

Abstract
Both central α-melanocyte-stimulating hormone and corticotropin-releasing hormone (CRH) have been implicated in feeding and neuroendocrine mechanisms. The anatomical overlap and functional similarities between these two neurotransmitter systems led to the hypothesis that CRH might act as one of the mediators of the central actions of the melanocortin system. By double-labeling in situ hybridization, a subpopulation of CRH neurons in the paraventricular nucleus of the hypothalamus (PVN) were shown to contain the melanocortin-4 receptor (MC4R), concentrated in the ventromedial part of the parvicellular PVN (up to 33%). Intracerebroventricular injection of melanocortin agonist MTII to conscious and freely moving rats induced a rapid induction of CRH gene transcription in the PVN. This effect was accompanied by a rise in plasma corticosterone levels in a dose- and time-dependent manner, with the maximum response observed 30 min after MTII injection. MTII (0.5 nmol)-induced increase in plasma corticosterone was attenuated by the selective MC4R antagonist HS014 (0.25-1.0 nmol) and nonselective CRH receptor antagonist α-helical-CRH9-41 (0.125-0.5 nmol) in a dose-dependent manner. Moreover, the anorectic effect of MTII was evaluated at 1, 2, and 24 hr after intracerebroventricular injection. Approximately half of the inhibitory effect of MTII (0.5 nmol) on food intake was reversed by pretreatment withα-helical-CRH9-41 at 0.25 and 0.5 nmol doses. Collectively, these results provide evidence that CRH acts as a downstream mediator of melanocortin signaling and contributes to the mechanisms by which the central melanocortin system controls feeding and neuroendocrine responses.
Keywords: melanocortin-4 receptors, MTII, corticotropin-releasing hormone, α-helical-CRH9-41, paraventricular nucleus of the hypothalamus, food intake, hypothalamo-pituitary-adrenal axis
Introduction
The central melanocortin system is defined by the agonist α-melanocyte-stimulating hormone (α-MSH), the antagonist Agouti-related protein (AGRP), and three melanocortin receptors (MCRs) that are expressed in the CNS: MC3R, MC4R, and MC5R. α-MSH is derived from the post-translational processing of the precursor protein proopiomelanocortin (POMC), which is primarily synthesized in the arcuate nucleus of the hypothalamus (Watson et al., 1978; Krieger, 1979; Gee et al., 1983), whereas AGRP is expressed exclusively in a subset of arcuate neurons, distinct from the cell population expressing POMC (Ollmann et al., 1997; Wilson et al., 1999).
The central melanocortin system has become the focus of much attention in recent years because of its critical role in the control of eating and related behaviors. For example, intracerebroventricular infusions of the agonists α-MSH and MTII (a synthetic melanocortin agonist of MC3R and MC4R) suppress food intake in several rodent models, as well as in nonhuman primates (Poggioli et al., 1986; Fan et al., 1997; Rossi et al., 1998; Thiele et al., 1998; Murphy et al., 2000; Koegler et al., 2001; Wirth et al., 2001). These anorectic effects of α-MSH and MTII can be blocked by coadministration of the physiological antagonist AGRP (Rossi et al., 1998). Moreover, central administration of AGRP alone potently stimulates food intake (Hagan et al., 2000; Kim et al., 2000; Lu et al., 2001; Wirth and Giraudo, 2001). Consistent with these pharmacological findings, genetic manipulations resulting in overexpression of AGRP or deficiency of POMC synthesis lead to hyperphagia and obesity (Ollmann et al., 1997; Krude et al., 1998; Yaswen et al., 1999). There is evidence that the α-MSH anorexigenic and AGRP orexigenic effects are integrated by MC4R. A targeted disruption of the MC4R gene in mice eliminates the anorectic effects of MTII (Marsh et al., 1999). In addition to the feeding effect, several pharmacological studies have suggested a role for the central melanocortin system in the regulation of the activity of the hypothalamo-pituitary-adrenal (HPA) axis (Calogero et al., 1988; Ludwig et al., 1998; Von Frijtag et al., 1998; Dhillo et al., 2002).
Although the neural mechanisms by which melanocortins control feeding and regulate the HPA axis remain mostly unknown, evidence suggests that CRH may be one of the candidates relaying melanocortin signaling. First, like α-MSH, central administration of corticotropin-releasing hormone (CRH) and CRH-related peptides induces anorectic effects (Rivest et al., 1989; Heinrichs and Richard, 1999; Richard et al., 2002). Second, it is well established that CRH neurons in the paraventricular nucleus of the hypothalamus (PVN) play a pivotal role in the regulation of the HPA axis (Vale et al., 1981). In addition to the functional similarities, the melanocortin and CRH systems exhibit anatomical overlap. For instance, MC4R mRNA is expressed in the medial parvicellular subdivision of the PVN, in which CRH neurons are predominantly located (Bloom et al., 1982; Hwang and Guntz, 1997). Also, studies have revealed that CRH neurons in the PVN are innervated by α-MSH neuronal terminals (Liposits et al., 1988; Mihaly et al., 2002). On the basis of such a pattern of anatomical connections and the functional similarities between melanocortin and CRH systems, the present study hypothesizes that CRH may be involved in the mechanisms by which the central melanocortin system regulates feeding behavior and HPA axis. Thus, the first experiment examines the extent of colocalization of MC4R and CRH, whereas the second set of experiments examines the effects of activation of MC4R on CRH gene transcription. Finally, we explore the role of the endogenous CRH system in mediating the anorectic and neuroendocrine responses elicited by exogenous melanocortin agonist.
Materials and Methods
Animals
Adult male Sprague Dawley rats (Charles River Laboratories, Wilmington, MA) weighing 250-300 gm were housed in plastic cages. Animals had ad libitum access to water and food (Rodent Chow 5008; Ralston Purina, St. Louis, MO) and were maintained on a 12 hr light schedule (on 6:00 A.M. to 6:00 P.M.), with constant temperature and humidity. Animals were allowed to acclimate to these housing conditions for 1 week before experiments began. All procedures described were approved by the University of Michigan Committee on Use and Care of Animals.
Surgery
After 1 week of habituation to the housing conditions, rats were anesthetized with sodium pentobarbital (50 mg/100 gm body weight, i.p.) for stereotaxic surgery. Animals were mounted on the stereotaxic instrument. To achieve the flat skull position, the incisor bar was adjusted until the heights of lambda and bregma were equal. Stainless-steel guide cannulas (26 gauge; Plastics One, Roanoke, VA) were implanted into the lateral ventricles, +1.5 mm lateral and -0.8 mm posterior to bregma, and 3.5 mm ventral to the surface of the skull. Guide cannulas were fixed to the skull with three screws and dental cement. A dummy cannula was placed into the guide cannula, protruding 1 mm below the tip of the guide cannula, to prevent blockage. After intracerebroventricular cannulation, rats were housed in individual cages. Animals were handled daily and habituated to the injection procedure for a minimum of 10 d to minimize stress. Placement of cannulas in the lateral ventricle was functionally confirmed by injection of 1 μl of angiotensin II (10 μm) on the fifth or sixth day after surgery. Only rats that drank >5 ml of tap water within 10 min after injection were used for the microinjection experiments.
Microinjection
Melanocortin agonist MTII (Phoenix Pharmaceuticals, Belmont, CA), MC4 receptor selective antagonist HS 014 (Phoenix Pharmaceuticals), and CRH receptor antagonist α-helical-CRH9-41 (Peninsula Laboratories, San Diego, CA) were freshly dissolved in 0.9% sterile saline before use. All intracerebroventricular injections were performed on conscious, unstrained, freely moving rats in their home cages. Injections were made over 1 min using a 33 gauge stainless injector connected to a 10 μl glass syringe, which was operated by an infusion pump set to dispense 2 μl of solution per minute. The injector was inserted and extended 1 mm beyond the tip of the guide cannula. Drug solutions or vehicle were infused in a volume of 2 μl delivered over 1 min. An additional minute was allowed for diffusion and prevention of backflow through the needle track before the injector was withdrawn.
Experimental protocol
Experiment 1: Expression of MC4R mRNA in CRH-containing neurons in the PVN. Three animals that were naive to any experimental procedure were used to determine the degree of colocalization of MC4R mRNA and CRH mRNA in the PVN. Animals were killed by decapitation in the early light cycle. Brains were rapidly removed and frozen in isopentane-dry ice bath at -40°C and stored at -80°C. Sections (10 μm) were cut on a cryostat through the hypothalamus. Tissue sections were stored at -80°C until processing for dual in situ hybridization.
Experiment 2: Effects of melanocortin agonist MTII on CRH gene transcription. Four groups of rats (n = 5-6 per group) were used. Two groups received intracerebroventricular saline injection, and the other two groups were injected with 1 nmol of MTII. Intracerebroventricular injections were performed in the early light cycle (8:00-10:30 A.M.). Animals were killed by decapitation at 15 or 30 min after injection. Brains were removed, frozen, sectioned, and stored as described above until processing for intronic in situ hybridization.
