

Abstract
The transcription factor Rel/nuclear factor (NF)-κB is known for its fundamental role in regulating immune and inflammatory responses. In the brain, constitutive NF-κB activity has been detected exclusively in neurons, and a large diversity of stimuli have been reported to induce NF-κB activity. Yet the function of this transcription factor in the nervous system remains unclear, and its role in neuroprotection or neurodegeneration is open to debate. Recently it was suggested that κB-driven gene expression in neurons is controlled by Sp1-like factors. To clarify such controversy, we have characterized here a novel mouse model in which the entire NF-κB-dependent transcriptional response is abolished in the forebrain. Calcium-calmodulin-dependent kinase II α promoter-driven tetracycline transactivator was used for regulated expression of a transdominant negative mutant of inhibitor κBα (super-repressor) together with a green fluorescent protein tracer. Inhibition of expression of a κB-dependent lacZ transgene was shown in triple transgenic mice, which correlated with the loss of κB-specific DNA binding. In transgenic organotypic hippocampal slice cultures, expression of the super-repressor led to strong cell death after neurotoxic insults. These data demonstrate for the first time that neuron-restricted ablation of NF-κB-driven gene expression increases neurodegeneration. This might lead to the path for new treatments of neurodegenerative diseases.
Keywords: transcription factor NF-κB, brain, tetracycline, neuroprotection, organotypic hippocampal slice cultures, kainate, ferrous sulfate, transgenic mice
Introduction
Nuclear factor (NF)-κB is a ubiquitously expressed, inducible factor that regulates the expression of a wide variety of genes and is involved in cell survival, growth, stress responses, and immune and inflammatory processes (Karin and Lin, 2002). The five mammalian NF-κB members, p50, p52, p65, or RelA, c-Rel, and RelB, share an N-terminal 300 amino acid Rel homology domain, which allows DNA binding, dimerization, and nuclear localization. In most cells, NF-κB/Rel complexes are sequestered in the cytoplasm by interaction with inhibitor (I) κB proteins. Nuclear translocation of NF-κB can be induced by multiple stimuli that trigger activation of an IκB kinase (IKK) complex, which in turn phosphorylates specific serines within the IκBs, leading to their ubiquitination and proteosomal degradation. Released NF-κB/Rel complexes migrate to the nucleus to function as transcription factors.
In certain regions of the brain, especially the cortex and hippocampus, constitutive nuclear NF-κB activity has been reported in neurons (Kaltschmidt et al., 1994; Schmidt-Ullrich et al., 1996; Bhakar et al., 2002). A large diversity of stimuli [glutamate, kainate, membrane depolarization, NGF, tumor necrosis factor, interleukin-1 (IL-1), amyloid β (Aβ), and secreted amyloid precursor protein (sAPP)] can induce NF-κB activity both in neurons and in glial cells. In vivo, nuclear NF-κB activity has been observed in neurodegenerative disorders and rodent models of seizure and stroke (for review, see Mattson and Calmandola, 2001; Kaltschmidt and Kaltschmidt, 2003). Analysis of knock-out mice for individual NF-κB genes has provided limited information about NF-κB in the nervous system, the function of which remains obscure. P50-deficient mice revealed both neuroprotective and neurodegenerative properties of p50 after challenge with different damaging insults (Schneider et al., 1999; Yu et al., 1999; Kassed et al., 2002). The role of NF-κB in the control of neuronal survival and cell death is still a matter of debate, (for review, see O'Neill and Kaltschmidt, 1997; Denk et al., 2000), in part because of the fact that two, non-mutually exclusive mechanisms must be considered: an indirect effect via modulation by glial cells or a direct role in neurons. In conventional knock-out mice, no specific cell type is addressed, making it impossible to discriminate between a role for NF-κB in glia or neurons. Therefore, we generated transgenic mice in which NF-κB activity is inhibited in forebrain neurons in a tetracycline-dependent manner and used them to address the role of NF-κB in neuronal survival.
Materials and Methods
Transgenic mice. Details of the inducible transgenic construct are available on request. Briefly, the human (h) IκBα-AA mutant has been described (Whiteside et al., 1995). The internal ribosomal entry site (IRES) from the encephalomyocarditis virus (Ghattas et al., 1991) was mutated in its last ATG. A thermoresistant green fluorescent protein (GFP) containing a substitution of valine 163 for alanine that improved proper folding of the protein and increased the fluorescence at 37°C was fused to the mutated IRES (Baubet et al., 2000). Two independent inducible hIκBα-AA-IRES-GFP transgenic lines were established, lines 282 and 3624, that gave the same pattern and level of expression of the super-repressor and the GFP. Data presented are from double transgenic mice (4 weeks to 4 months of age) generated from intercrosses between line 282 and calcium-calmodulin-dependent kinase II α (CAMKII)-tetracycline transactivator (tTA) mice (Mayford et al., 1996). Triple transgenic mice were obtained from intercrosses between CAMK-tTA/282 double transgenic mice and p105lacZ line 189-4 transgenic mice (Schmidt-Ullrich et al., 1996). Mice were kept in a specific antigen-free animal house facility and genotyped by PCR. Doxycycline (DOX) was given to the mice in drinking water at 2 mg/ml in 2.5% sucrose for a minimum of 12 d.
