

Abstract
Increased expression of certain extracellular matrix (ECM) molecules after CNS injury is believed to restrict axonal regeneration. The chondroitin sulfate proteoglycans (CSPGs) are one such class of ECM molecules that inhibit neurite outgrowth in vitro and are upregulated after CNS injury. We examined growth responses of several classes of axons to this inhibitory environment in the presence of a cellular fibroblast bridge in a spinal cord lesion site and after a growth factor stimulus at the lesion site (fibroblasts genetically modified to secrete NGF). Immunohistochemical analysis showed dense labeling of the CSPGs NG2, brevican, neurocan, versican, and phosphacan at the host-lesion interface after spinal cord injury (SCI). Furthermore, robust expression of NG2, and to a lesser extent versican, was also observed throughout grafts of control and NGF-secreting fibroblasts. Despite this inhibitory milieu, several axonal classes penetrated control fibroblast grafts, including dorsal column sensory, rubrospinal, and nociceptive axons. Axon growth was amplified more in the presence of NGF-secreting grafts. Confocal microscopy demonstrated that axon growth was, paradoxically, preferentially associated with NG2-rich substrates in both graft types. NG2 expression also increased after sciatic nerve injury, wherein axons successfully regenerate. Cellular sources of NG2 in SCI and peripheral nerve lesion sites included Schwann cells and endothelial cells. Notably, these same cellular sources in lesion sites produced the cell adhesion molecules L1 and laminin, and these molecules all colocalized. Thus, axons grow along substrates coexpressing both inhibitory and permissive molecules, suggesting that regeneration is successful when local permissive signals balance and exceed inhibitory signals.
Keywords: regeneration, chondroitin sulfate proteoglycans, extracellular matrix, spinal cord injury, inhibition, cell adhesion
Introduction
Failure of axonal regeneration after CNS injury is thought to result in part from a nonpermissive extracellular milieu surrounding the lesion site. Several inhibitory extracellular matrix (ECM) molecules, including the chondroitin sulfate proteoglycans (CSPGs) (Fitch and Silver, 1997; Lemons et al., 1999; Moon et al., 2002) and keratan sulfate proteoglycans (Jones and Tuszynski, 2002), are strongly upregulated after CNS injury. Other inhibitory components of the lesioned CNS include myelin-associated glycoprotein (MAG) (McKerracher et al., 1994; Filbin, 1995), Nogo (for review, see Fouad et al., 2001; Woolf, 2003), and semaphorins (Pasterkamp et al., 2001; De Winter et al., 2002).
The role of CSPGs in reducing axon growth after injury has been studied extensively. In vitro, CSPGs restrict neurite outgrowth, and their inhibitory properties have been attributed either to the core protein (Dou and Levine, 1994; Milev et al., 1994;Schmalfeldt et al., 2000; Ughrin et al., 2003) or to associated chondroitin sulfate side chains (Braunewell et al., 1995; Yamada et al., 1997; Fidler et al., 1999; Ughrin et al., 2003). After CNS injuries, several CSPGs are differentially increased over postinjury periods, including NG2, neurocan, brevican, versican, and phosphacan (Levine, 1994; Haas et al., 1999; McKeon et al., 1999; Asher et al., 2000, 2002; Thon et al., 2000; Plant et al., 2001, 2002; Jones et al., 2002, 2003; Moon et al., 2002; Tang et al., 2003). In addition, upregulation of a general CSPG marker correlates with aborted axon growth (Davies et al., 1997), and degradation of associated chondroitins after brain injury (Moon et al., 2001) and spinal cord injury (SCI) (Bradbury et al., 2002) partially enhances axon growth and functional recovery. Combined, these data suggest that CSPGs inhibit axonal regeneration after CNS lesions and that their targeted reduction can improve regeneration.
In contrast, injured CNS axons can grow to some extent without targeted reduction of CSPGs. For example, axons cross the host-lesion interface and extend into cellular grafts of various cell types placed into SCI lesion sites, including fibroblasts (Tuszynski et al., 1994; Blesch et al., 1998; Franzen et al., 1999), Schwann cells (Paino and Bunge, 1991; Xu et al., 1995; Weidner et al., 1999), or stem cells (Liu et al., 1998; Lu et al., 2002), despite distinct upregulation of several CSPGs at the lesion site (Plant et al., 2001; Jones et al., 2002, 2003; Tang et al., 2003).
Thus, although CSPGs inhibit axon growth, this inhibition is not absolute and other factors appear to compensate for the presence of CSPGs. The present study sought to elucidate mechanisms that may underlie and, conversely, balance CSPG-mediated effects on axonal regeneration after CNS injury. Confocal analysis reveals that after SCI, multiple axonal populations surprisingly associate with CSPG-rich substrates in a cellular bridge at the lesion site. Notably, cellular sources of CSPGs in the lesion site also produce L1 and laminin. Thus, simultaneous expression of permissive substrate molecules could potentially balance CSPG-mediated inhibition at CNS lesion sites, enabling axon growth to proceed.
Materials and Methods
Animal subjects and surgery. Subjects of this study were adult female Fischer 344 rats weighing 160-200 gm. National Institutes of Health guidelines for laboratory animal care and safety were followed strictly. Animals had ad libitum access to food and water throughout the study. All surgery was performed under anesthesia with a combination (2 ml/kg) of ketamine (25 mg/ml), rompun (1.3 gm/ml), and acepromazine (0.25 mg/ml). A total of 67 rats were used in this study as described below and included the following: subjects that received (1) cervical dorsal column spinal cord lesions (n = 44), (2) cervical dorsolateral funiculus spinal cord lesions (n = 8), and (3) sciatic nerve crush lesions (n = 4), and (4) intact animals (n = 11).
Retroviral construction and transduction of fibroblast cellular bridges. To provide cellular bridges for axon growth in spinal cord lesion sites, primary fibroblasts were prepared and genetically modified to produce either the reporter gene green fluorescent protein (GFP) or human NGF. Primary cultures of Fischer 344 rat fibroblasts were generated from skin biopsies and cultivated under cell culture conditions as described previously (Tuszynski et al., 1994; Grill et al., 1997). Retroviral vectors for transduction of primary rat fibroblasts were prepared as described previously (Tuszynski et al., 1996). Briefly, the retroviral vector expressing GFP was obtained by pSP65-hGFP-T by XbaI-HindIII digestion. The ends were filled with Klenow and ligated into the HpaI-digested vector pLXSN. The GFP transgene was constitutively expressed by the wild-type viral 5′ long terminal repeat (LTR) promoter, and a dominant selectable marker imparting neomycin resistance in cells was expressed by the SV40 promoter (Miller et al., 1993). The cDNA for the prepro-form of β-NGF was also placed into a Maloney murine leukemia virus expression cassette, and NGF was constitutively expressed by the viral LTR. Plasmids were introduced into the ecotropic producer cell line Psi-2 by calcium phosphate transfection. Supernatants from Psi-2 cells were used to infect the amphotropic PA317 cell line. G418-resistant clones that expressed full-length recombinant protein were identified, and viral titers of 106-107 colony-forming units were obtained. In vitro production of NGF from infected fibroblasts was determined using a two-site enzyme-linked immunoassay sensitive to 5 pg/ml (NGF antibody provided by Dr. J. Conner, University of California-San Diego, La Jolla, CA).
Cervical dorsal column spinal cord lesions. To study the association of regenerating axons with the inhibitory ECM, a dorsal column spinal cord lesion was performed at the C3 level, as described previously (Weidner et al., 2001). Briefly, rats were deeply anesthetized and C3 laminectomies were performed. A tungsten wire knife (Kopf Instruments, Tujunga, CA) was stereotaxically positioned at the spinal dorsal midline and then moved 0.6 mm left laterally and lowered 1.1 mm from the dorsal spinal cord surface. The knife blade was extruded 2.25 mm toward the midline, forming a 1.5-mm-wide wire arc. The arc was raised 2 mm and simultaneously met by a blunt glass rod that added compression from above to ensure full transection of axons in the dorsal columns bilaterally. After the lesion was completed, 2.0 μl (5.0 × 104 cells/μl) of control GFP-expressing primary rat syngenic fibroblasts was injected into 16 animals to provide a cellular bridge that has been reported previously (Blesch et al., 1999; Blesch and Tuszynski, 2001; Lu et al., 2001) to support axon growth into the lesion site. In addition, 16 animals received implants of primary rat syngenic fibroblasts that were genetically modified to produce human NGF, as described previously (see above). Before implantation, these cells secreted 27.1 ng of human NGF per 106 cells per day. Animals that received lesion only were sutured closed without receiving cellular implants. Animals were killed at the following postinjury time points: 3 d (GFP, n = 4; NGF, n = 4), 7 d (GFP, n = 4; NGF, n = 4), 14 d (GFP, n = 8; NGF, n = 8), and 28 d (GFP, n = 4; NGF, n = 4); lesion only, 14 d (n = 4); nonlesioned, intact animals were also examined (n = 7).
To evaluate growth responses of ascending dorsal column sensory axons after dorsal column lesions, the transganglionic label cholera toxin B subunit (CTB) (List Biologic, Campbell, CA) was injected into both sciatic nerves 3 d before death, as described previously (Bradbury et al., 1999). Briefly, 2 μl of a 1% solution of CTB dissolved in sterile water was injected into each sciatic nerve using a Hamilton syringe.
Cervical dorsolateral funiculus spinal cord lesions. To examine responses of lesioned rubrospinal tract axons to the inhibitory ECM, the dorsolateral funiculus was transected in eight rats. Briefly, a C3 laminectomy was performed and a Kopf tungsten wire knife was positioned stereotaxically at the spinal dorsal midline. The knife was then moved 0.6 mm to the left lateral midline and lowered to a depth of 1.1 mm from the dorsal spinal cord surface. The tip of the knife was extruded 2.25 mm in a lateral direction, forming a 1.5-mm-wide wire arc that was raised 2 mm and simultaneously met by a blunt glass rod to compress from above, ensuring full transection of rubrospinal axons in the dorsolateral funiculus. After the lesion was completed, 2.0 μl (5.0 × 104 cells/μl) of control GFP-expressing rat fibroblasts was injected into the lesion epicenter using a Picospritzer II (General Valve, Fairfield, NJ). Eight animals received implants of GFP-expressing fibroblasts and were perfused either 14 d (n = 4) or 28 d (n = 4) later. Trophic factors were not administered in this paradigm.
