increase in mEPSC frequency. The α1 adrenoceptor antagonist prazosin (10 μm) completely abolished the excitatory effects of NA, uncovering an inhibitory action (mEPSC frequency decreased to 35.2 ± 5.2% of control values; p < 0.01; n = 12) (Fig. 3A,F). These data are consistent with previous results demonstrating an α1-mediated increase in spontaneous IPSCs in PVN neurons that masks a weaker inhibition of GABA release via α2 adrenoceptor (Han et al., 2002). Our results suggest a similar role for NA at excitatory synapses. These findings demonstrate that NA acts predominantly via presynaptic α1 adrenoceptors to increase glutamate release from afferent nerve terminals. This increase in mEPSC frequency likely acts in concert with the previously defined actions of this catecholamine in sustaining the elevated activity of these cells.
Figure 3.
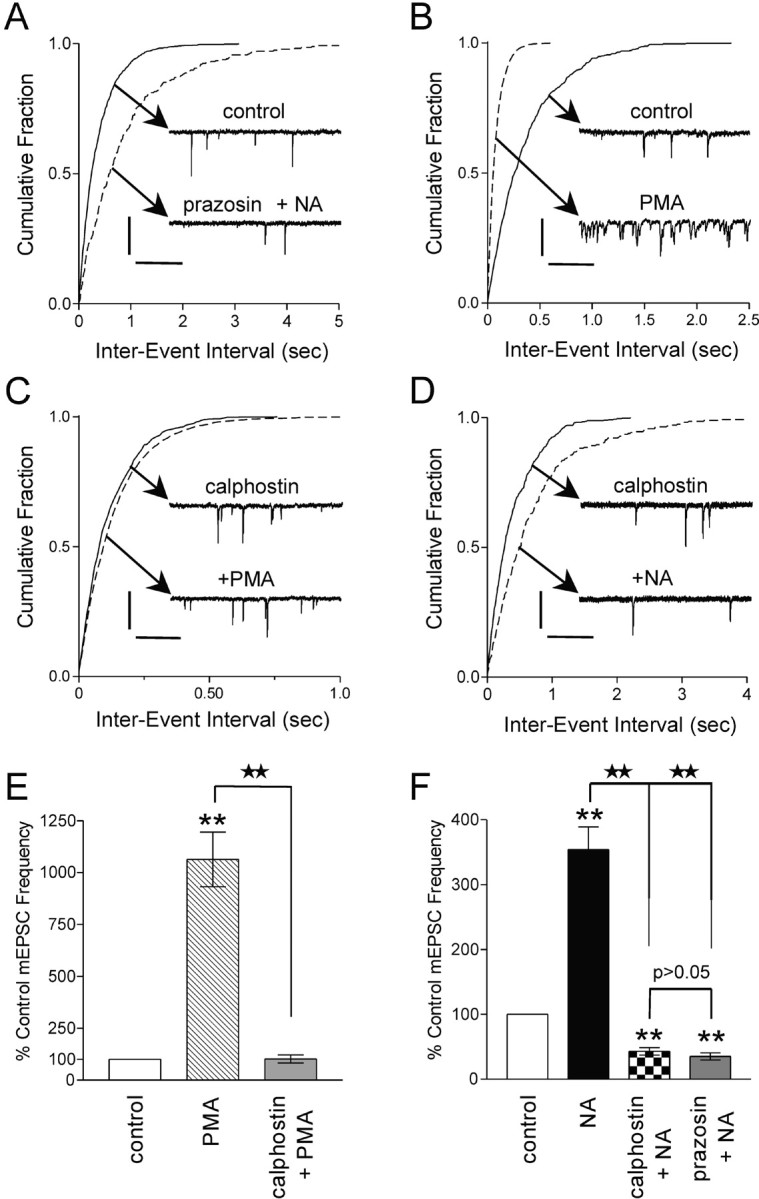
Increase in mEPSC frequency isα1 adrenoceptor mediated and PKC dependent. A, Cumulative fraction of mEPSC inter-event intervals before and during prazosin (10 μm) plus NA (100 μm) application. Inset are voltage-clamp traces depicting a decrease in the mEPSC frequency in response to NA when the α1 adrenoceptor is blocked (p < 0.01). B, Cumulative fraction of mEPSC inter-event intervals before and during PMA (1 μm) application. Inset are voltage-clamp traces depicting an increase in the mEPSC frequency in response to PMA (p < 0.01). C, Cumulative fraction of mEPSC inter-event intervals in response to PMA (1 μm) in the presence of calphostin C (100 nm). Inset are voltage-clamp traces depicting that no change in mEPSC frequency in response to PMA in the presence of calphostin C (p > 0.05) is observed. D, Cumulative fraction of mEPSC inter-event interval in response to NA (100 μm), in the presence of calphostin C (100 nm). Inset are voltage-clamp traces depicting a decrease in mEPSC frequency in response to NA in calphostin C (p < 0.05). E, Summary bar graph showing that PMA alone increases mEPSC frequency (1063.5 ± 131.4% of control; p < 0.01; n = 5) but fails to increase mEPSC frequency in the presence of calphostin C (102.1 ± 19.5% of control; n = 5; p > 0.05). F, Summary bar graph showing that NA decreases the frequency of mEPSCs in the presence of calphostin C (43.0 ± 5.7% of control; p < 0.01; n = 5) or prazosin (35.2 ± 5.2% of control; p < 0.01; n = 12). Calibration: A, 20 pA, 250 msec; B, 25 pA, 100 msec; C, 20 pA, 250 msec; D, 15 pA, 250 msec. Stars indicate comparing treatment with treatment. Asterisks indicate comparing treatment with control.