Western blot analysis and antibodies. Whole-cell extracts from 293T transfected cells or brains were prepared as reported (Schmidt-Ullrich et al., 1996). Immunoblots were incubated with rabbit polyclonal antibodies at 1:400 dilution and revealed with the Pierce enhanced chemiluminescence system. Serum against IκBα (#C-21) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA), and serum against GFP from was from BD Clontech (Ozyme, Saint-Quentin, France).
GFP detection and β-galactosidase staining. GFP fluorescence was visualized under a MZFLIII fluorescence binocular scope (Leica) for whole-brain sections or under an Axiophot microscope with an epiluminescent system (Zeiss) for 15 μm unfixed brain tissue sections. For immunolocalization of GFP, 4% paraformaldehyde-fixed 15 μm brain tissue sections were permeabilized in 2% BSA, pH 7.4/0.02% Triton X-100 for 1 hr and then incubated with polyclonal antibody against GFP (Medical and Biological Laboratories, Biovalley, Marne la Vallée, France) at 1:50 dilution. Secondary antibody (biotinylated goat anti-rabbit IgG) was applied at 1:100 dilution and revealed with streptavidin-alkaline phosphatase conjugate. 4-chloro-5-bromo-3-indoyl-β-galactoside (X-Gal) staining was performed as described (Schmidt-Ullrich et al., 1996).
Electrophoretic mobility shift assay. Nuclear extracts were prepared and band-shift assays were performed as reported (Schmidt-Ullrich et al., 1996), using the κB site derived from the promoter of the major histocompatibility complex (MHC) class I H-2 Kb gene as a probe.
Organotypic hippocampal slice cultures. Organotypic hippocampal slice cultures were prepared and maintained according to Stoppini et al. (1991). Briefly, hippocampi from transgenic mice (postnatal day 5) were cut perpendicularly to the longitudinal axis into 400-μm-thick slices by using a McIlwain tissue chopper (Mickle Laboratory Engineering, Gomshall, Surrey, UK) and kept in ice-cold MEM HBSS, pH 7.3, containing 25 mm HEPES and 2 mml-glutamine. Only complete and well preserved slices, selected under a binocular scope, were put on top of Millipore membranes (diameter 30 mm, pore size 0.4 μm), transferred to six-well plates containing 750 μl culture medium (50% MEM HBSS, 25% HBSS, 25% heat-inactivated horse serum, 2 mml-glutamine, 5 mm NaHCO3) per well (refreshed twice a week), and incubated in humidified 95% air/5% CO2 atmosphere at 37°C for 4 weeks.
Analysis of cell death by propidium iodide fluorescence. Two hours before FeSO4 or kainate (KA) treatment, propidium iodide (PI) was added at 0.1 mg/ml to the culture medium. Slices were then transferred into culture medium supplemented with 200 μm FeSO4 or 500 μm KA, 0.01 mg/ml PI for the indicated time (0, 1, 2, 4, 8, or 24 hr). After fixation, the extent of neuronal degeneration was quantified by the uptake of PI into the damaged cells (Macklis and Madison, 1990). The PI fluorescence was monitored by using an inverted confocal laser scanning microscope (LSM 5 Pascal, Zeiss, Oberkochen, Germany) equipped with a plan-Neofluar 5× objective. Relative cell death was calculated from each region of interest as follows: relative percentage of cell death = (Fexp - Fmin)/(Fmax - Fmin) × 100. Fexp is the fluorescence of the experimental test condition, Fmax is maximum fluorescence, and Fmin is the background fluorescence. Statistical analysis was done with ANOVA followed by a Bonferroni's post hoc test. The KA experiments were repeated three times with several animals (n = 9 tTA/hIκBα-AA mice; n = 6 hIκBα-AA controls). Boc-Asp (OMe)-fluoromethylketone (Boc-D-FMK), a general caspase inhibitor (D'Mello et al., 1998) (Merck, Fontenay sous bois, France), was added at 100 μm 1 hr before kainate treatment in organotypic hippocampal slice cultures from tTA/hIκBα-AA double transgenic mice. The number of dead cells was counted in the CA3 region. Statistical analysis was done with ANOVA followed by Scheffe′s post hoc test. Data are shown as mean ± SEM (10 independent determinations).
