

Abstract
The precise cause of neuronal death in Huntington's disease (HD) is unknown. Although no single specific protein-protein interaction of mutant huntingtin has emerged as the pathologic trigger, transcriptional dysfunction may contribute to the neurodegeneration observed in HD. Pharmacological treatment using the histone deacetylase inhibitor sodium butyrate to modulate transcription significantly extended survival in a dose-dependent manner, improved body weight and motor performance, and delayed the neuropathological sequelae in the R6/2 transgenic mouse model of HD. Sodium butyrate also increased histone and Specificity protein-1 acetylation and protected against 3-nitropropionic acid neurotoxicity. Microarray analysis showed increased expression of α- and β-globins and MAP kinase phosphatase-1 in sodium butyrate-treated R6/2 mice, indicative of improved oxidative phosphorylation and transcriptional regulation. These findings strengthen the hypothesis that transcriptional dysfunction plays a role in the pathogenesis of HD and suggest that therapies aimed at modulating transcription may target early pathological events and provide clinical benefits to HD patients.
Keywords: Huntington's disease, therapy, sodium butyrate, histones, transcription, microarray analysis, neuroprotection, R6/2 transgenic mice
Introduction
Huntington's disease (HD) is a progressive and fatal neurological disorder that is caused by an expanded CAG repeat in a gene coding for a protein of unknown function, huntingtin. There are no current drug therapies proven to help ameliorate or abrogate the disease process in HD. Although the exact cause of the selective neuronal death in HD remains unknown, it has been postulated that aberrant protein-protein interactions, including aggregation of the mutant huntingtin protein, may be toxic to neurons and lead to oxidative stress, mitochondrial dysfunction, apoptosis, energy metabolism defects, and excitotoxicity (Beal, 2000; Friedlander, 2003). The seminal event in this cascade may be transcriptional dysregulation initiated by direct binding of the mutant huntingtin protein or cleaved products to a number of transcription factors, disrupting the normal pattern of gene transcription and resulting in functional and degenerative changes (Cha, 2000; McCampbell et al., 2000; Steffan et al., 2000, 2001; Sugars and Rubinsztein, 2003). Changes in gene expression occur very early in polyglutamine transgene models of disease (Cha, 2000). Gene expression profiling has shown that mutant huntingtin protein selectively affects the pattern of gene expression (Luthi-Carter et al., 2000). These mRNA changes may affect neuronal functioning. A key strategy in the treatment of HD may therefore be to modulate transcription that leads to the altered signaling cascades underlying the HD pathology.
Transcription is regulated by complex interactions between many proteins, among them transcription factors and histones that ultimately affect the actions of DNA polymerase II on individual genes. Many of these interactions, in turn, are regulated by covalent modifications such as acetylation, methylation, and phosphorylation. Histone acetylation modulates a subset of genes and is determined by interplay between histone acetyltransferases and histone deacetylases (HDACs).
Mutant huntingtin can bind to histone acetyltransferase domains and reduce this activity, resulting in a reduction in histone acetylation and in repressed gene transcription (Steffan et al., 2000). Drugs that prevent the resulting histone deacetylation can help restore transcription in the presence of mutant huntingtin (McCampbell et al., 2001; Steffan et al., 2001). These compounds, known as HDAC inhibitors, affect histones as well as transcription factors that are regulated by acetylation. HDAC inhibitors promote transcriptional activation by relaxing the DNA conformation. They are selective in that only 2-5% of genes are affected (Van Lint et al., 1996). Because HDAC inhibitors induce growth arrest in cell proliferation models, HDAC inhibitors are currently under development as anti-cancer drugs (Butler et al., 2000; Richon et al., 2000; Vigushin and Coombes, 2002). The most widely studied compounds have been sodium butyrate, phenylbutyrate, trichostatin A, and suberoylanilide hydroxamic acid (SAHA). The butyrates, however, have been the best clinically studied compounds and are known to readily reach the brain (Egorin et al., 1999).
Both SAHA and sodium butyrate slow photoreceptor neuron degeneration and ameliorate lethality in a Drosophila model of HD (Steffan et al., 2001). Using cyclodextrin as a carrier, SAHA has also been reported to increase histone levels and improve motor performance in transgenic HD mice (Hockly et al., 2003).
Therefore, we investigated the effects of sodium butyrate on the clinical and neuropathological phenotype of the R6/2 transgenic mouse model of HD, on histone and Specificity protein-1 (Sp1) acetylation, and on gene expression in the R6/2 brain.
Materials and Methods
Animals. The Bedford Veterans Affairs Medical Center Laboratories have maintained a stable colony of R6/2 HD mice for >6 years, with founders originating from The Jackson Laboratory (Bar Harbor, ME). Male transgenic HD mice of the R6/2 strain were bred with females from their background strain (B6CBAFI/J). Offspring were genotyped using a PCR assay on tail DNA. We standardized criteria to ensure homogeneity of the cohorts within the testing groups (Dedeoglu et al., 2002). Mice were randomized from 28 litters all within 3 d from the same “f” generation. Body weights were taken at 20 d, and mice were equally distributed according to weight within each cohort. Mice that were <7 gm at 20 d were excluded from the experiments. The animals were housed five per cage under standard conditions with ad libitum access to water and food. The mice were handled under the same conditions by one investigator. Because we have not observed gender differences in survival in the R6/2 transgenic HD mouse model (Ferrante et al., 2000), female mice were used in the experimental paradigms. These experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by both the Veterans Affairs and Boston University Animal Care committees.
Intraperitoneal dosing. Based on previous studies (Egorin et al., 1999), a dose-response study was performed, treating groups of wild-type mice (n = 10) and littermate R6/2 mice (n = 20) with a 100, 200, 400, 600, 1200, 5000, and 10,000 mg/kg daily intraperitoneal injection (100 μl) of sodium butyrate (Acros Organics-Fisher Scientific, Houston, TX) dissolved in PBS and made fresh daily. Control groups were treated with PBS injection or untreated. Approximately 240 mice were used for behavioral and survival analyses.
Clinical assessment. Both motor performance and body weight were measured throughout the study. Training sessions were given on days 21 and 22 to acclimate the mice to the rotarod apparatus (Columbus Instruments, Columbus, OH). Motor performance (constant rotation at 16 rpm) was assessed weekly from 23 to 63 d of age and twice weekly from 63 d of age in the sodium butyrate-treated, PBS-treated R6/2 mice and in wild-type littermate control mice. Three 60 sec trials were given during each session and averaged. Body weights were recorded twice weekly at the same time of day in all groups.
Survival. R6/2 mice were assessed for morbidity and mortality twice daily, midmorning and late afternoon. Motor performance and ability to feed were closely monitored and used as the basis for determining when to euthanize the mice, the criterion being the point in time at which the HD mice were unable to right themselves after being placed on their back. In addition, deaths occurred overnight and were recorded the next morning. Two independent observers confirmed the criterion for killing (R.J.F. and J.K.K.).
Histone acetylation assay. At 42 d, groups of 10 R6/2 mice and littermate wild-type control mice were treated daily with the most optimal dose of sodium butyrate (1.2 gm/kg), as determined by survival studies, or PBS via intraperitoneal injections for 2 weeks. The mice were euthanized at 56 d of age, and the brains were rapidly frozen and stored at -80°C. Histone acetylation was determined using a Western blot analysis method reported previously (Warrell et al., 1998). Histones were isolated from sodium butyrate-treated and untreated whole brains. Western blot analysis for histone acetylation was performed using acetylated histone 3 (H3) and acetylated histone 4 (H4) antibodies (Upstate Biotechnology, Lake Placid, NY). Protein levels were determined via Coomassie protein assay (Pierce, Rockford, IL). Each of the sample measurements was conducted twice, with the investigator performing the assays (J.K.K.) blind to genotype and treatment group.
Sp1 acetylation immunoprecipitation and Western blot analysis. At 42 d of age, groups (n = 6) of R6/2 mice were treated daily with intraperitoneal injections of 1.2 gm/kg sodium butyrate or PBS for 2 weeks. The mice were euthanized, and tissue lysates were obtained by homogenizing each brain sample with 100 mm Tris buffer, pH 7.4, containing 1% Triton X-100, 150 mm NaCl, 1 mm sodium orthovanadate, 5 mm sodium fluoride, 3 mm PMSF, 3 mm DTT, 0.5 μg/ml leupeptin, and 10 μg/ml aprotinin. To monitor Sp1 acetylation in vivo, tissue lysates were precleared by the addition of 30 μl of protein A-Sepharose (50% v/v slurry) for 1 hr at 4°C and incubated with 2 μg of Sp1 antibody for 2-4 hr. Twenty-five microliters of protein A-Sepharose was added to lysates and left for 1 hr at 4°C. All beads were collected by centrifugation and washed twice with lysis buffer and once with PBS. The samples were boiled and divided into equal aliquots before separation on SDS-PAGE. Samples were electrophoresed under reducing conditions on 8% polyacrylamide gels. Proteins were then transferred to nitrocellulose membrane (Bio-Rad, Hercules, CA). Nonspecific binding was inhibited by incubation in Tris-buffered saline-Tween 20 (TBST) (50 mm Tris HCl, pH 8.0, 0.9% NaCl, 0.1% Tween 20) containing 5% nonfat dry milk for 0.5 hr. Primary antibodies against Sp1 (PEP2; Santa Cruz Biotechnology, Santa Cruz, CA) were diluted at 1:1000 in 1% milk TBST and exposed to membranes overnight at 4°C. Proteins immunoreactive to acetyl lysine were detected using acetyl lysine-specific antibodies (1:1000 dilution; Upstate Biotechnology). Immunoreactive proteins were detected according to an enhanced chemiluminescent protocol (Amersham Biosciences, Piscataway, NJ). The investigators performing assays (J.L., H.R.) were blind to the treatment groups.