To evaluate growth of injured rubrospinal tract axons after dorsolateral funiculus lesions, animals received injections of biotinylated-dextran amine (BDA) 10,000 molecular weight (Molecular Probes, Eugene, OR) into the red nucleus 14 d before perfusion. Three hundred nanoliters of a 10% BDA solution were injected through pulled-glass micropipettes (40 μm internal diameter) into each of two sites distributed evenly over the rostrocaudal extent of the right red nucleus, using a Picospritzer II at the following stereotaxic coordinates: anteroposterior, -0.57, -0.62; mediolateral, +0.08, +0.07; dorsoventral, -0.72, -0.67 (Paxinos and Watson, 1998). The labeled rubrospinal tract axons were visualized using streptavidin Alexa 488 (at 1:300; Molecular Probes), as described below.
Sciatic nerve crush. To investigate alterations of the extracellular matrix in a milieu that is known to support axonal regeneration successfully, four rats underwent bilateral sciatic nerve lesions. The sciatic nerve at the midfemoral level was dissected free, and a crush lesion was delivered by applying firm pressure with fine jewelry forceps for 20 sec. A suture was placed in adjacent muscle tissue to mark the lesion site. Animals were perfused 3 d later. Nonlesioned, intact animals were used as controls (n = 4).
Tissue processing and immunohistochemistry. After induction of deep anesthesia, animals were transcardially perfused with 4% paraformaldehyde (PFA) in 0.1 m phosphate buffer. Spinal cords or sciatic nerves were removed, postfixed overnight in 4% PFA, and placed in phosphate buffer containing 30% sucrose at 4°C. Spinal cords were sagittally sectioned on a cryostat set at 35 μm. One in seven sections was mounted on gelatin-coated glass slides for Nissl staining. Remaining sections were serially collected into 24-well plates for immunohistochemical labeling.
To determine the distribution of individual CSPG family members at the host-lesion interface, within fibroblast grafts placed in the lesion site, and in relation to injured and growing axons, immunolabeling was performed for NG2, brevican, neurocan, versican, and phosphacan. We reported previously that NG2 is a major CSPG family member deposited at sites of SCI and that its deposition peaks 7 d after injury (Jones et al., 2003); thus, in the present experiment, we examined its distribution 3, 7, 14, and 28 d after injury using the rabbit polyclonal anti-rat NG2 antibody (at 1:1000) (generous gift from B. Stallcup, Burnham Institute, La Jolla, CA). We also reported previously that expression of the CSPG components brevican, neurocan, and versican peak 14 d after SCI and that phosphacan is significantly elevated 28 d after injury (Jones et al., 2003). In the present experiment, we examined the distribution of these components at their respective time points of peak expression using the following monoclonal antibodies (Abs): neurocan (1F6 Ab at 1:6000 dilution; University of Iowa, Developmental Studies Hybridoma Bank, Iowa City, IA), brevican (RB18 Ab at 1:400 dilution; gift from Y. Yamaguchi, Burnham Institute, La Jolla, CA), versican (12C5 Ab at 1:8000 dilution; University of Iowa, Developmental Studies Hybridoma Bank), and phosphacan (3F8 Ab at 1:6000 dilution; University of Iowa, Developmental Studies Hybridoma Bank).
The distribution of other molecules that could influence axon growth in the injury site was also examined in grafts 14 and 28 d after SCI, including the cell adhesion molecules L1 and laminin, using monoclonal antibodies anti-L1 (2C2 Ab at 1:500 dilution; gift from M. Grumet, Rutgers University, Newark, NJ) and anti-laminin (2E8 Ab at 1:75 dilution; University of Iowa, Developmental Studies Hybridoma Bank). Finally, Schwann cell penetration of grafts was characterized using the mouse monoclonal nonmyelinating Schwann cell antibody 27C7 (at 1:200 dilution; gift from K. Wewetzer, University of Freiburg, Freiburg, Germany) 3, 7, 14, and 28 d after SCI. All sections were processed free-floating, and endogenous peroxidase activity was blocked with 0.6% hydrogen peroxide as described previously (Grill et al., 1997). Nonspecific antibody reactions were blocked with 5% horse serum (for monoclonal antibodies) or 5% goat serum (for polyclonal antibodies) for 1 hr at room temperature. Sections were incubated overnight at 4°C in primary antibody. After washing in Tris-buffered saline (TBS), sections were incubated with biotin-conjugated IgG anti-mouse secondary antibody (1:200 dilution; Vector Laboratories, Burlingame, CA) for 1 hr at room temperature followed by 1 hr incubation in avidin-biotinylated peroxidase complex (1:100 dilution; Elite Kit, Vector Laboratories) at room temperature. A primary antibody omission control was included to exclude nonspecific binding of the secondary antibody. Diaminobenzidine (0.05%) with nickel chloride (0.04%) was used as a chromagen, with reactions sustained for 3 min at room temperature. The sections were mounted on gelatin-coated slides, dehydrated, and coverslipped with DPX mounting medium (BDH Chemicals, Poole, UK).
Immunofluorescent double labeling was performed to identify the association of different axonal populations, cell types, and cell adhesion molecules with the expression of CSPG molecules. Studies focused specifically on co-association of axonal markers with NG2 because only the latter molecule and versican were expressed within grafts, and NG2 expression was predominant (see Results). After nonspecific antibody reactions were blocked with 5% goat serum for 1 hr at room temperature, free-floating sections were incubated overnight at 4°C with either the mouse monoclonal anti-NG2 antibody described above (at 1:800 dilution) or a rabbit polyclonal anti-rat NG2 antibody (at 1:1000 dilution; B. Stallcup) and simultaneously with one of the following antibodies: neurofilament (NF) antibody as a general axonal marker (RT97 monoclonal antibody at 1:250 dilution; Boehringer Mannheim, Mannheim, Germany), rabbit calcitonin gene-related peptide antibody (CGRP) (polyclonal at 1:8000 dilution; Chemicon, Temecula, CA), nonmyelinating Schwann cell antibody (27C7 monoclonal at 1:200 dilution; K. Wewetzer), Schwann cell marker (S-100 monoclonal at 1:2000 dilution; Dako, Carpenteria, CA), endothelial cell antibody (RECA1 monoclonal at 1:100 dilution; Serotec, Raleigh, NC), L1 antibody (2C2 monoclonal at 1:500 dilution; M. Grumet), and laminin antibody (2E8 monoclonal at 1:75 dilution; Developmental Studies Hybridoma Bank, Iowa City, IA). Sections were washed with TBS, incubated with Alexa 488 fluorophore goat anti-rabbit antibody (at 1:150 dilution; Molecular Probes) for 2.5 hr at room temperature and with Alexa 594 fluorophore goat anti-mouse antibody (at 1:150 dilution; Molecular Probes). Sections were washed with TBS, mounted on uncoated slides, and coverslipped with Fluoromount G (Southern Biotechnology, Birmingham, AL). Primary antibody omission controls were performed to exclude nonspecific binding. Fluorescent visualization was performed using an Olympus confocal microscope with an omnichrome series 43 Argon-krypton laser and appropriate filter sets (Olympus America, Melville, NY). Fluorescent bleed-through controls were tested using tissue stained solely with Alexa 488 to verify lack of detection in the 594 channel and conversely to verify that tissue stained solely with the 594 fluorophore was not detected in the 488 channel.
To examine the association of CTB-labeled dorsal column axons with NG2, sections were double labeled with CTB antibody (goat polyclonal at 1:5000 dilution; List Biologic) and NG2 antibody as described above (at 1:800 dilution) using a Cy5 fluorophore secondary donkey anti-goat antibody (Jackson ImmunoResearch, West Grove, PA).
Immunoblotting. Amounts of NG2 in the intact spinal cord, injured spinal cord, and injured spinal cord containing grafts of either GFP-expressing or NGF-secreting cells 2 weeks after injury (n = 4 animals per group) were compared by immunoblotting. In lesioned subjects, the cord sample consisted of a 5-mm-long block of tissue centered at the injury site, encompassing both the full lesion site and host tissue adjoining the lesion. Samples from intact animals were centered at the same spinal level as lesioned subjects (C3) and were also 5 mm in length. Tissue was standardized to 250 μg/μl (wet weight) with ice-cold extraction buffer (150 mm NaCl, 50 mm Tris, pH 8.0, 1.0% NP-40, 0.5% Na deoxycholate, 0.1% SDS, 1 mm PMSF) and homogenized on ice. Matched amounts of samples were run on 4-12% SDS-polyacrylamide gels (Invitrogen, Carlsbad, CA). Gels were blotted onto 0.45 μm pore nitrocellulose membranes (Fisher Scientific, Pittsburgh, PA). Nitrocellulose blots were blocked with 5% milk in PBS for 1 hr, incubated overnight at room temperature with polyclonal rabbit anti-rat NG2 antibody (at 1:500) (Goretzki et al., 1999), washed with PBS, incubated for 1 hr with a horseradish peroxidase-conjugated goat anti-rabbit IgG secondary antibody (1:100,000; Jackson ImmunoResearch), washed with PBS, and visualized using a SuperSignal chemiluminescence system (Pierce, Rockford, IL). Individual gels from each subject were scanned using a Umax transmitted light scanner, and pixel density per gel was measured using NIH Image software, as described previously (Blesch et al., 1999). Mean group values were compared using ANOVA with a significance level of 95%, and individual group differences were assessed by post hoc Fischer's. Data are presented as mean ± SEM.
Results
Grafts survive in the lesion site and support the growth of several classes of axons; NGF gene delivery increases axon growth
To examine specific interactions of growing axons with inhibitory ECM molecules after SCI, cells that support axonal regeneration were implanted into C3 dorsal column lesion sites, and immunolabeling for CSPG family members was performed. Nissl staining revealed that both control (GFP expressing) and NGF-secreting fibroblast grafts survived injection, filled the wire knife lesion cavity, and supported growth of host axons (Fig. 1) (descriptions below). NF labeling indicated that control cell grafts attracted the growth of moderate numbers of host axons and that NGF-secreting grafts were densely penetrated by axons (Fig. 1) (see Fig. 4). Thus, this lesion-grafting paradigm provided a model in which the association of growing axons with alterations in the extracellular matrix could be examined.
Figure 1.