Experiment 3: Effects of melanocortin agonist MTII on circulating corticosterone levels. For the time course of corticosterone in response to central administration of MTII, 44 rats were used. Forty animals received intracerebroventricular injection of saline (n = 20) or 1 nmol of MTII (n = 20). Injections were made in the early light cycle (8:30-11:00 A.M.). This time was chosen because in the morning, basal activity of the hypothalamo-pituitary-adrenal axis is low, and the HPA is also highly responsive to both activating and inhibitory stimuli (Dallman et al., 1994). Saline-treated and MTII-treated animals were then randomly assigned to four groups and killed 2.5, 15, 30, and 120 min after injection. An additional group of animals (n = 4) was killed at time 0 without injection and served as baseline controls. Trunk blood was collected into heparin-treated tubes. Plasma was separated by centrifugation (3000 rpm for 15 min) and frozen until corticosterone radioimmunoassays were conducted.
The dose-effect on circulating corticosterone was determined by injecting 0, 0.1, 0.5, or 1.0 nmol doses of MTII between 8:30 and 11:00 A.M. On the basis of the time course data, a blood sample was taken by tail-nick 30 min after intracerebroventricular injection. Animals were mildly restrained while tail-nick was conducted. A tail vein was cut with a razor blade, and blood was collected in a heparinized capillary tube for the determination of plasma corticosterone levels.
Experiment 4: Effects of a CRH receptor antagonist and a selective melanocortin MC4R antagonist on MTII-induced plasma corticosterone response. In our initial experiments, we used 1 nmol of MTII to induce an increase in circulating corticosterone levels. This high-dose effect, however, could not be attenuated with several doses of the nonselective CRH antagonist α-helical-CRH9-41. Considering the potential ceiling effect of MTII and the agonistic property of α-helical-CRH9-41 for CRH receptors at high doses (Menzaghi et al., 1994), we therefore lowered the dosage of MTII to 0.5 nmol instead of trying to further elevate the dosage of α-helical-CRH9-41 for the subsequent experiments. To examine the role of CRH in the MTII-induced changes in plasma corticosterone levels, six groups of animals (n = 4-16 per group) were pretreated with α-helical-CRH9-41 at 0, 0.125, 0.25, and 0.5 nmol 10 min before injection of 0.5 nmol of MTII or saline. Blood samples were taken by tail-nick at 30 min after MTII injection. Furthermore, to determine whether the actions of MTII on the activity of the HPA axis were mediated by the MC4R, four additional groups of animals (n = 4-6 per group) were infused with the selective MC4R antagonist HS014 at 0.25, 0.5, or 1.0 nmol 10 min before injection of 0.5 of nmol MTII or saline. Blood samples were taken by tail-nick at 30 min after MTII injection. Experiments for HS014 and α-helical-CRH9-41 shared saline-saline and saline-MTII treatment groups.
Experiment 5: Effects of a CRH receptor antagonist on MTII-induced suppression of food intake. For the feeding study, spontaneous food intake and body weight were measured for each rat 2 d before injection. These two parameters were counterbalanced across different treatment groups. Food was removed 1 hr before the dark cycle. Intracerebroventricular injections were performed at 15-20 min before the dark cycle (between 5:40 and 6:00 P.M.). To examine whether α-helical-CRH9-41 could attenuate the MTII-induced anorectic effects, we first replicated the dose-response relationship between MTII and food intake. Subsequently, a moderate effective dose of MTII (0.5 nmol) was used to induce anorectic effects. α-helical-CRH9-41 at 0, 0.25, and 0.5 nmol doses was infused to block CRH receptors 10 min before injection of 0.5 nmol of MTII or saline (five groups; n = 8-16 per group). After injection, a preweighed chow hopper was placed in the home cages at the onset of the dark cycle (6:00 P.M.). Food intake was measured by weighing the remaining pellets and the spillage at 1, 2, and 24 hr after injection. A red light was provided during the measurement of food consumption in the dark cycle. To minimize disruption of food accessibility, preweighed food was provided in two sets of containers for each animal.
Single-labeling in situ hybridization for CRH heteronuclear RNA
Antisense 35S-labeled cRNA probes for rat CRH heteronuclear RNA (hnRNA) (530 mer, complementary to the intron sequence) were generated with 35S-UTP and 35S-CTP using the standard transcription system. Brain sections were fixed in 4% paraformaldehyde for 1 hr and rinsed in 2× SSC (300 mm NaCl, 30 mm Na citrate, pH 7.2). Sections were acetylated in 0.1 m triethanolamine, pH 8.0, with 0.25% acetic anhydride (for 10 min) and dehydrated through a graded series of alcohol (50-100%). 35S-labeled cRNA probes were diluted to 3× 104 cpm/μl in 50% hybridization buffer (50% formamide, 10% dextran sulfate, 3× SSC, 50 mm sodium phosphate buffer, pH 7.4, 1× Denhardt's solution, 0.1 mg/ml yeast tRNA, and 10 mm DTT). Brain sections were hybridized with 70 μl of the diluted probes at 55°C overnight. Sections were rinsed in 2× SSC and incubated in RNase A buffer containing 200 μg/ml RNase A for 1 hr at 37°C followed by a series of washes of increasing stringency (2×, 1×, 0.5×, and 0.1× SSC, for 5 min each at room temperature). Finally, the sections were placed in 0.1× SSC at 65°C for 1 hr, rinsed in distilled water, and dehydrated in a graded series of alcohol. Brain sections were exposed to x-ray film for 14 d.
Levels of CRH hnRNA were evaluated by analyzing film autoradiography. Films were visualized under a CCD camera (Model XC-77; Sony, Tokyo, Japan), and brain section images were captured and analyzed with the AIS image analysis system (Imaging Research, Ontario, Canada). Signals were expressed as optical density levels above threshold. The threshold level was defined as 3.5 SDs above the mean optical density of a fiber tract region. Results were expressed as integrated optical density, which is the product of the signal intensity and number of pixels above the threshold within the defined brain region. Equivalent planes of coronal brain sections through the PVN were ensured for analysis between animals.
Double-labeling in situ hybridization for colocalization of MC4R mRNA and CRH mRNA
cRNA probes complementary to either the rat MC4R mRNA (1040 mer; courtesy of Ira Gantz, University of Michigan, Ann Arbor, MI) or the rat CRH mRNA were labeled with 35S-UTP and 35S-CTP (for the MC4R probe) or digoxigenin (dig)-UTP (for the CRH probe) using standard transcription methods. Brain sections were hybridized with a mixture of 35S-MC4R and dig-CRH probes at 55°C overnight. Sections were rinsed in 2× SSC, treated with RNase A (200 μg/ml) for 1 hr at 37°C, and washed in 2×, 1×, 0.5×, and 0.1× SSC (for 5 min each). Sections were placed in 0.1× SSC at 65°C for 1 hr followed by immunohistochemical staining for visualization of digoxigenin-labeled CRH probe. Brain sections were treated with a blocking solution (0.1 m phosphate buffer containing 0.5% Triton X-100 and 0.25% carageenan, pH 7.5) for 4 hr, and then incubated overnight with an antibody against digoxigenin and conjugated to alkaline phosphatase (sheep anti-dig-AP, and Fab fragments, Boehringer Mannheim, Indianapolis, IN), diluted 1:15,000. After rinsing twice in both 0.1 m phosphate buffer and 0.1 m Tris buffer (30 min each), sections were incubated with color reaction buffer containing 0.45% nitroblue tetrazolium chloride (Boehringer Mannheim), 0.35% 5-bromo-4-chloro-3-indoylphosphate 4-toluidine salt (Boehringer Mannheim), 5% polyvinyl alcohol, and 0.24% levamizole. Color reaction was completed in 3 hr. Sections were rinsed in water and incubated with 0.1 m glycine buffer, pH 2.2, containing 0.5% Triton X-100 for 10 min. Finally, sections were fixed in 2.5% glutaraldehyde for 2 hr. After rinsing in water and dehydrating in a graded series of alcohol, sections were dipped in liquid emulsion (Ilford KD-5; Polysciences, Warrington, PA), air-dried, and stored in a dark box at 4°C. After 14 d of exposure to emulsion, sections were developed, fixed, dehydrated, and coverslipped in a xylene-based mounting medium (Permount; Fisher Scientific, Houston, TX). CRH mRNA labeled with nonradioactive dig-probe was visualized as a purple-blue precipitate, and MC4R mRNA labeled with radioactive probe was visualized as silver grains. For evaluation of the colocalization of MC4R mRNA and CRH mRNA, six consecutive sections through the PVN were analyzed. Signal specificity was ensured either by hybridization with sense-strand probes or pretreatment of brain sections with RNase A (200 μg/ml at 37°C for 60 min).
Plasma corticosterone analysis
Plasma corticosterone was assayed using a highly specific corticosterone antibody developed in our laboratory. Briefly, 10 μl duplicate samples of plasma were heated at 70°C for 30 min to denature corticosterone-binding protein and incubated overnight with corticosterone antibody and [3H] corticosterone (Amersham, Arlington Heights, IL). Free and bound corticosterone were separated by incubation with charcoal for 15 min.
Statistical analysis
All results were analyzed by a one-way or two-way ANOVA followed by Bonferroni-Dunn post hoc testing. Significance levels were taken as p < 0.05.