The α1 adrenoceptor activates phospholipase C through the Gq family of G-proteins, resulting in phosphatidylinositol metabolism, the release of stored Ca2+, and activation of PKC (for review, see Zhong and Minneman, 1999). PKC activation mobilizes the reserve pool of vesicles (Gillis et al., 1996; Stevens and Sullivan, 1998), increases the sensitivity of the release process for Ca2+ (Brager et al., 2002), and facilitates the fusion of vesicles (Scepek et al., 1998; Yawo, 1999). These mechanisms may act individually or in concert to increase neurotransmitter release. Phorbol esters, such as PMA, activate PKC by binding to the C1 domain in an ATP-dependent manner (Liu and Heckman, 1998). The nonphysiological activation of PKC by lipid-soluble phorbol esters has been used widely for discerning the effects of the kinase in a number of neuronal and neuroendocrine preparations (Hilfiker and Augustine, 1999). To test the hypothesis that phorbol esters mimic the effects of NA, we examined mEPSC frequency in response to PMA (1 μm) (Fig. 3B). mEPSC frequency dramatically increased (1063.5 ± 131.4% of control; n = 5; p < 0.01) (Fig. 3E) in response to a 10 min PMA application. We observed no change in mEPSC amplitude (data not shown). To confirm that this increase in release was a result of the activation of PKC, the slice was incubated for 30 min with the broad-spectrum PKC inhibitor calphostin C (100 nm) (Hasuo et al., 2002), and the PMA application was repeated (Fig. 3C). In the presence of calphostin C, PMA treatment had no effect on mEPSC frequency (102.1 ± 19.5% of control; n = 5; p > 0.05) (Fig. 3E). These data suggest that PMA acts presynaptically to activate PKC and increase the release of glutamate.
Experiments were conducted to test whether PKC activation was necessary for the NA-mediated increase in glutamate release. Slices were preincubated for 30 min with calphostin C (100 nm), and NA was applied at a dose (100 μm) that normally elicits a robust increase in mEPSC frequency (Fig. 3D). Calphostin C completely abolished the NA-induced increase in mEPSC frequency and unmasked an inhibitory effect (mEPSC frequency decreased to 43.0 ± 5.7% of control; p < 0.01; n = 5) (Fig. 3F).
Phorbol ester and NA-induced PKC activation can attenuate inhibitory mGluR activity
Membrane-binding assays have established a robust distribution of mGluRs in the hypothalamus (Meeker et al., 1994). In particular, electrophysiological evidence demonstrates an important role for group III mGluRs in regulating glutamate release at magnocellular synapses (Schrader and Tasker, 1997; Oliet et al., 2001). We confirmed this finding with the demonstration that bath application of the group III mGluR agonist l-AP4 (25 μm) decreased the frequency of mEPSCs in magnocellular neurons (36.9 ± 8.5%; n = 3; p < 0.05) (Fig. 4A,C). No apparent postsynaptic effects of the agonist were observed (data not shown).
Figure 4.
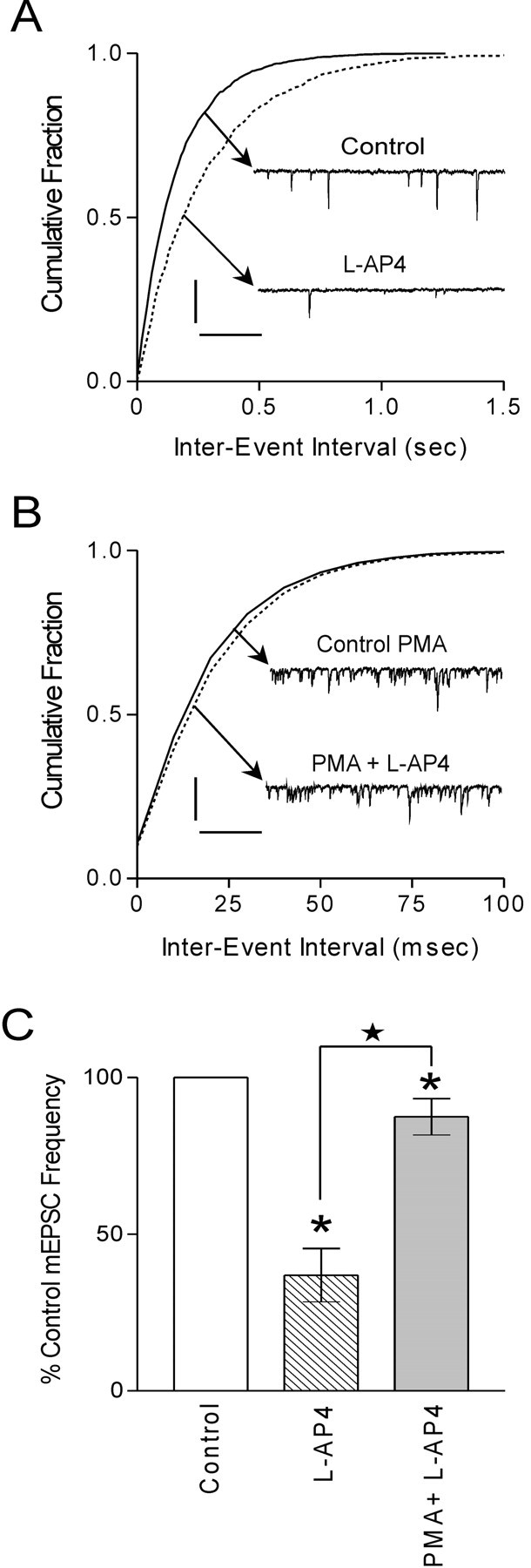
PKC activation attenuates mGluR-mediated inhibition. A, Cumulative fraction of mEPSC inter-event interval before and during l-AP4 (25 μm) application. Inset are voltage-clamp traces depicting a decrease in the mEPSC frequency in response to l-AP4 (p < 0.01). B, Cumulative fraction of mEPSC inter-event interval in response to l-AP4 (25 μm) in the presence of PMA (1 μm). Inset are voltage-clamp traces depicting a small decrease in mEPSC frequency in response to l-AP4 in PMA (p > 0.05). C, Summary bar graph demonstrates that l-AP4 decreases mEPSC frequency (36.9 ± 8.5% of control; p < 0.05; n = 3). In the presence of PMA, l-AP4 exhibits a small decrease in mEPSC frequency (87.5 ± 5.8% of control; p < 0.05; n = 5). The decrease in mEPSC frequency in l-AP4 is significantly different from that observed in PMA plus l-AP4 (p < 0.05). Calibration: A, 20 pA, 250 msec; B, 25 pA, 100 msec. Stars indicate comparing treatment with treatment. Asterisks indicate comparing treatment with control.