Nissl and neurofilament staining of GFP-expressing and NGF-secreting fibroblast grafts in spinal cord lesion site. A, Twenty-eight days after SCI, the spinal cord lesion site contains GFP-expressing fibroblasts and exhibits excellent host-graft integration. B, Similarly, NGF-secreting fibroblasts completely fill the lesion site and are well integrated. Sagittal section: left is rostral; arrows indicate host-lesion interface. Scale bars: A, B, 220 μm. C, Twenty-eight days after SCI, GFP-expressing grafts are penetrated by moderate numbers of NF-labeled axons that cross the host-graft interface. D, NGF-secreting grafts at the same time point demonstrate greater axonal density within the graft. Scale bars: C, D, 24 μm.
Figure 4.

Axons at the host-lesion interface and within cell bridges specifically associated with NG2 substrates. A, NF-labeled axons penetrate the host-lesion interface and extend into GFP-expressing cell grafts 28 d after SCI. Scale bars: A-C, 24 μm. B-F, NF-labeled axons are highly colocalized with NG2-expressing substrates within cell grafts. Scale bars: D-F, 14 μm. G, Axon density is markedly increased in all NGF-expressing cell grafts, as reported previously (Tuszynski et al., 1996), and NG2 immunolabeling is also present throughout these cell grafts (H). Scale bars: G-I, 24 μm. I-L, Neurofilament-labeled axon growth in NGF-secreting grafts is also specifically associated with NG2-labeled substrates. Scale bars: J-L, 14 μm. Arrows indicate examples of NF-labeled axons colocalizing with NG2. Micrographs are from the center of the lesion-graft site and are located ∼250 μm from the host-lesion interface.
NG2 is expressed in the injured spinal cord and throughout cellular bridges in lesion sites
Previous quantitative, biochemical analyses reported that NG2 is a major and likely the most abundant CSPG family member expressed after SCI (Jones et al., 2002). Supporting and extending previous observations, we found that NG2 is densely expressed in both host tissue and throughout grafts in the lesion site from 3 to 28 d after SCI (Fig. 2). Compared with modest baseline levels of expression (Fig. 2I), NG2 immunolabeling markedly increased in host tissue surrounding the lesion site as soon as 3 d after injury (Fig. 2A,B), continued to increase at 7 and 14 d, and sustained high levels of labeling by 28 d (Fig. 2C-H). The extent of NG2 expression in the host spinal cord surrounding the lesion site was similar in recipients of GFP-expressing and NGF-secreting fibroblast grafts and did not differ from patterns that we reported previously when performing spinal cord lesions alone without grafts (Jones et al., 2002).
Figure 2.

NG2 is upregulated in host tissue and within cell grafts at sites of SCI. A, Three days after SCI, NG2 expression is markedly upregulated in host tissue surrounding the lesion and GFP-expressing fibroblast implants. The cell graft itself exhibits little NG2 immunolabeling at this time point. Arrows indicate host-graft interface. Scale bars: A-J, 220 μm. B, A similar labeling pattern is present in subjects with NGF-secreting fibroblast grafts 3 d after injury. C, D, Seven days after injury, NG2 immunolabeling is detectable throughout the GFP-expressing grafts and is denser within NGF-secreting grafts. E-H, Fourteen and 28 d after SCI, NG2 immunolabeling remains evident at the host-lesion interface. NG2 cellular profiles are present throughout GFP-expressing grafts and more extensively label NGF-secreting grafts. Insets in G and H indicate that NG2-labeled profiles create a loose, lattice-like network within grafts and are denser in NGF-secreting grafts. Scale bar for insets, 22 μm. I, Intact spinal cord, showing low constitutive levels of NG2 expression in both white and gray matter. J, Primary antibody omission control (1o Ab Omission). K, NG2 immunoblotting of spinal cord extracts from intact animals, and 2 weeks after C3 lesions, animals with lesions only, GFP-expressing cells in the lesion site (GFP), or NGF-secreting cells in the lesion site (NGF). L, Quantification of relative levels of NG2 expression on immunoblots using NIH Image software (n = 4 animals per group). Mean ± SEM. Asterisks denote significant differences from intact subjects on post hoc Fischer's (p < 0.001).
Notably, production of NG2 was also extensive and dense throughout cell grafts implanted in the lesion site. Three days after injury, NG2 labeling was present primarily in graft regions that were adjacent to the host-lesion interface. Specifically, NG2 labeling was not detected within the center of either GFP- or NGF-producing grafts (Fig. 2A,B). Seven days after SCI, NG2 labeling had become distributed throughout grafts (Fig. 2C,D); at this time, the NG2 immunolabeling was denser in NGF-secreting grafts compared with GFP grafts. Fourteen and 28 d after SCI, NG2 labeling remained moderate in intensity throughout GFP-expressing grafts (Fig. 2E,G) and continued to increase in NGF-secreting grafts (Fig. 2F,H). Immunoblotting of freshly dissected spinal cords confirmed that levels of NG2 protein significantly increased 2 weeks after C3 lesions and that such increases were greatest in subjects that received cell implants in the lesion site (ANOVA; p < 0.001, with significant post hoc differences between all lesioned groups compared with intact animals) (Fig. 2K,L).
Brevican, neurocan, versican, and phosphacan are also expressed in injured host tissue surrounding lesion and grafts; only versican is expressed within grafts
We reported previously that expression of brevican, neurocan, and versican peaks 14 d after a C3 wire knife spinal cord lesion and that phosphacan expression is significantly elevated 28 d after injury (Jones et al., 2003). We therefore chose these time points in the present study to examine interactions of growing axons with expression of these CSPG components. As reported previously, immunoreactivity for brevican (Fig. 3A,B), neurocan (Fig. 3C,D), and versican (Fig. 3E,F) was upregulated in host spinal cord tissue surrounding the lesion site 14 d after injury. Similarly, phosphacan expression was increased after injury (Fig. 3G,H). No differences were observed in expression of these CSPGs in the host spinal cord in recipients of control or NGF-secreting cell grafts when compared with nongrafted, lesioned subjects of our previous studies. Within GFP-expressing and NGF-secreting cell grafts, only versican expression could be detected by immunolabeling. Fourteen days after SCI, versican immunolabeling filled both GFP-expressing and NGF-secreting fibroblast grafts (Fig. 3E,F). Notably, there were no clear differences in levels of versican labeling intensity when comparing GFP-expressing and NGF-secreting cell grafts, in distinction to the clearly observed differences in NG2 labeling intensity between the two graft types described above.
Figure 3.
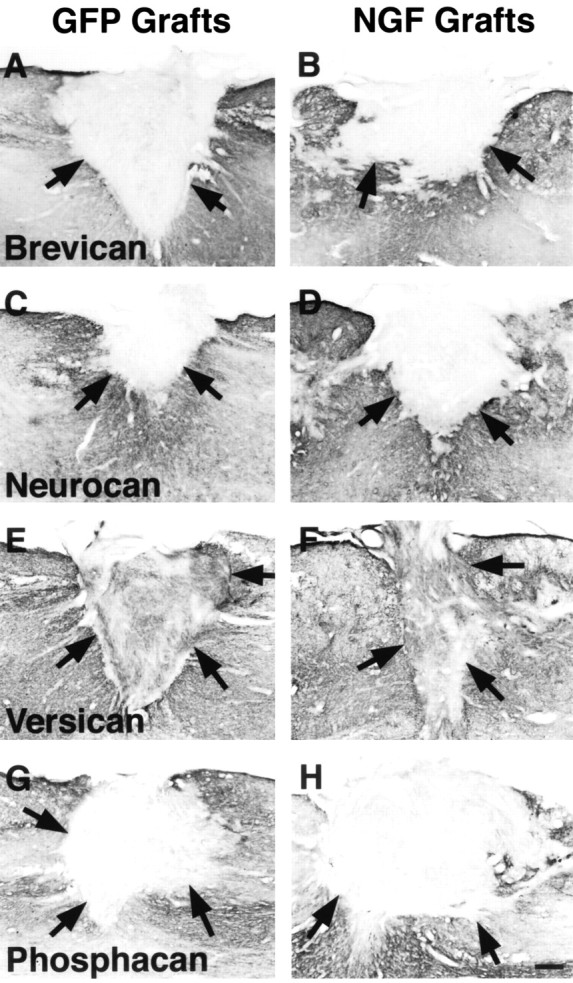
Several CSPG family members are upregulated at sites of SCI; only versican is expressed within cell grafts. A-D, Although brevican and neurocan immunolabeling are readily detected in host tissue surrounding a 14 d SCI site in both GFP-expressing (A, C) and NGF-secreting grafts (B, D), they are not detected within either cell graft types in the lesion cavity. A previous study reported that brevican and neurocan expression peak in host tissue 14 d after SCI (Jones et al., 2003). Scale bars: A-H, 220 μm. E, F, Notably, versican immunolabeling is present in both host and graft tissue 14 d after SCI in GFP-expressing (E) and NGF-secreting cell grafts (F). A previous study reported that versican expression peaks in host tissue 14 d after SCI (Jones et al., 2003). No clear differences in levels of versican labeling intensity are present comparing GFP-expressing and NGF-secreting cell grafts. G, H, Phosphacan labeling is readily detectable in host tissue but not in either GFP-expressing (G) or NGF-secreting cell grafts (H) 28 d after SCI. Phosphacan expression is significantly elevated 28 d after SCI (Jones et al., 2003).
Axons grow across the CSPG-rich lesion interface and specifically associate with CSPG substrates within grafts
Using axon-specific immunolabels and transganglionic axonal tracing, we observed extensive axonal penetration beyond the host-lesion interface and into grafts occupying the lesion cavity (Figs. 1, 4, 5, 6, 7). Notably, axons growing within grafts were specifically associated with CSPG-rich substrates. As stated above, NF-labeled axons penetrated GFP-expressing cell grafts in the lesion site 14 and 28 d after injury (Fig. 4A,D), and these penetrating axons colocalized extensively with cellular profiles expressing NG2 (Fig. 4C-F). There was a considerable increase in the density of NF labeling within NGF grafts (Fig. 4G), as reported previously (Tuszynski et al., 1996), and growing axons within these grafts were also closely associated with NG2-labeled substrates (Fig. 4I,L).
Figure 5.

