

Abstract
Noradrenaline and α-adrenoceptors have been implicated in the modulation of pain in various behavioral conditions. Noradrenergic neurons and synaptic inputs are present in neuronal circuits critical for pain modulation, but their actions on neurons in those circuits and consequently the mechanisms underlying noradrenergic modulation of pain remain unclear. In this study, both recordings in vitro and behavioral analyses in vivo were used to examine cellular and behavioral actions mediated by α1- and α2-adrenoceptors on neurons in the nucleus raphe magnus. We found that α1- and α2-receptors were colocalized in the majority of a class of neurons (primary cells) that inhibit spinal pain transmission and are excited during opioid analgesia. Activation of the α1-receptor depolarized whereas α2-receptor activation hyperpolarized these neurons through a decrease and an increase, respectively, in potassium conductance. Blockade of the excitatory α1-receptor or activation of the inhibitory α2-receptor significantly attenuated the analgesia induced by local opioid application, suggesting that α1-receptor-mediated synaptic inputs in these primary cells contribute to their excitation during opioid analgesia. In the other cell class (secondary cells) that is thought to facilitate spinal nociception and is inhibited by analgesic opioids, only α1-receptors were present. Blocking the α1-receptor in these cells significantly reduced the hyperalgesia (increased pain) induced by opioid abstinence. Thus, state-dependent activation of α1-mediated synaptic inputs onto functionally distinct populations of medullary pain-modulating neurons contributes to opioid-induced analgesia and opioid withdrawal-induced hyperalgesia.
Keywords: α1-adrenoceptors, α2-adrenoceptors, opioid, analgesia, hyperalgesia, pain, nucleus raphe magnus
Introduction
Noradrenaline (NA), a principal neurotransmitter in the CNS, is involved in various CNS functions, including modulation of pain in both acute and chronic conditions, such as opioid withdrawal and inflammatory pain states (Raja, 1995; Maldonado, 1997). Its central pain-modulating actions are mediated primarily by a well studied brain circuit that involves NA synaptic connections in the rostral ventromedial medulla (RVM) and A5 and A7 catecholamine cell groups in the pontine tegmentum. Neurons in the RVM, a critical supraspinal component of the CNS pain-modulating network, project directly to the spinal dorsal horn and control spinal pain transmission (Fields and Basbaum, 1999). Noradrenergic neurons in the A5 and A7 cell groups also modulate spinal pain transmission through their direct spinal projections (Burnett and Gebhart, 1991; Nuseir and Proudfit, 2000). The RVM does not contain noradrenergic cells (Basbaum, 1992) but receives a dense noradrenergic projection from the A5 and A7 neurons (Tanaka et al., 1996; Meng et al., 1997). These noradrenergic inputs have been demonstrated to affect pain modulation by RVM neurons (Haws et al., 1990; Nuseir et al., 1999).
Previous studies on the effect of NA inputs on RVM cell activity used either iontophoretic application or local microinjection of α-receptor agonists or antagonists in animals in vivo. These studies examined the α-adrenergic effect on spontaneous activity of RVM cells, and inconsistent results were reported (Proudfit, 1988; Haws et al., 1990). One potential confound is that the RVM has functionally distinct cell groups (Pan et al., 1997; Fields and Basbaum, 1999); however, the cellular distribution of α-receptor subtypes and their synaptic actions in the different cell groups are unknown.
We have characterized the cell types and neuronal circuit in the nucleus raphe magnus (NRM), a main component of the RVM, and have demonstrated how μ opioids produce antinociception by acting on two distinct NRM cell types, primary cells and secondary cells (Pan et al., 1990, 1997). μ opioid agonists in the NRM inhibit GABA synaptic inputs to primary cells and thus indirectly excite these cells, leading to inhibition of spinal pain transmission. In addition, μ agonists hyperpolarize secondary cells, reducing their facilitating action on spinal nociception. These behaviorally relevant cellular mechanisms mediating the pain-modulating actions of the two NRM cell types provide a unique target system to investigate the role of α-adrenoceptors in pain modulation. In the present study, we characterized the cellular actions of α1- and α2-receptors in the two types of NRM cells in vitro. We then examined the contribution of these actions to opioid-induced different pain states in rats in vivo.
Materials and Methods
All procedures involving the use of animals conformed to guidelines set by the University of Texas-M. D. Anderson Cancer Center Animal Care and Use Committee.
Brain slice preparations. Both adult (200-300 gm) and neonatal (9-14 d) male Wistar rats were used in this study. Neonatal rats were used to better visualize individual neurons in a slice with a Nomarski microscope for patch-clamp recording. The physiological and pharmacological properties of neurons from these young rats are indistinguishable from those of adult rats (Pan et al., 1997). Rats were anesthetized with halothane and then killed by decapitation. The brain was removed, and coronal brainstem slices (200 or 300 μm thick) containing the NRM were cut in a vibratome in cold (4°C) physiological saline. A single slice was submerged in a shallow recording chamber and perfused with preheated (35°C) physiological saline. The content of the physiological saline solution was (in mm): 126 NaCl, 2.5 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 2.4 CaCl2, 11 glucose, 25 NaHCO3, saturated with 95% O2 and 5% CO2.
Recordings. Both intracellular recording and whole-cell patch-clamp recording techniques were used to complement the advantages and limitations of the two recording configurations. No significant difference in results was observed between the two recording methods. For intracellular recordings, NRM neurons were penetrated with glass microelectrodes filled with potassium chloride (2 m) having a resistance of 40-80 MΩ. Membrane potentials or currents were recorded with a single-electrode voltage-clamp amplifier using switching frequencies between 3 and 6 kHz. The setting time of the clamp after a 10 mV step was typically 3-5 msec. Visualized whole-cell patch-clamp recordings were made from NRM neurons with a glass pipette (resistance 3-5 MΩ) filled with a solution containing (in mm): 126 potassium gluconate, 10 NaCl, 1 MgCl2, 11 EGTA, 10 HEPES, 2 ATP, 0.25 GTP, pH adjusted to 7.3 with KOH. A seal resistance of 2 GΩ or above and an access resistance of 15 MΩ or less were considered acceptable. Series resistance was optimally compensated.
