

Abstract
Cannabinoids have been shown to critically modulate cholinergic neurotransmission in the hippocampus, yet opposing effects of cannabinoid receptor 1 (CB1R) agonists on hippocampal synaptic acetylcholine (ACh) efflux have been reported. This study shows that administration of a synthetic CB1R agonist results in a biphasic, dose-dependent, effect on hippocampal ACh: a low (0.5 mg/kg, i.p.) and a high (5 mg/kg, i.p) dose of WIN55,212-2 induces a transient stimulation and a prolonged inhibition of hippocampal ACh efflux, respectively. Both effects of WIN55,212-2 are mediated through CB1 receptors coupled to Gi but involve different neuroanatomical sites. Thus, intrahippocampal infusion of the CB1R antagonist SR141716A or pertussis toxin blocked the inhibition of hippocampal ACh release induced by the high dose of WIN55,212-2, but was without effect on the stimulatory action of the low dose. In contrast, this latter effect was blocked by SR141716A or pertussis toxin infused, in dual microdialysis experiments, in the septum, in which the majority of cholinergic cell bodies projecting to the hippocampus reside. The stimulatory and inhibitory effects of WIN55,212-2 on hippocampal ACh involve dopamine D1 and D2 receptor activation, respectively, given that pretreatment with D1 and D2 receptor antagonists prevents the respective actions of WIN55,212-2. We propose that the in vivo observed biphasic effects of CB1R agonists on hippocampal ACh release result from a differential, functional association of anatomicaly distinct subpopulations of CB1-Gi coupled receptors to neurotransmitter systems that have opposing effects on ACh release. This concept could provide a theoretical framework to understand endocannabinoids as state-dependent modulators of neuronal activity.
Keywords: cannabinoids, acetylcholine, hippocampus, biphasic, microdialysis, septum, D1 dopamine receptors, D2 dopamine receptors
Introduction
Endocannabinoids in the CNS fine-tune information flow in neuronal networks associated with sensory integration and memory processing. Behavioral studies using SR141716A (Rinaldi-Carmona et al., 1995), the selective antagonist of the cannabinoid receptor 1 (CB1R) that is the prominent cannabinoid receptor in the CNS, and studies with CB1 knock-out mice suggest a role for endocannabinoids in cognitive functions (Reibaud et al., 1999; Lichtman et al., 2002). The hippocampus, a region that plays a crucial role in these processes, displays a high density of CB1Rs (Tsou et al., 1998) and high endocannabinoid levels (Di Marzo et al., 2000a).
At the molecular level, activity-dependent release of endocannabinoids, which act as retrograde signaling molecules, mediates depolarization-induced suppression of inhibition or excitation in this region (Wilson et al., 2001; Ohno-Shosaku et al., 2002). Exogenous cannabinoids also modulate memory, hippocampal neuronal activity, and synaptic plasticity (Childers et al., 1993; Lichtman et al., 1995; Hampson and Deadwyler, 1999). The in vitro effects of cannabinoid agonists have been well characterized in hippocampal slices, in which they decrease long-term potentiation and depression and inhibit neurotransmitter release, in particular that of acetylcholine (ACh) (for review, see Schlicker and Kathmann, 2001; Davies et al., 2002).
The septohippocampal cholinergic projection is considered a key neurotransmitter system in encoding learning and memory, and compounds that affect hippocampal ACh homeostasis have important therapeutic potential for various CNS pathologies. However, the in vivo neuromodulatory role of pharmacologically active cannabinoids on septohippocampal cholinergic activity remains poorly characterized. Surprisingly, in freely moving animals, opposite effects of CB1R agonists on hippocampal ACh have been reported. Gessa and colleagues (Gessa at al., 1997; Carta et al., 1998) showed that exogenously applied natural [Δ-9-tetrahydrocannabinol (THC)] or synthetic (WIN55,212-2) CB1R agonists attenuate hippocampal ACh efflux (Gessa at al., 1997; Carta et al., 1998), whereas Acquas et al. (2000, 2001) showed stimulation of hippocampal ACh release with the same compounds.
Both stimulatory and inhibitory effects after acute CB1R agonist administration have been reported in the context of other neurobiological responses (Chaperon and Thiebot, 1999). Interestingly, it has been shown that low doses of cannabinoids stimulate, whereas high doses inhibit, these functions. To infer the neurochemical profile and the potential therapeutic interest of direct or indirect cannabinoid agonists that would act by regulating hippocampal ACh release, we used an in vivo microdialysis approach to determine (1) whether the effects of the synthetic CB1R agonist WIN55,212-2 on hippocampal ACh also followed a biphasic pattern and (2) the nature of this dual regulation. Indeed, no valid unifying hypothesis exists at this point to explain the biphasic effects of cannabinoid compounds. These could be attributable to many different underlying mechanisms, such as the existence of diverse biochemical substrates (e.g., receptors) triggering opposing intracellular events or of activity-dependent, multiple-state, cell-signaling pathways and neuroanatomical circuits involved in the response to cannabinoids. We thus investigated whether the stimulatory and inhibitory components of the cholinergic response to WIN55,212-2 are mediated through the same or distinct (1) molecular entities (receptors, G-proteins), (2) neuroanatomical sites of action, or (3) neurotransmitter systems regulating septohippocampal cholinergic activity.
Materials and Methods
Animals. All studies were performed according to the guidelines set forth by the National Institutes of Health and implemented by the Animal Care and Use Committee of Eli Lilly and Company. Male Wistar rats (250-300 gm; Harlan Sprague Dawley, Indianapolis, IN) were used for experiments. They were housed in a vivarium for at least 1 week before use under standard laboratory conditions; water and food were available ad libitum during this period.
Surgical procedures. Two days before the microdialysis experiments, the rats were anesthetized with a mixture of chloral hydrate and pentobarbital (170 and 36 mg/kg in 30% propylene glycol and 14% ethanol), placed in a stereotaxic apparatus, and implanted with a guide cannula [Bioanalytical Systems (BAS), West Lafayette, IN] in the hippocampus [coordinates: anteroposterior (AP), -5.2; mediolateral (ML), 5.2; dorsoventral (DV), -3.8] according to Paxinos and Watson (1982). Twenty-four hours before testing, a 4 mm concentric microdialysis probe (model BR-4; BAS) was inserted through the guide cannula.
For dual-probe microdialysis experiments, the rats were also implanted, under the same conditions, with a guide cannula (BAS) in the medial septal area (coordinates: AP, 0.5; ML, 1.0; DV, -6.5; angle 9°) according to Paxinos and Watson (1982), as described by Moor et al. (1994). Twenty-four hours before testing, a 2 mm concentric microdialysis probe (model BR-2; BAS) was inserted through the guide cannula. The correct location of the probes was verified histologically at the end of the experiment. Typical probe placements in the hippocampus and the septum are illustrated in Figure 1.
Figure 1.
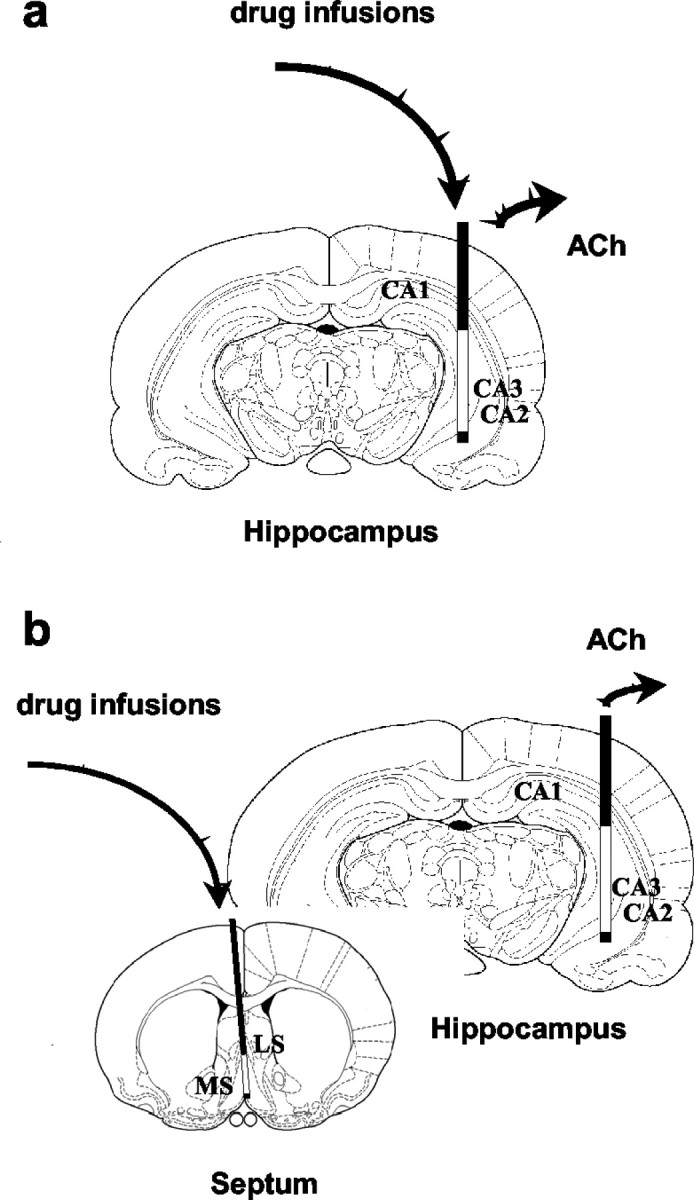
Placements of microdialysis probes in single- and dual-microdialysis experiments. The effects of systemically administered cannabinoids on ACh efflux in the hippocampus were measured with in vivo microdialysis. Dialysates collected through a 4 mm probe implanted in the hippocampus were analyzed for ACh by HPLC coupled to electrochemical detection (a, b). In some experiments, drugs were also infused in either the hippocampus through the ACh sampling microdialysis probe (a) or the septum through an additional 2 mm probe implanted in that region for drug delivery (b) (dual-microdialysis experiments).
In vivo microdialysis ACh measurements. ACh determination in hippocampal dialysates was performed as described previously (Damsma et al., 1988) with some modifications (Tzavara et al., 2003). On the day of the experiment, a modified Ringer's solution (in mm: 147.0 NaCl, 3.0 KCl, 1.3 CaCl2, 1.0 MgCl2, 1.0 Na2HPO4 · 7H2O, and 0.2 NaH2PO4 · H2O, pH 7.25) supplemented with 0.1 μm neostigmine was perfused at a rate of 2.4 μl/min in the hippocampus or in the hippocampus and the septum for dual-microdialysis experiments. Neostigmine was omitted from the solution perfused in the septum because the probe was used only for drug delivery (Moor et al., 1994). Samples were collected every 15 min and analyzed immediately, on-line, with HPLC coupled to electrochemical detection, with a 150 × 3 mm ACH-3 column [Environmental Sciences Associates (ESA), Chelmsford, MA] maintained at 35°C. The mobile phase [100 mm di-sodium hydrogen phosphate, 2 mm 1-octanesulfonic acid, and 50 μl/l of a microbicide reagent (MB; ESA), pH 8.0, adjusted with phosphoric acid] was delivered by an HPLC pump (ESA) at 0.4 ml/min. The potentiostat used for electrochemical detection (model Coulochem II; ESA) was connected with a solid phase reactor for ACh (model ACH-SPR; ESA) and with an analytical cell with platinum target (model 5041; ESA).