mGluRs themselves are also subject to regulation by intracellular messengers. Specifically, some studies propose the inhibition of group III mGluRs by PKC (Macek et al., 1999; Sorensen et al., 2002). To test the hypothesis that mGluR function would be compromised after the activation of PKC, we applied PMA (1 μm) and then l-AP4 (25 μm) 15 min later. As demonstrated above, PMA application elicited a robust increase in mEPSC frequency. In the presence of PMA, the effect of l-AP4 on mEPSC frequency was blunted (87.5 ± 5.8% of control; n = 5; p < 0.05) (Fig. 4B,C). This response was significantly different from l-AP4 alone (p < 0.05). These data suggest that PKC activation attenuates the decrease in glutamate release normally observed in response to activation of group III mGluRs on the presynaptic terminal.
We next examined whether activation of PKC by NA would also attenuate mGluR function. After a return to baseline of the NA-induced increase in mEPSC frequency, l-AP4 was applied at a dose that had previously decreased mEPSC frequency (25 μm) (Fig. 5A). Under these conditions, the effect of l-AP4 on mEPSC frequency was significantly attenuated (l-AP4 after NA; 82.1 ± 3.7% of control; p < 0.01; n = 5) (Fig. 5C) compared with l-AP4 alone (p < 0.01) (Fig. 4A,C, comparison not shown). Finally, we tested whether activation of the α1 adrenoceptor by NA was necessary for the inhibition of group III mGluRs. After the application of NA in the presence of prazosin (to block α1 adrenoceptors), l-AP4 clearly decreased the frequency of mEPSCs (42.9 ± 2.8% of control; p < 0.01; n = 7) (Fig. 5B,C). This was significantly different from the effects of l-AP4 after NA alone (p < 0.01) but was not different from l-AP4 alone (p > 0.05) (Fig. 4C, comparison not shown).
Figure 5.
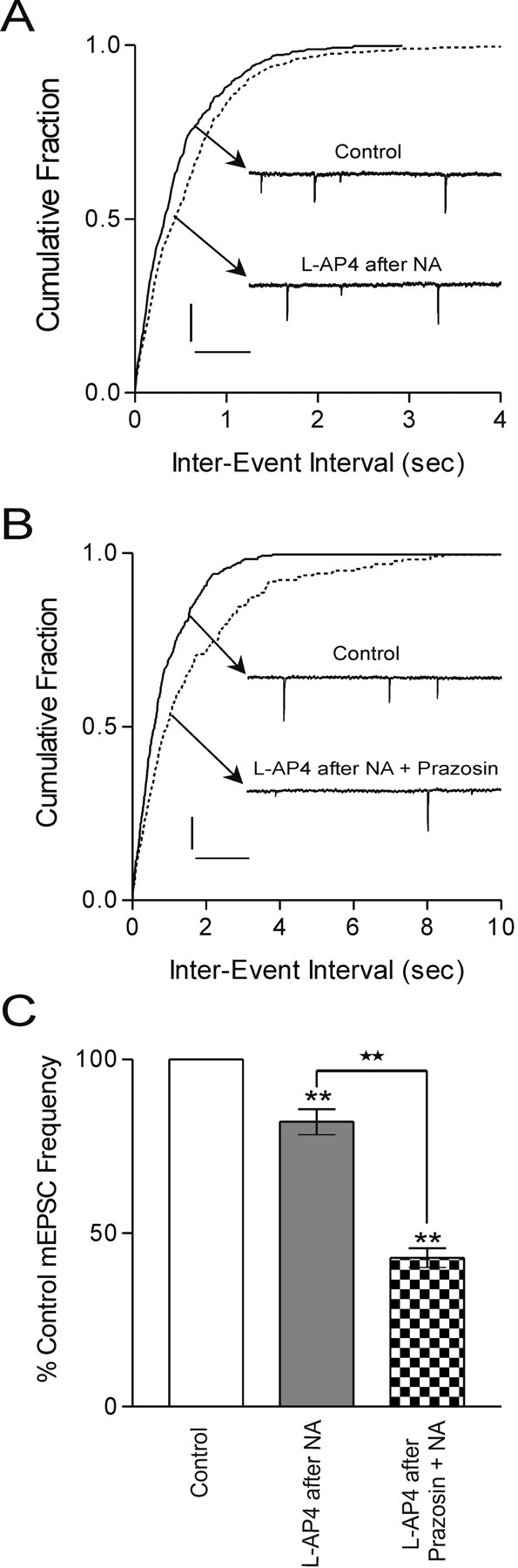
α1 adrenoceptor activation attenuates mGluR-mediated inhibition. A, Cumulative fraction of mEPSC inter-event interval before and during l-AP4 (25 μm) after application of NA (100 μm). Inset are voltage-clamp traces depicting a small decrease in mEPSC frequency in response to l-AP4 after NA (p < 0.05). B, Cumulative fraction of mEPSC inter-event interval before and during l-AP4 (25 μm) after application of prazosin (10 μm) plus NA (100 μm). Inset are voltage-clamp traces depicting a clear decrease in mEPSC frequency in response to l-AP4 after NA when the α1 adrenoceptor is blocked (p < 0.01). C, Summary bar graph showing a decrease in the frequency of mEPSCs with l-AP4 after NA (82.1 ± 3.7% of control; p < 0.01; n = 5) and with l-AP4 after prazosin plus NA (42.9 ± 2.8% of control; p < 0.01; n = 7). The decrease in mEPSC frequency after l-AP4 after NA is significantly different from that observed in l-AP4 after prazosin plus NA (p < 0.01). Calibration: A, 20 pA, 250 msec; B, 25 pA, 200 msec. Stars indicate comparing treatment with treatment. Asterisks indicate comparing treatment with control.