3-Nitropropionic acid administration. At 6 weeks, groups of 10 R6/2 mice were treated with daily intraperitoneal injections of 1.2 gm/kg sodium butyrate or PBS for 2 weeks. At the start of week 2, 3-nitropropionic acid (3-NP) (Sigma, St. Louis, MO) was dissolved in PBS, pH adjusted to 7.4, made fresh daily, and injected intraperitoneally nine times at 12 hr intervals (75 mg/kg) in R6/2 mice. The mice were euthanized 8-10 hr after the last 3-NP injection, transcardially perfused with buffered 4% paraformaldehyde, and processed for histopathologic evaluation. Glycerol-cryoprotected brains were frozen-sectioned at 50 μm and stained with cresyl violet; quantitative analysis of lesion volumes was performed in serial sections as described below.
Neuropathological evaluation of sodium butyrate treatment. Beginning at 21 d, R6/2 transgenic mice and wild-type littermate control mice were treated intraperitoneally with the most optimal dose of sodium butyrate (1.2 gm/kg, daily), as determined by survival studies, or with PBS injections. Groups of 10 animals from each treatment paradigm were deeply anesthetized and transcardially perfused with 4% buffered paraformaldehyde at 90 d of age. Forty mice were used for neuropathological analysis, as described previously (Ferrante et al., 2002a). Serially cut tissue sections were stained for Nissl substance and immunostained for huntingtin (monoclonal huntingtin antibody, 1:1000 dilution; Chemicon, Temecula, CA) and for acetylated histone 3 and histone 4 (dilution, 1:1000; Upstate Biotechnology), using a previously reported conjugated secondary antibody method in murine brain tissue samples (Ferrante et al., 2002a). Specificity for the antisera used in this study was examined in each immunochemical experiment to assist with the interpretation of the results. Preabsorption with excess target proteins, omission of the primary antibodies, and omission of secondary antibodies were performed to determine the amount of background generated from the detection assay.
Stereology and quantitation. Serial-cut coronal tissue sections beginning from the most rostral segment of the neostriatum to the level of the anterior commissure (interaural 5.34 mm relative to bregma 1.54 mm to interaural 3.7 mm relative to bregma 0.10 mm) were used for huntingtin aggregate analysis. Unbiased stereological counts of huntingtin-positive aggregates (≥1.0 mm) were obtained from the neostriatum in 10 mice each from sodium butyrate-treated (1.2 gm/kg) and PBS-treated R6/2 mice at 90 d using Neurolucida Stereo Investigator software (Microbrightfield, Colchester, VT). The total areas of the rostral neostriatum were defined in serial sections in which counting frames were randomly sampled. The optical dissector method was used to estimate the number of huntingtin-positive aggregates. Striatal neuron areas were analyzed by microscopic videocapture using a Windows-based image analysis system for area measurement (Optimas Bioscan, Edmonds, WA). The software automatically identifies and measures profiles. All computer-identified cell profiles were manually verified as neurons and exported to Microsoft (Seattle, WA) Excel. Cross-sectional areas were analyzed using Statview (Brain Power, Calabasas, CA).
Transgene expression. Transgene expression was determined in whole brains by Western blot analysis of groups (n = 6) of sodium butyrate-treated (1.2 gm · kg-1 · d-1) and untreated R6/2 mice. Brain tissue was homogenized in a protease inhibitor mixture buffer (Complete; Roche, Mannheim, Germany). Twenty micrograms of protein (supernatant fraction) were resolved on 10% SDS-polyacrylamide gel (Bio-Rad), transferred to a polyvinylidene difluoride membrane (Bio-Rad), and probed with 1C2 antibody (Euromedex, Mundolsheim, France) specific for regions of expanded huntingtin, using a horseradish peroxidase-conjugated anti-rabbit secondary antibody, followed by chemiluminescent detection (Western Lightening; NEN); the protein was then run with parallel blots probed against α-tubulin to normalize for gel loading.
Microarray gene expression analysis. Beginning at 6 weeks of age, four mice were treated for 2 weeks with daily intraperitoneal injections (1.2 gm/kg) of sodium butyrate. Mice were euthanized within 60 min of the last injection, and cortical and striatal brain tissues were dissected separately and snap frozen on dry ice. In parallel, samples from four untreated R6/2 and four wild-type mice were collected in the same manner. One microarray sample was prepared from 10 μg of total RNA from each tissue and hybridized to one U74Av2 array (Affymetrix, Santa Clara, CA). Four independent pair-wise comparisons were performed using MAS 5.0 software (Affymetrix) to evaluate gene expression changes between sodium butyrate-treated R6/2 and untreated R6/2 animals and between untreated R6/2 and untreated wild-type animals. Difference calls were scored, and only mRNAs that received difference calls in the same direction (increased or decreased) in at least three of the four pair-wise comparisons were included for presentation in the present study. Probability statistics for changes in the expression of these same mRNAs showed corresponding values of p < 0.003 in all individual array comparisons called “increased” or “decreased.”
Real-time PCR. Reverse transcription (RT) of 1 μg of total RNA was conducted with a SuperScript First-Strand Synthesis System for RT-PCR (Invitrogen, San Diego, CA) using random hexamer primers according to the manufacturer's instructions. Quantitative real-time PCR studies used a Bio-Rad iCycler to follow the amplification of cDNA products by using SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA) through 50 PCR cycles (95°C for 30 sec, 57°C for 1 min, 72°C for 1.5 min). Specific amplification of the target sequences was determined by melt curve analysis and DNA sequencing. Primer pair sequences were as follows: preproenkephalin (XM 131313), GTGTCCAGGCCCGAGTTC and TCTCCCGTTCCCAGTAGCTC; mitogen-activated protein (MAP) kinase phosphatase-1 (MKP-1) (NM 013642), AAGCAGAGGCGGAGTATCAT and TAGTTCAGGGCACTGTTCGT; α-globin (NM 008218), CCACCCTGCCGATTTCA and CCGCAGAAGGCAGCTTAAC; β-globin (NM 008220), CTTGGACCCAGCGGTACTTT and CCCAGCACAATCACGATCA; β-actin (X03672), AGGTATCCTGACCCTGAAG and GCTCATTGTAGAAGGTGTGG. Expression of the mRNAs of interest was calculated using the equation
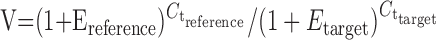 |
to correct for potential differences in RNA input and PCR primer efficiencies (Livak and Schmittgen, 2001). For this equation, V indicates the relative value of the target gene normalized to reference (β-actin), E indicates primer efficiency, and Ct indicates the threshold crossing cycle number. Differences between genotype and treatment groups were assessed using an unpaired, two-tailed Student's t test.
Northern blotting. Two micrograms of total RNA from brain samples of sodium butyrate-treated and untreated R6/2 and wild-type mice (n = 4 each) were separated on a 1.2% agarose-3% formaldehyde gel in 1× morpholinopropanesulfonate buffer, electrophoretically transferred to a nylon membrane (Genescreen II) in 1× Tris-acetate-EDTA, and dried in a standard utility oven at 65°C. cDNA probes were prepared from IMAGE clones (α-globin 555069, β-globin 318205, MKP-1 4235972; Invitrogen, Gaithersburg, MD) or cDNAs described in previous studies (enkephalin and β-actin probe sequences) (Luthi-Carter et al., 2002). Hybridizations and washes were conducted as described previously (Luthi-Carter et al., 2002). Blots were quantitated using a Molecular Dynamics (Sunnyvale, CA) PhosphorImager and its accompanying ImageQuant software. Signals for target mRNAs were expressed as ratios to β-actin signals (on the same blot), and the data were analyzed by unpaired two-tailed Student's t test.
Statistics. Data are expressed as means ± SEM. Statistical comparisons of rotarod data, weight data, and histology data were compared by ANOVA or repeated-measures ANOVA. Survival data were analyzed by Kaplan-Meier survival curves.