Results
Colocalization of MC4R and CRH in the PVN
The PVN consists of a number of distinct subdivisions, including five parvicellular and three magnocellular subdivisions (Swanson and Kuypers, 1980; Swanson et al., 1986). MC4R mRNA exhibited heterogeneity of distribution in the PVN with strong signals in the dorsal and medial parvicellular subdivisions. MC4R mRNA was also detectable in the anterior, ventral, and lateral parvicellular and magnocellular subdivisions of the PVN (Fig. 1). As reported previously (Swanson et al., 1986), CRH neurons are present predominantly in the medial parvicellular subdivision. CRH cells were also observed in the anterior parvicellular and anterior magnocellular subdivisions. Dual in situ hybridization histochemistry showed that a subset of CRH cells labeled for the MC4R were distributed in the anterior, ventromedial, and dorsal parvicellular subdivisions of the PVN (Figs. 2, 3). Overall, 10-15% of CRH-containing neurons in the PVN expressed the MC4R mRNA. However, up to 33% of the CRH neurons in the ventromedial part of the parvicellular PVN contained the MC4R. In contrast, the dorsolateral part of the parvicellular PVN had a low level of MC4R mRNA.
Figure 1.
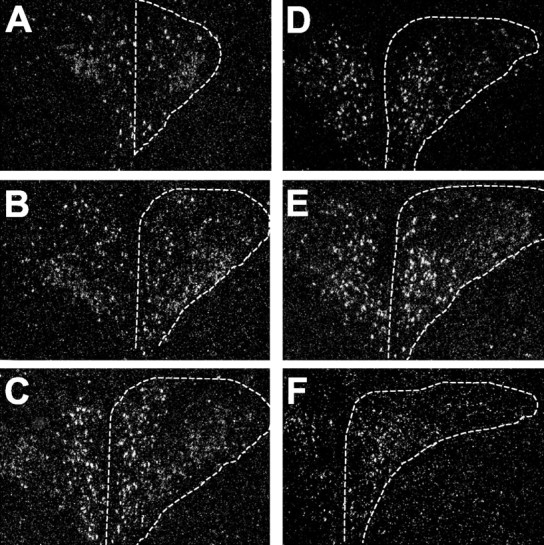
Dark-field emulsion autoradiograms showing the distribution of MC4R mRNA in the PVN of the rat hypothalamus. In situ hybridizations were performed using an MC4R mRNA probe on a series on rostral (A) to caudal (F) sections through the PVN. The sections are 100 μm apart. Locations of the PVN are indicated with dashed lines. MC4R mRNA-expressing cells are distributed in the anterior, dorsal, ventral, medial, and posterior parvicellular subdivisions of the PVN. Scattered MC4R-containing neurons were also noted in the magnocellular subdivision of the PVN.
Figure 2.
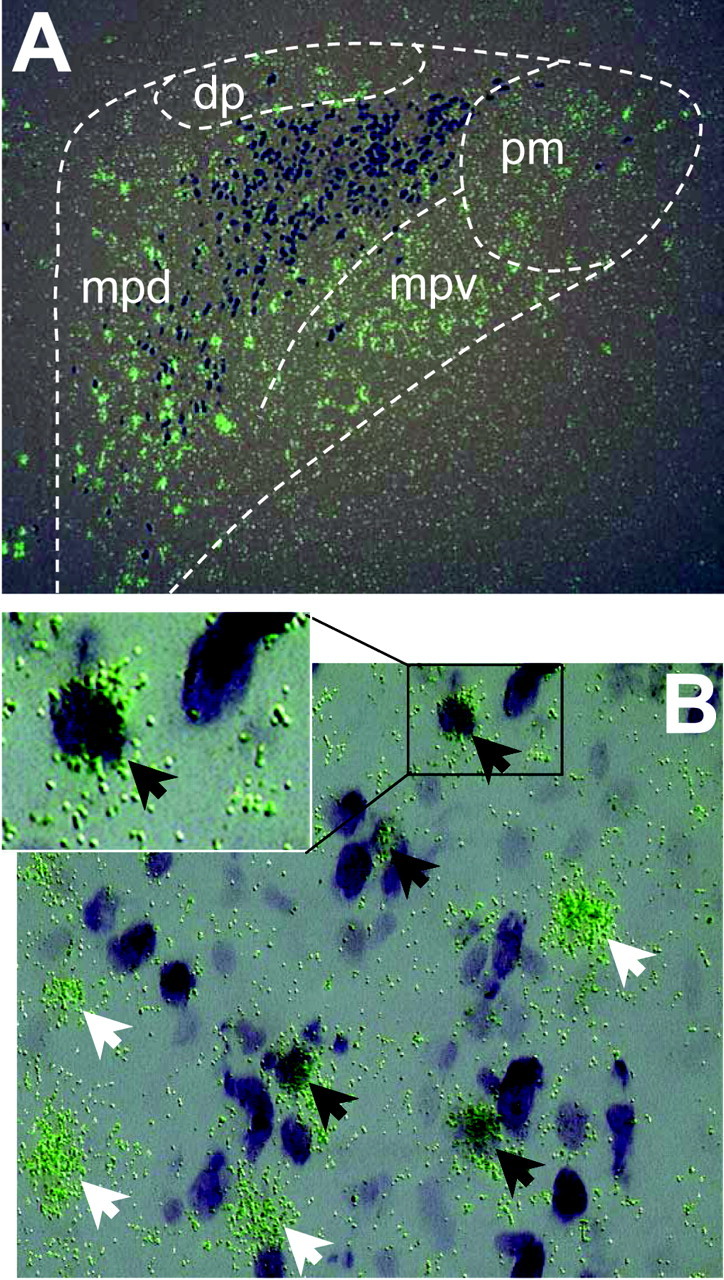
Colocalization of MC4R mRNA and CRH in the PVN. A, Microscopic images of dual in situ hybridization histochemistry of MC4R mRNA (35S-labeled riboprobe, clusters of green grains) and CRH mRNA (digoxigenin-labeled riboprobe, dark purple cells). Subdivisions of the PVN: dp, dorsal parvicellular; mpd, medial parvicellular, dorsal aspect; mpv, medial parvicellular, ventral aspect; pm, posterior magnocellular. B, High magnification of microscopic images showing double-labeled cells. Black arrows indicate cells double-labeled for MC4R mRNA and CRH mRNA. White arrows indicate cells labeled for MC4R mRNA only.
Figure 3.
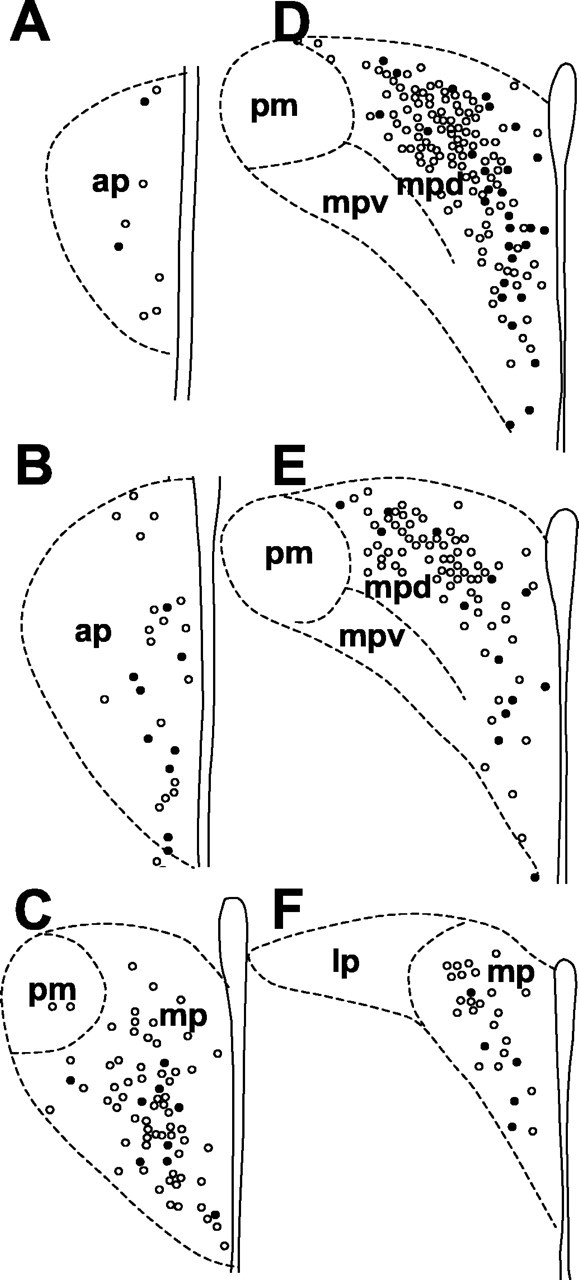
Schematic drawing indicating the distribution of CRH neurons that express MC4R mRNA in the paraventricular nucleus of the hypothalamus (A-F in a rostrocaudal sequence). Open circles indicate cells single labeled for CRH mRNA, and closed circles represent cells double labeled for both MC4R and CRH mRNA. ap, Anterior parvicellular; lp, lateral parvicellular; mp, medial parvicellular; mpd, medial parvicellular, dorsal aspect; mpv, medial parvicellular, ventral aspect; pm, posterior magnocellular.
Melanocortin signaling and CRH gene expression
To determine whether activation of the MC4R stimulated CRH gene transcription, we used a CRH hnRNA probe to detect its intronic sequence. Introns are removed rapidly post-transcriptionally before translation, so they will report on levels of de novo gene transcription. Levels of CRH hnRNA in the PVN were determined after infusion of melanocortin agonist MTII or vehicle into the lateral ventricle of conscious rats. We found that CRH hnRNA levels increased rapidly and dramatically after intracerebroventricular infusion of 1 nmol MTII, whereas animals injected with vehicle exhibited low expression of CRH hnRNA (Fig. 4A,B). An eightfold induction in CRH hnRNA was observed 15 min after injection of MTII (58.5 ± 15.8 vs 460 ± 187.6; vehicle vs MTII; p < 0.01). This induction appeared to be short-lived lived because CRH hnRNA levels declined 30 min after injection (56 ± 26.9 vs 297 ± 139.0; vehicle vs MTII; p < 0.05).