Results
Because the NF-κB family is multigenic and the knock-out of p65 is embryonic lethal, the only way to ablate NF-κB activity in vivo is to express in a conditional manner a transdominant inhibitor of NF-κB. We generated tissue-restricted NF-κB knock-out mice through targeting of a dominant-negative form of IκBα to the forebrain. This protein, the two N-terminal serines of which have been mutated into alanines, cannot be degraded and therefore acts as a super-repressor of NF-κB activity. In its presence, every NF-κB/Rel complex is sequestered in the cytoplasm and prevented from DNA binding and consequently from activating transcription (Whiteside et al., 1995). To avoid potential lethality problems, the super-repressor was conditionally expressed using the tetracycline system in a binary combination (Kistner et al., 1996) (Fig. 1A). In the inducible transgenic mouse, the operator sites of the tetracyclin bacterial operon drive the expression of a polycistronic messenger RNA encoding the dominant-negative human form of IκBα and a thermoresistant GFP separated by an IRES, which allows reinitiation of translation. The IRES has been mutated to reduce its efficiency and render the GFP reporter molecule a real tracer of the super-repressor expression. Transient transfection experiments were used to compare the expression levels of the mutant IκBα and GFP proteins obtained with the inducible system in the presence or absence of DOX (an analog of tetracycline that crosses the blood-brain barrier) and with the classic cytomegalovirus (CMV) enhancer-promoter. Figure 1B shows that DOX-dependent expression of the super-repressor is superior to that observed after transfection of CMV-hIκBα-AA, whereas drug-dependent expression of the GFP is much less than that obtained with the CMV-GFP construct. Direct visualization of cells expressing the mutant IκBα via the GFP could thus be achieved without any toxic side effects.
Figure 1.

Generation of a conditional forebrain-specific knock-out of NF-κB activity. A, The tetracycline-controlled system: scheme of the transgenic lines used. B, In the inducible transgenic construct, GFP is a tracer of the expression of the super-repressor. 293T cells were transiently transfected with hIκBα-AA or GFP under the control of a classic CMV promoter-enhancer or with the inducible transgenic construct containing both genes, hIκBα-AA and GFP, in a polycistronic unit under the control of a tetracyclin-dependent promoter, with or without a CMV expression vector for the tetracycline transactivator in the presence or absence of DOX at 1 μg/ml. Total protein extracts (60 μg) were analyzed by Western blot with antibodies against GFP or IκBα. C, Strong expression of the super-repressor is observed in double transgenic tTA/hIκBα-AA mice only in the absence of DOX. Shown is Western blot analysis of total protein extracts (120 μg) from brains of double transgenic mice treated or not with DOX, single transgenic, or nontransgenic mice. D, Restricted spatial distribution of the GFP in the brain of double transgenic tTA/hIκBα-AA mice is DOX dependent. Fluorescence images show the GFP visualized after coronal cut (top panel) or after sagittal tissue section (15 μm) (middle and bottom panels) of unfixed double transgenic brains. Note the absence of GFP in the hindbrain. Hi, Hippocampus; Cx, cortex; Cb, cerebellum.
Inducible transgenic animals were crossed with transactivating transgenic mice carrying the Tet repressor-derived mammalian transcriptional activator (tetR-VP16 or tTA) under the control of the CaMKIIα promoter, ensuring neuronal forebrain-specific expression (Mayford et al., 1996). In the resulting tTA/hIκBα-AA double transgenic mice, high level expression of the super-repressor was detected in total brain extracts by Western blot analysis, which was completely suppressed by DOX leading to a situation similar to single transgenic (hIκBα-AA) or wild-type mice (Fig. 1C). In the absence of the drug, GFP expression was consistently induced in hippocampal principal cells, cortex, and olfactory lobe of double transgenics (Fig. 1D) (and data not shown). As expected, no GFP expression could be detected in the hindbrain of these animals. Thus the system was functional regarding the drug-dependent and restricted spatial expression of the super-repressor.
We next asked whether NF-κB activity was genuinely inhibited in the forebrain of tTA/hIκBα-AA double transgenic mice. We first verified by electrophoretic mobility shift assay (EMSA) whether NF-κB DNA binding activity was suppressed in the hippocampus of double transgenic mice (Fig. 2A). Two NF-κB-specific complexes identified as p50/50 and p50/p65 (data not shown) were detected in hippocampal extracts from single transgenic or double transgenic mice treated with DOX, whereas only the p50/p50 complex was visible in extracts from untreated double transgenic mice. Therefore, expression of the super-repressor specifically prevented the appearance of nuclear p50/p65 heterodimers. To demonstrate that this inhibition of DNA binding activity led to repression of κB-dependent gene expression, κB-lacZ/tTA/hIκBα-AA triple transgenic mice were generated. In these mice, nuclear β-galactosidase is produced only in the presence of active NF-κB complexes. The strong β-galactosidase staining observed in the presence of DOX in the triple transgenics revealed constitutive NF-κB activity in a pattern identical to that of κB-lacZ single transgenics (Schmidt-Ullrich et al., 1996). It disappeared in the hippocampus and epithalamus and was much reduced in the cortex when the drug was absent and the super-repressor was expressed (as assessed by immunostaining for GFP) (Fig. 2B), indicating that NF-κB activity was inhibited. We conclude that in our mouse model NF-κB activity is efficiently ablated; however, these double transgenic mice do not present any obvious morphological defects, and no effect on neuronal survival could be observed in unstressed animals analyzed between 4 weeks and 6 months after birth (data not shown).