Results
The dose-response effects of intraperitoneal injection of sodium butyrate at 100, 200, 400, 600, and 1200 mg · kg-1 · d-1 on survival in HD R6/2 transgenic mice are shown in Figure 1A. Intraperitoneal administration of sodium butyrate at 1200 mg · kg-1 · d-1 significantly extended survival in R6/2 mice by 20.8% (PBS-treated, 101.3 ± 3.3 d; sodium butyrate-treated, 122.1 ± 8.5 d; F(6,120), 16.28; p ≤ 0.001). Significant prolongations of survival were also observed to a lesser extent at the 200, 400, and 600 mg · kg-1 · d-1 doses, with no effects observed with intraperitoneal sodium butyrate treatment at 100 mg · kg-1 · d-1 (100 mg · kg-1 · d-1 sodium butyrate-treated R6/2 mice, 106.2 ± 2.4 d; F(6,120), 1.81; p < 0.072; 200 mg · kg-1 · d-1 sodium butyrate-treated R6/2 mice, 111.0 ± 5.4 d; F(6,120), 6.59; p < 0.01; 400 mg · kg-1 · d-1 sodium butyrate-treated R6/2 mice, 110.8 ± 4.4 d; F(6,120), 6.23; p < 0.01; 600 mg · kg-1 · d-1 sodium butyrate-treated R6/2 mice, 114.5 ± 5.1 d; F(6,120), 9.74; p < 0.01). The mice became moribund and died between 1 and 11 d at 5 gm · kg-1 · d-1 sodium butyrate, with sudden death occurring within 0.5-2 hr at 10 gm · kg-1 · d-1 sodium butyrate intraperitoneal injection after treatment was initiated at 21 d.
Figure 1.

Survival, body weight, and motor performance analysis in sodium butyrate (SB)-treated R6/2 mice. Cohorts of R6/2 mice (n = 20) were treated from day 21 with an intraperitoneal injection of 100, 200, 400, 600, 1200, 5000, and 10,000 mg · kg-1 · d-1 sodium butyrate. Kaplan-Meier probability of survival analyses of sodium butyrate treatments in R6/2 mice and PBS-treated R6/2 mice are presented in A. Survival was significantly extended in all sodium butyrate dosing paradigms except the 100 mg · kg-1 · d-1 dose, with the greatest increase at the 1200 mg · kg-1 · d-1 dose. The data for the 5000 and 10,000 mg · kg-1 · d-1 doses resulted in marked rapidly coursing morbidity and mortality at on set of treatment and are not included. Effects of intraperitoneal sodium butyrate treatment on body weight in R6/2 HD transgenic mice are shown in B. A significant reduction in body weight loss was observed only after 12 weeks of age. Effects of intraperitoneal sodium butyrate treatment on rotarod performance (C) significantly improved motor performance in R6/2 HD transgenic mice throughout the temporal sequence of the experiment at each of the doses tested. *p < 0.01; **p < 0.001.
Intraperitoneal sodium butyrate treatment (400, 600, and 1200 mg · kg-1 · d-1) significantly improved rotarod performance throughout the entire measurement period (5-17 weeks) in contrast to PBS-treated R6/2 mice (PBS-treated R6/2 mice, 36.1 ± 7.5 sec; 400 mg · kg-1 · d-1 sodium butyrate-treated R6/2 mice, 47.5 ± 4.8 sec; 600 mg · kg-1 · d-1 sodium butyrate-treated R6/2 mice, 49.8 ± 4.3 sec; 1200 mg · kg-1 · d-1 sodium butyrate-treated R6/2 mice, 55.5 ± 2.9 sec; F(4,40), 12.83; 1200 mg · kg-1 · d-1 sodium butyrate-treated vs PBS-treated, p < 0.001). The data represent combined means from 5 to 14 weeks (Fig. 1B). The percentile increase in rotarod performance at 90 d for the 400, 600, and 1200 mg/kg doses was 24.0, 27.5, and 34.9%, respectively, compared with PBS-treated R6/2 mice.
Unlike other treatment regimens reported in R6/2 mice, in which reduced weight loss was observed early in the disease process (6-7 weeks) (Ferrante et al., 2000, 2002a; Dedeoglu et al., 2003), significant weight differences were only observed after 11 weeks of treatment and continued until death (Fig. 1C). The weight curves of sodium butyrate-treated and PBS-treated mice closely paralleled one another, maintaining a plateau at 19-20 gm until death ensued within each treatment group (Fig. 1C). Significant differences in body weight occurred as an epiphenomenon of survival extension and not as a primary independent event related to sodium butyrate treatment.
At 90 d, marked neuroprotection was observed. Similar to previous studies, there was an 18.9% reduction in brain weight in unsupplemented R6/2 mice compared with wild-type littermate control mice. In contrast, there was only a 5.6% brain weight loss in the R6/2 mice treated with the most efficacious dose of sodium butyrate, 1.2 gm · kg-1 · d-1 (wild-type littermate mice, 447 ± 12 mg; PBS-treated R6/2 mice, 363 ± 24; 1.2 gm/kg sodium butyrate-treated R6/2 mice, 422 ± 17; F(3,30), 12.54; sodium butyrate vs PBS, p < 0.001; sodium butyrate vs wild type, p < 0.53). Serial-cut coronal tissue sections revealed gross brain atrophy, bilateral ventricular hypertrophy, and flattening of the medial aspect of the striatum in the PBS-treated R6/2 brains (Fig. 2). Sodium butyrate treatment (1.2 gm · kg-1 · d-1) ameliorated these gross neuropathological sequelae in R6/2 mice compared with untreated mice at this time point. Whereas marked striatal neuron atrophy was present in untreated R6/2 mice at 90 d, the neuroprotective effects of sodium butyrate treatment (1.2 gm · kg-1 · d-1) significantly reduced striatal neuron atrophy in R6/2 mice by onefold (wild-type littermate control, 138.9 ± 12.6 μm2; sodium butyrate-treated R6/2 mice, 113.5 ± 14.2 μm2; PBS-treated R6/2 mice, 57.1 ± 23.7 μm2; F(3,30), 15.03; sodium butyrate vs PBS; p < 0.01) (Fig. 2).
Figure 2.
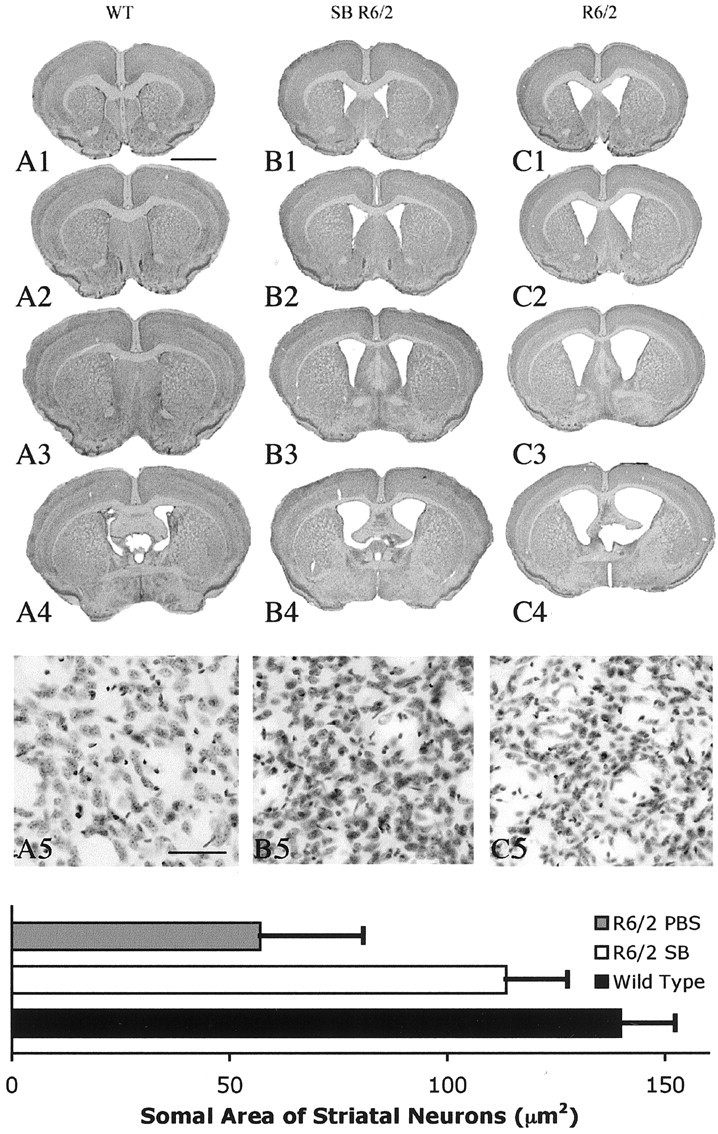
Gross brain and histopathological neuroprotection with sodium butyrate (SB) treatment. Photomicrographs of coronal serial step sections from the rostral neostriatum through the level of the anterior commissure in a wild-type littermate mouse (A1-A4), a sodium butyrate-treated (1.2 gm · kg-1 · d-1) R6/2 HD transgenic mouse (B1-B4), and a PBS-treated (C1-C4) R6/2 HD transgenic mouse at 90 d are shown. There was gross atrophy of the brain in the PBS-treated R6/2 mouse along with ventricular hypertrophy (C1-C4) compared with the wild-type littermate control mouse (A1-A4). In contrast, the sodium butyrate-treated R6/2 mouse brain (B1-B4) showed reduced gross brain atrophy and ventricular enlargement compared with the PBS-treated R6/2 mouse (C1-C4). Corresponding Nissl-stained tissue sections from the dorsomedial aspect of the neostriatum in a wild-type littermate control (A5), sodium butyrate-treated R6/2 mouse (B5), and PBS-treated R6/2 mouse (C5) are also shown. There was marked neuronal atrophy in the PBS-treated R6/2 mouse, with significantly less neuronal atrophy (p < 0.01) in the sodium butyrate-treated R6/2 mouse compared with this PBS-treated R6/2 mouse. The histogram shows means and SDs of somal areas of striatal neuronsquantitated in each group of mice (n = 10) (see Materials and Methods). Scale bars: A1-A4, B1-B4, C1-C4, 2 mm; A5, B5, C5, 100 μm.