CGRP-labeled sensory axons specifically associate with NG2-expressing substrates. A, CGRP-labeled nociceptive sensory axons penetrate the host-lesion interface and extend into GFP-expressing cell grafts 28 d after SCI. Scale bars: A-C, 24 μm. B, C, CGRP and NG2 labeling colocalize within GFP-expressing control grafts. D-G, High-magnification three-dimensional confocal microscopy demonstrates strict association of CGRP-labeled axons with NG2-expressing substrates. F, Composite Z-stack micrograph of merged CGRP (D) and NG2 (E) images. G, A rotated side view of the Z-stack composite in F demonstrates that the CGRP-labeled axon is orientated directly on the surface of the NG2-expressing substrate. Scale bars: D-G, 19 μm. H, All NGF-secreting cell grafts contain increased numbers of CGRP-labeled axons, as reported previously (Tuszynski et al., 1996). I, J, CGRP-labeled axon growth in NGF-secreting grafts is also associated specifically with NG2-expressing substrates. Scale bars: H-J, 24 μm. Arrows indicate examples of CGRP-labeled fibers associated with NG2-expressing substrates.
Figure 6.

Dorsal column sensory axons specifically associate with NG2-labeled substrates. A, CTB-traced dorsal column sensory axons penetrate the 28 d SCI host-lesion interface and extend throughout GFP-expressing fibroblast grafts. Scale bars: A-C, 24 μm. B, C, CTB-labeled axons highly colocalize with NG2-labeled substrates. Arrows indicate examples of CTB-labeled axons specifically associated with NG2-expressing substrates.
Figure 7.
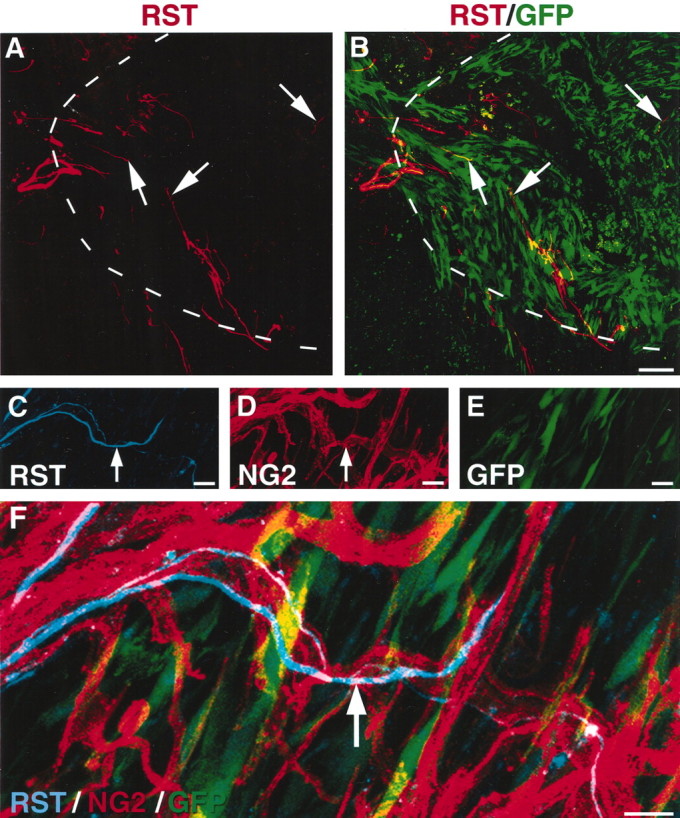
Rubrospinal tract axons specifically associate with NG2-labeled substrates. A, B, Rubrospinal tract (RST) axons penetrate the host-lesion interface and extend throughout GFP-expressing grafts 28 d after SCI. Dashed lines indicate host-lesion interface. Arrows indicate extensive rubrospinal tract growth into GFP-expressing grafts. Scale bars: A, B, 21 μm. C-F, High-magnification micrographs of rubrospinal tract growth inside GFP-expressing grafts. Note specific colocalization of rubrospinal tract axons with NG2-expressing substrate (arrows). Rubrospinal tract growth is not associated with GFP-expressing fibroblasts. Scale bars: C-F, 13 μm.
Specific labeling for nociceptive NGF-responsive sensory axons within grafts confirmed a specific association of this axonal class with NG2-expressing substrates (Fig. 5). CGRP-labeled axons crossed the NG2-rich host-lesion interface and within grafts associated with NG2-labeled processes, as demonstrated by high-magnification confocal Z-stacks (Fig. 5D-G). Three-dimensional rotation of the Z-stack composite demonstrated further that the positioning of CGRP axons directly followed the surface of the NG2 substrate. CGRP-labeled axons were substantially increased in number within NGF-secreting cell grafts (Fig. 5H). Once again, this larger number of axons within NGF-secreting grafts was specifically associated with NG2 substrates (Fig. 5J).
Examining a different class of sensory axons, we also observed penetration of dorsal column sensory axons transganglionically labeled with CTB into both GFP-expressing and NGF-secreting cell grafts (Fig. 6). These axons also associated specifically with NG2-rich substrates within both graft types 14 and 28 d after injury (Fig. 6) (A greater number of dorsal column sensory axons were not observed within NGF-secreting grafts, as expected, because this axonal population exhibits NT-3 rather than NGF sensitivity) (McMahon et al., 1994; Michael et al., 1997).
Transected rubrospinal axons also regenerate across the CSPG-rich host-lesion interface and associate with CSPG substrates
To examine the interaction of a third population of spinal cord axons with the CSPG-rich milieu after SCI, rubrospinal tract lesions were performed as described above, and GFP-expressing cells were grafted into the lesion site. Fourteen and 28 d after injury, transected rubrospinal tract axons extended across the CSPG-rich host-lesion interface and penetrated GFP-expressing cell grafts (Fig. 7A,B). Yet again, growing rubrospinal axonsassociated specifically with NG2-rich substrates when examined on thin-slice,high-resolution confocal microscopy (Fig. 7C-F).
Schwann cells are sources of NG2 within cell grafts
Using the 27C7 nonmyelinating Schwann cell marker (Wewetzer et al., 1997), we assessed the migration of Schwann cells into fibroblast grafts over a 28 d period after dorsal column lesions (Fig. 8). In the intact spinal cord, no 27C7 immunolabeling was detected (Fig. 8I). Three days after SCI, 27C7 immunolabeling was observed on the outer perimeter of both GFP-expressing and NGF-secreting cell grafts (Fig. 8); the intensity was greater in NGF grafts. Seven days after SCI, 27C7 immunolabeling penetrated grafts more extensively, reaching the core of grafts and increasing additionally in intensity by 14 d after grafting (Fig. 8C,E). Immunolabeling intensity was consistently greater in NGF-secreting grafts (Fig. 8D,F). By 28 d, 27C7 labeling remained moderately intense throughout GFP-expressing cell grafts (Fig. 8G), yet continued to increase in NGF-secreting grafts (Fig. 8H).
Figure 8.
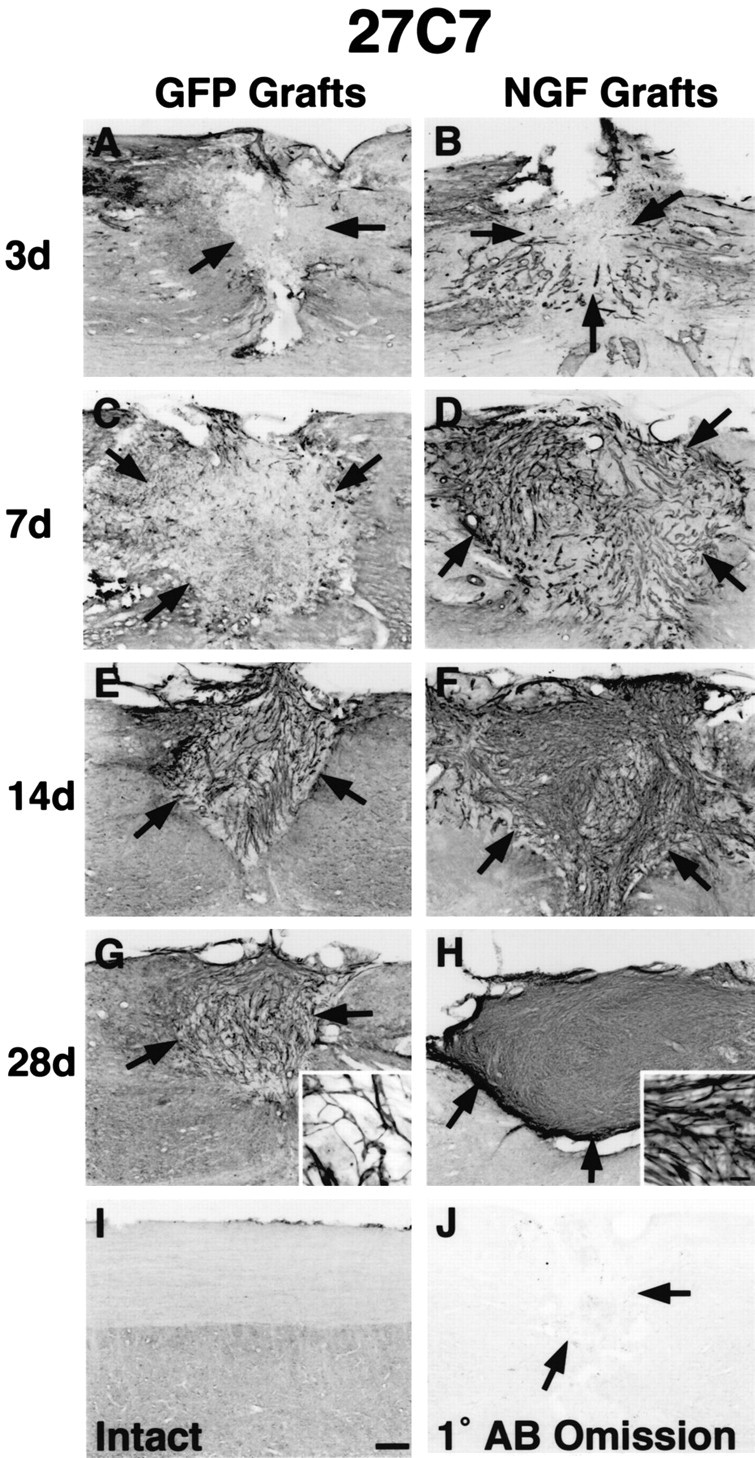
Schwann cells migrate into fibroblast grafts. 27C7 immunolabeling demonstrates increasing penetration of nonmyelinating Schwann cells into both GFP-expressing and NGF-secreting fibroblast grafts over time. A, Three days after injury, sparse 27C7 immunolabeling is present at host-lesion interface (arrows) but not within GFP-expressing grafts. Scale bars: A-J, 220 μm. B, Three days after injury in NGF-secreting grafts, 27C7 immunolabeling is also present at the host-lesion interface and in the graft regions adjacent to the interface, but not within the center of grafts. C, Seven days after SCI, modest 27C7 immunolabeling is present throughout GFP-expressing grafts and is greater in NGF-secreting grafts (D). E, F, Fourteen days after SCI, the density of 27C7 immunolabeling continues to increase in both GFP-expressing and NGF-secreting cell grafts. G, By 28 d, 27C7 immunolabeling within GFP-expressing grafts remains moderate, and the 27C7-labeled processes are loosely intertwined (inset). Scale bar: insets in G, H, 22 μm. H, Within NGF-secreting cell grafts, 27C7 levels have further increased 28 d after grafting, densely filling grafts (H, inset). I, 27C7 immunolabeling is not observed in the intact spinal cord. J, Primary antibody omission control (1° AB Omission).