Figure 2.
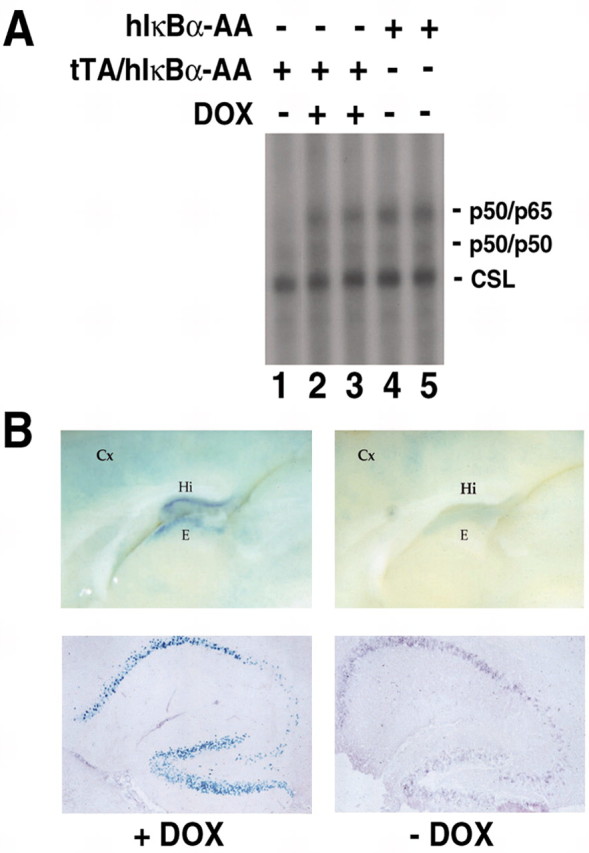
Expression of the super-repressor leads to complete elimination of NF-κB activity in vivo in a drug-dependent manner. A, Inhibition of NF-κB DNA binding activity in the hippocampus of double transgenic mice. For EMSA, 35 μg of total protein extracts from hippocampus of double transgenics treated or not with DOX or single transgenic mice were incubated with a double-stranded oligonucleotide corresponding to a canonical κB site located in the promoter of the MHC class I gene H-2 Kb. Lines indicate the different NF-κB complexes: the p50/p50 homodimers and the p50/p65 heterodimers. Note the steadiness of the internal control, the CSL complex that binds a half-κB site and is involved in the Notch pathway. B, Inhibition of κB-dependent lacZ reporter transgene expression in the forebrain of κB-lacZ/tTA/hIκBα-AA triple transgenic mice treated or not with DOX. Left and right top panels are whole-mounts of X-Gal staining. Bottom panels, X-Gal staining (blue) and immunohistochemistry of the GFP (purple) on sagittal sections demonstrate that inhibition of NF-κB reporter activity correlates with expression of transgenic hIκBα-AA mRNA. Hi, Hippocampus, Cx, cortex, E, epithalamus.
To assess whether neurons lacking NF-κB activity are more susceptible to neuronal stress, hippocampal slice cultures were established to analyze under standardized conditions in an organotypic context the kinetics of cell death in living neurons after exposure to FeSO4 or kainate, two agents reported to induce neurodegeneration (Weiss et al., 1990; Zhang et al., 1993). We preferred this ex vivo approach over in vivo state because it is highly reproducible, and it allowed us to circumvent the resistance to excitotoxic cell death of our transgenics in vivo resulting from enriched C57BL/6 background (Schauwecker and Steward, 1997). Because we have shown that the responses of tTA/hIκBα-AA double transgenics in the presence of DOX and hIκBα-AA single transgenic mice were similar (see above) (and our unpublished data), cell death experiments were performed with hippocampal slice cultures from tTA/hIκBα-AA double transgenic or hIκBα-AA single transgenic mice. Neuronal degeneration was visualized by the uptake of PI into damaged cells, thus precluding the use of DOX, which results in a diffuse yellow fluorescence interfering with that of PI. No significant cell death could be observed in untreated slice cultures regardless of the genotype considered (Fig. 3A-D); however, PI fluorescence values were slightly but significantly (p < 0.001) reduced in CA3 of nontreated double transgenic slices, revealing an increased neuroprotection of this specific region in the presence of high amounts of the super-repressor (Fig. 3B,D). Treatment with FeSO4 for 24 hr led to visible cell death and increased PI fluorescence values in the hippocampal area CA1 and dentate gyrus (GD) in both single and double transgenic slices. The neuronal damage clearly increased because of expression of the super-repressor in the double transgenics (Fig. 3A,B). To investigate the response to kainate, a time course of kainate-treatment was undertaken (Fig. 3C,D). Remarkably, decreased PI fluorescence values (p < 0.001) were observed in both CA3 and dentate gyrus briefly treated (1 hr) double transgenic slices. In these slices overexpressing the super-repressor, cell death was detected in the dentate gyrus after 2 hr, a time point when no neurodegeneration was visible in the controls. Then, neuronal damage in double transgenic slices increased with incubation time to reach a peak at 8 hr with maximal PI fluorescence values (p < 0.001) in the hippocampal regions CA1, CA3, and GD as shown by quantitative pseudocolor image (Fig. 3E). After 24 hr exposure to kainate, kainate-induced neuronal damage was much stronger in double transgenic slices, resulting in a complete loss of the CA3 region, diminished PI fluorescence values, and disappearance of almost all hippocampal cytoarchitecture. In contrast, the principal layers of the hippocampus CA1, CA3, and GD were still distinguishable and intensively stained by PI in control single transgenic slices. In slices of single transgenics, cell death rose continuously with a maximum of neurodegeneration obtained after 24 hr. Both the temporal course of degeneration and sensitivity to the excitotoxic stress are affected in double transgenic slices. These results indicate that the absence of NF-κB potentiates both FeSO4- and kainate-induced neuronal death and that NF-κB plays a key role in neuroprotection under stress conditions.