There is an early and progressive accumulation of huntingtin-immunoreactive aggregates in R6/2 mice (Ferrante et al., 2000). In contrast to most other compounds that have been efficacious in the R6/2 transgenic mice, sodium butyrate treatment resulted in no significant reduction in huntingtin-positive striatal aggregates or ubiquitin-positive inclusions at 90 d of age compared with age-matched PBS-treated R6/2 mice (huntingtin aggregates in sodium butyrate-treated R6/2 mice, 5.02 × 106 ± 1.07; PBS-treated R6/2 mice, 5.31 × 106 ± 1.21; F(2,22), 1.23; p < 0.27) (Fig. 3). We reported a similar dissociation between efficacy and aggregate suppression using mithramycin, an aureolic acid antibiotic that binds to G-C-rich DNA sequences (Ferrante et al., 2002b). In common with sodium butyrate, mithramycin may also act to modulate transcription by displacing transcriptional activators that bind to G-C-rich regions of promoters (D. M. Miller et al., 1987; Chatterjee et al., 2001).
Figure 3.

Huntingtin and ubiquitin immunoreactivity in sodium butyrate-treated R6/2 mice. Huntingtin immunostained tissue sections from the neostriatum of a PBS-treated R6/2 transgenic mouse (A) and sodium butyrate-treated R6/2 HD transgenic mouse (B) euthanized at 90 d of age are shown. Sodium butyrate (1.2 gm · kg-1 · d-1) and PBS treatments were started at 21 d. There were no significant differences (p < 0.27) in the number and size of huntingtin aggregates and inclusions between treated and untreated mice. Similarly, no differences were observed in ubiquitin-positive inclusions between PBS-treated and sodium butyrate-treated R6/2 mice (C and D, respectively). Scale bar: (in B) A-D, 100 μm.
At 8 weeks of age, Western blot analysis showed hypoacetylation of H3 and H4 in R6/2 mice compared with wild-type mice, with a marked increase in H3 and H4 acetylation in sodium butyrate-treated R6/2 mice (1.2 gm · kg-1 · d-1) (Fig. 4). We did not observe any difference in increased acetylation of H3 and H4 between the sodium butyrate doses at 600 mg · kg-1 · d-1 and 1.2 gm · kg-1 · d-1. Immunocytochemical detection of acetylated H3 and H4 confirmed these findings. There was robust H3 and H4 acetylation immunoreactivity in brain sections of wild-type mice, reduced tissue H3 and H4 immunoreactivity in R6/2 mice, and greatly enhanced H3 and H4 immunostaining in sodium butyrate-treated R6/2 mice (Fig. 5).
Figure 4.

Western blot of acetylated H3 and H4 in sodium butyrate (sb)-treated R6/2 mice. R6/2 mice were treated with 1.2 gm · kg-1 · d-1 sodium butyrate for 2 weeks starting at 42 d and euthanized at 56 d. Hypoacetylation of H3 and H4 immunoreactivities were present in R6/2 mice compared with wild-type littermate control mice (WT). Sodium butyrate-treated R6/2 mice showed a marked increase in acetylated H3 and H4 activity. Protein levels were determined using Coomassie protein assay.
Figure 5.
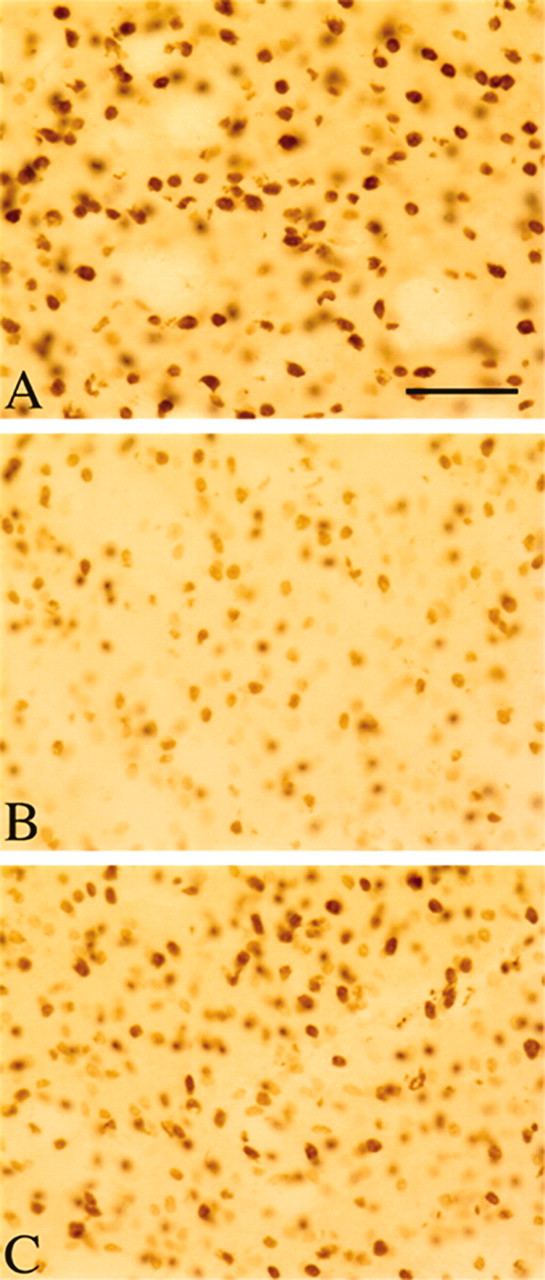
Striatal tissue immunohistochemistry of acetylated histone 4 in sodium butyrate-treated R6/2 mice. At 90 d of age, robust acetylated histone 4 immunohistochemistry was present in wild-type littermate control striatal tissue specimens (A), with hypoacetylation in the R6/2 mice (B). C, Sodium butyrate treatment (1.2 gm · kg-1 · d-1) increased acetylation of histone 4 in R6/2 mice. Scale bar: (in A) A-C, 100 μm.
We reported previously that Sp1 acetylation is significantly augmented in sodium butyrate-treated wild-type B6CBA mice at 8 weeks of age after 2 weeks of treatment (Ryu et al., 2003a). Similarly, Sp1 acetylation levels were increased in brains of sodium butyrate-treated R6/2 mice (1.2 gm · kg-1 · d-1), as determined by immunoprecipitation using an Sp1 antibody followed by immunoblotting using acetyl lysine antibody (Ac-Sp1) or Sp1 antibody alone. The basal levels of Sp1 did not change with sodium butyrate treatment (Fig. 6).
Figure 6.

The histone deacetylase inhibitor sodium butyrate (SB) enhances Sp1 acetylation in vivo. Cohorts of R6/2 mice (n = 6) were treated daily with sodium butyrate (1.2 gm/kg) or PBS intraperitoneal injections for 2 weeks. Sp1 acetylation levels from homogenized brains of PBS- and sodium butyrate-treated R6/2 mice were determined by immunoprecipitation (IP) using an Sp1 antibody followed by immunoblotting using acetyl lysine (Ac-lysine) antibody (Ac-Sp1) or Sp1 antibody alone (Sp1). Sp1 acetylation was increased in sodium butyrate-treated R6/2 mice. Note that levels of Sp1 did not change with sodium butyrate treatment.
Sodium butyrate treatment for 2 weeks (1.2 gm · kg-1 · d-1), with administration of 3-NP during continuous sodium butyrate treatment started the second week, resulted in marked neuroprotection from 3-NP striatal damage in R6/2 mice compared with 3-NP- and PBS-treated R6/2 mice (Fig. 7). Only 2 of 10 3-NP-sodium butyrate-treated R6/2 mice had small bilateral lesions, whereas 7 of 10 3-NP-PBS-treated R6/2 mice had small to large bilateral striatal lesions. Histopathological evaluation of 3-NP-induced striatal lesion volumes, as determined by Nissl staining, showed significantly less tissue damage in sodium butyrate-treated mice than in PBS-treated R6/2 mice (sodium butyrate-treated 3-NP R6/2 mice, 2.94 ± 0.38 mm3; PBS-treated 3-NP R6/2 mice, 11.03 ± 2.57 mm3; p < 0.001). There was a 73.3% reduction in lesion volume in the sodium butyrate-treated R6/2 mice.
Figure 7.

Sodium butyrate neuroprotection from 3-NP acid toxicity in R6/2 mice. Groups of R6/2 mice (n = 10) were treated for 2 weeks with 1.2 gm · kg-1 · d-1 sodium butyrate or PBS starting at 42 d. 3-NP was administered at the beginning of the second week for 4.5 d along with PBS and sodium butyrate treatments. Sodium butyrate treatment prevented 3-NP striatal-induced damage in R6/2 mice (A) compared with PBS-treated R6/2 mice (B). Histopathological evaluation of 3-NP-induced striatal lesions showed bilateral striatal lesions (areas of pallor) in PBS-treated R6/2 mice (B). Scale bar: (in A) A, B, 2 mm.