Figure 4.
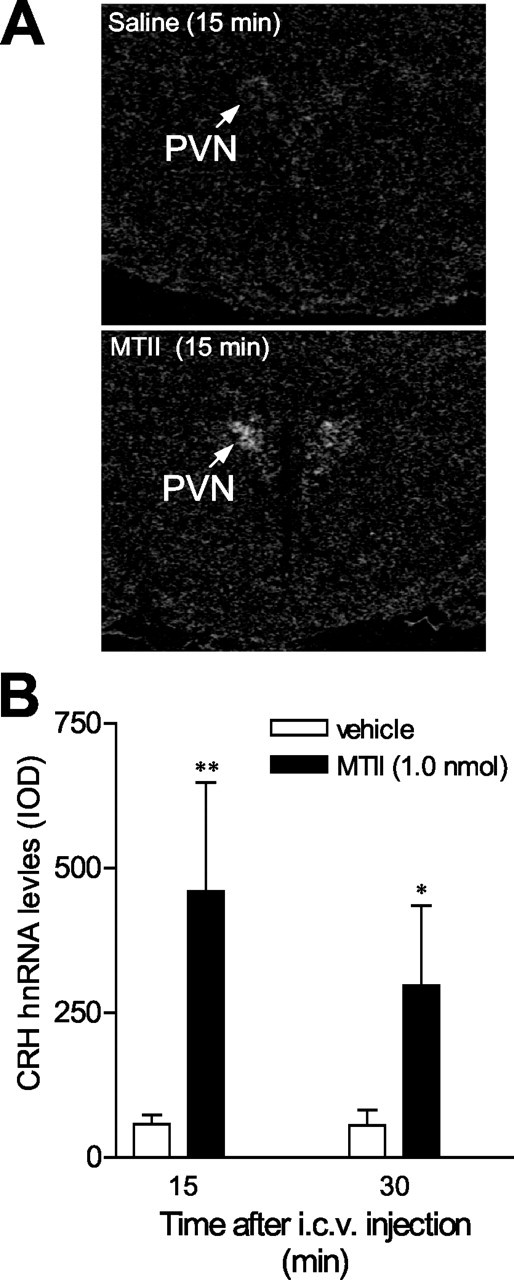
Changes in CRH heteronuclear RNA levels in the PVN after intracerebroventricular (i.c.v.) infusion of melanocortin agonist MTII (1 nmol) or vehicle into the lateral ventricle of conscious, freely moving rats. CRH hnRNA levels were detected using an intronic in situ hybridization. A, Dark-field film autoradiographs showing induction of CRH hnRNA by melanocortin agonist MTII (bottom) compared with the vehicle treatment (top). B, Induction of CRH hnRNA after intracerebroventricular injection of 1 nmol of MTII (n = 5-6 per group). *p < 0.05, **p < 0.01 versus vehicle-treated controls. IOD, Integrated optical density.
Functional interaction between melanocortin and CRH systems
Evidence from our colocalization and expression data described above (Results) suggests a functional link between CRH and melanocortin systems. We therefore decided to investigate consequences of melanocortin-induced activation of CRH expression with respect to two physiological functions, namely the hypothalamo-pituitary-adrenal axis activity and food intake.
Melanocortin signaling and corticosterone secretion
It is well established that stimulation of CRH expression in the PVN and peptide release in the median eminence lead to HPA axis activation (Rivest et al., 1989; Heinrichs and Richard, 1999; Richard et al., 2002). Thus, one physiological consequence of PVN CRH activation would be its stimulatory actions on the pituitary-adrenal axis. First, we examined the time course of the effects of MTII on circulating corticosterone. The plasma corticosterone level was analyzed at 2.5, 15, 30, and 120 min after intracerebroventricular injection of 1 nmol MTII. ANOVA conducted for plasma corticosterone concentrations revealed a significant interaction between treatment and time (F(1,38) = 8.9; p < 0.01). As shown in Figure 5A, plasma corticosterone levels increased rapidly from baseline values of 1.45 ± 0.80 μg/dl to a maximum of 25.9 ± 3.65 μg/dl 30 min after injection of 1 nmol MTII and returned to the baseline level by 120 min (3.9 ± 2.19 μg/dl). MTII treatment significantly increased plasma corticosterone levels relative to vehicle treatment at 15 and 30 min after injection. A smaller increase in plasma corticosterone levels was observed at 15 min after vehicle injection but subsided rapidly at 30 min, reflecting a small and short-lived (<30 min) stress response to intracerebroventricular injection in conscious animals.
Figure 5.
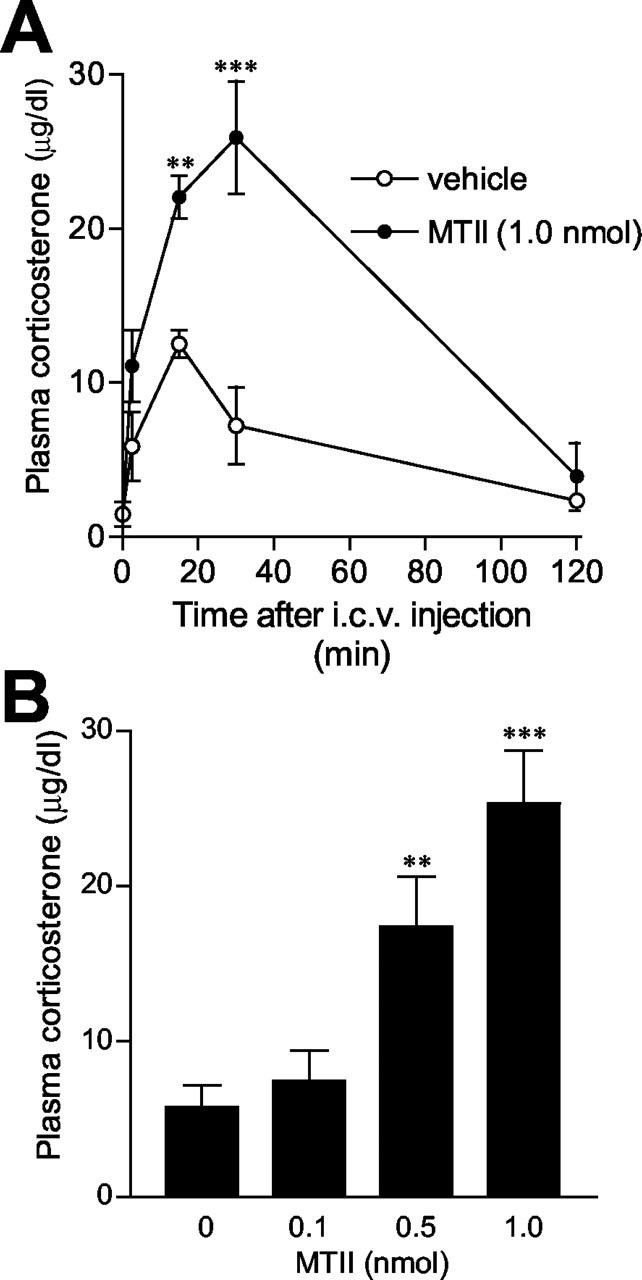
A, Time course of plasma corticosterone levels in response to intracerebroventricular (i.c.v.) injection of MTII (1 nmol) or vehicle to conscious, freely moving rats. Corticosterone levels increased rapidly after infusion of MTII and reached the peak at 30 min after injection. A smaller, short-lived increase in corticosterone levels was also observed after vehicle injection. Data are expressed as the mean ± SEM (n = 5-6 per group). B, Dose-effect of MTII. MTII dose-dependently increased plasma corticosterone levels. Data are expressed as mean ± SEM (n = 6 for vehicle; n = 4 for 0.1 nmol of MTII; n = 5 for 0.5 nmol of MTII; n = 6 for 1.0 nmol of MTII). **p < 0.01, ***p < 0.001 versus vehicle-treated controls.
The dose-response for MTII is shown in Figure 5B. One-way ANOVA revealed that MTII dose-dependently increased circulating corticosterone concentrations at 30 min after injection compared with the vehicle condition (F(3,17) = 12.12; p < 0.0005). MTII at 0.5 and 1.0 nmol doses significantly increased plasma corticosterone levels. The magnitude of the increase induced by 1 nmol MTII was comparable with the time course study with the same dose at the 30 min time point. On the basis of our preliminary observations, we chose to use a 0.5 nmol dose of MTII for additional experiments. Figure 6A shows the effect of pretreatment with the selective MC4 receptor antagonist HS014 on the MTII-induced rise in circulating corticosterone levels. ANOVA revealed a significant effect of treatment (F(5,36) = 11.462; p < 0.0001). Post hoc analyses indicated that HS014 at 0.25, 0.5, and 1.0 nmol significantly antagonized the MTII (0.5 nmol)-induced increase in plasma corticosterone levels (0.25 nmol, p < 0.01; 0.5 nmol, p < 0.05; 1.0 nmol, p < 0.0001). HS014 alone had no effect compared with the vehicle-vehicle condition (p = 0.269). Figure 6B shows the effect of pretreatment with different doses of α-helical-CRH9-41 on the MTII-elicited increase in circulating corticosterone levels. ANOVA revealed a significant effect of treatment (F(5,37) = 9.146; p < 0.0001). Post hoc analyses indicated that pretreatment with the doses of 0.25 and 0.5 nmol of α-helical-CRH9-41 significantly attenuated the effect of MTII on plasma corticosterone levels (0.25 nmol, p < 0.05; 0.5 nmol, p < 0.001). α-helical-CRH9-41 alone produced no significant effect on circulating corticosterone levels, compared with the vehicle-vehicle condition (p = 0.649).