Drugs. WIN55,212-2 was purchased from Tocris Cookson (Ballwin, MO). SR141716A was provided by Eli Lilly and Company. Pertussis toxin (PTX), SCH23390, and S-raclopride were purchased from Sigma (St. Louis, MO). For systemic administration WIN55,212-2 and SR141716A were dissolved in saline containing 2% DMSO and 2% cremophor EL and were injected intraperitoneally at a volume of 3 ml/kg. For intrahippocampal or intraseptal administration, the drugs were dissolved in perfusion solution that contained 0.5% DMSO and 0.5% cremophor EL and were infused through the microdialysis probe at a rate of 2.4 μl/min for 30 min. Standard drug concentrations used for local administration experiments in this study were 100 μm for both WIN55,212-2 and SR141716A. It should be noted that the actual concentration of the compounds reaching the brain parenchyma is a mere fraction (∼1% in vivo recovery) of their concentration in the perfusion solution. Thus, the estimated concentration of the compounds applied topically is in the low micromolar range, i.e., in the same order of magnitude as those used in in vitro experiments (Gifford and Ashby, 1996). PTX was infused 24 hr before the experiment (Gronier and Rasmussen, 1999) through a combined infusion-microdialysis probe (model IBR-2 for septum and model IBR-4 for hippocampus; BAS) that was inserted through the guide cannula; 100 ng in a total volume of 0.001 ml were delivered per infusion with a flow rate of 300nl/min by using a 0.05 ml Hamilton syringe connected to a Univentor (Zejtun, Malta) microdialysis syringe pump. SCH23390 and S-raclopride were injected subcutaneously at a volume of 1 ml/kg 15 min before WIN55,212-2.
Statistics. Data (n = 4-7 rats per group) are expressed as multifold change from baseline, which is the average of the five basal values before any manipulation and were analyzed either with two-way [i.e., treatment (between-subjects variable) × time (within-subjects variable)] or three-way (treatment 1 × treatment 2 × time) ANOVA, followed by Duncan's test.
Results
There were no statistically significant differences in the basal values of ACh among the different experimental groups. Interestingly, there were no differences in hippocampal ACh in either animals under single- compared with dual-microdialysis schedule or in animals that received intraseptal or intrahippocampal PTX infusions compared with non-infused animals. Therefore, basal ACh values were pooled and are presented together. Basal ACh levels in the hippocampus were 192 ± 18 fmol ACh per 15 min.
Dose-dependent effects of WIN55,212-2 on ACh efflux in the hippocampus
The effects of a single systemic administration of WIN55,212-2 (0.5, 1, or 5 mg/kg, i.p.) on ACh efflux in the hippocampus are shown in Figure 2. The low dose of WIN55,212-2 (0.5 mg/kg, i.p.) significantly increased ACh efflux in the hippocampus compared with vehicle injection. ACh was increased, up to a peak value of 190%, 15 min after the injection of WIN55,212-2 (0.5 mg/kg, i.p.) and returned to baseline levels 1 hr later. On the contrary, the high dose of WIN55,212-2 (5 mg/kg, i.p.) significantly decreased ACh efflux in the hippocampus compared with vehicle injection. ACh was decreased (to a nadir value of 50%) 30 min after the injection of WIN55,212-2 (5 mg/kg, i.p.) and remained decreased through the end of the 3 hr observation period. In an additional set of experiments in which we monitored the effects of the high dose of WIN55,212-2 (5 mg/kg, i.p.) on hippocampal ACh for a longer period, ACh levels returned to baseline after 6 hr (data not shown). Interestingly, the intermediate dose of 1 mg/kg WIN55,212-2 had no effect on hippocampal ACh, suggesting that opposing networks and/or homeostatic inter-regulations mediate enhancing and suppressing effects of cannabinoid agonists on cholinergic neurotransmission.
Figure 2.
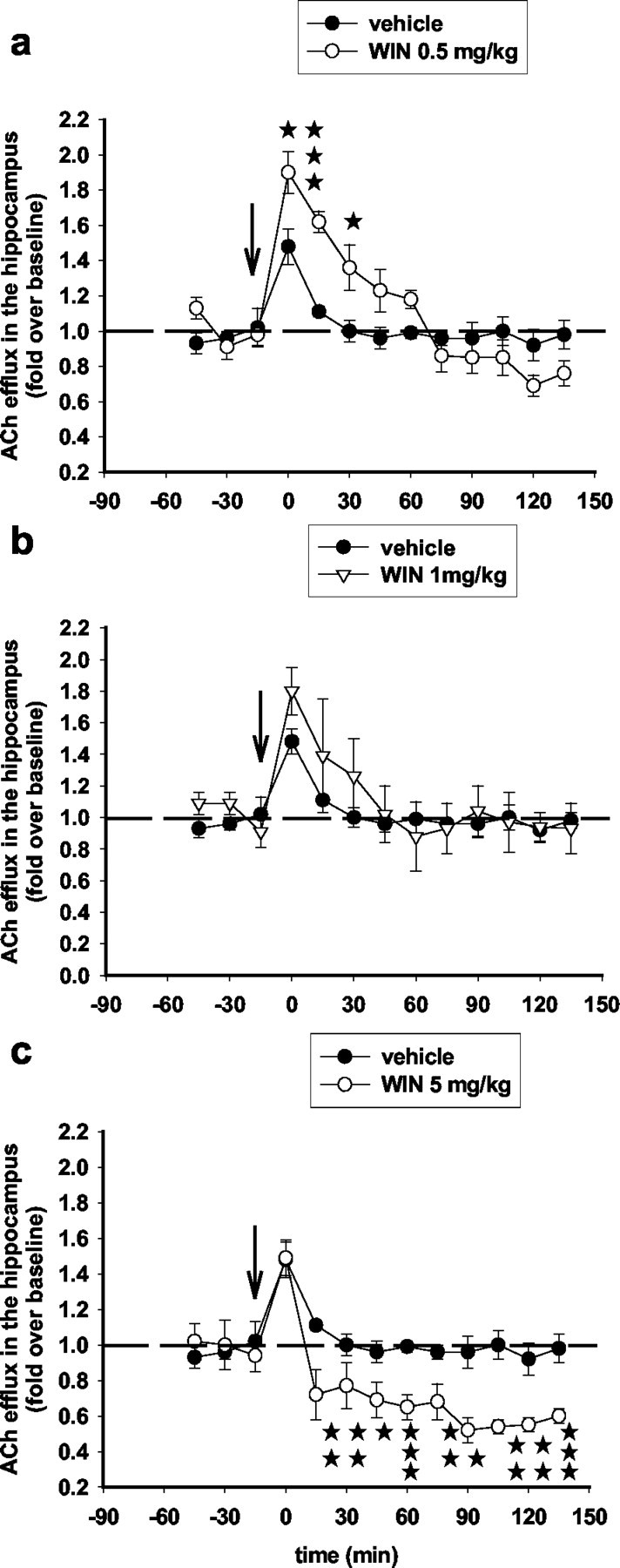
Biphasic, dose-dependent effects of WIN55,212-2 on ACh efflux in the hippocampus. Systemic administration (arrow) of 0.5 mg/kg WIN55,212-2 (intraperitoneally) increased ACh efflux in the hippocampus (F(12,108) = 6.46; p < 0.001; two-way ANOVA) (a), 1 mg/kg WIN55,212-2 had no effect (F(10,60) = 0,53; NS; two-way ANOVA) (b), and 5 mg/kg WIN55,212-2 inhibited (F(12,96) = 1.88; p < 0.05; two-way ANOVA) (c) ACh efflux in the hippocampus. Data (expressed as fold over baseline established before any treatment) represent mean ± SEM of n = 5-6 animals per group. *p < 0.05, **p < 0.01, and ***p < 0.001 for WIN55,212-2 versus vehicle-treated animals, at each time point.
Systemic administration of SR141716A (0.1 mg/kg, i.p.) attenuates the effects of WIN55,212-2 (0.5 and 5 mg/kg) on ACh efflux in the hippocampus
To investigate whether the effects of the low (0.5 mg/kg) and high (5 mg/kg) doses of WIN55,212-2 are mediated through CB1 receptors, the CB1 receptor antagonist SR141716A was administered intraperitoneally at a dose of 0.1 mg/kg together with WIN55,212-2. At this dose, SR141716A had no effect on hippocampal ACh (data not shown) when administered alone. Figure 3 shows that SR141716A reversed the effects of the low (0.5 mg/kg) and high (5 mg/kg) doses of WIN55,212-2, suggesting that both effects are mediated through CB1 receptors. Coadministration of SR141716A significantly attenuated the increase in hippocampal ACh induced by the low dose of WIN55,212-2 (0.5 mg/kg) (Fig. 3a). SR141716A also attenuated the decrease in hippocampal ACh induced by the high dose of WIN55,212-2 (5 mg/kg) (Fig. 3b).
Figure 3.
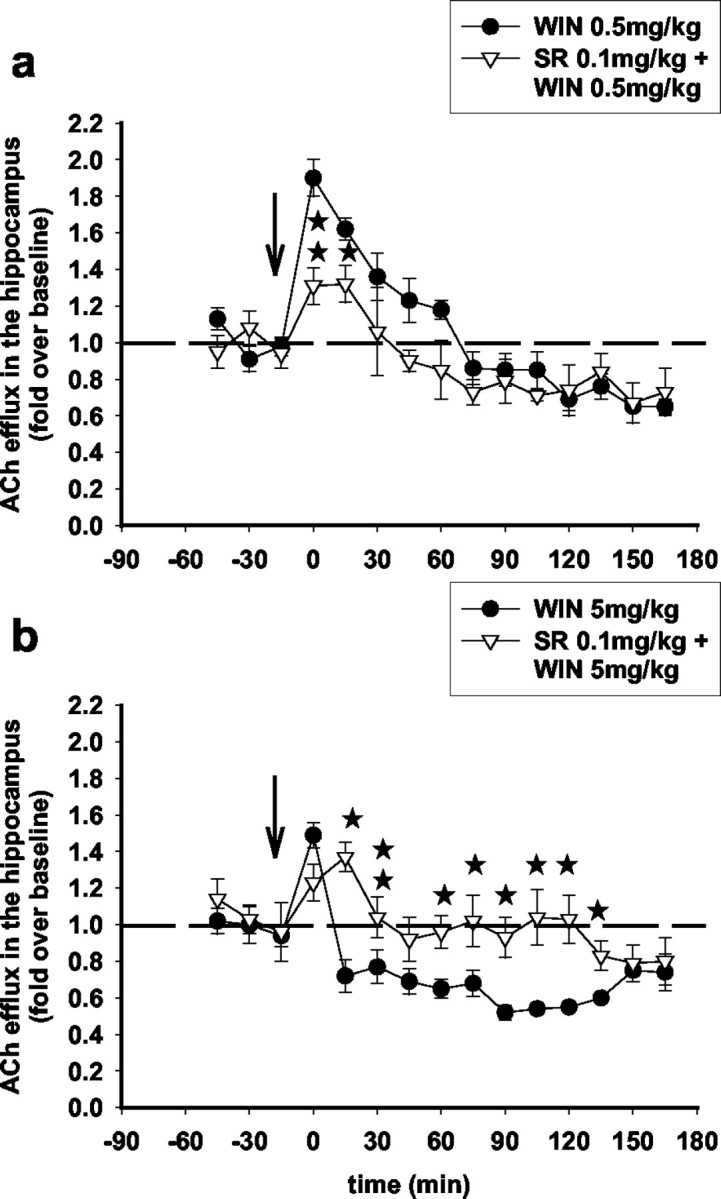
The CB1 antagonist SR141716A, administered systemically, attenuates both the stimulatory and inhibitory effects of WIN55,212-2 on ACh efflux in the hippocampus. Systemic administration of SR141716A [0.1 mg/kg, i.p.; administered simultaneously with WIN55,212-2 (arrow)] attenuated the stimulatory effects of WIN55,212-2 (0.5 mg/kg) (a) (F(10,90) = 2.4; p < 0.05; three-way ANOVA) and the inhibitory effects of WIN55,212-2 (5 mg/kg) (b) (F(10,70) = 2.06; p < 0.05; three-way ANOVA) on ACh efflux in the hippocampus. Data (expressed as fold over baseline established before any treatment) represent mean ± SEM of n = 4-6 animals per group. *p < 0.05 and **p < 0.01 for animals treated with SR141716A and WIN55,212-2 versus animals treated with WIN55,212-2 only, at each time point.