Huntingtin expression is regulated by exogenous promoters in the R6/2 transgenic mice. Treatments modulating transcription could work by suppressing the expression of the huntingtin transgene. Western blot analysis showed no differences in transprotein expression levels between sodium butyrate-treated R6/2 mice and PBS-treated R6/2 mice (Fig. 8).
Figure 8.

Huntingtin (Htt) expression in sodium butyrate (SB)-treated R6/2 mice. Transgene expression was determined in whole-brain samples by Western blot analysis of sodium butyrate-treated (1.2 gm · kg-1 · d-1) R6/2 mice, PBS-treated R6/2 mice, and wild-type mice. No differences in huntingtin expression levels in sodium butyrate-treated R6/2 mice were observed compared with PBS-treated mice. Parallel blots probed against α-tubulin were run to normalize for gel loading.
Microarray gene expression profiling was performed in striatal and cortical tissues from sodium butyrate-treated and untreated R6/2 and wild-type mice. Consistent with previous data on R6/2 mice, 69 probe sets reported changes in R6/2 striatum compared with wild-type striatum. These included decreases in preproenkephalin (two probe sets), ryanodine receptor, insulin-like growth factor-binding protein 5 (IGFBP5), neuronatin, and T-box brain gene 1, as well as increases in apolipoprotein D and β2 microglobulin, as observed previously. Eighteen changes in gene expression in R6/2 cerebral cortex attributable to genotype alone included decreases in complexin II, troponin C, myosin light chain, and IGFBP5. There was a selective change in gene expression in response to sodium butyrate treatment. We observed 20 mRNA changes in sodium butyrate-treated R6/2 mice (Table 1). We focused on the most robust changes, in which there were 100% possible change calls in both striatum and cortex. Among the mRNAs in R6/2 mice with these inclusion criteria, three mRNAs were detected in all eight of the array comparisons. These specific mRNAs were α- and β-globin and MKP-1 (Table 2). The increased expression of these mRNAs was confirmed using real-time PCR and Northern blotting (Fig. 9). Although sodium butyrate treatment altered gene expression in R6/2 mice, some gene expression changes reported previously in R6/2 mice were not corrected. The expression of preproenkephalin mRNA, normally decreased in R6/2 mice, was unaffected by treatment. These findings represent a selective change in gene expression in response to sodium butyrate treatment and do not alter global mRNA expression. Moreover, there is not a uniform upregulation of mRNAs that are normally decreased by the disease process.
Table 1.
Changes in gene expression in sodium butyrate-treated R6/2 mice
|
Brain region |
GenBank accession number |
Probe ID |
mRNA |
Direction of change |
Percentage of change calls |
|---|---|---|---|---|---|
| Striatum plus cortex | X61940 | 104598_at | MKP-1 | Increase | 100 |
| Striatum plus cortex | V00714 | 94781_at | Hemoglobin, α adult chain 1 | Increase | 100 |
| Striatum plus cortex | J00413 | 101869_s_at | Hemoglobin, β adult major chain | Increase | 100 |
| Cortex | V00722 | 103534_at | Hemoglobin, β 1 | Increase | 75 |
| Cortex | V00727 | 160901_at | c-fos oncogene | Increase | 100 |
| Cortex | AI854404 | 160131_at | Angiomotin like 2 | Increase | 100 |
| Cortex | AI845584 | 93285_at | MAPK phosphatase-3 (MKP-3) | Increase | 75 |
| Cortex | M88354 | 93381_at | Arginine vasopressin | Increase | 75 |
| Striatum | D45859 | 101836_at | Protein phosphatase 1B, β isoform | Increase | 75 |
| Striatum | AB028272 | 96254_at | DNAJ-heat shock protein 40 | Increase | 75 |
| Striatum plus cortex | C79248 | 94689_at | Unknown | Decrease | 75 |
| Cortex | L31397 | 103031_g_at | Dynamin | Decrease | 75 |
| Cortex | M18775 | 102431_at | Microtubule-associated protein tau | Decrease | 75 |
| Cortex | AV330064 | 161114_i_at | Unknown | Decrease | 75 |
| Striatum | AJ007909 | 98525_f_at | Erythroid differentiation regulator | Decrease | 100 |
| Striatum | AI451558 | 97161_at | Unknown | Decrease | 75 |
| Striatum | AI153421 | 96215_f_at | Unknown | Decrease | 75 |
| Striatum | D00073 | 95350_at | Transthyretin | Decrease | 75 |
| Striatum | AA711516 | 96302_at | Unknown | Decrease | 75 |
| Striatum
|
AI183202
|
92724_at
|
Heterogeneous nuclear ribonucleoprotein A1
|
Decrease
|
75
|
Samples from sodium butyrate-treated R6/2, untreated R6/2, and wild-type mice (n = 4 from each group) were analyzed for differential mRNA expression by microarray. Array data are expressed as increased or decreased based on a minimum difference call cutoff of three of four independent comparisons using samples from individual mice (see Materials and Methods). Most important in the gene array findings was that three known genes (α-and β-globin and MKP-1) were highly increased in expression. These increases in mRNAs were specific to both the striatum and neocortex and were detected in all eight array comparisons.
Table 2.
Changes in α - and β -globin and MPK-1 gene expression in sodium butyrate-treated R6/2 mice
|
|
|
NaBu Txd R6/2 versus R6/2 |
Untreated R6/2 versus WT |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| mRNA
|
Region
|
Array
|
PCR
|
Northern blot
|
Array
|
PCR
|
Northern blot
|
||||
| MKP-1 | Striatum | I | 181%a | NC | 75% | ||||||
| Cortex | I | 204%e | NC | 81% | |||||||
| α-Globin | Striatum | I | 206%c | NC | 120% | ||||||
| Cortex | I | 159%c | NC | 99% | |||||||
| β-Globin | Striatum | I | 215%b | NC | 104% | ||||||
| Cortex | I | 206%d | D | 39%a | |||||||
| Preproenkephalin
|
Striatum
|
NC
|
101%
|
98%
|
D
|
52%b
|
62%a
|
||||
Samples from sodium butyrate (NaBu)-treated R6/2, untreated R6/2, and wild-type (WT) mice (n = 4 from each group) were analyzed for differential mRNA expression by microarray, real-time PCR, and Northern blotting. Array data are expressed as follows: I, Increased; D, decreased; NC, no change (based on a minimum difference call cutoff of three of four independent comparisons using samples from individual mice; see Materials and Methods). PCR and Northern blot data are presented as mean values expressed as a percentage of the baseline group (either untreated R6/2 or wild-type mice) after normalization to β-actin signal (see Materials and Methods). aSignificantly different from comparison group (p < 0.05); bsignificantly different from comparison group (p < 0.02); csignificantly different from comparison group (p < 0.01); dsignificantly different from comparison group (p < 0.001); esignificantly different from comparison group (p < 0.0001).
Figure 9.

MKP-1 is increased by sodium butyrate treatment. Northern blot analysis of 2 μg of total RNA from brain samples of sodium butyrate-treated (1.2 gm · kg-1 · d-1) and untreated R6/2 mice (n = 4 in each group) confirmed that sodium butyrate treatment increases the expression of MKP-1 mRNA compared with both untreated R6/2 mice and wild-type (WT) littermate control mice. A β-actin hybridization signal on the same blot (shown) was used as a loading control. Quantitative and statistical data are presented in Table 2.
Discussion
Despite great progress, a causal pathway from the HD gene mutation to neuronal dysfunction and death has not yet been established. Early molecular events likely trigger cascades of damage and compensatory responses, leading to dysfunctional neurons that are susceptible to other insults, such as oxidative injury, excitotoxic stress, inflammatory and proapoptotic signals, and energy depletion (Beal, 2000; Friedlander, 2003). Neuroprotective therapies targeted at specific molecular mechanisms have the potential to dramatically delay the onset and slow the progression of disease in transgenic mouse models of HD (Chen et al., 2000; Ferrante et al., 2000, 2002a, b; Andreassen et al., 2001a, b; Dedeoglu et al., 2002, 2003; Karpuj et al., 2002; Hockly et al., 2003). Several pilot clinical trials in HD patients have been initiated recently from the findings observed in mouse trials. Experimental evidence increasingly points to a proximal toxicity residing in mutant huntingtin or its cleaved products and their pathological interactions with other proteins, including transcription factors (Cha, 2000; Sugars and Rubinsztein, 2003). Thus, therapies aimed at transcriptional modulation might target early events in HD pathogenesis and ameliorate secondary pathologic cascades.