The preceding results indicate that NG2 expression within the lesioned spinal cord, and within grafts placed in the lesion cavity, correlates temporally with Schwann cell migration into these regions. To identify whether, in fact, Schwann cells are cellular sources of NG2 within lesion sites of the nervous system, immunofluorescent confocal microscopy was performed on sections double labeled for 27C7 and NG2 and separately on sections double labeled for the S-100 Schwann cell marker (Brockes et al., 1979) and NG2 (Fig. 9). We also examined Schwann cell regulation of NG2 production in a sciatic nerve lesion model to determine whether findings in CNS injury models parallel changes in peripheral nerve injury models. In the intact sciatic nerve, weak amounts of NG2 immunolabeling were observed (Fig. 9B,E). Colocalization studies showed very little association of NG2 labeling with either 27C7-labeled (Fig. 9A-C) or S-100-labeled (Fig. 9D-F) cellular profiles in the intact sciatic nerve; however, 3 d after nerve crush, levels of NG2 immunolabeling increased considerably in the sciatic nerve, and specific colocalization was readily apparent between NG2 and both 27C7 (Fig. 9G-I) and S-100 (Fig. 9J-L). Thus, reactive Schwann cells in the PNS produce NG2 after injury. Scant additional amounts of NG2 immunoreactivity appeared to be associated with sources other than Schwann cells after nerve lesion, suggesting that NG2 can be produced by multiple cellular sources after peripheral injury (Fig. 9I). Paralleling these findings in the injured peripheral nerve, double-labeling experiments in animals with spinal cord injury demonstrated specific colocalization of 27C7 (Fig. 9M-O) and S-100 (Fig. 9P-R) with NG2 labeling in both GFP-expressing and NGF-secreting fibroblast grafts 14 and 28 d after SCI. S-100 is known to label astrocytes (Ghandour et al., 1981) in addition to Schwann cells, but astrocytes do not infiltrate fibroblast grafts appreciably (Grill et al., 1997). Colocalization studies with GFP-expressing fibroblasts and NG2 showed no overlap (Fig. 7D-F), excluding fibroblasts as the cellular source of NG2 in the graft. Thus, Schwann cells migrating into sites of CNS injury-grafting produce NG2.
Figure 9.

NG2 is produced by Schwann cells and endothelial cells after sciatic nerve injury and in the spinal cord lesion site. A, 27C7 immunolabeling in the intact sciatic nerve. Few Schwann cells are nonmyelinating. B, E, Little NG2 immunolabeling is detected in the intact sciatic nerve. C, Merged images of 27C7-NG2 immnuolabeling demonstrate no colocalization of these markers in the intact sciatic nerve (arrows). Scale bars: A-C, 11 μm. D, S-100 immunolabeling in the intact sciatic nerve. F, Merged images of S-100-NG2 immunolabeling further confirm no colocalization of NG2 with Schwann cells in the intact sciatic nerve (arrows). Scale bars: D-F, 9 μm. G, 27C7-labeled Schwann cells are prominent 3 d after sciatic nerve crush. Section is located 5 mm distal to crush site. H, K, NG2 immunolabeling is also increased 3 d after sciatic nerve lesion. I, Merged image demonstrates strict colocalization of 27C7-NG2 labeling (arrows). Scale bars: G-I, 11 μm. J, S-100-labeled Schwann cells in the crushed sciatic nerve. L, Merged images of S-100/NG2 labeling further confirm colocalization of NG2 with Schwann cells in the injured sciatic nerve (arrows). Scale bars: J-L, 11 μm. M-O, 27C7-labeled Schwann cells within GFP-expressing cell grafts in 28 d spinal cord lesion cavity also strictly colocalize with NG2 labeling, indicating that Schwann cells migrating into the lesioned CNS also produce NG2. Scale bars: M-O, 11 μm. P-R, S-100-labeled Schwann cells also strictly colocalize with NG2 labeling within GFP-expressing cell grafts. Scale bars: P-R, 11 μm. S, NG2 immunolabeling is present along tubular processes within cell grafts in the lesion cavity (GFP-expressing graft 28 d after implantation). T, The endothelial cell marker RECA1 identifies blood vessels in the same graft section. U, Merged image of A and B demonstrates colocalization of RECA1-NG2, indicating that NG2 is also produced by, or specifically deposited along, endothelial cells comprising blood vessels. Scale bars: S-U, 24 μm.
Endothelial cells are an additional cellular source of NG2
NG2 labeling also outlined larger, tubular structures throughout fibroblast grafts that resembled vasculature (Fig. 9S). Immunolabeling with the endothelial cell marker RECA1 confirmed that neo-vascularization occurred within grafts (Fig. 9T) and that NG2 labeling also colocalized specifically with RECA1 (Fig. 9U). Thus, endothelial cells are an additional source of NG2 within grafts in the lesion cavity. NG2-RECA1-specific immunolabeling was also observed in the injured sciatic nerve (data not shown).
The cell adhesion molecules L1 and laminin are also coexpressed with NG2
To examine Schwann cell production of molecules other than CSPG components that might influence axon growth within sites of SCI, L1 and laminin immunolabeling were performed. After SCI, the cell adhesion molecule L1 was expressed on long thin cellular structures that resembled Schwann cell processes throughout both GFP-expressing and NGF-secreting fibroblast grafts (Fig. 10). L1 immunoreactivity was present on more cellular processes in NGF-secreting cell grafts than in GFP-expressing fibroblasts 14 and 28 d after dorsal column SCI (Fig. 10A,B). High-magnification colocalization studies indicated that L1 and NG2 labeling were highly colocalized on cells within grafts (Fig. 10C-E). Double labeling also indicated that L1 was highly colocalized with the Schwann cell marker S-100 within grafts (Fig. 10F-H). L1 is also a known cellular marker for nonmyelinating Schwann cells (Martini and Schachner, 1986).
Figure 10.
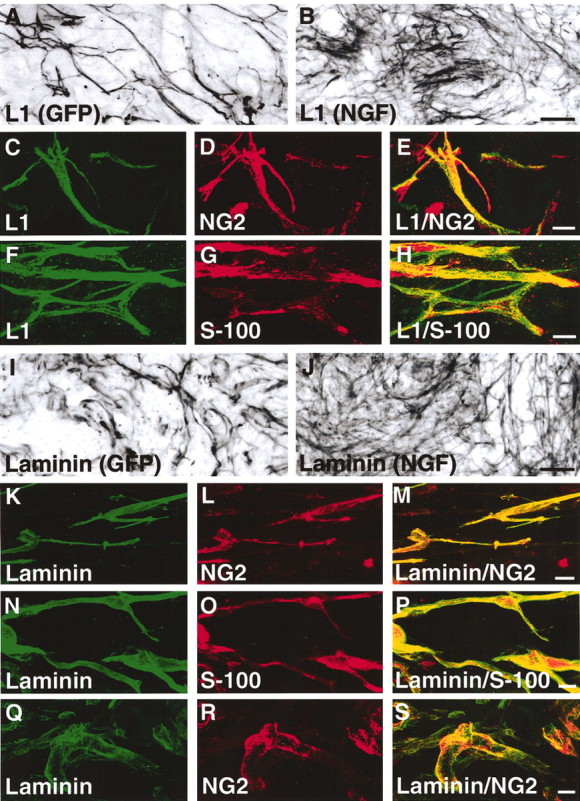
L1 and laminin are coexpressed in cell grafts and colocalize with NG2. A, Immunolabeling for the cell adhesion molecule L1 is present in 28 d GFP-expressing cell grafts and is markedly increased in NGF-secreting grafts (B). L1 labeling is distributed along long cellular processes that are suggestive of Schwann cells. Scale bars: A, B, 44 μm. L1 labeling (C) and NG2 labeling (D) consistently colocalize within all cell grafts (E) (28 d GFP graft pictured). Scale bars: C-E, 9 μm. L1 labeling (F) also colocalizes with the Schwann cell marker S-100 (G, H) within all cell grafts (28 d GFP graft pictured), confirming L1 production by NG2-producing Schwann cells. Scale bars: F-H, 9 μm. Laminin immunolabeling (I) is also present in GFP-expressing and NGF-secreting cell grafts in 28 d lesion cavity (J). Scale bars: I, J, 44 μm. Laminin labeling (K) and NG2 labeling (L) also consistently colocalize (M, merged image) within all cell grafts (28 d GFP graft pictured). Scale bars: K-M, 10 μm. Laminin labeling (N) also colocalizes with the Schwann cell marker S-100 (O, P) within all cell grafts (2 d GFP graft pictured), confirming Laminin production by NG2 producing Schwann cells. Scale bars: N-P, 9 μm. Laminin (Q) and NG2 labeling (R) also colocalize on (S, merged image) vascular structures (28 d GFP graft pictured). Scale bars: Q-S, 10 μm.
Similar to patterns of L1 immunolabeling, laminin expression was observed throughout grafts and was more dense within NGF-secreting grafts than GFP-expressing grafts 14 and 28 d after SCI (Fig. 10I,J). Consistent with patterns of L1 labeling, laminin structures within grafts also strictly colocalized with NG2-labeled structures (Fig. 10K-M, Q-S). Strict colocalization of laminin labeling with NG2 was observed on cellular profiles suggestive of both Schwann cells (Fig. 10K-M) and blood vessels (Fig. 10Q-S). Laminin labeling also strictly colocalized with S-100 labeling within grafts in the lesion site (Fig. 10N-P). Laminin is a known cellular marker for Schwann cells (Cornbrooks et al., 1983). Thus, both Schwann cells and endothelial cells within grafts were identified as cellular substrates that coexpress inhibitory CSPGs as well as the permissive cell adhesion molecules L1 and laminin in the injured CNS.