Figure 6.
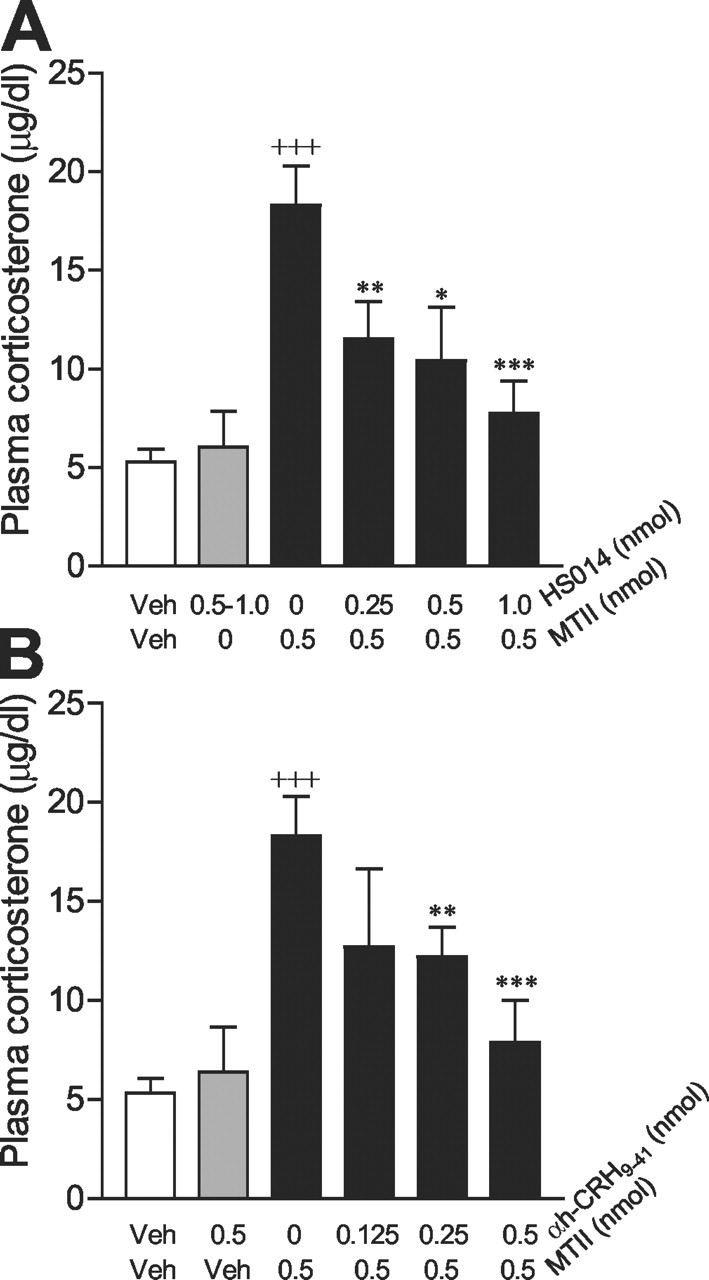
Effects of blockade of MC4Rs and CRH receptors on MTII-elicited effects on circulating corticosterone levels. A, Blockade of MC4Rs with the selective MC4R antagonist HS014. The treatment groups were as follows: vehicle (Veh) plus vehicle (n = 16), 0.5-1.0 nmol of HS014 plus vehicle (n = 6), vehicle plus 0.5 nmol of MTII (n = 8), 0. 25 nmol of HS014 plus 0.5 nmol of MTII (n = 4), 0.5 nmol of HS014 plus 0.5 nmol of MTII (n = 4), 1.0 nmol of HS014 plus 0.5 nmol of MTII (n = 6). B, Blockade of CRH receptors with the nonselective CRH receptor antagonist α-helical-CRH9-41 (αh-CRH 9-41). The treatment groups were as follows: vehicle plus vehicle (n = 16); 0.5 nmol of α-helical-CRH9-41 plus vehicle (n = 6); vehicle plus 0.5 nmol of MTII (n = 8); 0.125 nmol of α-helical-CRH9-41 plus 0.5 nmol of MTII (n = 4); 0.25 nmol of α-helical-CRH9-41 plus 0.5 nmol of MTII (n = 7); and 0.5 nmol of α-helical-CRH9-41 plus 0.5 nmol of MTII (n = 4). Data are expressed as mean ± SEM. +++p < 0.0001 versus vehicle-vehicle; *p < 0.05; **p < 0.01; ***p < 0.001 versus vehicle-0.5 nmol of MTII.
Melanocortin signaling and feeding
Because both the CRH and melanocortin systems regulate feeding behavior, we examined whether the endogenous CRH system was involved in the feeding effects induced by melanocortin signaling. As shown in Figure 7, injections of MTII at 0.1 and 1.0 nmol doses resulted in significant decreases in food intake over 2, 4, and 24 hr periods, compared with the vehicle condition. A moderate dose of MTII (0.5 nmol) was used to induce anorexia, and the role of CRH in MTII-induced anorexia was further characterized (Fig. 8). We found that MTII (0.5 nmol) significantly decreased spontaneous food intake over 1, 2, and 24 hr periods. Although the CRH receptor antagonist α-helical-CRH9-41 alone had no effect on food intake, it significantly attenuated the suppressive effect of MTII on food intake. Repeated measures of ANOVA for the drug, time, and interaction effects were as follows: F(3,41) = 20.4, p < 0.0001; F(3,41) = 552.7, p < 0.001; F(3,41) = 12.0, p < 0.001. α-helical-CRH9-41 at 0.25 and 0.5 nmol doses significantly attenuated MTII-induced suppression of food intake at 1, 2, and 24 hr (Fig. 8) after injection compared with the vehicle-vehicle group. However, neither dose completely abolished the anorectic effects of MTII at any time points. Food intake over 24 hr in the groups treated with α-helical-CRH9-41 (0.25 and 0.5 nmol) and MTII (0.5 nmol) was significantly lower than vehicle-treated controls (75% of the vehicle-vehicle condition; p < 0.01). Approximately half of the anorectic effect of MTII was reversed by the CRH receptor antagonist.
Figure 7.
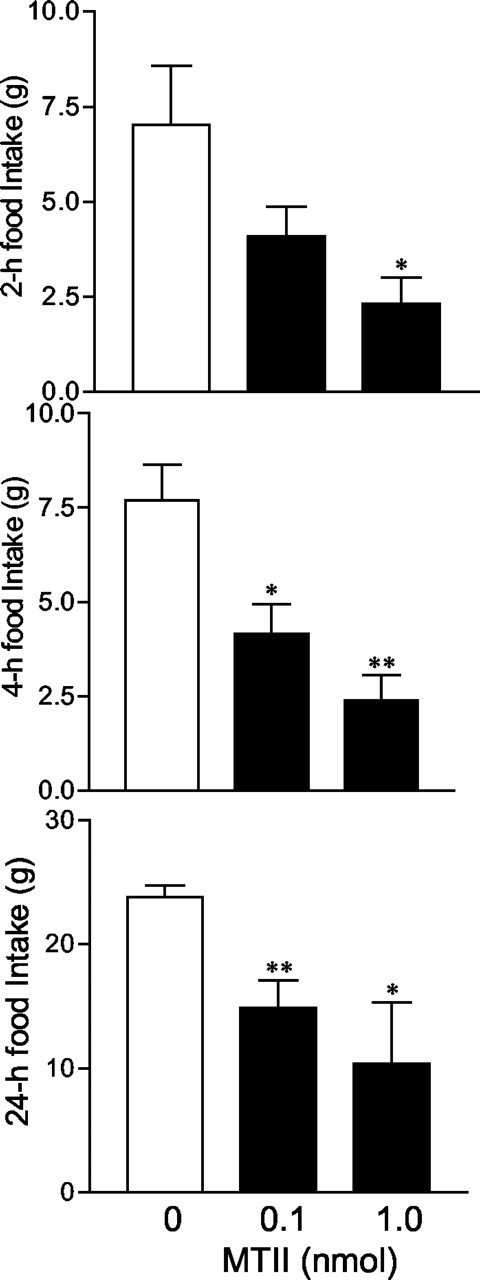
Effect of MTII on spontaneous food intake. Central administration of MTII suppresses food intake (n = 5 per group). *p < 0.05; **p < 0.01 versus the vehicle-treated group.
Figure 8.