Blockade of group III mGluRs after NA1 does not affect priming
We next tested whether pharmacological inhibition of group III mGluRs during the second NA application would alter the priming response. If synaptic priming occurs because of different activation states of the mGluR (where less glutamate release during NA1 is attributed to a functional mGluR and more glutamate release during NA2 is attributed to mGluR inactivation), then inhibiting the mGluR during NA2 should have little effect on priming. The priming experiment was repeated, but instead, the group III mGluR antagonist MAP4 (250 μm) was bath applied after NA1 (Fig. 6A). The initial application of NA increased the frequency of mEPSCs to 276.3 ± 38.9% of control (p < 0.01; n = 7) (Fig. 6A,B). After the recovery of mEPSC frequency to control levels, the synapses were rechallenged with NA in the presence of MAP4. This resulted in an additional amplification in the mEPSC frequency to 427.0 ± 70.5% of control (p < 0.01; n = 7) (Fig. 6A,B). The responses elicited by NA1 and NA2 are significantly different (p < 0.01) (Fig. 6B). Consistent with the results from earlier experiments, we observed an increase in mEPSC amplitude in response to the second application of NA (Fig. 6C). The mEPSC amplitude increased from 107.0 ± 2.4% (p > 0.05) of control to 150.6 ± 7.9% (p < 0.01; n = 5) (Fig. 6C) of control for NA1 and NA2, respectively. These data collectively suggest that mGluRs are inactivated after NA application. Consequently, they are unavailable to curtail the release of glutamate when the synapses are rechallenged with NA.
Figure 6.

Blockade of group III mGluRs after NA1 does not affect priming. A, The priming effect of NA is still observed when NA2 is applied in the presence of MAP4 (250 μm). NA2 (2 min, 100 μm) shows a greater increase in mEPSC frequency than NA1 (2 min, 100 μm). B, Summary bar graph of increases in mEPSC frequency (NA1, 276.3 ± 38.9% of control; p < 0.01; NA2, 427.0 ± 70.5% of control; p < 0.01; n = 7). The mEPSC frequencies from NA1 and NA2 are significantly different (p < 0.01). C, Summary bar graph showing effects of NA on mEPSC amplitude (NA1, 107.0 ± 2.4% of control; p > 0.05; NA2, 150.6 ± 7.9%; p < 0.01; n = 5). The mEPSC amplitudes from NA1 and NA2 are significantly different (p < 0.01). Stars indicate comparing treatment with treatment. Asterisks indicate comparing treatment with control.
Group III mGluRs mediate priming
Finally, we tested the inhibitory contribution of group III mGluRs during the initial response to NA. If synaptic priming is caused by differences in mGluR activity during NA1 and NA2, then inhibiting the mGluR for the duration of NA1 and NA2 application should potentiate release under both conditions, effectively eliminating the observation of “priming.” In the presence of MAP4, NA elicited a robust increase in mEPSC frequency (353.5 ± 61.1%; n = 4; p < 0.01) (Fig. 7A,B) but had no effect on mEPSC amplitude (95.4 ± 4.4%; n = 4; p > 0.05) (Fig. 7C). We failed to see any more potentiation when NA was applied a second time in the presence of MAP4. The effect on mEPSC frequency (376.5 ± 57%; p < 0.01) (Fig. 7A,B) and amplitude (93.5 ± 4.5%; p > 0.05) (Fig. 7C) by NA2 was not different from the response to the first application (p > 0.05) (Fig. 7B,C). The absence of any change in amplitude may be because of the continuous application of MAP4 for the duration of the experiment. The demonstration that inhibition of presynaptic mGluRs increases the amplitude of spontaneous EPSCs (Bandrowski et al., 2003) is consistent with this idea. We tested this possibility in a separate set of experiments by applying MAP4 in the presence of TTX. Under these conditions, MAP4 significantly increased the amplitude of mEPSCs (121.7 ± 1.9% of control; p < 0.01; n = 6; data not shown). In light of this evidence, it is plausible that the priming of both mEPSC frequency and amplitude results from the functional inactivation of presynaptic autoreceptors. Together, these data suggest that activation of α1 adrenoceptors by NA curtails the efficacy of group III mGluRs via a PKC-dependent mechanism to prime the glutamatergic synapse for subsequent exposure of the agonist (Fig. 8).
Figure 7.

Blockade of group III mGluRs abolishes NA-induced priming. A, In the presence of MAP4 (250 μm), two 2 min applications of NA (100 μm) separated by 15 min exhibit similar increases in mEPSC frequency. B, Summary bar graph of increases in mEPSC frequency (NA1, 353.5 ± 61.1% of control; p < 0.01; NA2, 376.5 ± 57.0% of control; p < 0.01; n = 4). The mEPSC frequencies from NA1 and NA2 are not significantly different (p > 0.05). C, Summary bar graph showing effects of NA on mEPSC amplitude (NA1, 95.4 ± 4.4% of control; p > 0.05; NA2, 93.4 ± 4.5%; p > 0.05; n = 4). The mEPSC amplitudes from NA1 and NA2 are not significantly different (p > 0.05). Asterisks indicate comparing treatment with control.