NG2 deposition widely precedes axonal growth
The preceding observations suggest that injured axons extend along NG2-expressing substrates at the lesion interface and within cell grafts. Alternatively, it is possible that axons penetrate the host-lesion interface before NG2 is deposited at sites of injury and that NG2-expressing Schwann cells subsequently associate with axons and locally secrete NG2. To address these possibilities, early postinjury time points were examined. We reported recently that NG2 expression appears as early as 24 hr after dorsal column lesions (Jones et al., 2002), whereas axons begin to penetrate fibroblast grafts placed at sites of injury between 3 and 7 d after injury (our unpublished observations). Thus, sections from animals 7 d after injury were double labeled for NG2-NF and examined using high-resolution confocal microscopy (Fig. 11). At this time point, most NF labeling was restricted to the region of the host-lesion interface and extended for only short distances within cell grafts, whereas NG2 labeling was present in most regions of the graft (Fig. 2). NG2 reaction product occupied a higher proportion of each field than NF labeling. The following three patterns of NG2 and NF labeling were observed. (1) Axons were associated with regions of NG2 deposition; notably, in many such cases, axons extended partially along the NG2 substrate, with remaining substrate present beyond the neurite, suggesting that the axon was in the active process of extending along the NG2 substrate (Fig. 11). (2) Regions of CSPG deposition were observed that lacked associated axons. This was the most common observation and indicated that CSPG deposition did not require the presence of a preceding, associated axon. (3) Axons were present that were not associated with CSPG labeling. This was the least common observation. Collectively, these findings at early time points after injury suggest that CSPG deposition primarily precedes axon growth and that most extending axons subsequently associate with CSPG-expressing substrates.
Figure 11.

NG2 deposition precedes axonal growth. Double immunolabeling for NG2 and neurofilament 7 d after injury in a GFP-expressing cell graft. A, NG2 is expressed throughout the extracellular matrix in the graft tissue adjoining the host-graft interface (dashed line; graft is to the right). NF-labeled axons penetrate the host-graft interface (B) and extend along NG2 substrates (C, arrow). Considerable regions of CSPG deposition lack associated axons, indicating that CSPG deposition does not require the presence of an associated axon (arrowheads). Scale bar, 24 μm.
Discussion
Findings of this study suggest that growing axons after injury in vivo are associated, paradoxically, with inhibitory CSPG substrates. Schwann cells and endothelial cells are identified as sources of these inhibitory CSPGs within a milieu of regrowing axons. Yet these same cells also produce the permissive cell adhesion molecules L1 and laminin, suggesting that successful axon growth after adult injury is the result of a balance of positive and negative signals located in highly localized sites: if a preponderance of positive signals is present, then growth will proceed even in the presence of inhibitory molecules.
Failure of axonal regeneration in the adult mammalian CNS has been attributed to various inhibitory molecules, including CSPGs (Davies et al., 1997; Lemons et al., 1999; Asher et al., 2000; Moon et al., 2001; Bradbury et al., 2002). The current study investigated whether spinal cord axons are capable of overcoming an extracellular milieu rich in CSPGs and indeed demonstrates growth of several axonal populations after injury, including CTB-labeled dorsal column sensory, rubrospinal, and nociceptive axons across a lesion-graft site and into a milieu that is rich in CSPGs. We also show extensive CSPG expression in the crushed sciatic nerve, a milieu in which injured axons regenerate (Forman and Berenberg, 1978). Although the inhibitory effects of CSPGs on axon growth have been demonstrated most clearly in vitro (Dou and Levine, 1994; Friedlander et al., 1994; Milev et al., 1994; McKeon et al., 1995; Yamada et al., 1997; Zuo et al., 1998b; Fidler et al., 1999; Schmalfeldt et al., 2000; Snow et al., 2002; Ughrin et al., 2003), it is likely that these molecules also inhibit axon growth in vivo. Upregulation of CSPGs and other inhibitory proteoglycans occurs as early as 1 d after SCI and is sustained for extended periods up to 6 months after injury (Jones and Tuszynski, 2002; Jones et al., 2002, 2003; Tang et al., 2003); however, axonal extension during nervous system development, and most likely, as the present study suggests, after injury in the adult, is a balance of attractive and repulsive forces in the local environment consisting of several ECM molecules, cell adhesion molecules, and growth factors (McKeon et al., 1995; Walsh and Doherty, 1996). Although local CSGPs contribute to inhibition of axon growth in the injured adult spinal cord, they are strictly colocalized with the cell adhesion molecules laminin and L1, which support growth. The presence of a growth factor such as NGF additionally tips the balance in favor of growth, significantly increasing axon penetration into grafts.
Nonmyelinating Schwann cells were sources of NG2 in both central and peripheral injury sites in this study. This is consistent with a recent report that AN2, the mouse homolog of rat NG2, colocalizes with p75 and L1 (immature and nonmyelinating Schwann cell markers) but not with MAG (a myelinating Schwann cell marker) in the developing sciatic nerve (Schneider et al., 2001). The latter study also reported that AN2 protein is downregulated in intact adult sciatic nerve. Martin et al. (2001) also reported that NG2 does not colocalize with p75 in the intact sciatic nerve, a finding consistent with our own observation that Schwann cells in unlesioned sciatic nerves do not express NG2, but they express readily detectable quantities 3 d after nerve crush (Fig. 9). Zuo and colleagues (1998a, 2002) also reported that general CSPG expression was upregulated in injured peripheral nerve. Thus, Schwann cells produce NG2 during development, downregulate expression in adulthood, and re-express NG2 after injury, constituting a significant source of an inhibitory ECM molecule despite their recognized role in supporting and guiding axon growth during development and regeneration.
Although several CSPGs are strongly re-expressed in the host spinal cord after injury, only NG2 and versican are expressed in the fibroblast grafts. We reported previously that NG2 appears to constitute the major CSPG species produced at sites of SCI (Jones et al., 2002); observations of the present study are consistent with these previous findings and demonstrate additionally that quantities of NG2 in animals with cell grafts significantly exceed amounts in subjects with lesions alone (Fig. 2). The broad expression of NG2 by oligodendrocyte progenitor cells (Nishiyama et al., 1996a,b; Jones et al., 2002), macrophages (Bu et al., 2001; McTigue et al., 2001; Zhang et al., 2001; Jones et al., 2002), and Schwann cells (Figs. 9, 10) (Schneider et al., 2001) after SCI most likely accounts for strong NG2 immunolabeling in both the host (macrophages, oligodendrocyte progenitor cells) and the graft tissue (Schwann cells). The greater amount of NG2 immunolabeling in NGF-secreting grafts compared with GFP-expressing grafts is likely because of the higher density of Schwann cells in the former graft type. Versican deposition, however, may be the result of an oligodendrocyte cell response to injury in the host tissue (Asher et al., 2002) and to fibroblasts (Zimmermann and Ruoslahti, 1989) within grafts. Expression of the CSPGs brevican, neurocan, and phosphacan is restricted to reactive astrocytes (Haas et al., 1999; McKeon et al., 1999; Thon et al., 2000; Jones et al., 2003), a cell type that does not infiltrate fibroblast grafts appreciably (Grill et al., 1997), likely accounting for the lack of expression of these CSPG molecules within grafts.
A potential caveat to the concept that a balance of attractive and repulsive factors influenced axonal growth in this experiment was the use of fibroblasts to constitute a cellular matrix in the injury site. Fibroblasts might secrete factors that alter neurite recognition of inhibitory components of the NG2 glycoprotein; however, this possibility is rendered improbable for two reasons. First, we observed a similar pattern of axon association with NG2-expressing surfaces when various substrates lacking fibroblasts are placed in the lesion site, including purified Schwann cells, stem cells, or type I collagen alone without cells (L. L. Jones, P. Lu, N. Weidner, and M. H. Tuszynski, unpublished observations); the common element in all cases is that Schwann cells migrate into the region (if not already grafted), secrete NG2, and associate with extending axons. Second, axons also associate with NG2 that is located in host tissue adjacent to the lesion boundary, where fibroblasts are not present. Thus, it is unlikely that responses of injured axons to NG2 were altered by a factor secreted by fibroblasts.
Many research reports that address the topic of axon growth after CNS injury conclude with the statement that the most effective approach to augmenting regeneration will likely require a combinatorial approach of providing the following: (1) axon growth substrates, (2) growth-promoting molecules such as growth factors, and (3) neutralization of inhibitory influences. The present findings lend substantial mechanistic support to this concept; however, these findings also raise a more subtle interpretation of combinatorial approaches to regeneration, suggesting that the provision of a growth-promoting stimulus of sufficient magnitude may counterbalance inhibition to a degree that will allow successful axon growth even in the absence of targeted reduction of inhibitors. Clearly, the issues raised by the preceding statement are complex. For example, inhibitory ECM molecules, which are produced adjacent to lesion sites, are most likely to influence axon sprouting and growth immediately in the region of injury. In contrast, inhibitory molecules associated with long, white matter tracts, including Nogo (Fouad et al., 2001) and MAG (McKerracher et al., 1994; Filbin, 1995), are more likely to restrict long-distance axon growth beyond the injury site through white matter tracts. Thus, local growth factor delivery at lesion sites may be sufficient to counterbalance inhibitory influences of the ECM but may not be sufficient to overcome inhibition of white matter remote from the injury site. In such a case, propelling axon growth for significant distances might require neutralizers of inhibition or extended gradients of growth factors; however, inhibition in intact white matter of the mature nervous system, as in the developing nervous system, may be necessary to maintain the topography of highly specific nervous system projections (for review, see Grumet et al., 1996; Margolis and Margolis, 1997; Rauch, 1997; Buffo et al., 2000). If so, then neutralization of inhibition in an attempt to enhance repair of injured systems could induce aberrant and dysfunctional sprouting of noninjured systems that are also exposed to neutralizers. For example, Buffo et al. (2000) reported that neutralization of the myelin-associated inhibitor Nogo in the intact cerebellum induced aberrant sprouting of local circuitry. Interestingly, evidence also exists that neurites that are actively growing in the CNS may be less sensitive to white matter inhibition, extending for long distances through white matter in which myelin-associated inhibitors have not been experimentally neutralized (Davies et al., 1997). Thus, additional study of these complex possibilities in animal models is required to more precisely define parameters that will optimize axonal regeneration in the adult CNS.