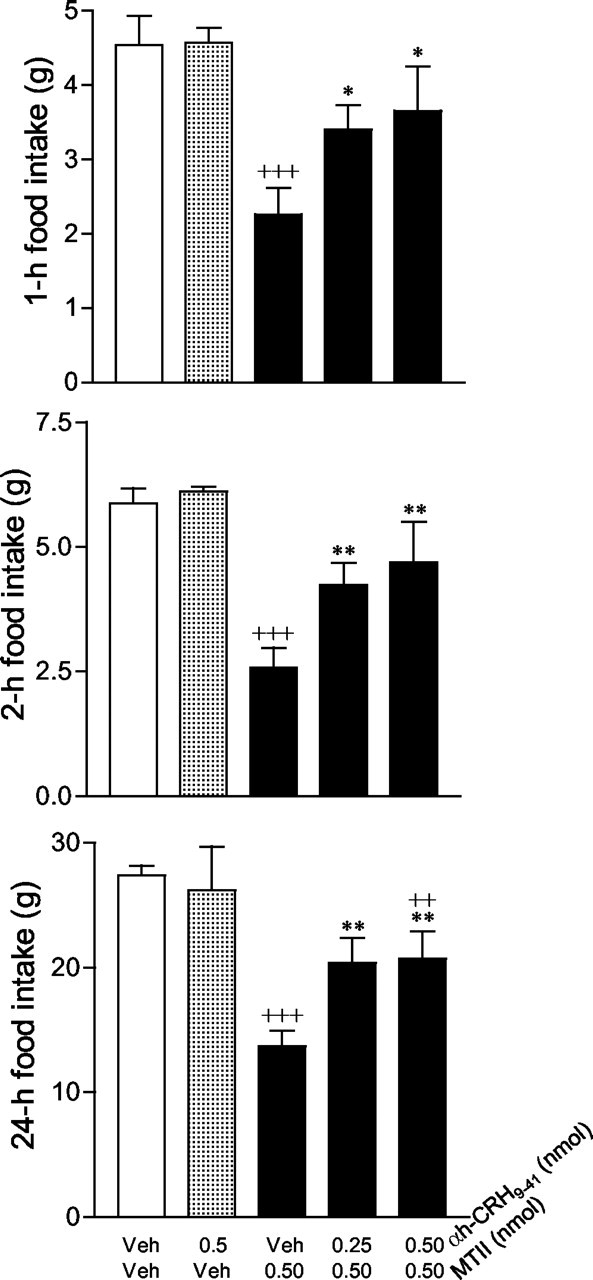
Effect of α-helical-CRH9-41 (αh-CRH 9-41) on MTII-induced suppression of food intake. The treatment groups were as follows: vehicle (Veh) plus vehicle (n = 12); 0.5 nmol of α-helical-CRH9-41 plus vehicle (n = 3); vehicle plus 0.5 nmol of MTII (n = 16); 0.25 nmol of α-helical-CRH9-41 plus 0.5 nmol of MTII (n = 8); and 0.5 nmol of α-helical-CRH9-41 plus 0.5 nmol of MTII (n = 8). Data represent the mean ± SEM, as measured by grams of food intake. +++p < 0.0001; ++p < 0.01 for comparisons with vehicle-vehicle; **p < 0.01; *p < 0.05 for comparisons with vehicle-0.5 nmol of MTII.
Discussion
We have described a range of evidence for a functional link between the central melanocortin and CRH systems. First, we showed that a subpopulation of CRH neurons contained MC4R in the PVN. Second, central administration of the melanocortin agonist MTII was observed to stimulate CRH gene transcription. Third, we confirmed and extended previous reports of the anorectic and neuroendocrine effects elicited by melanocortin agonists. We demonstrated that MTII activated the HPA axis, which appeared to be mediated by the MC4R, and it inhibited food intake in a time-related and dose-dependent manner. These effects were significantly attenuated by pretreatment with CRH antagonist α-helical-CRH9-41, indicating that CRH lies functionally downstream of melanocortin signaling and may represent one of the neurochemical pathways through which the melanocortin system regulates its feeding and neuroendocrine functions.
Interaction between melanocortin and CRH systems
Using double-labeling in situ hybridization, we have demonstrated for the first time that MC4R is expressed in a subset of CRH neurons in the PVN. These neurons are concentrated in the ventromedial part of the parvicellular PVN, in accordance with the prominent projections of α-MSH in this subdivision (Liposits et al., 1988; Bagnol et al., 1999; Mihaly et al., 2002). This coexistence of MC4R and CRH provides an anatomical basis for the possible direct interaction between endogenous α-MSH and PVN CRH neurons. Consistent with this interaction, we have found that genetic inactivation of MC4R in mice results in a dramatic decrease in CRH mRNA levels in the PVN (our unpublished data). However, previous pharmacological studies on the effect of α-MSH on CRH mRNA in the PVN have been contradictory. For example, one study found that CRH mRNA levels were unaffected by continuous infusion of α-MSH for 6d to rats with ad libitum access to food, whereas another reports that CRH mRNA expression was increased by intermittent treatment with α-MSH (every 6 hr for 64 hr) in fasted animals (Fekete et al., 2000; McMinn et al., 2000). Discrepancies between these studies may be attributable to the sizable pool of CRH mRNA, the dosage of α-MSH, and different chronic treatment regimens used in these studies.
In the present study, we used a heteronuclear RNA probe, which detected the intron sequence of CRH hnRNA, thus providing a reliable indicator of changes in de novo CRH gene transcription. CRH hnRNA levels increased dramatically 15 min after central administration of MTII and declined with increasing time. Considering that a subpopulation of CRH neurons contains MC4R, MTII may activate CRH gene transcription directly through the MC4R on these neurons. However, in contrast to the subdivision-specific colocalization pattern of MC4R and CRH, MTII-induced activation of CRH gene transcription in the PVN displayed a generalized effect without notable subdivisional preference. Consistent with our findings, Sarkar and colleagues (2002) recently reported that phosphorylated cAMP response element-binding protein, a neuronal activation marker, was induced in >50% of PVN CRH neurons by central administration of α-MSH. Thus, it is likely that the effects of melanocortin agonists on CRH neurons recruit additional mechanisms either via positive feedback within the PVN or through excitatory relaying nuclei. In fact, an ultrashort positive feedback of CRH on its own biosynthesis within the PVN has been proposed on the basis of observations of a CRH-induced increase in expression of CRH and CRH1 receptor in this region (Imaki et al., 1996; Mansi et al., 1996). Therefore, increased CRH biosynthesis and resultant release induced by MC4R activation might subsequently induce positive autoregulation and generalize its effect through the CRH1 receptor in a paracrine manner (Makino et al., 2002). It is also possible that melanocortin agonists may regulate PVN CRH gene transcription through those extra-PVN structures that contain melanocortin receptors and send excitatory inputs to the PVN, such as the amygdala.
Physiological correlates of melanocortin-CRH interaction
Central injections of the melanocortin agonist MTII to conscious, freely moving rats induced anorexia and increased the plasma concentrations of corticosterone in a time-related and dose-dependent manner. Although the MTII-induced anorexia has been thought to be mediated by the MC4R because of the inability of MTII to suppress food intake in MC4R-/- mice (Marsh et al., 1999), it was unknown which melanocortin receptor was involved in the melanocortin-induced neuroendocrine response. We have shown here that pretreatment with a selective MC4R antagonist, HS014, blocks MTII-elicited rise in plasma corticosterone levels, suggesting that MC4R mediates the MTII-induced activation of the HPA axis.
CRH has been well characterized as a primary factor regulating basal and stress-induced activity of the HPA axis (Rivest et al., 1989; Heinrichs and Richard, 1999; Richard et al., 2002). The combination of increased CRH gene transcription in the PVN and elevated plasma corticosterone levels elicited by melanocortin agonist MTII suggests that the activation of the CRH system might serve as one of the mechanisms by which melanocortins stimulate the pituitary-adrenal axis. To test this hypothesis, we used a CRH antagonist, α-helical-CRH9-41, to block CRH receptors before central administration of MTII. α-helical-CRH9-41 significantly attenuated the rise in plasma corticosterone levels induced by a moderate dose of MTII (0.5 nmol). However, in our initial experiment, α-helical-CRH9-41 failed to attenuate the effect elicited by a high dose of MTII (1 nmol), although MTII at this dose was observed to increase CRH gene transcription. One of the possibilities is that 1 nmol of MTII might induce the ceiling effect on circulating corticosterone levels, which made clear blockade impossible with the doses of α-helical-CRH9-41 used in this study. Indeed, we found that the magnitude of circulating corticosterone levels induced by 1 nmol of MTII was comparable with 30 min restraint stress (25-30 μg/dl). Another possibility is that MTII at high doses may directly influence the activity of the pituitary-adrenal axis. This notion is supported by our observations of α-MSH-immunoreactive fibers in the median eminence (Bagnol et al., 1999) and identification of melanocortin receptors in the pituitary (Lorsignol et al., 1999).
CRH is not only a major regulator of neuroendocrine responses, but it produces anorectic effects when administered centrally (Richard et al., 2002). CRH receptor antagonist α-helical-CRH9-41 has been shown to reverse CRH- and stress-induced suppression of feeding (Krahn et al., 1986; Heinrichs and Koob, 1992; Heinrichs et al., 1992; Menzaghi et al., 1993). In the present study, we found that blockade of CRH receptors with α-helical-CRH9-41 significantly attenuated MTII-induced anorexia. Central infusions of this compound alone had no effect on food intake, suggesting that its blockade of MTII-induced anorexia was not attributable to its intrinsic properties. These results suggested that altering endogenous CRH activity may serve as one of the mechanisms by which melanocortin agonists and antagonists regulate appetite. Although the precise sites for the melanocortin-CRH interaction in the control of appetite are unclear, our anatomical and expression data, along with findings from other researchers, suggest a role for the PVN. For example, α-MSH and MTII act as potent inhibitors of food intake when injected directly into the PVN (Giraudo et al., 1998; Kim et al., 2000). In addition, food deprivation decreases CRH mRNA levels in the PVN (Suemaru et al., 1986; Brady et al., 1990), and lesions of CRH neurons in this region markedly enhance feeding (Menzaghi et al., 1993). However, we cannot exclude the involvement of neural substrates other than CRH in the mediation of melanocortin-induced anorexia, because the effects of MTII were only attenuated and not completely returned to the control level by CRH receptor antagonist α-helical-CRH9-41. Using other CRH receptor antagonists that do not have agonistic effects might reveal an even higher contribution of CRH.