In the present experiments, we show that intraperitoneal administration of the HDAC inhibitor sodium butyrate significantly extends survival in the R6/2 model of HD. Survival is a powerful and relevant endpoint for neuroprotection that provides a context for other aspects of the phenotype, such as behavior and neuropathology, and enables a ready comparison of the relative potency of different treatments. Sodium butyrate treatment also improved motor performance, gross brain weight and atrophy, and striatal neuron atrophy. Although sodium butyrate treatment was neuroprotective, it did not prevent body weight loss, a finding similar to other transcriptionally active compounds (Ferrante et al., 2002b; Hockly et al., 2003). Sodium butyrate also failed to attenuate huntingtin aggregation. We observed this phenomenon previously using the transcription modulator mithramycin (Ferrante et al., 2002b). This suggests that neuroprotective therapies need not affect huntingtin aggregation and supports the view that insoluble aggregates may not be inherently toxic. The specificity of neuronal vulnerability in HD may be more dependent on soluble monomers or hetero-oligomers of huntingtin found in affected neurons (Sisodia, 1998; Kuemmerle et al., 1999) and in soluble protein-protein interactions, including interactions with transcription factors. In addition, brain tissue levels of acetylated H3 and H4 activity in Western blot analysis and by immunocytochemistry were increased in sodium butyrate-treated R6/2 mice, consistent with a direct neuroprotective effect related to acetylation in the brain. These findings demonstrate that sodium butyrate has significant efficacy in improving the neurological and neuropathological phenotype observed in the R6/2 transgenic model of HD, and they suggest that HDAC inhibitors may provide clinical benefit to HD patients, most likely by preventing the deleterious effects of mutant huntingtin on transcription.
Mutant huntingtin and other polyglutamine-containing proteins can directly interact with a number of transcription factors, resulting in altered coactivation, repression, or derepression (Cha, 2000). These include p53 and cAMP response element-binding protein (Steffan et al., 2000), TATA-binding protein (Nakamura et al., 2001), mSin3a (Steffan et al., 2000), and Sp1 (Dunah et al., 2002; Li et al., 2002). Although soluble interactions may be most toxic, many transcription factors are also directly sequestered into nuclear inclusions formed by polyglutamine-containing mutant huntingtin (Preisinger et al., 1999; McCampbell et al., 2000; Holbert et al., 2001, 2003; Nucifora et al., 2001; Steffan et al., 2001). HDAC inhibitors selectively increase transcription because of the specificity of acetylation targets (Struhl, 1998; Richon et al., 2000). Thus, they may only be able to reverse effects exerted through a portion of the transcriptional machinery. Sp1 is a transcription factor binding to G-C-rich DNA sites (D. M. Miller et al., 1987). Although Sp1 has been suggested to regulate the expression of housekeeping genes, it has also been reported to be involved in differentiation, proliferation, and other cellular functions (Krainc et al., 1998; Black et al., 1999; Ryu et al., 2003a, b). Polyglutamine expansion may result in neuronal death, in part, by directly affecting Sp1 (Dunah et al., 2002; Li et al., 2002). We reported previously that HDAC inhibitors increase Sp1 acetylation and provide neuroprotection in a model of oxidative stress (Ryu et al., 2003b). The augmentation of Sp1 acetylation in R6/2 mice treated with sodium butyrate may partially explain the observed neuroprotection.
In the present study, microarray analysis showed increased expression of MKP-1 and α- and β-globins in sodium butyrate-treated R6/2 mice. The MAP kinase (MAPK) system is an important intracellular signaling pathway that regulates transcription and other cellular effectors, such as phospholipase C, protein kinase C, cytoplasmic phospholipase-A2, and MKP-1 (Nishibe et al., 1990; Wood et al., 1992; Nemenoff et al., 1993). This regulatory network functions through phosphorylation and dephosphorylation reactions. MKP-1 is a tyrosine phosphatase that dephosphorylates MAPKs, thereby inactivating them. MAPKs are involved in maintaining multiple neuronal functions that are associated with HD pathogenesis, including synaptic vesicle biogenesis, glutamate release, neurite outgrowth and maintenance, and apoptosis (Brondello et al., 1999). It has been suggested that increased MKP-1 concentrations alter the steady-state activity of MAPK, improving cellular function without triggering entrance into the cell cycle (Bhalla and Iyengar, 1999; Bhalla et al., 2002). Histone acetylation induces MKP-1 transcription, elevating MKP-1 mRNA (Li et al., 2001). Because MKP-1 may regulate the transcriptional machinery, increased MKP-1 may contribute to the observed neuroprotection in sodium butyrate-treated R6/2 mice. We are currently examining whether overexpression of MKP-1 and ERK1 (extracellular signal-regulated kinase 1) inhibition ameliorates huntingtin pathology. Increased expression of MKP-1 has been reported in an in vitro model of polyglutamine-induced cell death in PC12 cells (Wu et al., 2002). Although the authors suggest that the induction of MKP-1 is associated with mutant huntingtin and cell death, increased MKP-1 expression may be a failed attempt at cellular recovery in their model rather than a pathological event.
α- and β-globins also showed increased expression in sodium butyrate-treated R6/2 mice. Enhanced globin gene expression has been reported in sodium butyrate-treated patients with thalassemia and for hemoglobin sickle cell anemia (A. A. Miller et al., 1987; Perrine et al., 1989). There is evidence that globins are expressed in neurons and that this expression is developmentally regulated (Ohyagi et al., 1994). In addition to myoglobin and hemoglobin, a third hemoprotein, neuroglobin, has been isolated recently from vertebrate brain (Mammen et al., 2002). Overexpression of this protein protects against ischemia in an in vivo model of stroke (Sun et al., 2003). Globins contain a heme-binding domain and participate in diverse processes such as oxygen transport, oxygen storage, and nitric oxide detoxification. A secondary consequence of the HD gene defect may be impaired energy metabolism that may lead to increased free radical generation and oxidative damage (Browne et al., 1999; Beal, 2000; Tabrizi et al., 2000; Bogdanov et al., 2001). Increased globin expression may provide neuroprotective properties that enhance oxidative phosphorylation in R6/2 mice, counterbalancing HD-related metabolic deficits.
3-NP is a mitochondrial toxin that results in irreversible inhibition of succinate dehydrogenase, inhibiting both the Krebs' cycle and Complex II activity of the electron transport chain. Complex II is an entrance pathway into the electron transport chain, where it reduces ubiquinone (coenzyme Q), an important electron carrier to Complex III. 3-NP produces excitotoxic-mediated striatal lesions in both humans and experimental animals, closely resembling the pathology observed in HD (Alston et al., 1977; Ludolph et al., 1991; Beal et al., 1993; Brouillet et al., 1995). 3-NP reduces cellular levels of ATP and produces profound energy deficits within cells, irreversibly inhibiting the Krebs' cycle and Complex II activity, resulting in an energy-deficient hypoxia (Hamilton and Gould, 1987; Novelli et al., 1988; Ludolph et al., 1992). Sodium butyrate enhances Sp1 acetylation and inhibits 3-NP-induced excitotoxicity in vivo (Ryu et al., 2003a, b). In the current experiments, increased acetylation of Sp1 induced by sodium butyrate treatment was associated with nearly complete protection from striatal 3-NP toxicity. Resistance to 3-NP toxicity in sodium butyrate-treated R6/2 mice may be attributable, in part, to improved oxidative metabolism via increased globin expression.
We show that sodium butyrate is neuroprotective in R6/2 transgenic mice. There are a number of possible explanations for this neuroprotection, and it is likely that more than one contributes. Sodium butyrate acts at the transcriptional level by increasing the acetylation of histones, thereby releasing constraints on the DNA template and reactivating a number of genes (Candido et al., 1978; Sealy and Chalkey, 1978). Using the percentage of increase in survival as a standard, sodium butyrate is among the most efficacious compounds yet tested. Because the butyrates have known oral and CNS bioavailability and because toxicity has been low and tolerability acceptable in both human and animal studies (Collins et al., 1995; Egorin et al., 1999), sodium butyrate and related compounds are promising neuroprotective agents for HD that warrant additional preclinical testing and possible consideration for clinical trials in the future. An important implication of the multiple levels of molecular pathology existing in HD is that it will most likely be possible to combine neuroprotective therapies to maximize efficacy. HDAC inhibitors, including the butyrates, may act more proximally and are candidates for combination with other neuroprotective compounds currently being developed for HD.
Footnotes
This work was supported by National Institutes of Health Grants NS35255 and AT00613 (S.M.H. and R.J.F.), AG13846 and AG12992 (R.J.F.), NS045242 (R.J.F., R.R.R., R.L.-C., and S.M.H.), and the Veterans Administration (H.R. and R.J.F.), the Huntington's Disease Society of America (R.J.F. and S.M.H.), the Hereditary Disease Foundation (S.M.H. and R.J.F.), and the Jerry McDonald Research Fund in Huntington's Disease (H.R. and R.J.F.). We thank Dr. M. Flint Beal for his constructive suggestions and careful review of this manuscript. Technical support was provided by Kerry Cormier and John Kuster.