Cell classification. All NRM cells recorded were classified into either a primary or secondary cell type according to the criteria described in our previous study (Pan et al., 1990). Briefly, primary cells have a wider action potential (>1 msec width at threshold), have a more negative resting membrane potential (-58 to -72 mV), and are insensitive to μ agonists. Secondary cells have a narrower action potential (<1 msec), often display spontaneous firing, and are hyperpolarized by μ agonists. Cells that could not be clearly classified into either type were not included in this study.
Behavioral experiments and microinjection. Adult male Wistar rats were maintained lightly anesthetized in a stereotaxic apparatus with a constant infusion of methohexital (10 mg/ml, i.v., at 0.8 ml/hr). A 26 gauge single guide cannula was aimed at the ventrolateral periaqueductal gray (PAG) [anteroposterior (AP): -7.8; lateral (L): ±0.8; ventral (V): -6.0] and a second, double-guide cannula was aimed at the NRM (AP: -11.0; L: 0; V: -10.7). Tail-flick latency to a radiant heat stimulus was measured every 2 min. The heat intensity was set to elicit stable baseline latencies between 2.5 and 3.5 sec (for analgesia tests) or 5 and 6 sec (for withdrawal tests). The cutoff time was 12 sec. After at least six baseline trials, drugs were delivered through a 33 gauge injector cannula with an infusion pump at a rate of 0.5 μl/min. NRM placement controls were accomplished by aiming the NRM guide cannula 1.5 mm above the target site. All cannula placements for both the PAG and the NRM were histologically verified afterward, and only those located within the designated area were included in the results. Morphine and naloxone were injected intravenously through an intravenous catheter.
Data analysis and materials. Membrane potentials and currents were measured with the software Chart (AD Instruments) or AxoGraph (Axon Instruments). Dose-response data were plotted and fitted with the logistic equation using KaleidaGraph (Synergy Software). Numerical data, presented as mean ± SEM, were analyzed and compared by Student's t tests or by an ANOVA for repeated measures and the Tukey-Kramer test of post hoc analysis with the software GB-Stat (Dynamic Microsystems). All drugs were applied through the bath solution. The following compounds were used: noradrenaline, phenylephrine, prazosin, UK14304, clonidine, yohimbine and idazoxan. All materials were purchased from Sigma (St. Louis, MO) or Research Biochemicals (Natick, MA).
Results
NA produces distinct effects in the two cell types
NRM cells were classified as either a primary or a secondary cell type according to the criteria described in Materials and Methods and in our previous studies (Pan et al., 1990, 1997).
In the majority of primary cells tested (20 of 34, 59%), application of NA (10 μm) produced a composite effect under current clamp (Fig. 1Aa). It induced an initial depolarization that then declined in amplitude in the continued presence of NA. Washout of NA caused a rebound depolarization before the membrane potential recovered to the pre-NA level. The average amplitude of the initial depolarization produced by 10 μm NA was 5.5 ± 0.7 mV from an average resting membrane potential of -62 mV with no spontaneous action potential induced (n = 20). The amount of depolarization decline varied from cell to cell, and in some cells it lead to a net hyperpolarization before washout of NA. In these cells, the membrane potential before wash ranged from 5.1 mV (depolarization) to -3.7 mV (hyperpolarization) with an average of 1.7 ± 0.6 mV (31% of initial depolarization; n = 20). The variability in depolarization decline appeared independent of the duration of NA application, because prolonged NA application failed to produce further decline in a given cell. Interestingly, the peak amplitude of the rebound depolarization after NA removal was significantly larger than that of the initial depolarization in the same cells (9.1 ± 0.6 mV; 166%; measured from pre-NA baseline potential; n = 20). TTX (1 μm) had no effect on the NA-induced composite response (n = 4), excluding the involvement of action potential-dependent, presynaptic effects by surrounding cells.
Figure 1.

NA produces distinct effects in the two cell types of the NRM. A, NA-induced composite effect on the membrane potential of a primary cell (a) and membrane depolarization through α1-receptors in another primary cell (b) under current clamp. B, NA-induced membrane depolarization accompanied by spontaneous firing of action potentials (truncated) and blocked by the α1 antagonist prazosin in a secondary cell.
In the other 14 primary cells (41%), NA (10 μm) produced a membrane depolarization without significant decline in amplitude and with no rebound depolarization after wash (Fig. 1Ab). The average amplitude of depolarization in these primary cells was 10.4 ± 1.1 mV (n = 14), significantly larger than the initial depolarization in the cells with a composite NA response (5.5 ± 0.7 mV; p < 0.01). The α1-adrenoceptor antagonist prazosin (1 μm) completely blocked the NA effect in these cells (control, 11.3 ± 1.9 mV; in prazosin, 0.1 ± 0.1 mV; n = 6) (Fig. 1Ab). It suggests that the NA-induced depolarization in these primary cells is mediated by the α1-receptor.
In secondary cells, by contrast, NA (10 μm) caused a membrane depolarization in all cells tested (7.7 ± 1.0 mV; n = 14) (Fig. 1B). In every cell, the depolarization was accompanied by spontaneous firing of action potentials and was completely blocked by the α1 antagonist prazosin (1 μm; n = 3) (Fig. 1B).
α1- and α2-adrenoceptors are colocalized in the primary cells
To determine the adrenoceptor subtypes that mediate the composite NA effect in those primary cells, we first examined the actions of selective α-adrenoceptor agonists. In primary cells that displayed a composite response to NA (Fig. 2A), the selective α1 agonist phenylephrine (PE) (10 μm) produced a membrane depolarization without significant amplitude decline or rebound depolarization seen in the NA effect (8.3 ± 1.3 mV; n = 9) (Fig. 2B). Furthermore, in these primary cells, application of the α2 agonist UK14304 (300 nm) produced a membrane hyperpolarization with an average amplitude of 6.2 ± 0.7 mV (n = 7) (Fig. 2C). The UK14304-induced hyperpolarization was completely blocked by the α2-receptor antagonist idazoxan (1 μm; n = 3) (Fig. 2C). Another α2-adrenoceptor agonist, clonidine (1 μm), also caused a membrane hyperpolarization in these cells (5.8 ± 0.9 mV; n = 4). When PE and UK14304 were tested in the same primary cells with a composite NA response (n = 5), both the α1-induced depolarization and the α2-induced hyperpolarization were observed (Fig. 2). These results suggest that most of the primary cells express both excitatory α1- and inhibitory α2-adrenoceptors, the activation of which causes membrane depolarization and hyperpolarization, respectively.