The biological actions of CRH are mediated via two receptor subtypes, CRH1 and CRH2. These two receptor subtypes exhibit distinct anatomical distribution and pharmacological specificity (Richard et al., 2002). Although the action of CRH on the HPA axis function has been proposed to be predominantly CRH1 mediated, it has been suggested that the feeding effects of CRH primarily involve CRH2 (Steckler and Holsboer, 1999). However, because the CRH antagonist used in the present study cannot distinguish these receptor subtypes, whether the feeding and neuroendocrine responses induced by melanocortin agonism are mediated by CRH through distinct CRH receptors will require further investigation.
Alteration of the CRH system and correlated feeding and neuroendocrine responses elicited by melanocortin agonist MTII, as indicated in the present study, raise a question. Is the feeding effect of MTII dependent on its effect on the HPA axis, or vice versa, or are they independent? The experiments here were not designed to address this question, because we chose different optimal times to investigate the responses of the HPA axis and food intake. We examined plasma corticosterone levels in response to central administration of MTII in the early light cycle, when basal activity of the HPA axis is low and the responsiveness of the HPA axis is maximal to a given stimulus, whereas the experiments for feeding were performed after the onset of the dark cycle, when animals normally eat. In view of the ability of α-MSH to decrease food intake in mice with adrenal gland deficiency (Yaswen et al., 1999), we would propose that the melanocortin-induced anorectic effects may not be dependent on its actions on the pituitary-adrenal axis.
Implications for eating and affective disorders
Hyperactivity of the HPA axis and overproduction of CRH have been implicated in anorexia nervosa and affective disorders such as stress, depression, and anxiety (Licinio et al., 1996; Arborelius et al., 1999). Here, we have shown evidence that the melanocortin system may be a key regulator of the HPA axis and CRH production. Consistent with our findings, the site at which MC4R is most expressed on CRH neurons (the ventromedial part of the parvicellular PVN) has been reported recently to play a dominant role in sustaining HPA hyperactivity in the repeated stress condition (Viau and Sawchenko, 2002). In addition, a role for the melanocortin system in human eating disorders is supported by the recent identification of association between defects in the human AGRP gene and MC4R gene with anorexia nervosa and obesity, respectively (Barsh et al., 2000; Vink et al., 2001). Given the comorbidity between eating disorders and stress-related disorders (Kennedy et al., 1994), a better understanding of the central melanocortin system in the modulation of eating and stress responses will provide insight into the etiology of these disorders.
In conclusion, the present study delineates an anatomical and functional relationship between the central melanocortin and CRH systems. Our observations provide direct evidence that melanocortin agonism functions as a rapid enhancer of CRH synthesis and that melanocortin agonists may mediate their effects on both feeding and HPA functions via activation of the CRH system.
Footnotes
This study was supported by a pilot feasibility grant from the University of Michigan Gastrointestinal Peptide Research Center, National Institutes of Health Grant P30-DK-34933 (X.Y.L.), and National Institute of Mental Health Grant MH-42251 (S.J.W.). We are grateful to Audrey Seasholtz and John Stead for helpful comments, and to Jim Stewart for his assistance in collecting blood samples.
Correspondence should be addressed to Xin-Yun Lu, Department of Pharmacology, University of Texas Health Science Center at San Antonio, San Antonio, TX 78229. E-mail: xylu@umich.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/237863-10$15.00/0
References
- Arborelius L, Owens MJ, Plotsky PM, Nemeroff CB ( 1999) The role of corticotropin-releasing factor in depression and anxiety disorders. J Endocrinol 160: 1-12. [DOI] [PubMed] [Google Scholar]
- Bagnol D, Lu XY, Kaelin CB, Day HE, Ollmann M, Gantz I, Akil H, Barsh GS, Watson SJ ( 1999) Anatomy of an endogenous antagonist: relationship between Agouti-related protein and proopiomelanocortin in brain. J Neurosci 19: RC26(1-7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barsh GS, Farooqi IS, O'Rahilly S ( 2000) Genetics of body-weight regulation. Nature 404: 644-651. [DOI] [PubMed] [Google Scholar]
- Bloom FE, Battenberg EL, Rivier J, Vale W ( 1982) Corticotropin releasing factor (CRF): immunoreactive neurones and fibers in rat hypothalamus. Regul Pept 4: 43-48. [DOI] [PubMed] [Google Scholar]
- Brady LS, Smith MA, Gold PW, Herkenham M ( 1990) Altered expression of hypothalamic neuropeptide mRNAs in food-restricted and food-deprived rats. Neuroendocrinology 52: 441-447. [DOI] [PubMed] [Google Scholar]
- Calogero AE, Gallucci WT, Gold PW, Chrousos GP ( 1988) Multiple feedback regulatory loops upon rat hypothalamic corticotropin-releasing hormone secretion. Potential clinical implications. J Clin Invest 82: 767-774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallman MF, Akana SF, Levin N, Walker CD, Bradbury MJ, Suemaru S, Scribner KS ( 1994) Corticosteroids and the control of function in the hypothalamo-pituitary-adrenal (HPA) axis. Ann NY Acad Sci 746: 22-31. [DOI] [PubMed] [Google Scholar]
- Dhillo WS, Small CJ, Seal LJ, Kim MS, Stanley SA, Murphy KG, Ghatei MA, Bloom SR ( 2002) The hypothalamic melanocortin system stimulates the hypothalamo-pituitary-adrenal axis in vitro and in vivo in male rats. Neuroendocrinology 75: 209-216. [DOI] [PubMed] [Google Scholar]
- Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD ( 1997) Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature 385: 165-168. [DOI] [PubMed] [Google Scholar]
- Fekete C, Legradi G, Mihaly E, Tatro JB, Rand WM, Lechan RM ( 2000) alpha-Melanocyte stimulating hormone prevents fasting-induced suppression of corticotropin-releasing hormone gene expression in the rat hypothalamic paraventricular nucleus. Neurosci Lett 289: 152-156. [DOI] [PubMed] [Google Scholar]
- Gee CE, Chen CL, Roberts JL, Thompson R, Watson SJ ( 1983) Identification of proopiomelanocortin neurones in rat hypothalamus by in situ cDNA-mRNA hybridization. Nature 306: 374-376. [DOI] [PubMed] [Google Scholar]
- Giraudo SQ, Billington CJ, Levine AS ( 1998) Feeding effects of hypothalamic injection of melanocortin 4 receptor ligands. Brain Res 809: 302-306. [DOI] [PubMed] [Google Scholar]
- Hagan MM, Rushing PA, Pritchard LM, Schwartz MW, Strack AM, Van Der Ploeg LH, Woods SC, Seeley RJ ( 2000) Long-term orexigenic effects of AgRP-(83—132) involve mechanisms other than melanocortin receptor blockade. Am J Physiol Regul Integr Comp Physiol 279: R47-R52. [DOI] [PubMed] [Google Scholar]
- Heinrichs SC, Koob GF ( 1992) Corticotropin-releasing factor modulates dietary preference in nutritionally and physically stressed rats. Psychopharmacology 109: 177-184. [DOI] [PubMed] [Google Scholar]
- Heinrichs SC, Richard D ( 1999) The role of corticotropin-releasing factor and urocortin in the modulation of ingestive behavior. Neuropeptides 33: 350-359. [DOI] [PubMed] [Google Scholar]
- Heinrichs SC, Cole BJ, Pich EM, Menzaghi F, Koob GF, Hauger RL ( 1992) Endogenous corticotropin-releasing factor modulates feeding induced by neuropeptide Y or a tail-pinch stressor. Peptides 13: 879-884. [DOI] [PubMed] [Google Scholar]
- Hwang BH, Guntz JM ( 1997) Downregulation of corticotropin-releasing factor mRNA, but not vasopressin mRNA, in the paraventricular hypothalamic nucleus of rats following nutritional stress. Brain Res Bull 43: 509-514. [DOI] [PubMed] [Google Scholar]
- Imaki T, Naruse M, Harada S, Chikada N, Imaki J, Onodera H, Demura H, Vale W ( 1996) Corticotropin-releasing factor up-regulates its own receptor mRNA in the paraventricular nucleus of the hypothalamus. Brain Res Mol Brain Res 38: 166-170. [DOI] [PubMed] [Google Scholar]