Correspondence should be addressed to Dr. Robert J. Ferrante, Geriatric Research Education and Clinical Center Unit 182B, Bedford Veterans Affairs Medical Center, 200 Springs Road, Bedford, MA 01730. E-mail: rjferr@bu.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/239418-10$15.00/0
References
- Alston TA, Mela L, Bright HJ ( 1977) 3-Nitropropionate, the toxic substance of Indigofera, is a suicide inactivator of succinate dehydrogenase. Proc Natl Acad Sci USA 74: 3767-3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreassen OA, Ferrante RJ, Huang HM, Dedeoglu A, Park L, Ferrante KL, Kwon J, Borchelt DR, Ross CA, Gibson GE, Beal MF ( 2001a) Dichloroacetate exerts therapeutic effects in transgenic mouse models of Huntington's disease. Ann Neurol 50: 112-117. [DOI] [PubMed] [Google Scholar]
- Andreassen OA, Ferrante RJ, Dedeoglu A, Beal MF ( 2001b) Lipoic acid improves survival in transgenic mouse models of Huntington's disease. NeuroReport 12: 3371-3374. [DOI] [PubMed] [Google Scholar]
- Beal MF ( 2000) Energetics in the pathogenesis of neurodegenerative diseases. Trends Neurosci 7: 298-304. [DOI] [PubMed] [Google Scholar]
- Beal MF, Brouillet E, Jenkins BG, Ferrante RJ, Kowall NW, Miller JM, Storey E, Srivastava R, Rosen BR, Hyman BT ( 1993) Neurochemical and histo-logic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J Neurosci 13: 4181-4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhalla US, Iyengar R ( 1999) Emergent properties of networks of biological signaling pathways. Science 283: 381-387. [DOI] [PubMed] [Google Scholar]
- Bhalla US, Ram PT, Iyengar R ( 2002) MAP kinase phosphatase as a locus of flexibility in a mitogen-activated protein kinase signaling network. Science 297: 1018-1023. [DOI] [PubMed] [Google Scholar]
- Black AR, Jensen D, Lin SY, Azizkhan JC ( 1999) Growth/cell cycle regulation of Sp1 phosphorylation. J Biol Chem 274: 1207-1215. [DOI] [PubMed] [Google Scholar]
- Bogdanov MB, Andreassen OA, Dedeoglu A, Ferrante RJ, Beal MF ( 2001) Increased oxidative damage to DNA in a transgenic mouse model of Huntington's disease. J Neurochem 79: 1246-1249. [DOI] [PubMed] [Google Scholar]
- Brondello JM, Pouyssegur J, McKenzie FR ( 1999) Reduced MAP kinase phosphatase-1 degradation after p42/p44MAPK-dependent phosphorylation. Science 286: 2514-2517. [DOI] [PubMed] [Google Scholar]
- Brouillet E, Hantraye P, Ferrante RJ, Dolan R, Kowall NW, Beal MF ( 1995) Chronic mitochondrial energy impairment produces selective striatal degeneration and abnormal choreiform movements in primates. Proc Natl Acad Sci USA 92: 7105-7109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne SE, Ferrante RJ, Beal MF ( 1999) Oxidative stress in Huntington's disease. Brain Pathol 9: 147-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler LM, Agus DB, Scher HI, Higgins B, Rose A, Cordon-Cardo C, Thaler HT, Rifkind RA, Marks PA, Richon VM ( 2000) Suberoylanilide hydroxamic acid, an inhibitor of histone deacetylase, suppresses the growth of prostate cancer cells in vitro and in vivo Cancer Res 60: 5165-5170. [PubMed] [Google Scholar]
- Candido EP, Reeves R, Davie JR ( 1978) Sodium butyrate inhibits histone deacetylation in cultured cells. Cell 14: 105-113. [DOI] [PubMed] [Google Scholar]
- Cha JH ( 2000) Transcriptional dysregulation in Huntington's disease. Trends Neurosci 23: 387-392. [DOI] [PubMed] [Google Scholar]
- Chatterjee S, Zaman K, Ryu H, Conforto A, Ratan RR ( 2001) Sequence-selective DNA binding drugs mithramycin A and chromomycin A3 are potent inhibitors of neuronal apoptosis induced by oxidative stress and DNA damage in cortical neurons. Ann Neurol 49: 345-354. [PubMed] [Google Scholar]
- Chen M, Ona VO, Li M, Ferrante RJ, Fink KB, Zhu S, Bian J, Guo L, Farrell LA, Hersch SM, Hobbs W, Vonsattel JP, Cha JH, Friedlander RM ( 2000) Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat Med 6: 797-801. [DOI] [PubMed] [Google Scholar]
- Collins AF, Pearson HA, Giardina P, McDonagh KT, Brusilow SW, Dover GJ ( 1995) Oral sodium phenylbutyrate therapy in homozygous beta thalassemia: a clinical trial. Blood 85: 43-49. [PubMed] [Google Scholar]
- Dedeoglu A, Kubilus JK, Jeitner TM, Matson SA, Bogdanov M, Kowall NW, Matson WR, Cooper AJ, Ratan RR, Beal MF, Hersch SM, Ferrante RJ ( 2002) Therapeutic effects of cystamine in a murine model of Huntington's disease. J Neurosci 22: 8942-8950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedeoglu A, Kubilus JK, Yang L, Ferrante KL, Hersch SM, Beal MF, Ferrante RJ ( 2003) Creatine therapy provides neuroprotection after onset of clinical symptoms in Huntington's disease transgenic mice. J Neurochem 85: 1359-1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunah AW, Jeong H, Griffin A, Kim YM, Standaert DG, Hersch SM, Mouradian MM, Young AB, Tanese N, Krainc D ( 2002) Sp1 and TAFII130 transcriptional activity disrupted in early Huntington's disease. Science 296: 2238-2243. [DOI] [PubMed] [Google Scholar]
- Egorin MJ, Yuan ZM, Sentz DL, Plaisance K, Eiseman JL ( 1999) Plasma pharmacokinetics of butyrate after intravenous administration of sodium butyrate or oral administration of tributyrin or sodium butyrate to mice and rats. Cancer Chemother Pharmacol 43: 445-453. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Andreassen OA, Jenkins BG, Dedeoglu A, Kuemmerle S, Kubilus JK, Kaddurah-Daouk R, Hersch SM, Beal MF ( 2000) Neuroprotective effects of creatine in a transgenic mouse model of Huntington's disease. J Neurosci 20: 4389-4397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante RJ, Andreassen OA, Dedeoglu A, Ferrante KL, Jenkins BG, Hersch SM, Beal MF ( 2002a) Therapeutic effects of coenzyme Q10 and remacemide in transgenic mouse models of Huntington's disease. J Neurosci 22: 1592-1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante RJ, Dedeoglu A, Kubilus JK, Sugars KL, Rubinsztein DC, Ryu H, Lee JH, Beal MF, Ratan RR ( 2002b) Therapeutic effects of mithramycin in R6/2 transgenic Huntington's disease mice. Soc Neurosci Abstr 28: 725. [Google Scholar]
- Friedlander RM ( 2003) Apoptosis and caspases in neurodegenerative diseases. N Engl J Med 348: 1365-1375. [DOI] [PubMed] [Google Scholar]
- Hamilton BF, Gould DH ( 1987) Nature and distribution of brain lesions in rats intoxicated with 3-nitropropionic acid: a type of hypoxic (energy deficient) brain damage. Acta Neuropathol 72: 286-297. [DOI] [PubMed] [Google Scholar]
- Hockly E, Richon VM, Woodman B, Smith DL, Zhou X, Rosa E, Sathasivam K, Ghazi-Noori S, Mahal A, Lowden PA, Steffan JS, Marsh JL, Thompson LM, Lewis CM, Marks PA, Bates GP ( 2003) Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington's disease. Proc Natl Acad Sci USA 100: 2041-2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holbert S, Denghien I, Kiechle T, Rosenblatt A, Wellington C, Hayden MR, Margolis RL, Ross CA, Dausset J, Ferrante RJ, Neri C ( 2001) The Gln-Ala repeat transcriptional activator CA150 interacts with huntingtin: neuropathologic and genetic evidence for a role in Huntington's disease pathogenesis. Proc Natl Acad Sci USA 98: 1811-1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holbert S, Dedeoglu A, Humbert S, Saudou F, Ferrante RJ, Neri C ( 2003) Cdc42-interacting protein 4 binds to huntingtin: neuropathologic and biological evidence for a role in Huntington's disease. Proc Natl Acad Sci USA 100: 2712-2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpuj MV, Becher MW, Springer JE, Chabas D, Youssef S, Pedotti R, Mitchell D, Steinman L ( 2002) Prolonged survival and decreased abnormal movements in transgenic model of Huntington disease, with administration of the transglutaminase inhibitor cystamine. Nat Med 8: 143-149. [DOI] [PubMed] [Google Scholar]
- Krainc D, Bai G, Okamoto S, Carles M, Kusiak JW, Brent RN, Lipton SA ( 1998) Synergistic activation of the N-methyl-d-aspartate receptor subunit 1 promoter by myocyte enhancer factor 2C and Sp1. J Biol Chem 273: 26218-26224. [DOI] [PubMed] [Google Scholar]
- Kuemmerle S, Gutekunst CA, Klein AM, Li XJ, Li SH, Beal MF, Hersch SM, Ferrante RJ ( 1999) Huntington aggregates may not predict neuronal death in Huntington's disease. Ann Neurol 46: 842-849. [PubMed] [Google Scholar]