Figure 8.

Cross-talk between metabotropic receptor signaling pathways. Schematic illustrates the putative interaction between the presynaptic α1 adrenoceptor and group III mGluRs. α1, α1 Adrenoceptor; PLC, phospholipase C; IP3, inositol triphosphate; DAG, diacylglycerol; PKC, protein kinase C; RRP, readily releasable pool; NA, noradrenaline.
Discussion
Our findings demonstrate that the activation of PKC via α1 adrenoceptors not only increases the frequency of mEPSCs but also functionally attenuates presynaptic group III mGluRs. Moreover, by removing mGluR-mediated feedback inhibition, α1 adrenoceptor activation effectively primes excitatory synapses that terminate on the magnocellular neuroendocrine cells of the PVN. Physiologically, this interaction between two distinct classes of presynaptic metabotropic receptors (α1 adrenoceptor and group III mGluR) may provide a means by which hormone output can be sustained or potentiated to meet homeostatic demands.
α1 Adrenoceptor-mediated activation of PKC increases glutamate release
We demonstrate here that activation of PKC by α1 adrenoceptors increases glutamate release from excitatory afferent fibers in PVN. This may result from distinct actions of this kinase at different steps in the release process, including the mobilization of neurotransmitter-filled vesicles from the reserve pool (Gillis et al., 1996; Stevens and Sullivan, 1998), an increase in the sensitivity of the release process for Ca2+ (Brager et al., 2002), and the facilitation of vesicle fusion (Scepek et al., 1998; Yawo, 1999). One, or a combination, of these mechanisms likely underlies the initial NA-induced increase in mEPSC frequency.
We also observe an increase in the amplitude of mEPSCs in response to the second application of NA. Changes in the amplitude of miniature events are normally associated with postsynaptic changes in receptor numbers (Conti and Weinberg, 1999). Accordingly, the increase in event amplitude may result from an interaction between postsynaptic α1 adrenoceptors and AMPA receptors, although studies examining a link between PKC and postsynaptic AMPA receptors suggest a depression of AMPA receptor function after PKC activation (Daw et al., 2000). Another possibility is that mEPSCs observed in response to the first application of NA act as trophic factors to increase postsynaptic spine density (McKinney et al., 1999) and thus may explain the increase in amplitude in response to the second application of NA. This effect, however, occurs over a much longer time frame, requiring many hours to develop. An alternate possibility is that changes in amplitude are conferred presynaptically. Excitatory synapses onto magnocellular neurons of the SON exhibit multiquantal release when the probability of release is elevated by high-frequency trains (Kombian et al., 2000). Our finding that mEPSC amplitude increases when group III mGluRs are blocked suggest that increases in amplitude in response to the second application of NA (when mGluRs are inactive) may be controlled presynaptically. The recent description of increases in spontaneous EPSC amplitude after inhibition of presynaptic mGluRs in the sensorimotor cortex (Bandrowski et al., 2003) is consistent with this idea.
PKC inhibition of mGluR activity
In addition to the short-term increases in neurotransmitter release, increasing evidence suggests that protein kinases may also serve as molecular substrates that regulate the initiation and maintenance of longer-term changes in synaptic efficacy. Our findings demonstrate that PKC serves as a molecular link between α1 adrenoceptors and group III mGluRs, leading to a long-lasting inhibition of mGluRs. Because these receptors play a pivotal role in gating excitatory input to magnocellular neurons (Schrader and Tasker, 1997; Oliet et al., 2001), their functional inactivation by PKC results in an amplification of glutamate release when the synapses are rechallenged by a second application of NA. Our findings are supported by the demonstration that PKC interacts with the group II and group III mGluRs to inhibit their function (Macek et al., 1999; Sorensen et al., 2002). The inhibition by PKC of group III mGluRs may result from an uncoupling of the receptor from its G-protein cascade (Macek et al., 1999) or by a direct phosphorylation of Ser862 on the intracellular C terminus by PKC (Sorensen et al., 2002). The demonstration that the endogenous ligand NA can inhibit group III mGluR function is consistent with previous findings from the hippocampus that adenosine analogs, which also increase PKC activity (Macek et al., 1998), inhibit mGluR function. Importantly, we now demonstrate that the inhibition of mGluRs conferred by PKC is long lasting, extending well beyond the time frame of α1 adrenoceptor activation. Exposing synapses to NA once increases the capacity of the terminals to release glutamate in response to subsequent exposure to the agonist. Essentially, the synapses have been primed.
Mechanism of mGluR inhibition of mEPSC release
Group III mGluRs can decrease neurotransmitter release by inhibiting presynaptic voltage-dependent calcium channels (Perroy et al., 2002). Because the TTX-insensitive release of glutamate in magnocellular neurons does not depend on extracellular Ca2+ (Inenaga et al., 1998), an interaction between mGluRs and presynaptic calcium channels is unlikely to be responsible for the effects observed here. Our findings, when coupled with the demonstration that glutamate release at mitral bulb synapses is also regulated by group III mGluRs in a manner that is downstream of Ca2+ influx (Schoppa and Westbrook, 1997), provide evidence for an alternative mechanism through which mGluRs may inhibit transmitter release. The demonstration that a subset of group III mGluRs is located at the site of vesicle fusion (Shigemoto et al., 1996) suggests that the effect we described may be attributable to a direct protein–protein interaction between the mGluR complex and a part of the vesicle release machinery (Scanziani et al., 1995).