Figure 2.

α1-mediated depolarization and α2-mediated hyperpolarization coexist in a primary cell. A, The composite effect induced by NA in a primary cell. B, Membrane depolarization induced by the α1-receptor agonist PE in the same cell as in A. C, Membrane hyperpolarization induced by the α2-receptor agonist UK14304 and its blockade by the α2-receptor antagonist idazoxan in the same cell as in A and B.
Because both α1- and α2-receptors were present in the primary cells, it is possible that the composite NA effect in these cells results from the complex interactions of the α1-mediated depolarization and the α2-mediated hyperpolarization when NA is applied. We then examined the NA effect after blocking either α1-or α2-receptors with selective antagonists. After addition of idaxozan (1 μm) to block α2-receptors in primary cells with the composite NA response, NA induced a membrane depolarization without significant amplitude decline or rebound depolarization (Fig. 3A). The initial depolarization was 5.6 ± 1.2 mV in control and 7.6 ± 1.3 mV in idaxozan (n = 6). On the other hand, when the α1-receptors were blocked by prazosin (1 μm), NA caused a membrane hyperpolarization instead of a composite depolarization (control, 7.0 ± 1.1 mV initial depolarization; in prazosin, -5.8 ± 0.6 mV hyperpolarization; n = 9) (Fig. 3B). These findings demonstrate that bath-applied NA simultaneously activates both of the colocalized α1- and α2-receptors, causing concomitant α1-induced depolarization and α2-induced hyperpolarization. The peak time for the NA-induced hyperpolarization was generally later than that for the initial depolarization (Fig. 3B). This supports the notion that the onset of α2 hyperpolarization contributes to the decline in depolarization amplitude during NA application. It is also likely that the rebound depolarization is related to the washout of α2-mediated hyperpolarization.
Figure 3.
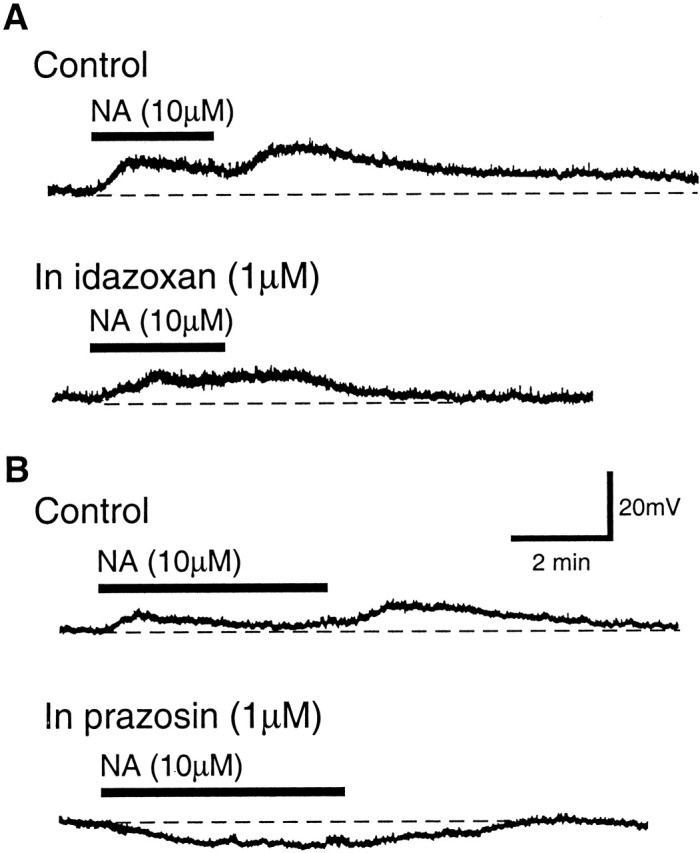
The composite NA effect is related to the coactivation of α1- and α2-receptors in the primary cell. A, NA effects on the membrane potential of a primary cell in control and in the presence of idazoxan to block the α2-receptor. B, NA effects on the membrane potential of another primary cell in control and in the presence of prazosin to block the α1-receptor. Note the hyperpolarization induced by NA in the presence of prazosin.
To further characterize and compare NA activation of the two adrenoceptors, dose-response curves were constructed separately for NA-induced depolarization (α1) and hyperpolarization (α2) in the presence of the corresponding antagonist. As shown in Figure 4A, NA effects on the two receptors had a similar dose range with a small difference in EC50 values. The NA-induced depolarization in idaxozan had an EC50 of 4.2 μm, whereas the EC50 for the NA-induced hyperpolarization in prazosin was 4.5 μm, suggesting that NA has a slightly higher affinity for the α1-receptor over the α2-receptor.
Figure 4.