Conclusions
Axons crossing a region of CNS injury and growing into a cellular bridge specifically associate with surfaces expressing inhibitory ECM molecules. A balance of inhibition and stimulation are likely to account for these findings, suggesting that highly localized responses to a variety of growth cues ultimately determine the success or failure of axonal regeneration.
Footnotes
This work was supported by the National Institutes of Health, the Veterans Administration, the Hollfelder Foundation, and the Heumann Foundation. We thank Dr. D. Miller for the Maloney murine leukemia virus, Dr. J. Conner for performing the NGF ELISA, Dr. M. Grumet for the L1 antibody, Dr. K. Wewetzer for the 27C7 antibody, Dr. B. Stallcup for the NG2 antibodies, and Dr. Y. Yamaguchi for the brevican antibody.
Correspondence should be addressed to Dr. Mark H. Tuszynski, Department of Neurosciences-0626, University of California-San Diego, 9500 Gilman Drive, La Jolla, CA 92093. E-mail: mtuszyns@ucsd.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/239276-13$15.00/0
References
- Asher RA, Morgenstern DA, Fidler PS, Adcock KH, Oohira A, Braistead JE, Levine JM, Margolis RU, Rogers JH, Fawcett JW ( 2000) Neurocan is upregulated in injured brain and in cytokine-treated astrocytes. J Neurosci 20: 2427-2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asher RA, Morgenstern DA, Shearer MC, Adcock KH, Pesheva P, Fawcett JW ( 2002) Versican is upregulated in CNS injury and is a product of oligodendrocyte lineage cells. J Neurosci 22: 2225-2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blesch A, Tuszynski MH ( 2001) GDNF gene delivery to injured adult CNS motor neurons promotes axonal growth, expression of the trophic neuropeptide CGRP, and cellular protection. J Comp Neurol 436: 399-410. [DOI] [PubMed] [Google Scholar]
- Blesch A, Grill RJ, Tuszynski MH ( 1998) Neurotrophin gene therapy in CNS models of trauma and degeneration. Prog Brain Res 117: 473-484. [DOI] [PubMed] [Google Scholar]
- Blesch A, Uy HS, Grill RJ, Cheng JG, Patterson PH, Tuszynski MH ( 1999) Leukemia inhibitory factor augments neurotrophin expression and corticospinal axon growth after adult CNS injury. J Neurosci 19: 3556-3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury EJ, Khemani S, King VR, Priestley JV, McMahon SB ( 1999) NT-3 promotes growth of lesioned adult rat sensory axons ascending in the dorsal columns of the spinal cord. Eur J Neurosci 11: 3873-3883. [DOI] [PubMed] [Google Scholar]
- Bradbury EJ, Moon LD, Popat RJ, King VR, Bennett GS, Patel PN, Fawcett JW, McMahon SB ( 2002) Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature 416: 636-640. [DOI] [PubMed] [Google Scholar]
- Braunewell KH, Martini R, LeBaron R, Kresse H, Faissner A, Schmitz B, Schachner M ( 1995) Up-regulation of a chondroitin sulphate epitope during regeneration of mouse sciatic nerve: evidence that the immunoreactive molecules are related to the chondroitin sulphate proteoglycans decorin and versican. Eur J Neurosci 7: 792-804. [DOI] [PubMed] [Google Scholar]
- Brockes JP, Fields KL, Raff MC ( 1979) Studies on cultured rat Schwann cells. I. Establishment of purified populations from cultures of peripheral nerve. Brain Res 165: 105-118. [DOI] [PubMed] [Google Scholar]
- Bu J, Akhtar N, Nishiyama A ( 2001) Transient expression of the NG2 proteoglycan by a subpopulation of activated macrophages in an excitotoxic hippocampal lesion. Glia 34: 296-310. [DOI] [PubMed] [Google Scholar]
- Buffo A, Zagrebelsky M, Huber AB, Skerra A, Schwab ME, Strata P, Rossi F ( 2000) Application of neutralizing antibodies against NI-35/250 myelin-associated neurite growth inhibitory proteins to the adult rat cerebellum induces sprouting of uninjured Purkinje cell axons. J Neurosci 20: 2275-2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornbrooks CJ, Carey DJ, McDonald JA, Timpl R, Bunge RP ( 1983) In vivo and in vitro observations on laminin production by Schwann cells. Proc Natl Acad Sci USA 80: 3850-3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies SJ, Fitch MT, Memberg SP, Hall AK, Raisman G, Silver J ( 1997) Regeneration of adult axons in white matter tracts of the central nervous system. Nature 390: 680-683. [DOI] [PubMed] [Google Scholar]
- De Winter F, Oudega M, Lankhorst AJ, Hamers FP, Blits B, Ruitenberg MJ, Pasterkamp RJ, Gispen WH, Verhaagen J ( 2002) Injury-induced class 3 semaphorin expression in the rat spinal cord. Exp Neurol 175: 61-75. [DOI] [PubMed] [Google Scholar]
- Dou CL, Levine JM ( 1994) Inhibition of neurite growth by the NG2 chondroitin sulfate proteoglycan. J Neurosci 14: 7616-7628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidler PS, Schuette K, Asher RA, Dobbertin A, Thornton SR, Calle-Patino Y, Muir E, Levine JM, Geller HM, Rogers JH, Faissner A, Fawcett JW ( 1999) Comparing astrocytic cell lines that are inhibitory or permissive for axon growth: the major axon-inhibitory proteoglycan is NG2. J Neurosci 19: 8778-8788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filbin MT ( 1995) Myelin-associated glycoprotein: a role in myelination and in the inhibition of axonal regeneration? Curr Opin Neurobiol 5: 588-595. [DOI] [PubMed] [Google Scholar]
- Fitch MT, Silver J ( 1997) Activated macrophages and the blood-brain barrier: inflammation after CNS injury leads to increases in putative inhibitory molecules. Exp Neurol 148: 587-603. [DOI] [PubMed] [Google Scholar]
- Forman DS, Berenberg RA ( 1978) Regeneration of motor axons in the rat sciatic nerve studied by labeling with axonally transported radioactive proteins. Brain Res 156: 213-225. [DOI] [PubMed] [Google Scholar]
- Fouad K, Dietz V, Schwab ME ( 2001) Improving axonal growth and functional recovery after experimental spinal cord injury by neutralizing myelin associated inhibitors. Brain Res Brain Res Rev 36: 204-212. [DOI] [PubMed] [Google Scholar]
- Franzen R, Martin D, Daloze A, Moonen G, Schoenen J ( 1999) Grafts of meningeal fibroblasts in adult rat spinal cord lesion promote axonal regrowth. NeuroReport 10: 1551-1556. [DOI] [PubMed] [Google Scholar]
- Friedlander DR, Milev P, Karthikeyan L, Margolis RK, Margolis RU, Grumet M ( 1994) The neuronal chondroitin sulfate proteoglycan neurocan binds to the neural cell adhesion molecules Ng-CAM/L1/NILE and N-CAM, and inhibits neuronal adhesion and neurite outgrowth. J Cell Biol 125: 669-680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghandour MS, Langley OK, Labourdette G, Vincendon G, Gombos G ( 1981) Specific and artefactual cellular localizations of S 100 protein: an astrocyte marker in rat cerebellum. Dev Neurosci 4: 66-78. [DOI] [PubMed] [Google Scholar]
- Goretzki L, Burg MA, Grako KA, Stallcup WB ( 1999) High-affinity binding of basic fibroblast growth factor and platelet-derived growth factor-AA to the core protein of the NG2 proteoglycan. J Biol Chem 274: 16831-16837. [DOI] [PubMed] [Google Scholar]
- Grill R, Murai K, Blesch A, Gage FH, Tuszynski MH ( 1997) Cellular delivery of neurotrophin-3 promotes corticospinal axonal growth and partial functional recovery after spinal cord injury. J Neurosci 17: 5560-5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grumet M, Friedlander DR, Sakurai T ( 1996) Functions of brain chondroitin sulfate proteoglycans during developments: interactions with adhesion molecules. Perspect Dev Neurobiol 3: 319-330. [PubMed] [Google Scholar]
- Haas CA, Rauch U, Thon N, Merten T, Deller T ( 1999) Entorhinal cortex lesion in adult rats induces the expression of the neuronal chondroitin sulfate proteoglycan neurocan in reactive astrocytes. J Neurosci 19: 9953-9963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones LL, Tuszynski MH ( 2002) Spinal cord injury elicits expression of keratan sulfate proteoglycans by macrophages, reactive microglia, and oligodendrocyte progenitors. J Neurosci 22: 4611-4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones LL, Yamaguchi Y, Stallcup WB, Tuszynski MH ( 2002) NG2 is a major chondroitin sulfate proteoglycan produced after spinal cord injury and is expressed by macrophages and oligodendrocyte progenitors. J Neurosci 22: 2792-2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones LL, Margolis RU, Tuszynski MH ( 2003) The chondroitin sulfate proteoglycans neurocan, brevican, phosphacan and versican are differentially regulated following spinal cord injury. Exp Neurol 182: 399-411. [DOI] [PubMed] [Google Scholar]
- Lemons ML, Howland DR, Anderson DK ( 1999) Chondroitin sulfate proteoglycan immunoreactivity increases following spinal cord injury and transplantation. Exp Neurol 160: 51-65. [DOI] [PubMed] [Google Scholar]
- Levine JM ( 1994) Increased expression of the NG2 chondroitin-sulfate proteoglycan after brain injury. J Neurosci 14: 4716-4730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Himes BT, Tryon B, Moul J, Chow SY, Jin H, Murray M, Tessler A, Fischer I ( 1998) Intraspinal grafting of fibroblasts genetically modified by recombinant adenoviruses. NeuroReport 9: 1075-1079. [DOI] [PubMed] [Google Scholar]
- Lu P, Blesch A, Tuszynski MH ( 2001) Neurotrophism without neurotropism: BDNF promotes survival but not growth of lesioned corticospinal neurons. J Comp Neurol 436: 456-470. [DOI] [PubMed] [Google Scholar]
- Lu P, Jones LL, Snyder EY, Tuszynski MH ( 2003) Neural stem cells constitutively secrete neurotrophic factors and promote extensive host axonal growth after spinal cord injury. Exp Neurol 181: 115-129. [DOI] [PubMed] [Google Scholar]