CB1 receptors in the hippocampus mediate the inhibitory effects of WIN55,212-2 (5 mg/kg) but not the stimulatory effects of WIN55,212-2 (0.5 mg/kg) on ACh efflux in this region
To investigate whether the effects of the low (0.5 mg/kg) and high (5 mg/kg) doses of WIN55,212-2 are mediated locally in the hippocampus, SR141716A (100 μm) was infused in this region through the microdialysis probe for 30 min (15 min before and 15 min after the injection of WIN55,212-2). A 30 min infusion of SR141716A at this dose did not significantly alter hippocampal ACh efflux compared with vehicle infusion (data not shown).
Intrahippocampal infusion of SR141716A (100 μm) did not attenuate the increase in hippocampal ACh induced by the low dose of WIN55,212-2 (0.5 mg/kg), and, in fact, it slightly but significantly increased it (Fig. 4a). On the contrary, SR141716A infused locally in the hippocampus at this same dose attenuated the decrease in hippocampal ACh induced by the high dose of WIN55,212-2 (5 mg/kg) (Fig. 4b), suggesting that activation of CB1 receptors in the hippocampus inhibits local ACh efflux. To confirm this, we infused WIN55,212-2 (1, 10, and 100 μm) in the hippocampus through the microdialysis probe for 15 min. Figure 5 shows that intrahippocampal WIN55,212-2 infusions dose dependently inhibited ACh efflux. WIN55,212-2 at 10 and 100 μm but not at 1 μm effectively reduced ACh efflux to a nadir value of 65% (for the 100 μm infusion) (Fig. 5a). The inhibition in hippocampal ACh induced by the 100 μm WIN55,212-2 infusion was reversed by a 30 min SR141716A (100 μm) intrahippocampal infusion through the microdialysis probe (15 min before and throughout the 15 min infusion of WIN55,212-2) (Fig. 5b). These results show that inhibition of hippocampal ACh efflux by high doses of WIN55,212-2 is mediated through CB1 receptors operating locally in the hippocampus but suggest an extrahippocampal site of action for the stimulatory low doses of the cannabinoid.
Figure 4.
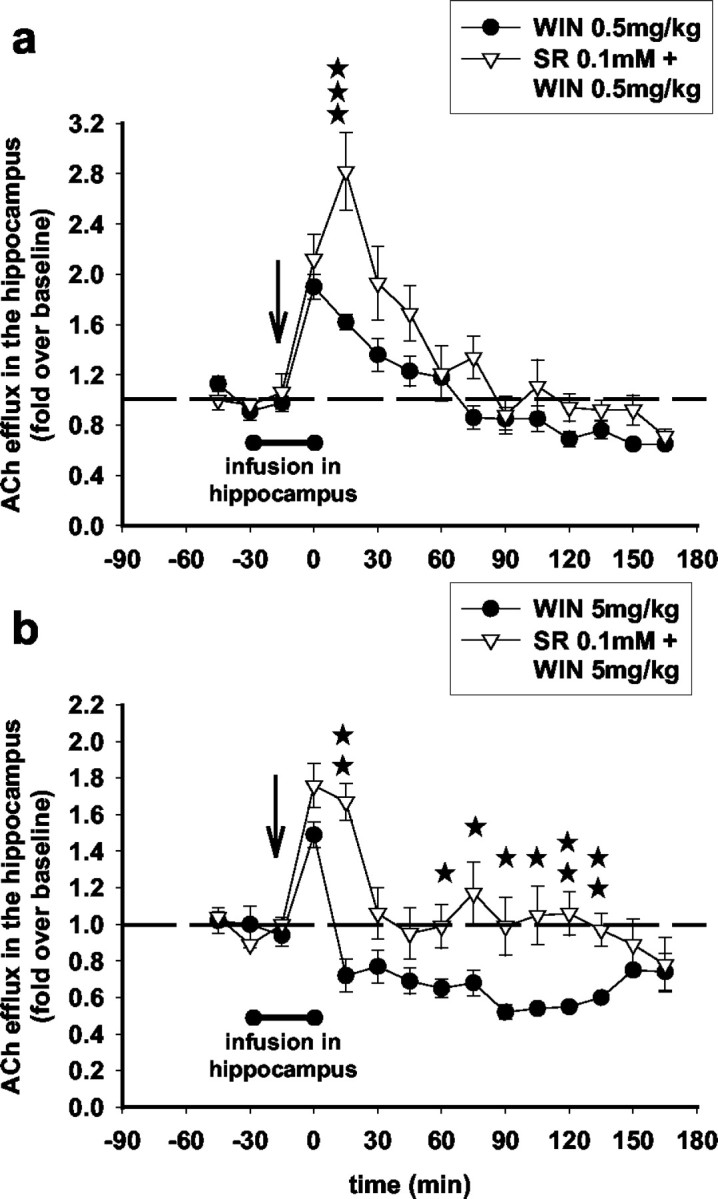
The CB1 antagonist SR141716A, infused locally in the hippocampus, attenuates the inhibitory but not the stimulatory effects of WIN55,212-2 on ACh efflux in this region. Intrahippocampal infusion of SR141716A [0.1 mm through the microdialysis probe for 30 min (filled bar) and 15 min before and 15 min after WIN55,212-2 injection (arrow)] did not attenuate the stimulatory effects of WIN55,212-2 (0.5 mg/kg) (a) (F(10,100) = 1.15; NS; three-way ANOVA) but reversed the inhibitory effects of WIN55,212-2 (5 mg/kg) (b) (F (10,120) = 2.09; p < 0.05; three-way ANOVA) on ACh efflux in the hippocampus. Data (expressed as fold over baseline established before any treatment) represent mean ± SEM of n = 4-6 animals per group. *p < 0.05, **p < 0.01, and ***p < 0.001 for animals infused with SR141716A and treated with WIN55,212-2 versus animals treated with WIN55,212-2 only, at each time point.
Figure 5.
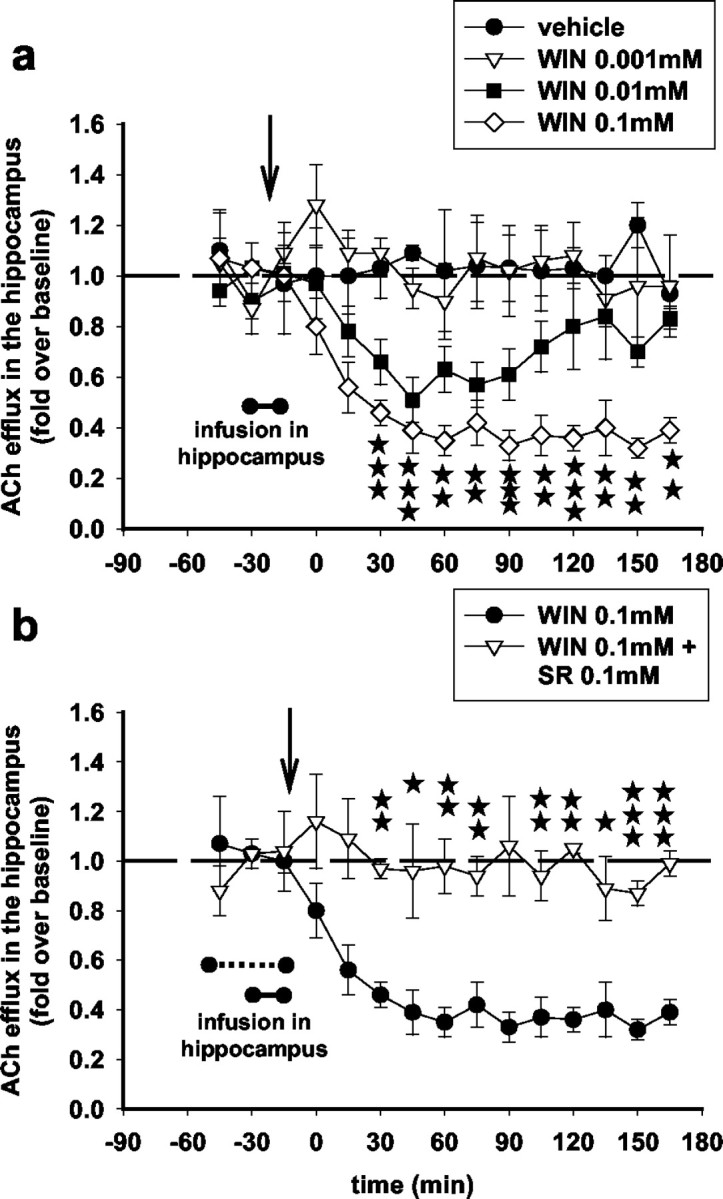
WIN55,212-2 infused locally in the hippocampus inhibits ACh efflux in this region; reversal by SR141716A. a, Dose-dependent inhibition (F(36,72) = 2.24; p < 0.01; two-way ANOVA) of hippocampal ACh efflux after intrahippocampal infusion of WIN55,212-2 for 15 min (filled bar). Data (expressed as fold over baseline established before any treatment) represent mean ± SEM of n = 5-6 animals per group. Levels of statistical significance are pictured for the 0.1 mm dose of WIN55,212-2 only for clarity of the figure; *p < 0.05, *p < 0.01, and ***p < 0.001 for animals infused with WIN55,212-2 0.1 mm versus animals infused with vehicle at each time point. b, Intrahippocampal infusion of SR141716A (dashed bar) for 30 min (15 min before and throughout the 15 min infusion of WIN55,212-2) reversed the inhibitory effects of WIN55,212-2 (0.1 mm) on ACh efflux in the hippocampus (F(12,156) = 1.84; p < 0.05; three-way ANOVA). Data (expressed as fold over baseline established before any treatment) represent mean ± SEM of n = 4-6 animals per group. *p < 0.05, **p < 0.01, and ***p < 0.001 for animals infused with SR141716A and WIN55,212-2 versus animals infused with WIN55,212-2 only, at each time point.