- Li J, Gorospe M, Hutter D, Barnes J, Keyse SM, Liu Y ( 2001) Transcriptional induction of MKP-1 in response to stress is associated with histone H3 phosphorylation-acetylation. Mol Cell Biol 23: 8213-8224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SH, Cheng AL, Zhou H, Lam S, Rao M, Li H, Li XJ ( 2002) Interaction of Huntington disease protein with transcriptional activator Sp1. Mol Cell Biol 22: 1277-1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD ( 2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25: 402-408. [DOI] [PubMed] [Google Scholar]
- Ludolph AC, He F, Spencer PS ( 1991) 3-Nitropropionic acid-exogenous animal neurotoxin and possible human striatal toxin. Can J Neurol Sci 18: 492-498. [DOI] [PubMed] [Google Scholar]
- Ludolph AC, Seelig M, Ludolph A ( 1992) 3-Nitropropionic acid decreases cellular energy levels and causes neuronal degeneration in cortical ex-plants. Neurodegeneration 1: 155-161. [Google Scholar]
- Luthi-Carter R, Strand A, Peters NL, Solano SM, Hollingsworth ZR, Menon AS, Frey AS, Spektor BS, Penney EB, Schilling G, Ross CA, Borchelt DR, Tapscott SJ, Young AB, Cha JH, Olson JM ( 2000) Decreased expression of striatal signaling genes in a mouse model of Huntington's disease. Hum Mol Genet 9: 1259-1271. [DOI] [PubMed] [Google Scholar]
- Luthi-Carter R, Hanson SA, Strand AD, Bergstrom DA, Chun W, Peters NL, Woods AM, Chan EY, Kooperberg C, Krainc D, Young AB, Tapscott SJ, Olson JM ( 2002) Dysregulation of gene expression in the R6/2 model of polyglutamine disease: parallel changes in muscle and brain. Hum Mol Genet 11: 1911-1926. [DOI] [PubMed] [Google Scholar]
- Mammen PP, Shelton JM, Goetsch SC, Williams SC, Richardson JA, Garry MG, Garry DJ ( 2002) Neuroglobin, a novel member of the globin family, is expressed in focal regions of the brain. J Histochem Cytochem 50: 1591-1598. [DOI] [PubMed] [Google Scholar]
- McCampbell A, Taylor JP, Taye AA, Robitschek J, Li M, Walcott J, Merry D, Chai Y, Paulson H, Sobue G, Fischbeck KH ( 2000) CREB-binding protein sequestration by expanded polyglutamine. Hum Mol Genet 9: 2197-2202. [DOI] [PubMed] [Google Scholar]
- McCampbell A, Taye AA, Whitty L, Penney E, Steffan JS, Fischbeck KH ( 2001) Histone deacetylase inhibitors reduce polyglutamine toxicity. Proc Natl Acad Sci USA 98: 15179-15184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AA, Kurschel E, Osieka R, Schmidt CG ( 1987) Clinical pharmacology of sodium butyrate in patients with acute leukemia. Eur J Cancer Clin Oncol 23: 1283-1287. [DOI] [PubMed] [Google Scholar]
- Miller DM, Polansky DA, Thomas SD, Ray R, Campbell VW, Sanchez J, Koller CA ( 1987) Mithramycin selectively inhibits transcription of G-C containing DNA. Am J Med Sci 294: 388-394. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Jeong SY, Uchihara T, Anno M, Nagashima K, Nagashima T, Ikeda S, Tsuji S, Kanazawa I ( 2001) SCA17, a novel autosomal dominant cerebellar ataxia caused by an expanded polyglutamine in TATA-binding protein. Hum Mol Genet 10: 1441-1448. [DOI] [PubMed] [Google Scholar]
- Nemenoff RA, Winitz S, Qian NX, Van Putten V, Johnson GL, Heasley LE ( 1993) Phosphorylation and activation of a high molecular weight form of phospholipase A2 by p42 microtubule-associated protein 2 kinase and protein kinase C. J Biol Chem 268: 1960-1964. [PubMed] [Google Scholar]
- Nishibe S, Wahl MI, Hernandez-Sotomayor SM, Tonks NK, Rhee SG, Carpenter G ( 1990) Increase of the catalytic activity of phospholipase C-gamma 1 by tyrosine phosphorylation. Science 250: 1253-1256. [DOI] [PubMed] [Google Scholar]
- Novelli A, Reilly JA, Lysko PG, Henneberry RC ( 1988) Glutamate becomes neurotoxic via the N-methyl-d-aspartate receptor when intracellular energy levels are reduced. Brain Res 451: 205-212. [DOI] [PubMed] [Google Scholar]
- Nucifora Jr FC, Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, Takahashi H, Tsuji S, Troncoso J, Dawson VL, Dawson TM, Ross CA ( 2001) Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science 291: 2423-2428. [DOI] [PubMed] [Google Scholar]
- Ohyagi Y, Yamada T, Goto I ( 1994) Hemoglobin as a novel protein developmentally regulated in neurons. Brain Res 635: 323-327. [DOI] [PubMed] [Google Scholar]
- Perrine SP, Miller BA, Faller DV, Cohen RA, Vichinsky EP, Hurst D, Lubin BH, Papayannopoulou T ( 1989) Sodium butyrate enhances fetal globin gene expression in erythroid progenitors of patients with Hb SS and beta thalassemia. Blood 74: 454-459. [PubMed] [Google Scholar]
- Preisinger E, Jordan BM, Kazantsev A, Housman D ( 1999) Evidence for a recruitment and sequestration mechanism in Huntington's disease. Philos Trans R Soc Lond B Biol Sci 354: 1029-1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richon VM, Sandhoff TW, Rifkind RA, Marks PA ( 2000) Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc Natl Acad Sci USA 97: 10014-10019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu H, Lee J, Olofsson BA, Mwidau A, Deodoglu A, Escudero M, Flemington E, Azizkhan-Clifford J, Ferrante RJ, Ratan RR ( 2003a) Histone deacetylase inhibitors prevent oxidative neuronal death independent of expanded polyglutamine repeats via an Sp1-dependent pathway. Proc Natl Acad Sci USA 100: 4281-4286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu H, Lee JH, Zaman K, Ferrante RJ, Ross BD, Neve R, Ratan RR ( 2003b) Sp1 and Sp3 are oxidative stress-inducible, anti-death transcription factors in cortical neurons. J Neurosci 23: 3597-3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sealy L, Chalkley R ( 1978) The effect of sodium butyrate on histone modification. Cell 14: 115-121. [DOI] [PubMed] [Google Scholar]
- Sisodia SS ( 1998) Nuclear inclusions in glutamine repeat disorders: are they pernicious, coincidental, or beneficial? Cell 95: 1-4. [DOI] [PubMed] [Google Scholar]
- Steffan JS, Kazantsev A, Spasic-Boskovic O, Greenwald M, Zhu YZ, Gohler H, Wanker EE, Bates GP, Housman DE, Thompson LM ( 2000) The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci USA 97: 6763-6768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, Kurokawa R, Housman DE, Jackson GR, Marsh JL, Thompson LM ( 2001) Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila Nature 413: 739-743. [DOI] [PubMed] [Google Scholar]
- Struhl K ( 1998) Histone acetylation and transcriptional regulatory mechanisms. Genes Dev 12: 599-606. [DOI] [PubMed] [Google Scholar]
- Sugars KL, Rubinsztein DC ( 2003) Transcriptional abnormalities in Huntington disease. Trends Genet 19: 233-238. [DOI] [PubMed] [Google Scholar]
- Sun Y, Jin K, Peel A, Mao XO, Xie L, Greenberg DA ( 2003) Neuroglobin protects the brain from experimental stroke in vivo Proc Natl Acad Sci USA 100: 3497-3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabrizi SJ, Workman J, Hart PE, Mangiarini L, Mahal A, Bates G, Cooper JM, Schapira AH ( 2000) Mitochondrial dysfunction and free radical damage in the Huntington R6/2 transgenic mouse. Ann Neurol 47: 80-86. [DOI] [PubMed] [Google Scholar]
- Van Lint C, Emiliani S, Verdin E ( 1996) The expression of a small fraction of cellular genes is changed in response to histone hyperacetylation. Gene Expr 5: 245-253. [PMC free article] [PubMed] [Google Scholar]
- Vigushin DM, Coombes RC ( 2002) Histone deacetylase inhibitors in cancer treatment. Anticancer Drugs 13: 1-13. [DOI] [PubMed] [Google Scholar]
- Warrell Jr RP, He LZ, Richon V, Calleja E, Pandolfi PP ( 1998) Therapeutic targeting of transcription in acute promyelocytic leukemia by use of an inhibitor of histone deacetylase. J Natl Cancer Inst 90: 1621-1625. [DOI] [PubMed] [Google Scholar]
- Wood KW, Sarnecki C, Roberts TM, Blenis J ( 1992) Ras mediates nerve growth factor receptor modulation of three signal-transducing protein kinases: MAP kinase, Raf-1, and RSK. Cell 68: 1041-1050. [DOI] [PubMed] [Google Scholar]
- Wu ZL, O'Kane TM, Scott RW, Savage MJ, Bozyczko-Coyne D ( 2002) Protein tyrosine phosphatases are upregulated and participate in cell death induced by polyglutamine expansion. J Biol Chem 77: 44208-44213. [DOI] [PubMed] [Google Scholar]