Figure 3.

Forebrain ablation of NF-κB activity leads to increased cell death in organotypic hippocampal slice cultures of double transgenic mice. A, FeSO4-induced neuronal damage. Slice cultures from double transgenic tTA/hIκBα-AA or single transgenic hIκBα-AA mice were treated for 24 hr without (controls, top panels) or with 200 μm FeSO4 (bottom panels). B, PI fluorescence values measured 0 or 24 hr after FeSO4 treatment. C, Time course of kainate-induced neurodegeneration. Slice cultures from double transgenic tTA/hIκBα-AA or single transgenic hIκBα-AA mice were treated with 500 μm kainate for the indicated times. D, PI fluorescence values measured in untreated and kainate-treated slices at different time points. Neuronal damage was visualized by red PI fluorescence imaging. CA1, Hippocampal region CA1; CA3, hippocampal region CA3. E, Pseudocolor quantification of cell death 8 hr after KA treatment.
To determine the features of the increased kainate-induced cell death observed in our system, we assessed whether apoptotic cascade activation was involved (Vincent et al., 2002). Figure 4 shows that Boc-D-FMK, a general caspase inhibitor, significantly inhibited kainate-mediated cell death in the CA3 region of slices from tTA/hIκBα-AA double transgenics by 50% (p < 0.02) and to a lesser extent in the dentate gyrus and CA1. Thus, the absence of neuronal NF-κB activity at least in the CA3 region led to increased caspase activation, a hallmark of programmed cell death.
Figure 4.
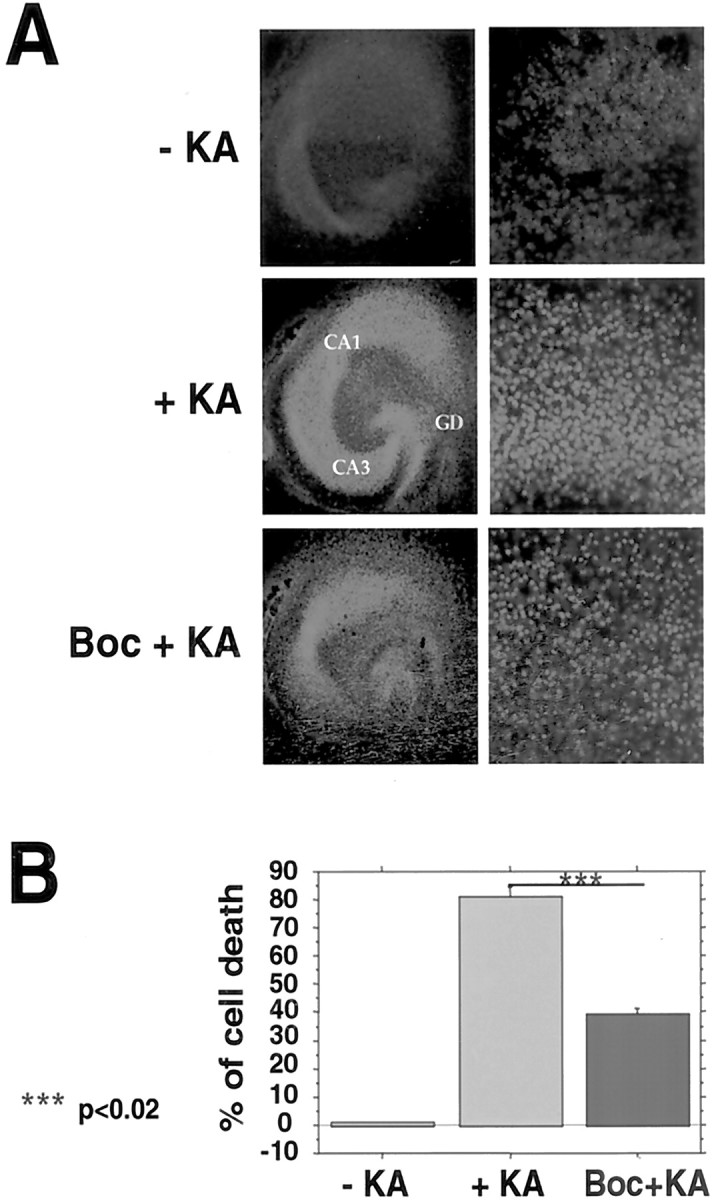
Cell death after kainate treatment is caused in part by increased apoptosis. A, Effect of the caspase inhibitor Boc-D-FMK on kainate-induced neurodegeneration. Double transgenic tTA/hIκBα-AA hippocampal slices were pretreated for 1 hr with 100 μm Boc-D-FMK and then stressed for 8 hr with 500 μm KA. Overviews of representative slices are shown on the left. Higher magnification views of the corresponding CA3 regions scored for dead cells are on the right. Neuronal damage was visualized by red PI fluorescence imaging. GD, Dentate gyrus; CA1, hippocampal region CA1; CA3, hippocampal region CA3. B, Quantification of the effect of Boc-D-FMK treatment in the CA3 region.