CB1 receptors in the septum mediate the stimulatory effects of WIN55,212-2 (0.5 mg/kg) but not the inhibitory effects of WIN55,212-2 (5 mg/kg) on hippocampal ACh efflux
The majority of the cell bodies of the cholinergic neurons that project to the hippocampus reside in the medial septum-diagonal band of Broca. Modulation of neurotransmitter release and/or receptor activation in the septal area affects ACh efflux in the hippocampus. To investigate whether the stimulatory effects of WIN55,212-2 (0.5 mg/kg) are mediated through CB1 receptors localized in the septal area, SR141716A (100 μm) was infused in this region through the microdialysis probe for 30 min (15 min before and 15 min after the injection of WIN55,212-2), and ACh efflux was measured in the hippocampus in dual-microdialysis experiments. A 30 min septal infusion of SR141716A at this dose did not significantly alter hippocampal ACh efflux compared with vehicle infusion (data not shown).
Infusion of SR141716A in the septum attenuated the increase in hippocampal ACh induced by the low dose of WIN55,212-2 (0.5 mg/kg) (Fig. 6a). On the contrary, SR141716A infused in the septum did not attenuate the decrease in hippocampal ACh induced by the high dose of WIN55,212-2 (5 mg/kg) (Fig. 6b). These results suggest that the stimulatory effects of the low dose of the WIN55,212-2 involve CB1 receptors of the septal area. To confirm this finding, we investigated whether intraseptal WIN55,212-2 administration alters hippocampal ACh efflux. WIN55,212-2 (100 μm) was infused in the septum for 15 min through the microdialysis probe, and ACh efflux was measured in the hippocampus in dual-microdialysis experiments. Intraseptal WIN55,212-2 infusion significantly increased ACh efflux in the hippocampus. ACh was increased, up to a peak value of 80%, 15 min after the infusion of WIN55,212-2 (100 μm) and returned to baseline levels 1 hr later (Fig. 7a), in a pattern similar to the one observed after systemic administration of a low dose of WIN55,212-2 (0.5 mg/kg) (Fig. 2a). A 30 min infusion of SR141716A (100 μm) in the medial septum (15 min before and throughout the 15 min infusion of WIN55,212-2), together with WIN55,212-2 (100 μm), prevented the increase in hippocampal ACh induced by the agonist (Fig. 7b).
Figure 6.
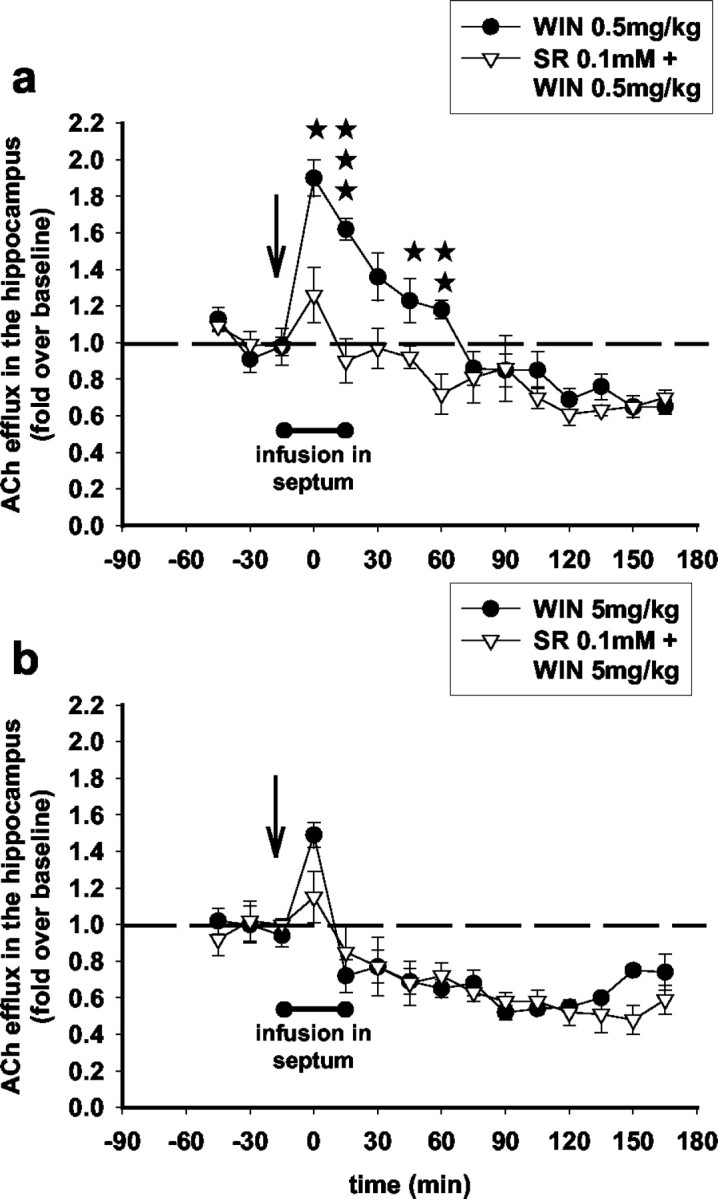
The CB1 antagonist SR141716A, infused locally in the septum, attenuates the stimulatory but not the inhibitory effects of WIN55,212-2 on ACh efflux in the hippocampus. Intrahippocampal infusion of SR141716A [0.1 mm through the microdialysis probe for 30 min (filled bar), 15 min before and 15 min after WIN55,212-2 injection (arrow)] reversed the stimulatory effects of WIN55,212-2 (0.5 mg/kg) (a) (F(10,110) = 1.98; p < 0.05; three-way ANOVA) but did not affect the inhibitory effects of WIN55,212-2 (5 mg/kg) (b) (F(10,150) = 1.16; NS; three-way ANOVA) on ACh efflux in the hippocampus. Data (expressed as fold over baseline established before any treatment) represent mean ± SEM of n = 4-5 animals per group. *p < 0.05, **p < 0.01, and ***p < 0.001 for animals infused with SR141716A and treated with WIN55,212-2 versus animals treated with WIN55,212-2 only, at each time point.
Figure 7.
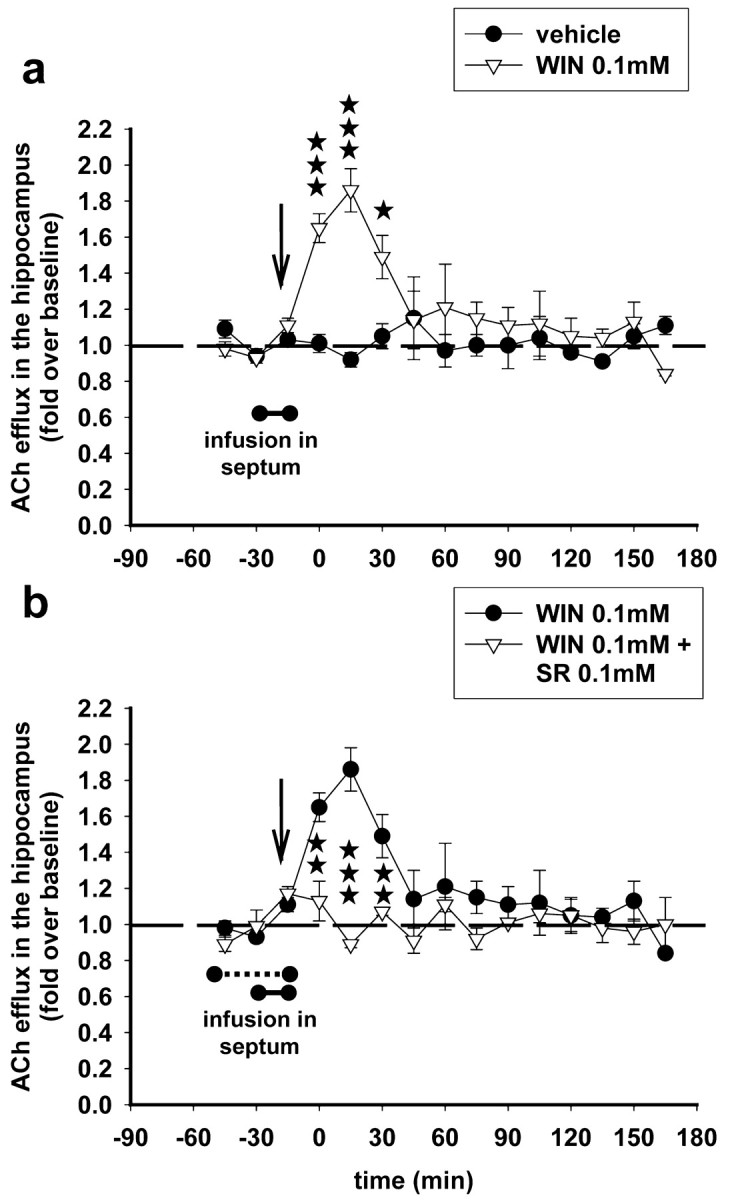
WIN55,212-2 infused locally in the septum stimulates ACh efflux in the hippocampus; reversal by SR141716A. a, Stimulation of hippocampal ACh efflux after intraseptal infusion of WIN55,212-2 (0.1 mm) for 15 min (filled bar) (F(11,66) = 2.77; p < 0.01; two-way ANOVA). Data (expressed as fold over baseline established before any treatment) represent mean ± SEM of n = 3-5 animals per group. *p < 0.05, *p < 0.01, and ***p < 0.001 for animals infused with WIN55,212-2 0.1 mm versus animals infused with vehicle at each time point. b, Intraseptal infusion of SR141716A (dashed bar) for 30 min (15 min before and throughout the 15 min infusion of WIN55,212-2) reversed the stimulatory effects of WIN55,212-2 (0.1 mm) on ACh efflux in the hippocampus (F(11,121) = 2.05; p < 0.05; three-way ANOVA). Data (expressed as fold over baseline established before any treatment) represent mean ± SEM of n = 3-5 animals per group. *p < 0.05, **p < 0.01, and ***p < 0.001 for animals infused with SR141716A and WIN55,212-2 versus animals infused with WIN55,212-2 only, at each time point.
In dual-microdialysis measurements, tetrodotoxin (TTX) (1 μm) was infused in the septum through the microdialysis probe for 60 min, at the end of the experiment, to ensure by pharmacological means that changes in neuronal activity initiated at the level of septum are indeed reflected in the ACh measurements in hippocampus. In agreement with previous observations (Moor et al., 1994), septal TTX infusion resulted in an initial increase in hippocampal ACh (for ∼30 min), followed by a prolonged, profound decrease in ACh efflux to almost undetectable levels (data not shown).