The role of mEPSCs in conveying information at CNS synapses has been hotly debated (for review, see Staley, 1999). Findings from a number of central nuclei suggest that these stochastic, action potential-independent events may not be mere noise. Increasing the frequency of mEPSCs has functional consequences for action potential generation in magnocellular neuroendocrine cells (Kombian et al., 2000) and cerebellar interneurons (Carter and Regehr, 2002). Additionally, a decrease in miniature GABAergic IPSCs has been linked in the hippocampus to the development of epilepsy (Hirsch et al., 1999). This increase in the background “noise” resulting from the activation of α1 adrenoceptors may in fact lead to an increase in the general excitability of these cells.
Physiological significance
Adaptive responses mediated by the hypothalamus generally require sustained activation until homeostasis is achieved. In an attempt to restore fluid homeostasis in response to acute challenges such as dehydration or hemorrhage, the magnocellular neuroendocrine cells exhibit a prolonged increase in firing rate (Wakerley et al., 1978). This increase in activity, in response to a single acute stimulus, may outlast the physiological perturbation by >24 hr in some cases (Wakerley et al., 1978). Furthermore, the second stimulus in a repeated hemorrhagic protocol elicits an increase in vasopressin release compared with the first stimulus (DeMaria et al., 1987; Lilly et al., 1989). Our findings that NA primes the excitatory synaptic input to magnocellular neuroendocrine cells offer a potential explanation for this observation. The synaptic mechanisms described here would act in concert with previously described changes in activity of these cells in response to changes in plasma osmolarity (Oliet and Bourque, 1993) to increase spiking activity.
Because mGluRs are vital in regulating the synaptic excitability of these cells (Schrader and Tasker, 1997; Oliet et al., 2001), it stands to reason that activating intracellular pathways that inhibit the activity of these receptors would provide an ideal solution for sustaining or potentiating glutamatergic input. That this switch may be initiated by NA, which is released in the hypothalamus during the response to physiological stressors, only serves to highlight the exceedingly clever design of neuronal circuitry that restores homeostasis.
Footnotes
This work was supported by operating grants from the Canadian Institutes of Health Research (CIHR) and the Alberta Heritage Foundation for Medical Research (AHFMR). J.S.B.is an AHFMR and CIHR scholar. We thank Drs. C.W. Bourque, M. Hirasawa, and Q. J. Pittman for advice on this manuscript.
Correspondence should be addressed to Jaideep S. Bains, Department of Physiology and Biophysics, University of Calgary, 3330 Hospital Drive, N.W., Alberta, T2N 4N1, Canada. E-mail: jsbains@ucalgary.ca.
Copyright © 2003 Society for Neuroscience 0270-6474/03/236223-09$15.00/0
References
- Anwyl R ( 1991) The role of the metabotropic receptor in synaptic plasticity. Trends Pharmacol Sci 12: 324–326. [DOI] [PubMed] [Google Scholar]
- Armstrong WE, Gallagher MJ, Sladek CD ( 1986) Noradrenergic stimulation of supraoptic neuronal activity and vasopressin release in vitro: mediation by an alpha 1-receptor. Brain Res 365: 192–197. [DOI] [PubMed] [Google Scholar]
- Bandrowski AE, Huguenard JR, Prince DA ( 2003) Baseline glutamate levels affect group I and II mGluRs in layer V pyramidal neurons of rat sensorimotor cortex. J Neurophysiol 89: 1308–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskys A, Malenka RC ( 1991) Agonists at metabotropic glutamate receptors presynaptically inhibit EPSCs in neonatal rat hippocampus. J Physiol (Lond) 444: 687–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourque CW, Renaud LP ( 1984) Activity patterns and osmosensitivity of rat supraoptic neurones in perfused hypothalamic explants. J Physiol (Lond) 349: 631–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brager DH, Capogna M, Thompson SM ( 2002) Short-term synaptic plasticity, simulation of nerve terminal dynamics, and the effects of protein kinase C activation in rat hippocampus. J Physiol (Lond) 541: 545–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AG, Regehr WG ( 2002) Quantal events shape cerebellar interneuron firing. Nat Neurosci 5: 1309–1318. [DOI] [PubMed] [Google Scholar]
- Conti F, Weinberg RJ ( 1999) Shaping excitation at glutamatergic synapses. Trends Neurosci 22: 451–458. [DOI] [PubMed] [Google Scholar]
- Crowley WR, Shyr SW, Kacsoh B, Grosvenor CE ( 1987) Evidence for stimulatory noradrenergic and inhibitory dopaminergic regulation of oxytocin release in the lactating rat. Endocrinology 121: 14–20. [DOI] [PubMed] [Google Scholar]
- Daftary SS, Boudaba C, Szabo K, Tasker JG ( 1998) Noradrenergic excitation of magnocellular neurons in the rat hypothalamic paraventricular nucleus via intranuclear glutamatergic circuits. J Neurosci 18: 10619–10628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daw MI, Chittajallu R, Bortolotto ZA, Dev KK, Duprat F, Henley JM, Collingridge GL, Isaac JT ( 2000) PDZ proteins interacting with C-terminal GluR2/3 are involved in a PKC-dependent regulation of AMPA receptors at hippocampal synapses. Neuron 28: 873–886. [DOI] [PubMed] [Google Scholar]