Discussion
In this study, we describe a new mouse model expressing in a tetracycline-dependent manner a transdominant negative mutant of IκBα (hIκBα-AA) together with tracer amounts of GFP in neurons of the forebrain. The dominant inhibitor affects all members of the NF-κB/Rel family and provides means of controlling the amplitude and duration of NF-κB inhibition. We have shown that in double transgenic mice, the expression of both super-repressor and GFP in the forebrain was dependent strictly on treatment by doxycycline and that a strong induction was observed without drug. The model has been validated further by the abolition of NF-κB DNA binding in the hippocampus of double transgenics and by the block of κB-dependent transcriptional response of a lacZ reporter transgene and of an endogenous target gene, such as NF-κB1 (data not shown). Interestingly in our system, only the p50/p65 heterodimers, reported to constitute both the inducible and constitutive NF-κB active species in the brain (Kaltschmidt et al., 1993; Kaltschmidt et al., 1994; Schmidt-Ullrich et al., 1996), are prevented from binding to DNA. DNA binding of p50 homodimers was not affected, which probably correlates with the weak affinity of IκBα for p50 homodimers. In contrast to these bona fide NF-κB complexes, it was suggested recently that κB-driven expression in neurons is the result of transacting Sp1-like factors (Mao et al., 2002). Here we show that in forebrain neurons all κB-driven expression could be inhibited by transgenic expression of super-repressor IκBα-AA, arguing against a major role of Sp1-like factors.
In our double transgenic mice no alteration of neuronal survival could be observed in unstressed adult animals, suggesting that neuronal NF-κB inhibition merely affects neuron viability at the resting state during adulthood. During development, increased apoptosis has been reported in vivo in embryonic day (E) 14 trigeminal ganglia neurons of p65-deficient embryos (Hamanoue et al., 1999) and in the hindbrain neuroepithelium of E11.5 double IKK1-/-/IKK2-/- mutant embryos, which are totally devoid of NF-κB (Li et al., 2000). E16 cortical neurons infected with adenovirus encoding IκBαM super-repressor also present a significant reduction of neuron viability in vitro (Bhakar et al., 2002). Developing neurons, which have not yet established connections, might exhibit a higher sensitivity to apoptosis compared with that of mature neurons. In the double transgenic slices, the slightly increased survival of the CA3 region, which exhibits specific glutamate receptor usage, might reflect the rescue in the absence of NF-κB of the double transgenic neurons by another neuroprotective pathway.
After challenge with damaging insults, such as FeSO4 or kainate exposure, ex vivo organotypic hippocampal slice cultures from double transgenic mice expressing the super-repressor in neurons exhibited a higher vulnerability to neuronal excitotoxicity than slices from single transgenic mice. This loss of neuroprotection involved apoptotic cascade activation. These findings demonstrate that neuron-restricted ablation of NF-κB-driven gene expression increases neurodegeneration in neurotoxicity paradigms. They support a critical biological role of NF-κB in such a pathological process. These data are consistent with the results of Mattson and coworkers, who showed that NF-κB contributes to neuronal cell protection after excitotoxic exposure in vitro (Mattson et al., 1997) or in vivo by using κB decoy DNA (Yu et al., 1999). Similarly the NF-κB-dependent protection against amyloid β peptide was also abrogated by overexpression of super-repressor IκBαM in cultured cerebellar neurons (Kaltschmidt et al., 1999). Surprisingly, our mouse model and mice deficient in p50 harbor a similar phenotype, i.e., increased damage of hippocampal neurons after excitotoxic damage (Yu et al., 1999), whereas p50 homodimers do not constitute the active NF-κB species in brain. Lack of NF-κB p50 also exacerbates degeneration of hippocampal neurons after chemical exposure in vivo (Kassed et al., 2002). In contrast, NF-κB inhibition by an inhibitory peptide in the rat striatum protects striatal cells from excitotoxic-induced apoptosis (Nakai et al., 2000), and in mice lacking p50 ischemic damage it is reduced significantly (Schneider et al., 1999). Specific suppression of NF-κB by the super-repressor partially protected hippocampal HT22 cells against haloperidol, a dopamine receptor antagonist, cell death in vitro (Post et al., 1998). IκBαM blockade of NF-κB activation by aspirin was also shown to be protective against neurotoxicity elicited by the glutamate (Grilli et al., 1996). The nature and site of the damaging insult, which determine potential interactions with other transcription factors such as AP1 or p53, might lead to different fates.