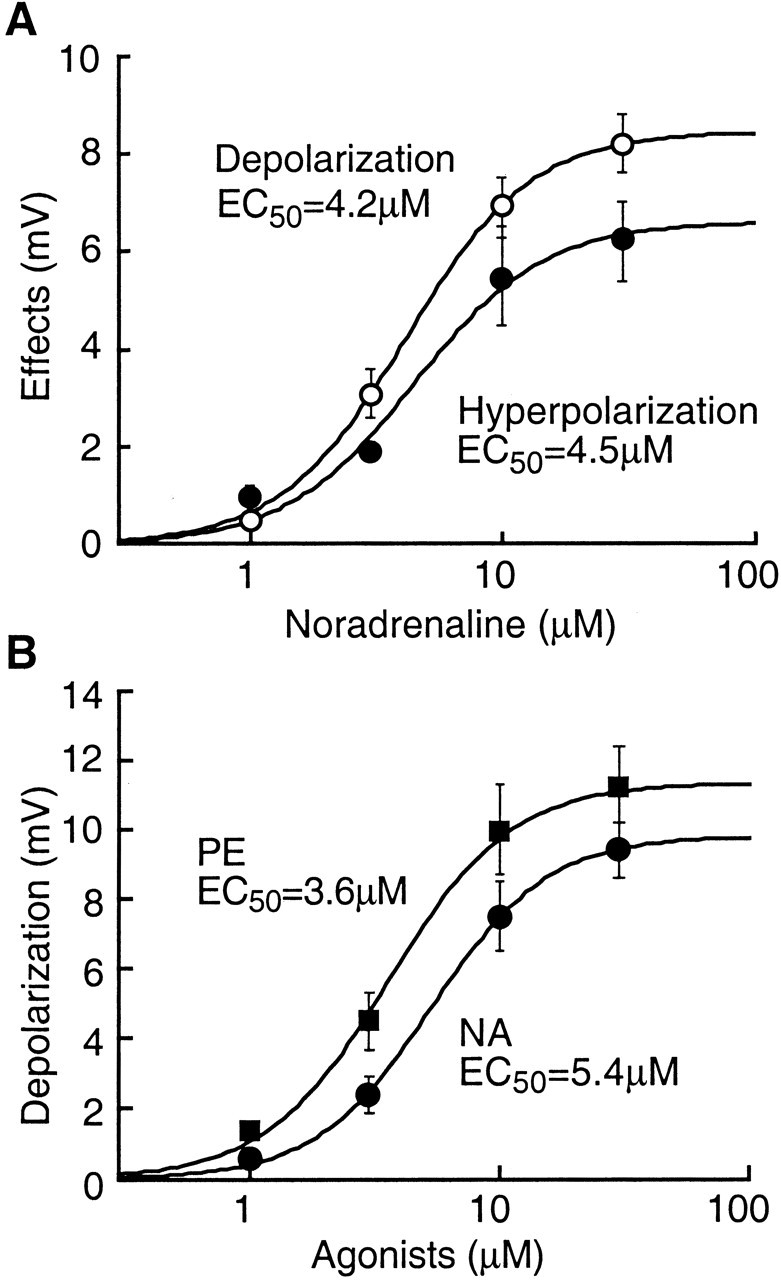
NA has similar potency at the α1- and α2-receptors. A, Dose-response curves for the NA-induced depolarization in idaxozan (α1-mediated effect, open circles) and for the NA-induced hyperpolarization in prazosin (α2-mediated effect, filled circles) in primary cells. B, Dose-response curves for the α1-receptor-mediated depolarization by NA (filled circles) and by PE (filled squares) in NRM cells without the composite NA response.
In cells without the composite NA response, the α1 agonist PE caused a membrane depolarization that was similar in primary (n = 3) and secondary (n = 5) cells, with an average amplitude of 8.5 ± 1.6 mV (n = 8); however, the α2 agonist UK14304 or clonidine had no effect on these cells (n = 7). This is consistent with the hypothesis that concomitant α2-mediated hyperpolarization contributes to the composite NA effect in the majority of primary cells as described above. For comparison, Figure 4B shows the dose-response curves for NA and PE in those cells without the composite NA response. The EC50 estimated for the NA and PE effects was 5.4 and 3.6 μm, respectively.
Potassium conductance is involved in both α1-mediated depolarization and α2-mediated hyperpolarization
In both cell types under whole-cell voltage clamp with a holding potential of -60 mV, PE induced an inward current of 40 ± 6pA with a decrease in membrane conductance (n = 17). In seven of these cells (7 of 17, 41%), the PE-induced inward current reversed its polarity near the potassium equilibrium potential (-100.5 ± 4.8 mV; n = 7) (Fig. 5A). The reversal potential was shifted to -59.6 ± 2.3 mV when the extracellular potassium concentration was increased to 10.5 mm (n = 3). These results suggest that the α1 depolarization is mediated, at least partially, by a decease in potassium conductance. In the remaining 10 cells, the PE-induced inward current did not reverse polarity at potentials as negative as -130 mV, indicating that other membrane conductances or intracellular factors are involved in the α1 effect. These observations are in agreement with our previous report on the ionic mechanism for the α1-receptor-induced depolarization in dorsal raphe neurons (Pan et al., 1994) and with those in other brain sites (Larkman and Kelly, 1992; McCormick, 1992).
Figure 5.
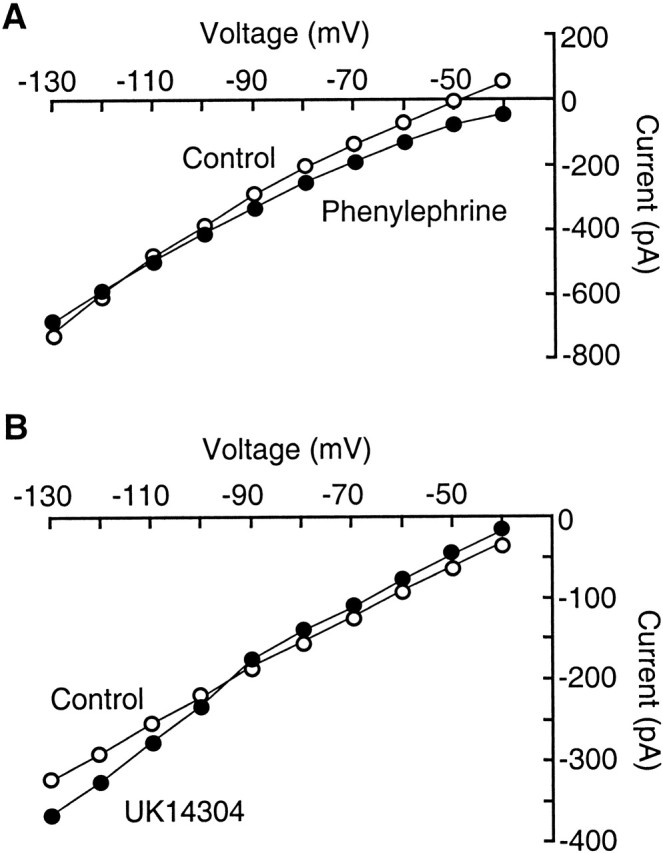
Potassium conductance is involved in both the α1-mediated depolarization and the α2-mediated hyperpolarization. A, Graph showing current-voltage relationship in control (open circles) and in the presence of phenylephrine (10 μm) in a primary cell under voltage clamp. B, Graph of current-voltage relationship in control (open circles) and in the presence of UK14304 (300 nm) in another primary cell.