- Day TA, Ferguson AV, Renaud LP ( 1984) Facilitatory influence of noradrenergic afferents on the excitability of rat paraventricular nucleus neurosecretory cells. J Physiol (Lond) 355: 237–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMaria EJ, Lilly MP, Gann DS ( 1987) Potentiated hormonal responses in a model of traumatic injury. J Surg Res 43: 45–51. [DOI] [PubMed] [Google Scholar]
- Dunwiddie TV, Masino SA ( 2001) The role and regulation of adenosine in the central nervous system. Annu Rev Neurosci 24: 31–55. [DOI] [PubMed] [Google Scholar]
- Gillis KD, Mossner R, Neher E ( 1996) Protein kinase C enhances exocytosis from chromaffin cells by increasing the size of the readily releasable pool of secretory granules. Neuron 16: 1209–1220. [DOI] [PubMed] [Google Scholar]
- Han SK, Chong W, Li LH, Lee IS, Murase K, Ryu PD ( 2002) Noradrenaline excites and inhibits GABAergic transmission in parvocellular neurons of rat hypothalamic paraventricular nucleus. J Neurophysiol 87: 2287–2296. [DOI] [PubMed] [Google Scholar]
- Hasuo H, Matsuoka T, Akasu T ( 2002) Activation of presynaptic 5-hydroxytryptamine 2A receptors facilitates excitatory synaptic transmission via protein kinase C in the dorsolateral septal nucleus. J Neurosci 22: 7509–7517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatton GI, Li Z ( 1998) Intrinsic controls of intracellular calcium and intercellular communication in the regulation of neuroendocrine cell activity. Cell Mol Neurobiol 18: 13–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilfiker S, Augustine GJ ( 1999) Regulation of synaptic vesicle fusion by protein kinase C. J Physiol (Lond) 515: 169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch JC, Agassandian C, Merchan-Perez A, Ben Ari Y, DeFelipe J, Esclapez M, Bernard C ( 1999) Deficit of quantal release of GABA in experimental models of temporal lobe epilepsy. Nat Neurosci 2: 499–500. [DOI] [PubMed] [Google Scholar]
- Inenaga K, Honda E, Hirakawa T, Nakamura S, Yamashita H ( 1998) Glutamatergic synaptic inputs to mouse supraoptic neurons in calcium-free medium in vitro J Neuroendocrinol 10: 1–7. [DOI] [PubMed] [Google Scholar]
- Jourdain P, Israel JM, Dupouy B, Oliet SH, Allard M, Vitiello S, Theodosis DT, Poulain DA ( 1998) Evidence for a hypothalamic oxytocin-sensitive pattern-generating network governing oxytocin neurons in vitro J Neurosci 18: 6641–6649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kombian SB, Hirasawa M, Mouginot D, Chen X, Pittman QJ ( 2000) Short-term potentiation of miniature excitatory synaptic currents causes excitation of supraoptic neurons. J Neurophysiol 83: 2542–2553. [DOI] [PubMed] [Google Scholar]
- Legendre P, Poulain DA ( 1992) Intrinsic mechanisms involved in the electrophysiological properties of the vasopressin-releasing neurons of the hypothalamus. Prog Neurobiol 38: 1–17. [DOI] [PubMed] [Google Scholar]
- Leibowitz SF, Eidelman D, Suh JS, Diaz S, Sladek CD ( 1990) Mapping study of noradrenergic stimulation of vasopressin release. Exp Neurol 110: 298–305. [DOI] [PubMed] [Google Scholar]
- Lilly MP, DeMaria EJ, Bruhn TO, Gann DS ( 1989) Potentiated cortisol response to paired hemorrhage: role of angiotensin and vasopressin. Am J Physiol 257: R118–R126. [DOI] [PubMed] [Google Scholar]
- Liu WS, Heckman CA ( 1998) The sevenfold way of PKC regulation. Cell Signal 10: 529–542. [DOI] [PubMed] [Google Scholar]
- MacDermott AB, Role LW, Siegelbaum SA ( 1999) Presynaptic ionotropic receptors and the control of transmitter release. Annu Rev Neurosci 22: 443–485. [DOI] [PubMed] [Google Scholar]
- Macek TA, Schaffhauser H, Conn PJ ( 1998) Protein kinase C and A3 adenosine receptor activation inhibit presynaptic metabotropic glutamate receptor (mGluR) function and uncouple mGluRs from GTP-binding proteins. J Neurosci 18: 6138–6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macek TA, Schaffhauser H, Conn PJ ( 1999) Activation of PKC disrupts presynaptic inhibition by group II and group III metabotropic glutamate receptors and uncouples the receptor from GTP-binding proteins. Ann NY Acad Sci 868: 554–557. [DOI] [PubMed] [Google Scholar]
- McKinney RA, Capogna M, Durr R, Gahwiler BH, Thompson SM ( 1999) Miniature synaptic events maintain dendritic spines via AMPA receptor activation. Nat Neurosci 2: 44–49. [DOI] [PubMed] [Google Scholar]
- Meeker RB, Greenwood RS, Hayward JN ( 1994) Glutamate receptors in the rat hypothalamus and pituitary. Endocrinology 134: 621–629. [DOI] [PubMed] [Google Scholar]
- Moos FC, Rossi K, Richard P ( 1997) Activation of N-methyl-d-aspartate receptors regulates basal electrical activity of oxytocin and vasopressin neurons in lactating rats. Neuroscience 77: 993–1002. [DOI] [PubMed] [Google Scholar]
- Mouginot D, Kombian SB, Pittman QJ ( 1998) Activation of presynaptic GABAB receptors inhibits evoked IPSCs in rat magnocellular neurons in vitro J Neurophysiol 79: 1508–1517. [DOI] [PubMed] [Google Scholar]
- Nakajima Y, Yamamoto T, Nakayama T, Nakanishi S ( 1999) A relationship between protein kinase C phosphorylation and calmodulin binding to the metabotropic glutamate receptor subtype 7. J Biol Chem 274: 27573–27577. [DOI] [PubMed] [Google Scholar]