In conclusion, we have shown that NF-κB has a neuroprotective function that is essential for the maintenance of brain homeostasis in adult mice. Because overactivation of ionotropic glutamate receptors has been involved as a pathophysiological factor in multiple acute and chronic neurological disorders such as seizure, stroke, and neurodegenerative diseases, these results might have important implications for the generation of new therapeutics.
Footnotes
This work was supported in part by grants from Institut National de la Santé et de la Recherche Médicale and Ligue Nationale contre le Cancer (équipe labellisée) to A.I., European Community to A.I. and C.K., Volkswagen-Stiftung to C.K., and Institut Pasteur (PTR38) to S.M. We thank E. Kandel for kindly providing us with the CAMK-tTA transgenic mice, C. Cimper for helpful histological assistance, and R. Kelly and R. Weil for critical reading of this manuscript. S.M. is particularly indebted to the Service Chirurgie 3 Hôpital d'Auxerre and Dr. I. Aupérin.
Correspondence should be addressed to Sylvie Mémet, Unité de Biologie Moléculaire de l'Expression Génique, Unité de Recherche Associée Centre National de la Recherche Scientifique 2582, Institut Pasteur, 75724 Paris Cedex 15, France. E-mail: symemet@pasteur.fr.
B. Goudeau's present address: Unité Mixte de Recherche 7000, Faculté deMédecine Pitié-Salpêtrière, 75013 Paris, France.
Copyright © 2003 Society for Neuroscience 0270-6474/03/239403-06$15.00/0
V.F. and B.K. contributed equally to this work.
References
- Baubet V, Le Mouellic H, Campbell AK, Lucas-Meunier E, Fossier P, Brulet P ( 2000) Chimeric green fluorescent protein-aequorin as bioluminescent Ca2+ reporters at the single-cell level. Proc Natl Acad Sci USA 97: 7260-7265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhakar AL, Tannis LL, Zeindler C, Russo MP, Jobin C, Park DS, MacPherson S, Barker PA ( 2002) Constitutive nuclear factor-κ B activity is required for central neuron survival. J Neurosci 22: 8466-8475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denk A, Wirth T, Baumann B ( 2000) NF-kappaB transcription factors: critical regulators of hematopoeisis and neuronal survival. Cytokine Growth Factor Rev 11: 303-320. [DOI] [PubMed] [Google Scholar]
- D'Mello S, Aglieco F, Roberts MR, Borodezt K, Haycock JW ( 1998) A DEVD-inhibited caspase other than CPP32 is involved in the commitment of cerebellar granule neurons to apoptosis induced by K+ deprivation. J Neurochem 70: 1809-1818. [DOI] [PubMed] [Google Scholar]
- Ghattas IR, Sanes JR, Majors JE ( 1991) The encephalomyocarditis virus internal ribosome entry site allows efficient coexpression of two genes from a recombinant provirus in cultured cells and in embryos. Mol Cell Biol 11: 5848-5859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grilli M, Pizzi M, Memo M, Spano P ( 1996) Neuroprotection by aspirin and sodium salicylate through blockade of NF-κB activation. Science 274: 1383-1385. [DOI] [PubMed] [Google Scholar]
- Hamanoue M, Middleton G, Wyatt S, Jaffray E, Hay RT, Davies AM ( 1999) p75-mediated NF-kappa B activation enhances the survival response of developing sensory neurons to nerve growth factor. Mol Cell Neurosci 14: 28-40. [DOI] [PubMed] [Google Scholar]
- Kaltschmidt B, Kaltschmidt C ( 2003) Nuclear Factor κB. In: Regulation and role in disease (Beyaert R, ed), pp 375-394. New York: Kluwer.