The α2 agonist UK14304 or clonidine (1 μm) caused an outward current of 7 ± 1 pA and an increase in membrane conductance in primary cells (n = 7) (Fig. 5B). The reversal potential of the current was -101.5 ± 6.1 mV, suggesting that the α2 hyperpolarization is caused by an increase in potassium conductance. This α2 effect has been well characterized in many types of neurons (Grudt et al., 1995; Pralong and Magistretti, 1995; Arima et al., 1998; Bai and Renaud, 1998).
Activation of α1- but not α2-adrenoceptors mediates μ opioid-induced analgesia
Considering the distinct pain-modulating actions of NRM primary cells and secondary cells (Pan et al., 1997; Fields and Basbaum, 1999), we next investigated the functional role of α-adrenoceptors in opioid analgesia using a microinjection technique in rats in vivo. In a lightly anesthetized rat, microinjection of the μ opioid receptor agonist [d-Ala2, N-Me-Phe4,-Gly-ol5]enkephalin (DAMGO) (0.05 μg in 0.25 μl) into the PAG and saline into the NRM increased the tail-flick latency quickly to the cutoff time of 12 sec, indicating a potent analgesic effect (Fig. 6A, open circles) (n = 5 rats). When the α1 antagonist prazosin (0.105 μg in 1 μl) was microinjected into the NRM, this PAG DAMGO-induced analgesia was significantly attenuated (Fig. 6A, filled circles) (n = 5 rats). Prazosin itself in NRM did not change the pain threshold (Fig. 6A, open squares) (n = 5 rats). NRM microinjection of PE (up to 1 μg in 1 μl), which would excite all α1-expressing cells nonselectively, also had no effect on the tail-flick latency (Fig. 6A, filled squares) (n = 5 rats). These findings suggest that activation of α1-receptors in the NRM is required for the analgesia induced by PAG DAMGO.
Figure 6.
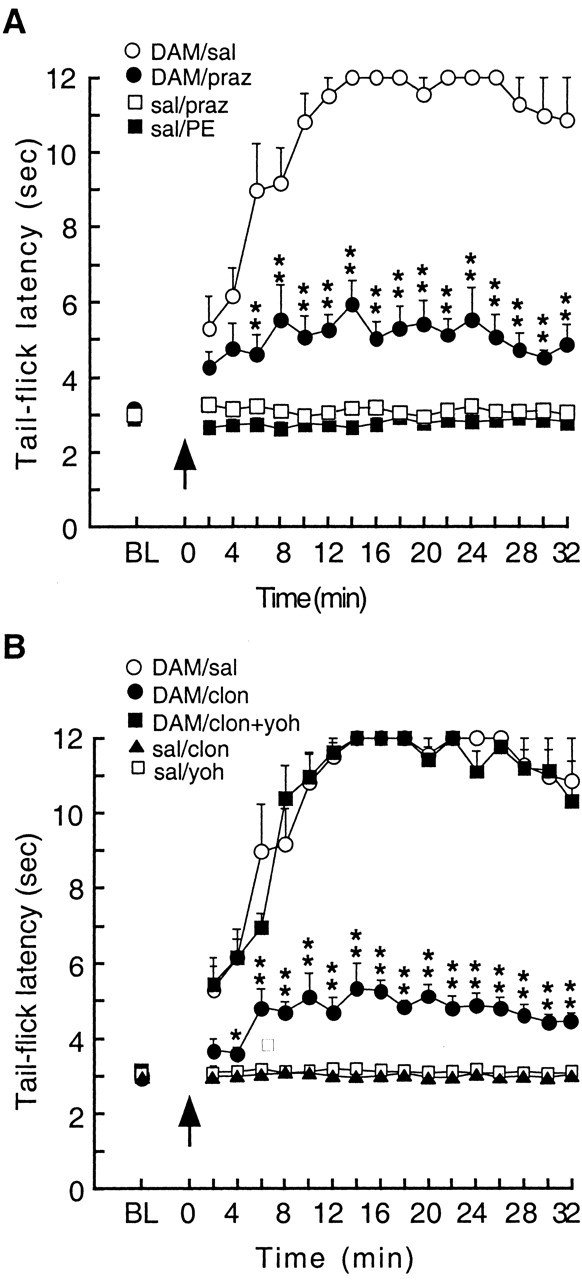
Activation of NRM α1-but not α2-receptors is required for the antinociceptive effect of opioids applied into the PAG. Tail-flick latencies were measured every 2 min before [baseline (BL), average of 6 trials] and after drug microinjections into the NRM and then into the PAG (arrow; time = 0). The cutoff time was 12 sec. A, Actions of α1-receptors. B, Actions of α2-receptors. Symbol labels represent drugs and their microinjection sites of PAG/NRM. DAM, DAMGO; sal, saline; praz, prazosin; PE, phenylephrine; clon, clonidine; yoh, yohimbine. n = 5 rats in each group. *p < 0.05; **p < 0.01 (ANOVA for repeated measure and the Tukey-Kramer test of post hoc analysis).
To determine the role of α2-receptors in PAG-elicited opioid analgesia, similar microinjection experiments were conducted with the α2 agonist and antagonist. Similar to the effect of the α1 antagonist, the α2 agonist (not antagonist) clonidine (0.267 μgin 1 μl) microinjected into the NRM significantly antagonized the analgesia (Fig. 6B, filled circles) (n = 5 rats). The clonidine effect was completely reversed by NRM coinjection of the α2 antagonist yohimbine (0.391 μgin1 μl) (Fig. 6B, filled squares) (n = 5 rats), suggesting a specific, α2-mediated effect. Clonidine or yohimbine alone in the NRM did not affect the animal's pain behavior (Fig. 6B, clonidine, filled triangles; yohimbine, open squares) (n = 5 rats in each group). Yohimbine in the NRM had no effect on PAG DAMGO-induced analgesia (n = 2 rats; data not shown). These results indicate that inhibition of the majority of NRM primary cells by activating their α2-receptors opposes the antinociception induced by PAG DAMGO.