Infusion of PTX (100 ng) into the hippocampus or the septum, respectively, attenuates the inhibitory effects of WIN55,212-2 (5 mg/kg) and the stimulatory effects of WIN55,212(0.5 mg/kg) on hippocampal ACh efflux
It has been shown in vitro that inactivation of Gi subunits of the G heterotrimeric proteins, to which CB1 receptors are commonly coupled, unravels an alternative functional coupling of these receptors to Gs subunits (Glass and Felder, 1997). This dual coupling of CB1 receptors to either inhibitory (Gi) or stimulatory (Gs) proteins has been proposed to account for the biphasic effects of CB1 compounds observed in vivo (Sulcova et al., 1998). To investigate whether inhibitory and stimulatory effects of WIN55,212-2 differentially involve Gi- versus Gs-proteins, Gi subunits were inactivated by PTX infusion (Gronier and Rasmussen, 1999) 24 hr before systemic administration of a low (0.5 mg/kg) or high (5 mg/kg) dose of WIN55,212-2. Because the inhibitory effects of the high dose of WIN55,212-2 on hippocampal ACh release are mediated locally in the hippocampus but the stimulatory effects of the low dose of the compound involve CB1 receptors of the septal area, PTX was infused in either the hippocampus or the septum in separate experiments.
Infusion of PTX (100 ng) into the hippocampus attenuated the effects of the high dose of WIN55,212-2 (5 mg/kg) on ACh efflux in this region but, as expected, did not affect the effects of the low dose of WIN55,212-2 (0.5 mg/kg) (Fig. 8).
Figure 8.
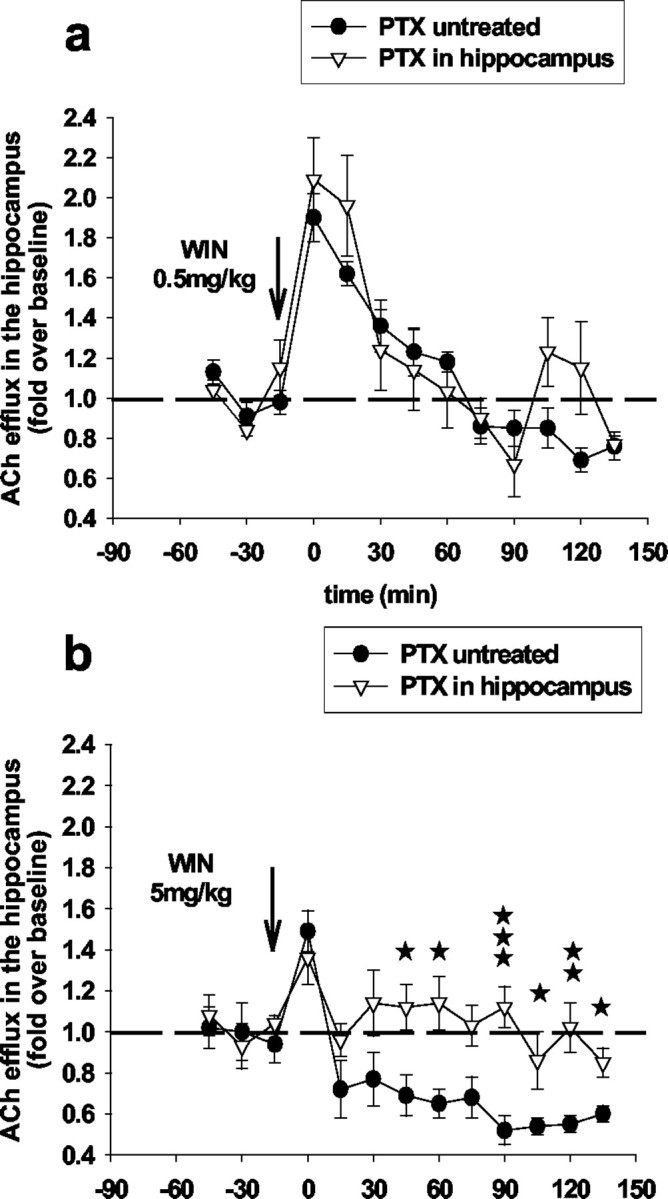
PTX induced inactivation of Gi in the hippocampus blocks the inhibitory but not the stimulatory effects of WIN55,212-2 on ACh efflux in this region. Intrahippocampal infusion of PTX (100 ng; 24 hr before the microdialysis experiment) did not attenuate the stimulatory effects of WIN55,212-2 (0.5 mg/kg) (a) (F(11,44) = 1.22; NS; two-way ANOVA) but reversed the inhibitory effects of WIN55,212-2 (5 mg/kg) (b) (F(11,33) = 329; p < 0.001; two-way ANOVA) on ACh efflux in the hippocampus. Data (expressed as fold over baseline established before any treatment) represent mean ± SEM of n = 4-5 animals per group. *p < 0.05, **p<0.01, and ***p < 0.001 for animals pretreated with PTX and treated with WIN55,212-2 versus animals treated with WIN55,212-2 only, at each time point.
Importantly, infusion of PTX (100 ng) into the septum completely reversed the effects of WIN55,212-2 (0.5 mg/kg) on ACh efflux in the hippocampus (Fig. 9), showing that both the stimulatory and inhibitory effects of the cannabinoid agonist are mediated through a common molecular substrate, the conventional coupling of CB1 receptors to Gi. The biphasic effects of the cannabinoid on hippocampal ACh efflux could be explained by a differential sensitivity of septal and hippocampal CB1 receptors to systemically administered WIN55,212-2 and/or a differential functional association of these anatomically distinct subpopulations of CB1-Gi-coupled receptors to neurotransmitter systems that have opposing effects on ACh release.
Figure 9.

PTX induced inactivation of Gi in the septum blocks the stimulatory effects of WIN55,212-2 on ACh efflux in the hippocampus. Intraseptal infusion of PTX (100 ng; 24 hr before the microdialysis experiment) reversed the stimulatory effects of WIN55,212-2 (0.5 mg/kg) on ACh efflux in the hippocampus (F(11,44) = 2.29; p < 0.01; two-way ANOVA). Data (expressed as fold over baseline established before any treatment) represent mean ± SEM of n = 4-5 animals per group. *p < 0.05 and **p < 0.01 for animals pretreated with PTX and treated with WIN55,212-2 versus animals treated with WIN55,212-2 only, at each time point.
The D1 antagonist SCH23390 and the D2 antagonist S-raclopride attenuate, respectively, the stimulatory effects of WIN55,212-2 (0.5 mg/kg) and the inhibitory effects of WIN55,212-2 (5 mg/kg) on ACh efflux in the hippocampus
Cholinergic activity in the septohippocampal pathway is regulated by dopamine (Durkin et al., 2000). Both positive and negative effects of dopamine on hippocampal ACh efflux, involving, respectively, D1 and D2 receptors, have been shown. To investigate whether the effects of the low and/or the high doses of WIN55,212-2 on ACh release in the hippocampus involve activation of dopaminergic circuits, the D1 receptor antagonist SCH23390 (0.3 mg/kg, s.c.) or the D2 receptor antagonist S-raclopride (1 mg/kg, s.c.) were injected 15 min before the injection of WIN55,212-2 (0.5 or 5 mg/kg). These doses of SCH23390 and S-raclopride have been shown to be efficacious in previous functional studies (Gruber et al., 2001). In addition to D1 receptors, SCH23390 shows high affinity in vitro for the 5-HT2 and 5-HT1C serotonergic receptors, although the doses required to induce serotonin receptor- mediated responses in vivo are >10-fold higher than those required to induce a D1-mediated response (Bourne 2001). To this end, it cannot be entirely excluded that the dose of SCH23390 used in the present study does not affect serotonergic receptors, as well.
The D1 antagonist SCH23390 (0.3 mg/kg, s.c.) attenuated the effects of 0.5 mg/kg WIN55,212-2 but not of 5 mg/kg WIN55,212-2 on ACh efflux in the hippocampus (Fig. 10). On the other hand, the D2 antagonist S-raclopride (1 mg/kg, s.c.) attenuated the effects of WIN55,212-2 (5 mg/kg), in accordance with previous data (Nava et al., 2000) (Fig. 11).
Figure 10.
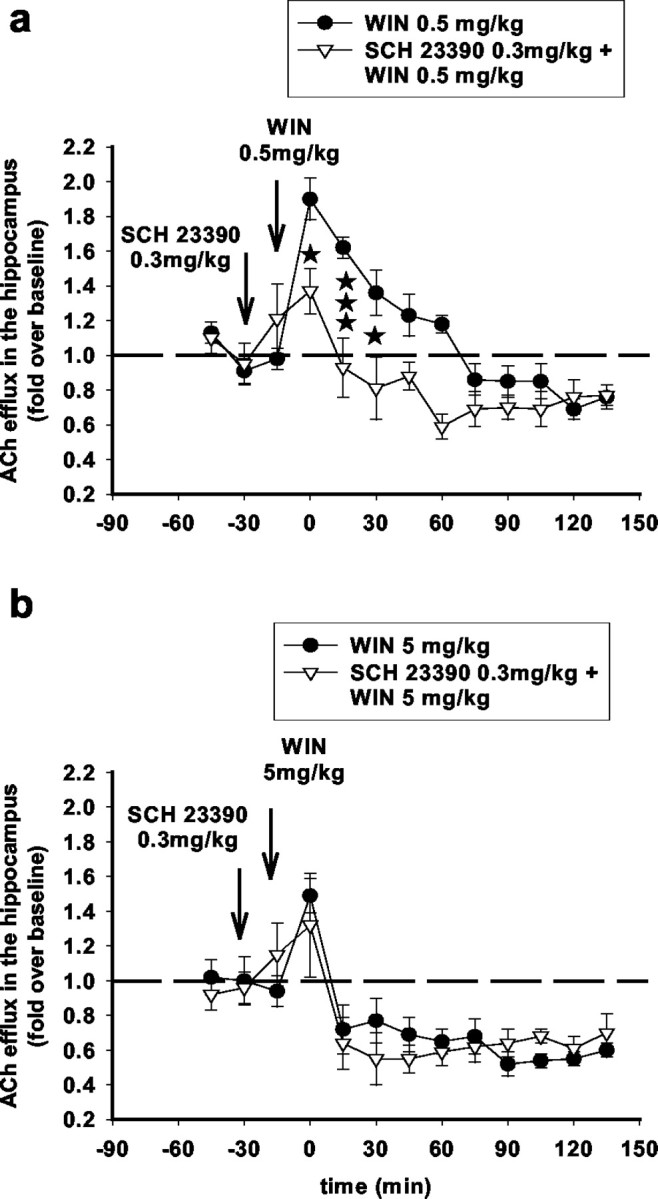
The D1 antagonist SCH23390 attenuates the stimulatory but not the inhibitory effects of WIN55,212-2 on ACh efflux in the hippocampus. SCH23390 administered subcutaneously at a dose of 0.3 mg/kg 15 min before WIN55,212-2 injection attenuated the stimulatory effects of WIN55,212-2 (0.5 mg/kg) (a) (F(1,9) = 7.74; p < 0.05; three-way ANOVA) but did not affect the inhibitory effects of WIN55,212-2 (5 mg/kg) (b) (F(1,5) = 3.75; NS; three-way ANOVA) on ACh efflux in the hippocampus. Data (expressed as fold over baseline established before any treatment) represent mean ± SEM of n = 4-6 animals per group. *p < 0.05 and ***p < 0.001 for animals treated with SCH23390 before WIN55,212-2 versus animals treated with WIN55,212-2 only, at each time point.