- Margolis RU, Margolis RK ( 1997) Chondroitin sulfate proteoglycans as mediators of axon growth and pathfinding. Cell Tissue Res 290: 343-348. [DOI] [PubMed] [Google Scholar]
- Martin S, Levine AK, Chen ZJ, Ughrin Y, Levine JM ( 2001) Deposition of the NG2 proteoglycan at nodes of Ranvier in the peripheral nervous system. J Neurosci 21: 8119-8128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini R, Schachner M ( 1986) Immunoelectron microscopic localization of neural cell adhesion molecules (L1, N-CAM, and MAG) and their shared carbohydrate epitope and myelin basic protein in developing sciatic nerve. J Cell Biol 103: 2439-2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeon RJ, Hoke A, Silver J ( 1995) Injury-induced proteoglycans inhibit the potential for laminin-mediated axon growth on astrocytic scars. Exp Neurol 136: 32-43. [DOI] [PubMed] [Google Scholar]
- McKeon RJ, Jurynec MJ, Buck CR ( 1999) The chondroitin sulfate proteoglycans neurocan and phosphacan are expressed by reactive astrocytes in the chronic CNS glial scar. J Neurosci 19: 10778-10788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKerracher L, David S, Jackson DL, Kottis V, Dunn RJ, Braun PE ( 1994) Identification of myelin-associated glycoprotein as a major myelin-derived inhibitor of neurite growth. Neuron 13: 805-811. [DOI] [PubMed] [Google Scholar]
- McMahon SB, Armanini MP, Ling LH, Phillips HS ( 1994) Expression and coexpression of Trk receptors in subpopulations of adult primary sensory neurons projecting to identified peripheral targets. Neuron 12: 1161-1171. [DOI] [PubMed] [Google Scholar]
- McTigue DM, Wei P, Stokes BT ( 2001) Proliferation of NG2-positive cells and altered oligodendrocyte numbers in the contused rat spinal cord. J Neurosci 21: 3392-3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael GJ, Kaya E, Averill S, Rattray M, Clary DO, Priestley JV ( 1997) TrkA immunoreactive neurones in the rat spinal cord. J Comp Neurol 385: 441-455. [PubMed] [Google Scholar]
- Milev P, Friedlander DR, Sakurai T, Karthikeyan L, Flad M, Margolis RK, Grumet M, Margolis RU ( 1994) Interactions of the chondroitin sulfate proteoglycan phosphacan, the extracellular domain of a receptor-type protein tyrosine phosphatase, with neurons, glia, and neural cell adhesion molecules. J Cell Biol 127: 1703-1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AD, Miller DG, Garcia JV, Lynch CM ( 1993) Use of retroviral vectors for gene transfer and expression. Methods Enzymol 217: 581-599. [DOI] [PubMed] [Google Scholar]
- Moon LD, Asher RA, Rhodes KE, Fawcett JW ( 2001) Regeneration of CNS axons back to their target following treatment of adult rat brain with chondroitinase ABC. Nat Neurosci 4: 465-466. [DOI] [PubMed] [Google Scholar]
- Moon LD, Asher RA, Rhodes KE, Fawcett JW ( 2002) Relationship between sprouting axons, proteoglycans and glial cells following unilateral nigrostriatal axotomy in the adult rat. Neuroscience 109: 101-117. [DOI] [PubMed] [Google Scholar]
- Nishiyama A, Lin XH, Giese N, Heldin CH, Stallcup WB ( 1996a) Co-localization of NG2 proteoglycan and PDGF alpha-receptor on O2A progenitor cells in the developing rat brain. J Neurosci Res 43: 299-314. [DOI] [PubMed] [Google Scholar]
- Nishiyama A, Lin XH, Giese N, Heldin CH, Stallcup WB ( 1996b) Interaction between NG2 proteoglycan and PDGF alpha-receptor on O2A progenitor cells is required for optimal response to PDGF. J Neurosci Res 43: 315-330. [DOI] [PubMed] [Google Scholar]
- Paino CL, Bunge MB ( 1991) Induction of axon growth into Schwann cell implants grafted into lesioned adult rat spinal cord. Exp Neurol 114: 254-257. [DOI] [PubMed] [Google Scholar]
- Pasterkamp RJ, Anderson PN, Verhaagen J ( 2001) Peripheral nerve injury fails to induce growth of lesioned ascending dorsal column axons into spinal cord scar tissue expressing the axon repellent Semaphorin3A. Eur J Neurosci 13: 457-471. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C ( 1998) The rat brain in stereotaxic coordinates, Ed 4. San Diego: Academic.
- Plant GW, Bates ML, Bunge MB ( 2001) Inhibitory proteoglycan immunoreactivity is higher at the caudal than the rostral Schwann cell graft-transected spinal cord interface. Mol Cell Neurosci 17: 471-487. [DOI] [PubMed] [Google Scholar]
- Rauch U ( 1997) Modeling an extracellular environment for axonal pathfinding and fasciculation in the central nervous system. Cell Tissue Res 290: 349-356. [DOI] [PubMed] [Google Scholar]
- Schmalfeldt M, Bandtlow CE, Dours-Zimmermann MT, Winterhalter KH, Zimmermann DR ( 2000) Brain derived versican V2 is a potent inhibitor of axonal growth. J Cell Sci 113: 807-816. [DOI] [PubMed] [Google Scholar]
- Schneider S, Bosse F, D'Urso D, Muller H, Sereda MW, Nave K, Niehaus A, Kempf T, Schnolzer M, Trotter J ( 2001) The AN2 protein is a novel marker for the Schwann cell lineage expressed by immature and nonmyelinating Schwann cells. J Neurosci 21: 920-933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow DM, Smith JD, Gurwell JA ( 2002) Binding characteristics of chondroitin sulfate proteoglycans and laminin-1, and correlative neurite outgrowth behaviors in a standard tissue culture choice assay. J Neurobiol 51: 285-301. [DOI] [PubMed] [Google Scholar]
- Tang X, Davies JE, Davies SJ ( 2003) Changes in distribution, cell associations, and protein expression levels of NG2, neurocan, phosphacan, brevican, versican V2, and tenascin-C during acute to chronic maturation of spinal cord scar tissue. J Neurosci Res 71: 427-444. [DOI] [PubMed] [Google Scholar]
- Thon N, Haas CA, Rauch U, Merten T, Fassler R, Frotscher M, Deller T ( 2000) The chondroitin sulphate proteoglycan brevican is upregulated by astrocytes after entorhinal cortex lesions in adult rats. Eur J Neurosci 12: 2547-2558. [DOI] [PubMed] [Google Scholar]
- Tuszynski MH, Peterson DA, Ray J, Baird A, Nakahara Y, Gage FH ( 1994) Fibroblasts genetically modified to produce nerve growth factor induce robust neuritic ingrowth after grafting to the spinal cord. Exp Neurol 126: 1-14. [DOI] [PubMed] [Google Scholar]
- Tuszynski MH, Gabriel K, Gage FH, Suhr S, Meyer S, Rosetti A ( 1996) Nerve growth factor delivery by gene transfer induces differential outgrowth of sensory, motor, and noradrenergic neurites after adult spinal cord injury. Exp Neurol 137: 157-173. [DOI] [PubMed] [Google Scholar]
- Ughrin YM, Chen ZJ, Levine JM ( 2003) Multiple regions of the NG2 proteoglycan inhibit neurite growth and induce growth cone collapse. J Neurosci 23: 175-186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh FS, Doherty P ( 1996) Cell adhesion molecules and neuronal regeneration. Curr Opin Cell Biol 8: 707-713. [DOI] [PubMed] [Google Scholar]
- Weidner N, Blesch A, Grill RJ, Tuszynski MH ( 1999) Nerve growth factor-hypersecreting Schwann cell grafts augment and guide spinal cord axonal growth and remyelinate central nervous system axons in a phenotypically appropriate manner that correlates with expression of L1. J Comp Neurol 413: 495-506. [DOI] [PubMed] [Google Scholar]
- Weidner N, Ner A, Salimi N, Tuszynski MH ( 2001) Spontaneous corticospinal axonal plasticity and functional recovery after adult central nervous system injury. Proc Natl Acad Sci USA 98: 3513-3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wewetzer K, Grothe C, Christ B, Seilheimer B ( 1997) Identification and characterization of differentiation-dependent Schwann cell surface antigens by novel monoclonal antibodies: introduction of a marker common to the non-myelin-forming phenotype. Glia 19: 213-226. [DOI] [PubMed] [Google Scholar]
- Woolf CJ ( 2003) No Nogo. Now where to go? Neuron 38: 153-156. [DOI] [PubMed] [Google Scholar]
- Xu XM, Guenard V, Kleitman N, Aebischer P, Bunge MB ( 1995) A combination of BDNF and NT-3 promotes supraspinal axonal regeneration into Schwann cell grafts in adult rat thoracic spinal cord. Exp Neurol 134: 261-272. [DOI] [PubMed] [Google Scholar]
- Yamada H, Fredette B, Shitara K, Hagihara K, Miura R, Ranscht B, Stallcup WB, Yamaguchi Y ( 1997) The brain chondroitin sulfate proteoglycan brevican associates with astrocytes ensheathing cerebellar glomeruli and inhibits neurite outgrowth from granule neurons. J Neurosci 17: 7784-7795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Tohyama K, Winterbottom JK, Haque NS, Schachner M, Lieberman AR, Anderson PN ( 2001) Correlation between putative inhibitory molecules at the dorsal root entry zone and failure of dorsal root axonal regeneration. Mol Cell Neurosci 17: 444-459. [DOI] [PubMed] [Google Scholar]
- Zimmermann DR, Ruoslahti E ( 1989) Multiple domains of the large fibroblast proteoglycan, versican. EMBO J 8: 2975-2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo J, Hernandez YJ, Muir D ( 1998a) Chondroitin sulfate proteoglycan with neurite-inhibiting activity is up-regulated following peripheral nerve injury. J Neurobiol 34: 41-54. [PubMed] [Google Scholar]
- Zuo J, Neubauer D, Dyess K, Ferguson TA, Muir D ( 1998b) Degradation of chondroitin sulfate proteoglycan enhances the neurite-promoting potential of spinal cord tissue. Exp Neurol 154: 654-662. [DOI] [PubMed] [Google Scholar]
- Zuo J, Neubauer D, Graham J, Krekoski CA, Ferguson TA, Muir D ( 2002) Regeneration of axons after nerve transection repair is enhanced by degradation of chondroitin sulfate proteoglycan. Exp Neurol 176: 221-228. [DOI] [PubMed] [Google Scholar]