Activation of α1-adrenoceptors mediates opioid abstinence-induced hyperalgesia
Our previous work demonstrated that during opioid abstinence-induced hyperalgesia, a state of increased pain sensitivity, μ-sensitive secondary cells in the NRM are predominantly active, indicative of their facilitating action on spinal nociception (Bederson et al., 1990; Kaplan and Fields, 1991; Pan et al., 2000). To determine whether α1-receptors are involve in this activation of secondary cells, the following experiments were performed in rats during the behavioral state of hyperalgesia induced by opioid abstinence.
Systemic application of morphine (2 mg/kg, i.v.) increased the tail-flick latency quickly to the cutoff time of 12 sec (Fig. 7). Subsequent application of naloxone (1 mg/kg, i.v.) immediately reversed the morphine-induced antinociception and further decreased the animal's pain threshold to levels below pre-morphine baseline, indicating a hyperalgesic state (Fig. 7, open circles). Microinjection of the α1 antagonist prazosin at the same dose as before (0.105 μg in 1 μl), into the NRM immediately after naloxone injection, significantly reduced the hyperalgesia (Fig. 7, filled circles). In contrast, the α2 agonist clonidine (0.267 μgin1 μl) in the NRM was without effect (Fig. 7, filled squares), indicating that the α2-expressing primary cells are not activated in this hyperalgesic state. Figure 8 illustrates the proposed mechanisms by which α-adrenoceptors regulate NRM cell activity during opioid analgesia and opioid withdrawal-induced hyperalgesia.
Figure 7.

Activation of NRM α1-receptors is required for the hyperalgesia during opioid withdrawal. After measurements of baseline tail-flick latencies, rats were injected with morphine (2 mg/kg, i.p.) followed by naloxone (1 mg/kg, i.p.) 26 min later to induce opioid abstinence (withdrawal)-induced hyperalgesia. NRM microinjections were made immediately after the naloxone injection. Symbol labels represent drugs microinjected into the NRM. sal, Saline; praz, prazosin; clon, clonidine. n = 5 rats in each group. *p < 0.05; **p < 0.01 (ANOVA for repeated measure and the Tukey-Kramer test of post hoc analysis).
Figure 8.

Diagram depicting the proposed roles of α-adrenoceptors in the NRM in opioid modulation of pain. During opioid analgesia, activation of noradrenergic inputs and α1-receptors contributes to excitation of primary cells, which inhibit spinal pain transmission, whereas secondary cells are inhibited by opioids through μ-receptors. Activation of α2-receptors inhibits the majority of primary cells expressing α2-receptors, reducing opioid analgesia. During opioid withdrawal, however, secondary cells are dominantly activated partly through α1-receptors and their activity increases spinal pain sensitivity, causing hyperalgesia. Most of the primary cells are inactive, as suggested by the observation that activation of α2-receptors by clonidine has no effect on the increased pain during opioid withdrawal.
Discussion
In the present study, we have demonstrated that the excitatory α1-adrenoceptor is present in both primary and secondary cell types in the NRM, but the inhibitory α2-adrenoceptor is only found in the majority of primary cells and is colocalized with the α1-receptor in these cells. Thus, simultaneous activation by NA of the colocalized α1- and α2-receptors in these primary cells causes a composite effect through their opposing modulation of potassium conductance. In addition, our behavioral results suggest that primary cells are activated, at least partially, by noradrenergic inputs through α1-receptors and that this α1-receptor-mediated activation of primary cells contributes to PAG DAMGO-induced analgesia. Our behavioral data also indicate that α1-mediated activation of secondary cells, but not the α2-expressing primary cells, contributes to the increased pain sensitivity during opioid withdrawal.
Roles of NRM α1- and α2-adrenoceptors in opioid modulation of pain
Unlike previous studies investigating tonic noradrenergic activity in the NRM and pain modulation in animals, the current study was designed to identify the role of α1- and α2-receptors in distinct pain-modulating cell types that are activated during opioid analgesia or during opioid withdrawal.
Our previous studies have shown that excitation of RVM Off-cells, or primary cells identified in vitro, mediates the analgesia induced by PAG DAMGO, consistent with their proposed inhibitory action on spinal pain transmission (Pan et al., 1997; Fields and Basbaum, 1999). Furthermore, we have demonstrated anatomically that Off-cells receive a dense noradrenergic input (Meng et al., 1997). The current finding that blockade of α1-receptors by prazosin considerably attenuates PAG DAMGO-induced analgesia indicates that activation of noradrenergic synaptic inputs acting on α1-receptors directly excites NRM primary cells (Off-cells) and is required for the antinociceptive effect of PAG opioids (Fig. 8). Further confirming this, selective inhibition of α2-expressing primary cells by clonidine significantly antagonizes this PAG opioid-elicited analgesia.
On the other hand, RVM On-cells, or secondary cells expressing μ-opioid receptors, have a pain-facilitating action and are inhibited by μ opioids during opioid analgesia (Pan et al., 1997; Fields and Basbaum, 1999). Increasing evidence suggests that activation of these μ-expressing medullary cells accounts at least partially for the increased pain sensitivity, or hyperalgesia, during opioid withdrawal and in other chronic pain states (Bederson et al., 1990; Kaplan and Fields, 1991; Pan et al., 2000; Porreca et al., 2001, 2002). These cells also receive a dense NA input (Meng et al., 1997). Our observation that α1-receptor blockade attenuates opioid withdrawal-induced hyperalgesia indicates that noradrenergic inputs excite NRM secondary cells through α1-receptors and that this excitation of secondary cells is required for the hyperalgesia during opioid withdrawal (Fig. 8). Selective inhibition of α2-expressing primary cells by clonidine, which antagonized the DAMGO analgesia, did not affect the hyperalgesia, suggesting that these primary cells do not play a significant role in the sensitized pain state. This is consistent with previous work indicating that RVM Off-cells (primary cells) are silent during acute opioid withdrawal (Bederson et al., 1990). Although α2-adrenoceptors in the NRM do not seem to be involved in the opioid withdrawal-induced hyperalgesia, α2-adrenoceptors in other brain areas have been implicated in the mediation or expression of other somatic symptoms during opioid withdrawal in animals and humans (Christie et al., 1997; Maldonado, 1997). For example, activation of α2-adrenoceptors in the bed nucleus of the stria terminalis inhibits opioid withdrawal-induced aversion (Delfs et al., 2000).