Figure 11.
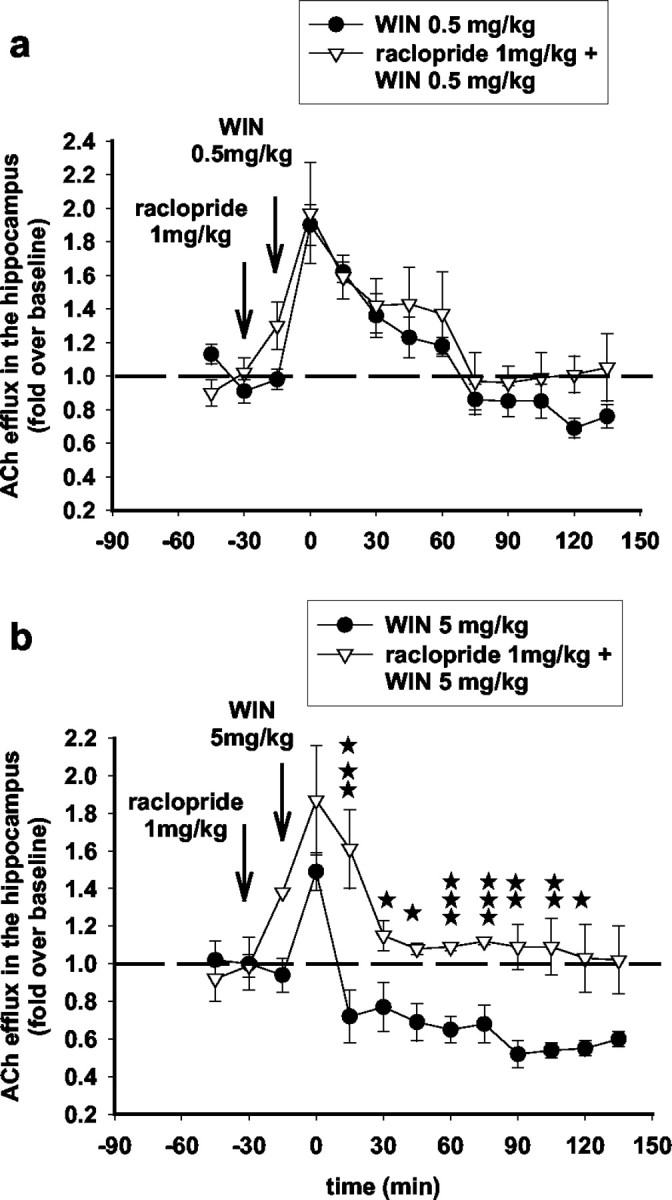
The D2 antagonist S-(-)-raclopride attenuates the stimulatory but not the inhibitory effects of WIN55,212-2 on ACh efflux in the hippocampus. Raclopride administered subcutaneously at a dose of 1 mg/kg 15 min before WIN55,212-2 injection did not affect the stimulatory effects of WIN55,212-2 (0.5 mg/kg) (a) (F(1,7) = 0.22; NS; three-way ANOVA) but reversed the inhibitory effects of WIN55,212-2 (5 mg/kg) (b) (F(1,6) = 8.24; p < 0.05; three-way ANOVA) on ACh efflux in the hippocampus. Data (expressed as fold over baseline established before any treatment) represent mean ± SEM of n = 4-6 animals per group. *p < 0.05, **p < 0.01, and ***p < 0.001 for animals treated with S-(-)-raclopride before WIN55,212-2 versus animals treated with WIN55,212-2 only, at each time point.
Although the behavior of the animals was not systematically monitored, it was evident that the 5 mg/kg dose of WIN55,212-2 caused a marked and long-lasting sedation in the animals. Systemic administration of SR141716A counteracted this behavioral suppression. On the other hand, local infusion of WIN55,212-2 in the hippocampus caused a pronounced and sustained decrease in hippocampal ACh without overtly affecting the behavior of the animals. Also, hippocampal infusion of SR141716A reversed the suppressing effect of the 5 mg/kg dose of WIN55,212-2 on ACh efflux but not on behavior. It was difficult to assess (with strictly behavioral parameters) the effects of combined administration of the D2 antagonist raclopride and the high dose of WIN55,212-2 on behavior, given that each drug alone caused sedation, and the animals after the combined drug administration appeared sedated as well. Clearly, however, raclopride completely reversed the WIN55,212-2-mediated reduction in ACh efflux. The same holds for administration of the D1 antagonist SCH23390, i.e., although rats treated with the low dose of WIN55,212-2 together with SCH23390 appeared overactive, and the D1 antagonist clearly abolished the increase in hippocampal acetylcholine induced by the low cannabinoid dose. Together, these results suggest that changes in ACh efflux in response to cannabinoid agents alone or in combination with dopaminergic compounds do not necessarily coincide with changes in behavioral (mainly motoric) outputs.
Discussion
Converging in vitro evidence supports the existence of separate neuronal CB1R populations displaying different sensitivity to cannabinoids (vide infra). Here we show that anatomically distinct CB1Rs mediate dose-dependent, opposite, functional in vivo responses, namely ACh efflux in hippocampus. These amphidromic responses involve the same structural entities (Gi-coupled CB1Rs) but different neuroanatomical sites and distinct dopaminergic components.
Biphasic effects of cannabinoids on hippocampal ACh are mediated through anatomically distinct CB1Rs coupled to Gi
Acute systemic WIN55,212-2 administration regulates septohippocampal cholinergic neurotransmission in a biphasic, dose-dependent pattern: a low dose of WIN55,212-2 stimulates and a high dose inhibits hippocampal ACh efflux. This suggests that opposing networks and/or homeostatic inter-regulations mediate enhancing and suppressing effects of cannabinoid agonists on cholinergic neurotransmission.
Considering the difference in route of administration, the herein determined stimulatory WIN55,212-2 dose corresponds to that used by Acquas et al. (2000), who report increased ACh efflux after systemic WIN55,212-2. Similarly, our inhibitory WIN55,212-2 dose is in the range of doses used by Gessa et al. (1997), who report a marked decrease of ACh after systemic WIN55,212-2. Thus, the apparent discordance between the aforementioned studies is not attributable to differences in administration route, probe type, or anesthesia but rather reflects the biphasic, dose-dependent effect of WIN55,212-2 on cholinergic neurotransmission as characterized here. These authors also report apparently contradictory, stimulatory, and inhibitory effects of cannabinoids on cortical ACh (Gessa et al., 1997; Acquas et al., 2000). Although our study was restricted in hippocampus, it is conceivable that the same biphasic regulation applies also for cholinergic neurotransmission in cortex.
Biphasic, dose-dependent effects of cannabinoid agonists were shown for other forebrain functional responses measured in vivo, namely hippocampal 2-deoxyglycose use (Margulies and Hammer, 1991), cortical evoked potentials (Turkanis and Karler, 1981), and locomotion (Sulcova et al., 1998; Sanudo-Pena et al., 2000). Interestingly, stimulatory and inhibitory doses of cannabinoids determined in these studies are comparable with those determined here, stimulatory doses being ∼10-fold lower than inhibitory ones. This suggests a biphasic, adaptive pattern for a cluster of possibly inter-regulated forebrain functions after cannabinoid administration.
Hypotheses evoked to explain in vivo biphasic effects of cannabinoids favor mostly the existence of two distinct, functionally opposing, structural modules (receptors/G-proteins) responsible for stimulatory and inhibitory actions. Although CB1Rs mediate most of pharmacological cannabinoid CNS actions (Chaperon and Thiebot, 1999), non-CB1/non-CB2 central effects reported for some agonists, including WIN55,212-2, suggest the existence of another putative cannabinoid receptor (Breivogel et al., 2001; Monory et al., 2002). Differential involvement of the putative cannabinoid receptor versus CB1R in modulating hippocampal glutamatergic and GABAergic neurotransmission was proposed (Hajos et al., 2001). Difference in net excitability of hippocampal synapses, attributable to preferential cannabinoid actions on glutamatergic versus GABAergic afferents, might account for biphasic effects in hippocampus. However, in other studies, CB1Rs alone mediate hippocampal inhibitory and excitatory synaptic regulation (Ohno-Shosaku et al., 2002). Here, the CB1R antagonist SR141716A blocked both stimulatory and inhibitory effects of WIN55,212-2, as observed previously (Gessa et al., 1997; Acquas et al., 2000). In most paradigms, SR141716A is ineffective in blocking non-CB1-mediated effects of WIN55,212-2 (Breivogel et al., 2001). Furthermore, THC, which is known to stimulate CB1Rs but does not activate the putative cannabinoid receptor, shares the ability of WIN55,212-2 to dually regulate ACh release (Carta et al., 1998; Acquas et al., 2001).
An alternative CB1R coupling to the stimulatory Gs-proteins was demonstrated in vitro, when conventional coupling to Gi was disrupted by PTX (Glass and Felder, 1997). Thus, under specific conditions, CB1R activation could enhance cAMP production (Maneuf and Brotchie, 1997) and hippocampal excitability in vitro (Okada et al., 1992). This dual CB1R coupling to either Gs- or Gi-proteins is proposed to explain amphidromic functional responses to cannabinoids (Sulcova et al., 1998; Sanudo-Pena et al., 2000); however, this hypothesis was not tested in vivo. Here, PTX-induced selective inactivation of Gi reversed both stimulatory and suppressing effects of WIN55,212-2. Overall, our results demonstrate that biphasic cannabinoid effects on hippocampal ACh solely involve Gi-coupled CB1Rs.