- Nissen R, Hu B, Renaud LP ( 1995) Regulation of spontaneous phasic firing of rat supraoptic vasopressin neurones in vivo by glutamate receptors. J Physiol (Lond) 484: 415–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliet SH, Bourque CW ( 1993) Mechanosensitive channels transduce osmosensitivity in supraoptic neurons. Nature 364: 341–343. [DOI] [PubMed] [Google Scholar]
- Oliet SH, Poulain DA ( 1999) Adenosine-induced presynaptic inhibition of IPSCs and EPSCs in rat hypothalamic supraoptic nucleus neurones. J Physiol (Lond) 520: 815–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliet SH, Piet R, Poulain DA ( 2001) Control of glutamate clearance and synaptic efficacy by glial coverage of neurons. Science 292: 923–926. [DOI] [PubMed] [Google Scholar]
- Perroy J, El Far O, Bertaso F, Pin JP, Betz H, Bockaert J, Fagni L ( 2002) PICK1 is required for the control of synaptic transmission by the metabotropic glutamate receptor 7. EMBO J 21: 2990–2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randle JC, Bourque CW, Renaud LP ( 1986a) Alpha 1-adrenergic receptor activation depolarizes rat supraoptic neurosecretory neurons in vitro Am J Physiol 251: R569–R574. [DOI] [PubMed] [Google Scholar]
- Randle JC, Mazurek M, Kneifel D, Dufresne J, Renaud LP ( 1986b) Alpha 1-adrenergic receptor activation releases vasopressin and oxytocin from perfused rat hypothalamic explants. Neurosci Lett 65: 219–223. [DOI] [PubMed] [Google Scholar]
- Sansig G, Bushell TJ, Clarke VR, Rozov A, Burnashev N, Portet C, Gasparini F, Schmutz M, Klebs K, Shigemoto R, Flor PJ, Kuhn R, Knoepfel T, Schroeder M, Hampson DR, Collett VJ, Zhang C, Duvoisin RM, Collingridge GL, van Der Putten H ( 2001) Increased seizure susceptibility in mice lacking metabotropic glutamate receptor 7. J Neurosci 21: 8734–8745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanziani M, Gahwiler BH, Thompson SM ( 1995) Presynaptic inhibition of excitatory synaptic transmission by muscarinic and metabotropic glutamate receptor activation in the hippocampus: are Ca 2+ channels involved? Neuropharmacology 34: 1549–1557. [DOI] [PubMed] [Google Scholar]
- Scanziani M, Salin PA, Vogt KE, Malenka RC, Nicoll RA ( 1997) Use-dependent increases in glutamate concentration activate presynaptic metabotropic glutamate receptors. Nature 385: 630–634. [DOI] [PubMed] [Google Scholar]
- Scepek S, Coorssen JR, Lindau M ( 1998) Fusion pore expansion in horse eosinophils is modulated by Ca 2+ and protein kinase C via distinct mechanisms. EMBO J 17: 4340–4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoepp DD ( 2001) Unveiling the functions of presynaptic metabotropic glutamate receptors in the central nervous system. J Pharmacol Exp Ther 299: 12–20. [PubMed] [Google Scholar]
- Schoppa NE, Westbrook GL ( 1997) Modulation of mEPSCs in olfactory bulb mitral cells by metabotropic glutamate receptors. J Neurophysiol 78: 1468–1475. [DOI] [PubMed] [Google Scholar]
- Schrader LA, Tasker JG ( 1997) Presynaptic modulation by metabotropic glutamate receptors of excitatory and inhibitory synaptic inputs to hypothalamic magnocellular neurons. J Neurophysiol 77: 527–536. [DOI] [PubMed] [Google Scholar]
- Shibuya I, Kabashima N, Ibrahim N, Setiadji SV, Ueta Y, Yamashita H ( 2000) Pre- and postsynaptic modulation of the electrical activity of rat supraoptic neurones. Exp Physiol 85: 145S–151S. [DOI] [PubMed] [Google Scholar]
- Shigemoto R, Kulik A, Roberts JD, Ohishi H, Nusser Z, Kaneko T, Somogyi P ( 1996) Target-cell-specific concentration of a metabotropic glutamate receptor in the presynaptic active zone. Nature 381: 523–525. [DOI] [PubMed] [Google Scholar]
- Sorensen SD, Macek TA, Cai Z, Saugstad JA, Conn PJ ( 2002) Dissociation of protein kinase-mediated regulation of metabotropic glutamate receptor 7 (mGluR7) interactions with calmodulin and regulation of mGluR7 function. Mol Pharmacol 61: 1303–1312. [DOI] [PubMed] [Google Scholar]
- Staley KJ ( 1999) Quantal GABA release: noise or not? Nat Neurosci 2: 494–495. [DOI] [PubMed] [Google Scholar]
- Stevens CF, Sullivan JM ( 1998) Regulation of the readily releasable vesicle pool by protein kinase C. Neuron 21: 885–893. [DOI] [PubMed] [Google Scholar]
- Tasker JG, Dudek FE ( 1991) Electrophysiological properties of neurones in the region of the paraventricular nucleus in slices of rat hypothalamus. J Physiol (Lond) 434: 271–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakerley JB, Poulain DA, Brown D ( 1978) Comparison of firing patterns in oxytocin- and vasopressin-releasing neurones during progressive dehydration. Brain Res 148: 425–440. [DOI] [PubMed] [Google Scholar]
- Yawo H ( 1999) Protein kinase C potentiates transmitter release from the chick ciliary presynaptic terminal by increasing the exocytotic fusion probability. J Physiol (Lond) 515: 169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong H, Minneman KP ( 1999) Alpha1-adrenoceptor subtypes. Eur J Pharmacol 375: 261–276. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG ( 2002) Short-term synaptic plasticity. Annu Rev Physiol 64: 355–405. [DOI] [PubMed] [Google Scholar]