The mechanisms underlying the activation of NRM noradrenergic inputs by PAG DAMGO and during opioid withdrawal are unclear. It is proposed that opioids in the PAG inhibit GABAergic neurons and thereby disinhibit PAG output neurons that project to and activate pain-inhibiting neurons in the RVM (Bellchambers et al., 1998; Fields and Basbaum, 1999). RVM-elicited antinociception is mediated partially by the subsequent activation, through direct projections, of spinally projecting noradrenergic neurons in the pontine A7 cell group, which also inhibit spinal nociception (Nuseir et al., 1999). Given that the RVM receives a dense noradrenergic innervation from the A7 cells (Tanaka et al., 1996) and that no such innervation has been reported from the PAG, it is possible that PAG opioids indirectly cause the activation of α1-adrenoceptors in the NRM by activating NRM-projecting A7 cells. During opioid withdrawal, however, RVM-projecting neurons in the PAG are not activated (Bellchambers et al., 1998). Noradrenergic neurons that are activated during opioid withdrawal include those in the A1 cell group of the caudal medulla and those in the locus coeruleus (LC) (Maldonado, 1997; Delfs et al., 2000). Both cell groups project to the NRM (Tanaka et al., 1996). Thus, it is possible that during opioid withdrawal, NRM α1-adrenoceptors are activated through activation of NRM-projecting A1 and LC noradrenergic cells.
Previous studies indicate that NRM microinjection of α1 agonists increases and α2 agonists decreases nociceptive responses, whereas microinjection of α1 and α2 antagonists has inconsistent effects on pain threshold (Proudfit, 1988; Haws et al., 1990; Galeotti et al., 1999; Holden et al., 1999). In the current study with anesthetized rats, we did not observe significant changes in baseline pain threshold by either agonists or antagonists of α-adrenoceptors. Because our cellular data indicated that α1 agonists in the NRM may have a hyperalgesic effect by causing more secondary cells to fire action potential, the lack of the PE effect on baseline pain threshold was unexpected; however, even when higher doses were used (up to 1 μg), still no significant PE effect was observed. The discrepancy with previous reports could be caused primarily by the level of anesthesia, which profoundly alters the tonic activity of NRM cells and associated baseline pain threshold in different experiment conditions. Nonselective α1-mediated activation of both primary and secondary cells also could confound and reduce secondary cell-mediated hyperalgesia. Finally, longer baseline tail-flick latency may be required to reveal a small hyperalgesic effect.
Functional significance of noradrenergic neurotransmission in pain modulation
One of the most significant roles of noradrenergic neurons in pain modulation is observed during opioid withdrawal. Abrupt cessation of opioid administration or acute application of opioid antagonists induces opioid withdrawal, which produces a series of aversive responses and symptoms, including an abnormal increase in pain sensitivity (hyperalgesia) in both humans and animals (Heishman et al., 1989; Kaplan and Fields, 1991; McNally and Akil, 2002; Raghavendra et al., 2002). Opioid withdrawal also causes hyperactivity of central noradrenergic neurons and increased noradrenaline levels in the brain (Maldonado, 1997). Increased noradrenergic activity has been implicated in various somatic symptoms of opioid withdrawal (Christie et al., 1997; Delfs et al., 2000). Thus, an increase in the activity of noradrenergic synaptic inputs to the NRM from neurons in the A1 cell group and the LC could mediate the hyperalgesia observed during opioid withdrawal. Our finding that an α1 antagonist administered in the NRM significantly attenuates opioid withdrawal-associated hyperalgesia indicates that α1-adrenoceptors may serve as a potential central target for the treatment of opioid withdrawal-related pain problems.
Footnotes
This work was supported by Grants DA14524 and DA01949 from the National Institute on Drug Abuse, National Institutes of Health.
Correspondence should be addressed to Dr. Zhizhong Z. Pan, Department of Symptom Research, Box 110, University of Texas-M. D. Anderson Cancer Center, 1515 Holcombe Boulevard, Houston, TX 77030. E-mail: zzpan@mdanderson.org.
Copyright © 2003 Society for Neuroscience 0270-6474/03/237950-08$15.00/0
References
- Arima J, Kubo C, Ishibashi H, Akaike N ( 1998) alpha2-Adrenoceptor-mediated potassium currents in acutely dissociated rat locus coeruleus neurones. J Physiol (Lond) 508: 57-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai D, Renaud LP ( 1998) Median preoptic nucleus neurons: an in vitro patch-clamp analysis of their intrinsic properties and noradrenergic receptors in the rat. Neuroscience 83: 905-916. [DOI] [PubMed] [Google Scholar]
- Basbaum A ( 1992) Anatomical studies of the noradrenergic projections to the spinal cord dorsal horn. In: Towards the use of noradrenergic agonists for the treatment of pai (Besson J, Guilbaud G, eds), pp 77-89. Amsterdam: Excerpta Medica.
- Bederson JB, Fields HL, Barbaro NM ( 1990) Hyperalgesia during naloxone-precipitated withdrawal from morphine is associated with increased on-cell activity in the rostral ventromedial medulla. Somatosens Mot Res 7: 185-203. [DOI] [PubMed] [Google Scholar]
- Bellchambers CE, Chieng B, Keay KA, Christie MJ ( 1998) Swim-stress but not opioid withdrawal increases expression of c-fos immunoreactivity in rat periaqueductal gray neurons which project to the rostral ventromedial medulla. Neuroscience 83: 517-524. [DOI] [PubMed] [Google Scholar]
- Burnett A, Gebhart GF ( 1991) Characterization of descending modulation of nociception from the A5 cell group. Brain Res 546: 271-281. [DOI] [PubMed] [Google Scholar]
- Christie MJ, Williams JT, Osborne PB, Bellchambers CE ( 1997) Where is the locus in opioid withdrawal? Trends Pharmacol Sci 18: 134-140. [DOI] [PubMed] [Google Scholar]
- Delfs JM, Zhu Y, Druhan JP, Aston-Jones G ( 2000) Noradrenaline in the ventral forebrain is critical for opiate withdrawal-induced aversion. Nature 403: 430-434. [DOI] [PubMed] [Google Scholar]
- Fields H, Basbaum A ( 1999) Central nervous system mechanisms of pain modulation. In: Textbook of pain, Ed 4 (Wall P, Melzack R, eds). London: Churchill Livingstone.