We also show that dual actions of WIN55,212-2 are mediated through distinct neuroanatomical CB1Rs. Suppressing, high-dose effects are mediated locally in hippocampus, whereas low-dose, excitatory effects are mediated in septum. Brain CB1Rs are localized in glutamatergic, dopaminergic, and cholinergic primary projecting neurons (Lu et al., 1999; Ong and Mackie, 1999; Hermann et al., 2002) and in GABAergic interneurons (Katona et al., 2000). These express low and high levels of CB1Rs, respectively (Marsicano and Lutz, 1999), suggesting that GABAergic circuits might be more sensitive to cannabinoids. Accordingly, WIN55,212-2 is an order of magnitude more potent in reducing GABAergic than glutamatergic neurotransmission in electrophysiological preparations (Hajos and Freund, 2002; Ohno-Shosaku et al., 2002). Consequently, the higher sensitivity of septal CB1 circuits reported here could be attributed to the prominence of GABAergic septal neurons in controlling cholinergic neurotransmission (Durkin et al., 2000). Indeed, blockade of septal GABAergic activity leads to cholinergic stimulation (Moor et al., 1998; Alreja et al., 2000). Interestingly, a primary reduction of cholinergic activity in septum can also induce a moderate increase in hippocampal ACh via deactivation of a negative, autoregulatory loop (Wu et al., 2000). Primary cholinergic reduction could result from CB1-mediated inhibition of brainstem-septal afferents (Woolf and Butcher, 1986) that, together with septohippocampal cholinergic collaterals (Leranth and Frotscher, 1989), are the source of septal ACh (Durkin et al., 2000). The inhibitory effects of WIN55,212-2, mediated locally in hippocampus and observed with higher doses, could result from activation of low-sensitivity CB1Rs localized in cholinergic terminals or in primary projections that modulate ACh release directly or indirectly.
It is established that opposing dopaminergic inputs dually regulate hippocampal ACh efflux. Whereas D2 agonists decrease hippocampal ACh (Day and Fibiger, 1994), D1 receptor activation enhances cholinergic activity (Day and Fibiger, 1994). Confirming previous findings (Nava et al., 2000), we show that D2 receptor activation mediates inhibitory, high-dose cannabinoid effects on ACh release. Furthermore, we show that D1 receptor activation is critical for the stimulatory actions of WIN55,212-2, consistent with a septal but not hippocampal site of action for low cannabinoid doses. D1-induced ACh enhancement is not mediated locally in hippocampus (G. G. Nomikos, unpublished observations) but involves most likely the septum, which expresses a high density of D1 versus D2 receptors (Zilles et al., 1991) and in which catecholaminergic afferents innervating cholinergic somata, directly or indirectly via GABAergic interneurons, have been identified (Lindvall and Stenevi, 1978; Moore, 1978; Milner, 1991; Durkin et al., 2000). Interestingly, both low (Tanda et al., 1997) and higher (Malone and Taylor, 1999) cannabinoid doses stimulate dopaminergic neuronal activity. Furthermore, other dopamine-dependent readouts, such as appetitive behavior, are regulated by cannabinoids in a biphasic manner (Martellotta et al., 1998). Thus, amphidromic cannabinoid effects appear to involve complex regulations of inter-dependent and possibly opposing dopaminergic circuits that respond with different sensitivity to cannabinoid signaling.
Implications of our findings
Cannabinoid-mediated inhibition of hippocampal ACh has been demonstrated repeatedly (Gifford and Ashby, 1996; Gessa et al., 1997; Nava et al., 2000; Kathmann et al., 2001) and has been associated with poor performance in learning-memory tasks (Nava et al., 2000; Mishima et al., 2002). Reduced ACh neurotransmission and subsequent cognitive impairment could be an important limit to possible therapeutic applications of cannabinoid agonists. We show that only high, but not low or moderate, although pharmacologically effective in, for example, pain models (Chaperon and Thiebot, 1999; Pertwee, 2001), doses suppress hippocampal ACh. Importantly, THC-induced memory impairment was associated to a D2-mediated decrease in hippocampal ACh (Nava et al., 2000). Inversely, low cannabinoid doses mobilize D1-mediated responses that are rather procognitive than detrimental to learning and memory (Hersi et al., 1995). In experimental animals, WIN55,212-2 administered over a range of doses can impair performance in a delay task (Hampson and Deadwyler, 2000). Nevertheless, a better understanding of the effects of low doses is warranted, because these have not been systematically evaluated in cognitive paradigms.
Thus, memory deficits induced by high cannabinoid doses could be attributed to a massive, nonselective CB1R occupancy, which disrupts the spatiotemporal specificity of endocannabinoid-mediated fine-tuning of synaptic plasticity, as proposed previously (Carlson et al., 2002). Still, studies with CB1 knock-out mice show that endocannabinoids operate in a phasic, highly dynamic manner that permits the renewal of mnemonic imprints (Reibaud et al., 1999; Marsicano et al., 2002; Varvel and Lichtman, 2002). Although, it is very difficult to evaluate the action of anandamide because it has a short half-time and is a partial agonist in many CB1R models, it is noteworthy that there is no cognitive dysfunction after administration of the endocannabinoid in animals (Lichtman et al., 1995). In fact, a low anandamide dose reversed cognitive deficits in diet-restricted animals (Hao et al., 2000), suggesting a state-dependent, neuromodulatory role of endocannabinoid systems. Because endocannabinoids are released locally “upon demand” (Di Marzo et al., 2000b), distinct neuroanatomical sites with high sensitivity to cannabinoid signaling, as the septal area identified here, are likely “hotspots” for activity-dependent, endocannabinoid fine-tuning of memory acquisition and extinction. Therefore, drugs that selectively target active endocannabinoid synapses, such as inhibitors of endocannabinoid uptake or endocannabinoid catabolism (Giuffrida et al., 2001; Goutopoulos and Makriyannis, 2002), could constitute effective medications for pain and motor disorders devoid of generalized side effects on memory.
Footnotes
Correspondence should be addressed to George G. Nomikos, Eli Lilly and Company, Lilly Corporate Center, DC0510, Indianapolis, IN 46285. E-mail: gnomikos@lilly.com.
Copyright © 2003 Society for Neuroscience 0270-6474/03/239374-11$15.00/0
References
- Acquas E, Pisanu A, Marrocu P, Di Chiara G ( 2000) Cannabinoid CB1 receptor agonists increase rat cortical and hippocampal acetylcholine release in vivo. Eur J Pharmacol 401: 179-185. [DOI] [PubMed] [Google Scholar]
- Acquas E, Pisanu A, Maroccu P, Goldberg SR, Di Chiara G ( 2001) D-9-tetrahydrocannabinol enhances cortical and hippocampal acetylcholine release in vivo: a microdialysis study. Eur J Pharmacol 419: 155-161. [DOI] [PubMed] [Google Scholar]
- Alreja M, Wu M, Liu W, Atkins JB, Leranth C, Shanabrough M ( 2000) Muscarinic tone sustains impulse flow in the septohippocampal GABA but not cholinergic pathway: implications for learning and memory. J Neurosci 20: 8103-8110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne JA ( 2001) SCH 23390: the first selective dopamine D1-like receptor antagonist. CNS Drug Rev 7: 399-414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breivogel CS, Griffin G, Di Marzo V, Martin BR ( 2001) Evidence for a new G protein coupled cannabinoid receptor in mouse brain. Mol Pharmacol 60: 155-163. [PubMed] [Google Scholar]
- Carlson G, Wang Y, Alger BE ( 2002) Endocannabinoids facilitate the induction of LTP in the hippocampus. Nat Neurosci 5: 723-724. [DOI] [PubMed] [Google Scholar]
- Carta G, Nava F, Gessa GL ( 1998) Inhibition of hippocampal acetylcholine release after acute and repeated D9-tetrahydrocannabinol in rats. Brain Res 809: 1-4. [DOI] [PubMed] [Google Scholar]
- Chaperon F, Thiebot MH ( 1999) Behavioral effects of cannabinoid agonists in animals. Crit Rev Neurobiol 13: 243-281. [DOI] [PubMed] [Google Scholar]
- Childers SR, Pacheco MA, Bennett BA, Edwards TA, Hampson RE, Mu J, Deadwyler SA ( 1993) Cannabinoid receptors: G-protein-mediated signal transduction mechanisms. Biochem Soc Symp 59: 27-50. [PubMed] [Google Scholar]
- Damsma G, Westerink BH, de Boer P, de Vries JB, Horn AS ( 1988) Basal acetylcholine release in freely moving rats detected by on-line transstriatal dialysis: pharmacological aspects. Life Sci 43: 1161-1168. [DOI] [PubMed] [Google Scholar]
- Davies SN, Pertwee RG, Riedel G ( 2002) Functions of cannabinoid receptors in the hippocampus. Neuropharmacology 42: 993-1007. [DOI] [PubMed] [Google Scholar]
- Day JC, Fibiger HC ( 1994) Dopaminergic regulation of septohippocampal cholinergic neurons. J Neurochem 63: 2086-2092. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Breivogel CS, Tao Q, Bridgen DT, Razdan RK, Zimmer AM, Zimmer A, Martin BR ( 2000a) Levels, metabolism, and pharmacological activity of anandamide in CB(1) cannabinoid receptor knockout mice: evidence for non-CB(1), non-CB(2) receptor-mediated actions of anandamide in mouse brain. J Neurochem 75: 2434-2444. [DOI] [PubMed] [Google Scholar]
- Di Marzo, Bisogno T, De Petrocellis L ( 2000b) Endocannabinoids: new targets for drug development. Curr Pharm Des 6: 1361-1380. [DOI] [PubMed] [Google Scholar]
- Durkin TP, Cazala P, Garcia R ( 2000) Transynaptic mechanisms controlling cholinergic neuronal activation in the septohippocampal and nBM-cortical pathways: differential roles in memory and attentional processes? In: The behavioral neuroscience of the septal region (Numan R, ed), pp 146-174. New York: Springer.