- Kaltschmidt C, Kaltschmidt B, Baeuerle PA ( 1993) Brain synapses contain inducible forms of the transcription factor NF-kappa B. Mech Dev 43: 135-147. [DOI] [PubMed] [Google Scholar]
- Kaltschmidt C, Kaltschmidt B, Neumann H, Wekerle H, Baeuerle PA ( 1994) Constitutive NF-κB activity in neurons. Mol Cell Biol 14: 3981-3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaltschmidt B, Uherek M, Wellmann H, Volk B, Kaltschmidt C ( 1999) Inhibition of NF-kappaB potentiates amyloid beta-mediated neuronal apoptosis. Proc Natl Acad Sci USA 96: 9409-9414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M, Lin A ( 2002) NF-kappaB at the crossroads of life and death. Nat Immunol 3: 221-227. [DOI] [PubMed] [Google Scholar]
- Kassed CA, Willing AE, Garbuzova-Davis S, Sanberg PR, Pennypacker KR ( 2002) Lack of NF-κB p50 exacerbates degeneration of hippocampal neurons after chemical exposure and impairs learning. Exp Neurol 176: 277-288. [DOI] [PubMed] [Google Scholar]
- Kistner A, Gossen M, Zimmermann F, Jerecic J, Ullmer C, Lubbert H, Bujard H ( 1996) Doxycycline-mediated quantitative and tissue-specific control of gene expression in transgenic mice. Proc Natl Acad Sci USA 93: 10933-10938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Estepa G, Mémet S, Israël A, Verma IM ( 2000) Complete lack of NF-κB activity in IKK1 and IKK2 double-deficient mice: additional defect in neurulation. Genes Dev 14: 1729-1733. [PMC free article] [PubMed] [Google Scholar]
- Macklis JD, Madison RD ( 1990) Progressive incorporation of propidium iodide in cultured mouse neurons correlates with declining electrophysiological status: a fluorescence scale of membrane integrity. J Neurosci Methods 31: 43-46. [DOI] [PubMed] [Google Scholar]
- Mao X, Morman AM, Barger SW ( 2002) Neuronal κB-binding factors consist of Sp1-related proteins. J Biol Chem 277: 44911-44919. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Calmandola S ( 2001) NF-kappaB in neuronal plasticity and neurodegerative disorders. J Clin Invest 107: 247-254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Goodman Y, Luo H, Fu W, Furukawa K ( 1997) Activation of NF-kappaB protects hippocampal neurons against oxidative stress-induced apoptosis: evidence for induction of manganese superoxide dismutase and suppression of peroxynitrite production and protein tyrosine nitration. J Neurosci Res 49: 681-697. [DOI] [PubMed] [Google Scholar]
- Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER ( 1996) Control of memory formation through regulated expression of a CaMKII transgene. Science 274: 1678-1683. [DOI] [PubMed] [Google Scholar]
- Nakai M, Qin ZH, Chen JF, Wang Y, Chase TN ( 2000) Kainic acid-induced apoptosis in rat striatum is associated with nuclear factor-kappaB activation. J Neurochem 74: 647-658. [DOI] [PubMed] [Google Scholar]
- O'Neill LA, Kaltschmidt C ( 1997) NF-κB: a crucial transcription factor for glial and neuronal cell function. Trends Neurosci 20: 252-258. [DOI] [PubMed] [Google Scholar]
- Post A, Holsber F, Behl C ( 1998) Induction of NF-κB activity during haloperidol-induced oxidative toxicity in clonal hippocampal cells: suppression of NF-κB and neuroprotection by antioxidants. J Neurosci 18: 8236-8246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauwecker PE, Steward O ( 1997) Genetic determinants of susceptibility to excitotoxic cell death: implications for gene targeting approaches. Proc Natl Acad Sci USA 94: 4103-4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt-Ullrich R, Mémet S, Lilienbaum A, Feuillard J, Raphael M, Israël A ( 1996) NF-κB activity in transgenic mice: developmental regulation and tissue specificity. Development 122: 2117-2128. [DOI] [PubMed] [Google Scholar]
- Schneider A, Martin-Villalba A, Weih F, Vogel J, Wirth T, Schwaninger M ( 1999) NF-kappa B is activated and promotes cell death in focal cerebral ischemia. Nat Med 5: 554-559. [DOI] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D ( 1991) A simple method for organotypic cultures of nervous tissue. J Neurosci Methods 37: 173-182. [DOI] [PubMed] [Google Scholar]
- Vincent VA, Robinson CC, Simsek D, Murphy GM ( 2002) Macrophage colony stimulating factor prevents NMDA-induced neuronal death in hippocampal organotypic cultures. J Neurochem 82: 1388-1397. [DOI] [PubMed] [Google Scholar]
- Weiss JH, Koh J, Baimbridge KG, Choi DW ( 1990) Cortical neurons containing somatostatin- or parvalbumin-like immunoreactivity are atypically vulnerable to excitotoxic injury in vitro. Neurology 40: 1288-1292. [DOI] [PubMed] [Google Scholar]
- Whiteside ST, Ernst MK, LeBail O, Laurent-Winter C, Rice NR, Israel A ( 1995) N- and C-terminal sequences control degradation of MAD3/IκBα in response to inducers of NF-κB activity. Mol Cell Biol 15: 5339-5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Zhou D, Bruce-Keller AJ, Kindy MS, Mattson MP ( 1999) Lack of the p50 subunit of nuclear factor-κB increases the vulnerability of hippocampal neurons to excitotoxic injury. J Neurosci 19: 8856-8865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Tatsumo T, Carney J, Mattson MP ( 1993) Basic FGF, NGF and IGF protects hippocampal neurons against iron-induced degeneration. J Cereb Blood Flow Metab 13: 378-388. [DOI] [PubMed] [Google Scholar]