- Galeotti N, Ghelardini C, Vinci MC, Bartolini A ( 1999) Role of potassium channels in the antinociception induced by agonists of alpha2-adrenoceptors. Br J Pharmacol 126: 1214-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grudt TJ, Williams JT, Travagli RA ( 1995) Inhibition by 5-hydroxytryptamine and noradrenaline in substantia gelatinosa of guinea-pig spinal trigeminal nucleus. J Physiol (Lond) 485: 113-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haws CM, Heinricher MM, Fields HL ( 1990) Alpha-adrenergic receptor agonists, but not antagonists, alter the tail-flick latency when microinjected into the rostral ventromedial medulla of the lightly anesthetized rat. Brain Res 533: 192-195. [DOI] [PubMed] [Google Scholar]
- Heishman SJ, Stitzer ML, Bigelow GE, Liebson IA ( 1989) Acute opioid physical dependence in postaddict humans: naloxone dose effects after brief morphine exposure. J Pharmacol Exp Ther 248: 127-134. [PubMed] [Google Scholar]
- Holden JE, Schwartz EJ, Proudfit HK ( 1999) Microinjection of morphine in the A7 catecholamine cell group produces opposing effects on nociception that are mediated by alpha1- and alpha2-adrenoceptors. Neuroscience 91: 979-990. [DOI] [PubMed] [Google Scholar]
- Kaplan H, Fields H ( 1991) Hyperalgesia during acute opioid abstinence: evidence for a nociceptive facilitating function of the rostral ventromedial medulla. J Neurosci 11: 1433-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkman PM, Kelly JS ( 1992) Ionic mechanisms mediating 5-hydroxytryptamine- and noradrenaline-evoked depolarization of adult rat facial motoneurones. J Physiol (Lond) 456: 473-490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado R ( 1997) Participation of noradrenergic pathways in the expression of opiate withdrawal: biochemical and pharmacological evidence. Neurosci Biobehav Rev 21: 91-104. [DOI] [PubMed] [Google Scholar]
- McCormick DA ( 1992) Cellular mechanisms underlying cholinergic and noradrenergic modulation of neuronal firing mode in the cat and guinea pig dorsal lateral geniculate nucleus. J Neurosci 12: 278-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally GP, Akil H ( 2002) Role of corticotropin-releasing hormone in the amygdala and bed nucleus of the stria terminalis in the behavioral, pain modulatory, and endocrine consequences of opiate withdrawal. Neuroscience 112: 605-617. [DOI] [PubMed] [Google Scholar]
- Meng XW, Budra B, Skinner K, Ohara PT, Fields HL ( 1997) Noradrenergic input to nociceptive modulatory neurons in the rat rostral ventromedial medulla. J Comp Neurol 377: 381-391. [PubMed] [Google Scholar]
- Nuseir K, Proudfit HK ( 2000) Bidirectional modulation of nociception by GABA neurons in the dorsolateral pontine tegmentum that tonically inhibit spinally projecting noradrenergic A7 neurons. Neuroscience 96: 773-783. [DOI] [PubMed] [Google Scholar]
- Nuseir K, Heidenreich BA, Proudfit HK ( 1999) The antinociception produced by microinjection of a cholinergic agonist in the ventromedial medulla is mediated by noradrenergic neurons in the A7 catecholamine cell group. Brain Res 822: 1-7. [DOI] [PubMed] [Google Scholar]
- Pan Z, Hirakawa N, Fields HL ( 2000) A cellular mechanism for the bidirectional pain-modulating actions of orphanin FQ/nociceptin. Neuron 26: 515-522. [DOI] [PubMed] [Google Scholar]
- Pan ZZ, Williams JT, Osborne PB ( 1990) Opioid actions on single nucleus raphe magnus neurons from rat and guinea-pig in vitro. J Physiol (Lond) 427: 519-532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan ZZ, Grudt TJ, Williams JT ( 1994) Alpha 1-adrenoceptors in rat dorsal raphe neurons: regulation of two potassium conductances. J Physiol (Lond) 478: 437-447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan ZZ, Tershner SA, Fields HL ( 1997) Cellular mechanism for anti-analgesic action of agonists of the kappa-opioid receptor. Nature 389: 382-385. [DOI] [PubMed] [Google Scholar]
- Porreca F, Burgess SE, Gardell LR, Vanderah TW, Malan Jr TP, Ossipov MH, Lappi DA, Lai J ( 2001) Inhibition of neuropathic pain by selective ablation of brainstem medullary cells expressing the μ-opioid receptor. J Neurosci 21: 5281-5288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porreca F, Ossipov MH, Gebhart GF ( 2002) Chronic pain and medullary descending facilitation. Trends Neurosci 25: 319-325. [DOI] [PubMed] [Google Scholar]
- Pralong E, Magistretti PJ ( 1995) Noradrenaline increases K-conductance and reduces glutamatergic transmission in the mouse entorhinal cortex by activation of alpha 2-adrenoreceptors. Eur J Neurosci 7: 2370-2378. [DOI] [PubMed] [Google Scholar]
- Proudfit HK ( 1988) Pharmacologic evidence for the modulation of nociception by noradrenergic neurons. Prog Brain Res 77: 357-370. [DOI] [PubMed] [Google Scholar]
- Raghavendra V, Rutkowski MD, DeLeo JA ( 2002) The role of spinal neuro-immune activation in morphine tolerance/hyperalgesia in neuropathic and sham-operated rats. J Neurosci 22: 9980-9989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raja SN ( 1995) Role of the sympathetic nervous system in acute pain and inflammation. Ann Med 27: 241-246. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Matsumoto Y, Murakami T, Hisa Y, Ibata Y ( 1996) The origins of catecholaminergic innervation in the rostral ventromedial medulla oblongata of the rat. Neurosci Lett 207: 53-56. [DOI] [PubMed] [Google Scholar]