- Gessa GL, Mascia MS, Casu MA, Carta G ( 1997) Inhibition of hippocampal acetylcholine release by cannabinoids: reversal by SR 141716A. Eur J Pharmacol 327: R1-R2. [DOI] [PubMed] [Google Scholar]
- Gifford AN, Ashby Jr CR ( 1996) Electrically evoked acetylcholine release from hippocampal slices is inhibited by the cannabinoid receptor agonist, WIN 55212-2, and is potentiated by the cannabinoid antagonist, SR 141716A. J Pharmacol Exp Ther 277: 1431-1436. [PubMed] [Google Scholar]
- Giuffrida A, Beltramo M, Piomelli D ( 2001) Mechanisms of endocannabinoid inactivation: biochemistry and pharmacology. J Pharmacol Exp Ther 298: 7-14. [PubMed] [Google Scholar]
- Glass M, Felder CC ( 1997) Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptor augments cAMP accumulation in striatal neurons: evidence for a Gs linkage to the CB1 receptor. J Neurosci 17: 5327-5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goutopoulos A, Makriyannis A ( 2002) From cannabis to cannabinergics: new therapeutic opportunities. Pharmacol Ther 95: 103-117. [DOI] [PubMed] [Google Scholar]
- Gronier B, Rasmussen K ( 1999) Pertussis toxin treatment differentially affects cholinergic and dopaminergic receptor stimulation of midbrain dopaminergic neurons. Neuropharmacology 38: 1903-1912. [DOI] [PubMed] [Google Scholar]
- Gruber SH, Nomikos GG, Mathe AA ( 2001) Dopamine receptor antagonists prevent the d-amphetamine-induced increase in calcitonin gene-related peptide levels in ventral striatum. J Neurosci Res 64: 606-611. [DOI] [PubMed] [Google Scholar]
- Hajos N, Freund TF ( 2002) Pharmacological separation of cannabinoid sensitive receptors on hippocampal excitatory and inhibitory fibers. Neuropharmacology 43: 503-510. [DOI] [PubMed] [Google Scholar]
- Hajos N, Ledent C, Freund TF ( 2001) Novel cannabinoid-sensitive receptor mediates inhibition of glutamatergic transmission in the hippocampus. Neuroscience 106: 1-4. [DOI] [PubMed] [Google Scholar]
- Hampson RE, Deadwyler SA ( 1999) Role of cannabinoid receptors in memory storage. Neurobiol Dis 5: 474-482. [DOI] [PubMed] [Google Scholar]
- Hampson RE, Deadwyler SA ( 2000) Cannabinoids reveal the necessity of hippocampal neural encoding for short-term memory in rats. J Neurosci 20: 8932-8942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao S, Avraham Y, Mechoulam R, Berry EM ( 2000) Low dose anandamide affects food intake, cognitive function, neurotransmitter and corticosterone levels in diet-restricted mice. Eur J Pharmacol 392: 147-156. [DOI] [PubMed] [Google Scholar]
- Hermann H, Marsicano G, Lutz B ( 2002) Coexpression of the cannabinoid receptor type 1 with dopamine and serotonin receptors in distinct neuronal subpopulations of the adult mouse forebrain. Neuroscience 109: 451-460. [DOI] [PubMed] [Google Scholar]
- Hersi AI, Rowe W, Gaudreau P, Quirion R ( 1995) Dopamine D1 receptor ligands modulate cognitive performance and hippocampal acetylcholine release in memory-impaired aged rats. Neuroscience 69: 1067-1074. [DOI] [PubMed] [Google Scholar]
- Kathmann M, Weber B, Schlicker E ( 2001) Cannabinoid CB1 receptor-mediated inhibition of acetylcholine release in the brain of NMRI, CD-1 and C57BL/6J mice. Naunyn Schmiedebergs Arch Pharmacol 363: 50-56. [DOI] [PubMed] [Google Scholar]
- Katona I, Sperlagh B, Magloczky Z, Santha E, Kofalvi A, Czirjak S, Mackie K, Vizi ES, Freund TF ( 2000) GABAergic interneurons are the targets of cannabinoid actions in the human hippocampus. Neuroscience 100: 797-804. [DOI] [PubMed] [Google Scholar]
- Leranth C, Frotscher M ( 1989) Organization of the septal region in the rat brain: cholinergic-GABAergic interconnections and the termination of hippocampus-septal fibers. J Comp Neurol 289: 304-314. [DOI] [PubMed] [Google Scholar]
- Lichtman AH, Dimen KR, Martin BR ( 1995) Systemic or intrahippocampal cannabinoid administration impairs spatial memory in rats. Psychopharmacology 119: 282-290. [DOI] [PubMed] [Google Scholar]
- Lichtman AH, Varvel SA, Martin BR ( 2002) Endocannabinoids in cognition and dependence. Prostaglandins Leukot Essent Fatty Acids 66: 269-285. [DOI] [PubMed] [Google Scholar]
- Lindvall O, Stenevi U ( 1978) Dopamine and norepinephrine neurons projecting to the septal area in the rat. Cell Tissue Res 190: 383-407. [DOI] [PubMed] [Google Scholar]
- Lu XR, Ong WY, Mackie K ( 1999) A light and electron microscopic study of the CB1 cannabinoid receptor in monkey basal forebrain. J Neurocytol 28: 1045-1051. [DOI] [PubMed] [Google Scholar]
- Malone DT, Taylor DA ( 1999) Modulation by fluoxetine of striatal dopamine release following Delta9-tetrahydrocannabinol: a microdialysis study in conscious rats. Br J Pharmacol 128: 21-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maneuf YP, Brotchie JM ( 1997) Paradoxical action of the cannabinoid WIN55, 212-2 in stimulated and basal cAMP accumulation in globus pallidus slices. Br J Pharmacol 120: 1397-1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies JE, Hammer RP ( 1991) D9-tetrahydrocannabinol alter cerebral metabolism in a biphasic, dose-dependent manner in rat brain. Eur J Pharmacol 202: 373-378. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Lutz B ( 1999) Expression of the cannabinoid receptor CB1 in distinct neuronal subpopulations in the adult mouse forebrain. Eur J Neurosci 11: 4213-4224. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, Hermann H, Tang J, Hofmann C, Zieglgansberger W, Di Marzo V, Lutz B ( 2002) The endogenous cannabinoid system controls extinction of aversive memories. Nature 418: 530-534. [DOI] [PubMed] [Google Scholar]
- Martellotta MC, Cossu G, Fattore L, Gessa GL, Fratta W ( 1998) Self-administration of the cannabinoid receptor agonist WIN 55, 212-2 in drug-naive mice. Neuroscience 85: 327-330. [DOI] [PubMed] [Google Scholar]
- Milner TA ( 1991) Cholinergic neurons in the rat septal complex: ultrastructural characterization and synaptic relations with catecholaminergic terminals. J Comp Neurol 314: 37-54. [DOI] [PubMed] [Google Scholar]
- Mishima K, Egashira N, Matsumoto Y, Iwasaki K, Fujiwara M ( 2002) Involvement of reduced acetylcholine release in Delta9-tetrahydrocannabinol-induced impairment of spatial memory in the 8-arm radial maze. Life Sci 72: 397-407. [DOI] [PubMed] [Google Scholar]
- Monory K, Tzavara ET, Lexime J, Ledent C, Parmentier M, Borsodi A, Hanoune J ( 2002) Novel, not adenylyl cyclase-coupled cannabinoid binding site in cerebellum of mice. Biochem Biophys Res Commun 292: 231-235. [DOI] [PubMed] [Google Scholar]
- Moor E, De Boer P, Beldhuis HJ, Westerink BH ( 1994) A novel approach for studying septo-hippocampal cholinergic neurons in freely moving rats: a microdialysis study with dual-probe design. Brain Res 648: 32-38. [DOI] [PubMed] [Google Scholar]
- Moor E, DeBoer P, Westerink BH ( 1998) GABA receptors and benzodiazepine binding sites modulate hippocampal acetylcholine release in vivo. Eur J Pharmacol 359: 119-126. [DOI] [PubMed] [Google Scholar]
- Moore RY ( 1978) Catecholamine innervation of the basal forebrain I. The septal area. J Comp Neurol 177: 665-684. [DOI] [PubMed] [Google Scholar]
- Nava F, Carta G, Battesi AM, Gessa GL ( 2000) D2 dopamine receptors enable D-9-tetrahydrocannabinol induced memory impairment and reduction of hippocampal extracellular acetylcholine concentration. Br J Pharmacol 130: 1201-1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Tsubokawa H, Mizushima I, Yoneda N, Zimmer A, Kano M ( 2002) Presynaptic cannabinoid sensitivity is a major determinant of depolarization-induced retrograde suppression at hippocampal synapses. J Neurosci 22: 3864-3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada M, Urae A, Mine K, Shoyama Y, Iwasaki K, Fujiwara M ( 1992) The facilitating and suppressing effects of D9-tetrahydrocannabinol on the rise in intrasynaptosomal Ca2+ concentration in rats. Neurosci Lett 140: 55-58. [DOI] [PubMed] [Google Scholar]
- Ong WY, Mackie K ( 1999) A light and electron microscopic study of the CB1 cannabinoid receptor in primate brain. Neuroscience 92: 1177-1191. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C ( 1982) The rat brain in stereotaxic coordinates. New York: Academic.
- Pertwee RG ( 2001) Cannabinoid receptors and pain. Prog Neurobiol 63: 569-611. [DOI] [PubMed] [Google Scholar]
- Reibaud M, Obinu MC, Ledent C, Parmentier M, Bohme GA, Imperato A ( 1999) Enhancement of memory in cannabinoid CB1 receptor knockout mice. Eur J Pharmacol 379: R1-R2. [DOI] [PubMed] [Google Scholar]
- Rinaldi-Carmona M, Barth F, Heaulme M, Alonso R, Shire D, Congy C, Soubrie P, Breliere JC, Le Fur G ( 1995) Biochemical and pharmacological characterisation of SR141716A, the first potent and selective brain cannabinoid receptor antagonist. Life Sci 56: 1941-1947. [DOI] [PubMed] [Google Scholar]
- Sanudo-Pena MC, Romero J, Seale GE, Fernandez-Ruiz JJ, Walker JM ( 2000) Activational role of cannabinoids on movement. Eur J Pharmacol 391: 269-274. [DOI] [PubMed] [Google Scholar]
- Schlicker E, Kathmann M ( 2001) Modulation of transmitter release via presynaptic cannabinoid receptors. Trends Pharmacol Sci 22: 565-572. [DOI] [PubMed] [Google Scholar]
- Sulcova E, Mechoulam R, Fride E ( 1998) Biphasic effects of anandamide. Pharmacol Biochem Behav 59: 347-352. [DOI] [PubMed] [Google Scholar]
- Tanda G, Pontieri FE, Di Chiara G ( 1997) Cannabinoid and heroin activation of mesolimbic dopamine transmission by a common mu1 opioid receptor mechanism. Science 276: 2048-2050. [DOI] [PubMed] [Google Scholar]
- Tsou K, Brown S, Sanudo-Pena MC, Mackie K, Walker JM ( 1998) Immunohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience 83: 393-411. [DOI] [PubMed] [Google Scholar]
- Turkanis SA, Karler R ( 1981) Excitatory and depressant effects of delta 9-tetrahydrocannabinol and cannabidiol on cortical evoked responses in the conscious rat. Psychopharmacology (Berl) 75: 294-298. [DOI] [PubMed] [Google Scholar]
- Tzavara ET, Davis RJ, Perry KW, Li X, Salhoff C, Bymaster FP, Witkin JM, Nomikos GG ( 2003) The CB1 receptor antagonist SR141716A selectively increases monoaminergic neurotransmission in the medial pre-frontal cortex: implications for therapeutic actions. Br J Pharmacol 138: 544-553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varvel SA, Lichtman AH ( 2002) Evaluation of CB1 receptor knockout mice in the Morris water maze. J Pharmacol Exp Ther 301: 915-924. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Kunos G, Nicoll RA ( 2001) Presynaptic specificity of endocannabinoid signaling in the hippocampus. Neuron 31: 453-462. [DOI] [PubMed] [Google Scholar]
- Woolf NJ, Butcher LL ( 1986) Cholinergic systems in the rat brain. III. Projections from the pontomesencephalic tegmentum to the thalamus, tectum, basal ganglia, and basal forebrain. Brain Res Bull 16: 603-637. [DOI] [PubMed] [Google Scholar]
- Wu M, Shanabrough M, Leranth C, Alreja M ( 2000) Cholinergic excitation of septohippocampal GABA but not cholinergic neurons: implications for learning and memory. J Neurosci 20: 3900-3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilles K, Werner L, Qu M, Schleicher A, Gross G ( 1991) Quantitative auto-radiography of 11 different transmitter binding sites in the basal forebrain region of the rat: evidence of heterogeneity in distribution patterns. Neuroscience 42: 473-481. [DOI] [PubMed] [Google Scholar]