- Kennedy SH, Kaplan AS, Garfinkel PE, Rockert W, Toner B, Abbey SE ( 1994) Depression in anorexia nervosa and bulimia nervosa: discriminating depressive symptoms and episodes. J Psychosom Res 38: 773-782. [DOI] [PubMed] [Google Scholar]
- Kim MS, Rossi M, Abusnana S, Sunter D, Morgan DG, Small CJ, Edwards CM, Heath MM, Stanley SA, Seal LJ, Bhatti JR, Smith DM, Ghatei MA, Bloom SR ( 2000) Hypothalamic localization of the feeding effect of agouti-related peptide and alpha-melanocyte-stimulating hormone. Diabetes 49: 177-182. [DOI] [PubMed] [Google Scholar]
- Koegler FH, Grove KL, Schiffmacher A, Smith MS, Cameron JL ( 2001) Central melanocortin receptors mediate changes in food intake in the rhesus macaque. Endocrinology 142: 2586-2592. [DOI] [PubMed] [Google Scholar]
- Krahn DD, Gosnell BA, Grace M, Levine AS ( 1986) CRF antagonist partially reverses CRF- and stress-induced effects on feeding. Brain Res Bull 17: 285-289. [DOI] [PubMed] [Google Scholar]
- Krieger DT ( 1979) Physiological significance of hypophysiotrophic and pituitary hormones on brain. Eur J Clin Invest 9: 107-110. [DOI] [PubMed] [Google Scholar]
- Krude H, Biebermann H, Luck W, Horn R, Brabant G, Gruters A ( 1998) Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat Genet 19: 155-157. [DOI] [PubMed] [Google Scholar]
- Licinio J, Wong ML, Gold PW ( 1996) The hypothalamic-pituitary-adrenal axis in anorexia nervosa. Psychiatry Res 62: 75-83. [DOI] [PubMed] [Google Scholar]
- Liposits Z, Sievers L, Paull WK ( 1988) Neuropeptide-Y and ACTH-immunoreactive innervation of corticotropin releasing factor (CRF)-synthesizing neurons in the hypothalamus of the rat. An immunocytochemical analysis at the light and electron microscopic levels. Histochemistry 88: 227-234. [DOI] [PubMed] [Google Scholar]
- Lorsignol A, Vande Vijver V, Ramaekers D, Vankelecom H, Denef C ( 1999) Detection of melanocortin-3 receptor mRNA in immature rat pituitary: functional relation to gamma3-MSH-induced changes in intracellular Ca2+ concentration? J Neuroendocrinol 11: 171-179. [DOI] [PubMed] [Google Scholar]
- Lu XY, Nicholson JR, Akil H, Watson SJ ( 2001) Time course of short-term and long-term orexigenic effects of Agouti-related protein (86-132). NeuroReport 12: 1281-1284. [DOI] [PubMed] [Google Scholar]
- Ludwig DS, Mountjoy KG, Tatro JB, Gillette JA, Frederich RC, Flier JS, Maratos-Flier E ( 1998) Melanin-concentrating hormone: a functional melanocortin antagonist in the hypothalamus. Am J Physiol 274: E627-633. [DOI] [PubMed] [Google Scholar]
- Makino S, Hashimoto K, Gold PW ( 2002) Multiple feedback mechanisms activating corticotropin-releasing hormone system in the brain during stress. Pharmacol Biochem Behav 73: 147-158. [DOI] [PubMed] [Google Scholar]
- Mansi JA, Rivest S, Drolet G ( 1996) Regulation of corticotropin-releasing factor type 1 (CRF1) receptor messenger ribonucleic acid in the paraventricular nucleus of rat hypothalamus by exogenous CRF. Endocrinology 137: 4619-4629. [DOI] [PubMed] [Google Scholar]
- Marsh DJ, Hollopeter G, Huszar D, Laufer R, Yagaloff KA, Fisher SL, Burn P, Palmiter RD ( 1999) Response of melanocortin-4 receptor-deficient mice to anorectic and orexigenic peptides. Nat Genet 21: 119-122. [DOI] [PubMed] [Google Scholar]
- McMinn JE, Wilkinson CW, Havel PJ, Woods SC, Schwartz MW ( 2000) Effect of intracerebroventricular alpha-MSH on food intake, adiposity, c-Fos induction, and neuropeptide expression. Am J Physiol Regul Integr Comp Physiol 279: R695-703. [DOI] [PubMed] [Google Scholar]
- Menzaghi F, Heinrichs SC, Pich EM, Tilders FJ, Koob GF ( 1993) Functional impairment of hypothalamic corticotropin-releasing factor neurons with immunotargeted toxins enhances food intake induced by neuropeptide Y. Brain Res 618: 76-82. [DOI] [PubMed] [Google Scholar]
- Menzaghi F, Howard RL, Heinrichs SC, Vale W, Rivier J, Koob GF ( 1994) Characterization of a novel and potent corticotropin-releasing factor antagonist in rats. J Pharmacol Exp Ther 269: 564-572. [PubMed] [Google Scholar]
- Mihaly E, Fekete C, Lechan RM, Liposits Z ( 2002) Corticotropin-releasing hormone-synthesizing neurons of the human hypothalamus receive neuropeptide Y-immunoreactive innervation from neurons residing primarily outside the infundibular nucleus. J Comp Neurol 446: 235-243. [DOI] [PubMed] [Google Scholar]
- Murphy B, Nunes CN, Ronan JJ, Hanaway M, Fairhurst AM, Mellin TN ( 2000) Centrally administered MTII affects feeding, drinking, temperature, and activity in the Sprague-Dawley rat. J Appl Physiol 89: 273-282. [DOI] [PubMed] [Google Scholar]
- Ollmann MM, Wilson BD, Yang YK, Kerns JA, Chen Y, Gantz I, Barsh GS ( 1997) Antagonism of central melanocortin receptors in vitro and in vivo by agouti-related protein. Science [Erratum (1998) 281: 1615] 278:135-138. [DOI] [PubMed] [Google Scholar]
- Poggioli R, Vergoni AV, Bertolini A ( 1986) ACTH-(1-24) and alpha-MSH antagonize feeding behavior stimulated by kappa opiate agonists. Peptides 7: 843-848. [DOI] [PubMed] [Google Scholar]
- Richard D, Lin Q, Timofeeva E ( 2002) The corticotropin-releasing factor family of peptides and CRF receptors: their roles in the regulation of energy balance. Eur J Pharmacol 440: 189-197. [DOI] [PubMed] [Google Scholar]
- Rivest S, Deshaies Y, Richard D ( 1989) Effects of corticotropin-releasing factor on energy balance in rats are sex dependent. Am J Physiol 257: R1417-R1422. [DOI] [PubMed] [Google Scholar]
- Rossi M, Kim MS, Morgan DG, Small CJ, Edwards CM, Sunter D, Abusnana S, Goldstone AP, Russell SH, Stanley SA, Smith DM, Yagaloff K, Ghatei MA, Bloom SR ( 1998) A C-terminal fragment of Agouti-related protein increases feeding and antagonizes the effect of alpha-melanocyte stimulating hormone in vivo Endocrinology 139: 4428-4431. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Legradi G, Lechan RM ( 2002) Intracerebroventricular administration of alpha-melanocyte stimulating hormone increases phosphorylation of CREB in TRH- and CRH-producing neurons of the hypothalamic paraventricular nucleus. Brain Res 945: 50-59. [DOI] [PubMed] [Google Scholar]
- Steckler T, Holsboer F ( 1999) Corticotropin-releasing hormone receptor subtypes and emotion. Biol Psychiatry 46: 1480-1508. [DOI] [PubMed] [Google Scholar]
- Suemaru S, Hashimoto K, Hattori T, Inoue H, Kageyama J, Ota Z ( 1986) Starvation-induced changes in rat brain corticotropin-releasing factor (CRF) and pituitary-adrenocortical response. Life Sci 39: 1161-1166. [DOI] [PubMed] [Google Scholar]
- Swanson LW, Kuypers HG ( 1980) The paraventricular nucleus of the hypothalamus: cytoarchitectonic subdivisions and organization of projections to the pituitary, dorsal vagal complex, and spinal cord as demonstrated by retrograde fluorescence double-labeling methods. J Comp Neurol 194: 555-570. [DOI] [PubMed] [Google Scholar]
- Swanson LW, Sawchenko PE, Lind RW ( 1986) Regulation of multiple peptides in CRF parvocellular neurosecretory neurons: implications for the stress response. Prog Brain Res 68: 169-190. [DOI] [PubMed] [Google Scholar]
- Thiele TE, van Dijk G, Yagaloff KA, Fisher SL, Schwartz M, Burn P, Seeley RJ ( 1998) Central infusion of melanocortin agonist MTII in rats: assessment of c-Fos expression and taste aversion. Am J Physiol 274: R248-254. [DOI] [PubMed] [Google Scholar]
- Vale W, Spiess J, River C, River J ( 1981) Characterization of a 41-residue ovine hypothalamic peptide that stimulates secrection of corticotropin and beta-endorphin. Science 213: 1394-1397. [DOI] [PubMed] [Google Scholar]
- Viau V, Sawchenko PE ( 2002) Hypophysiotropic neurons of the paraventricular nucleus respond in spatially, temporally, and phenotypically differentiated manners to acute vs repeated restraint stress. J Comp Neurol 445: 293-307. [DOI] [PubMed] [Google Scholar]
- Vink T, Hinney A, van Elburg AA, van Goozen SH, Sandkuijl LA, Sinke RJ, Herpertz-Dahlmann BM, Hebebrand J, Remschmidt H, van Engeland H, Adan RA ( 2001) Association between an agouti-related protein gene polymorphism and anorexia nervosa. Mol Psychiatry 6: 325-328. [DOI] [PubMed] [Google Scholar]
- Von Frijtag JC, Croiset G, Gispen WH, Adan RA, Wiegant VM ( 1998) The role of central melanocortin receptors in the activation of the hypothalamus-pituitary-adrenal-axis and the induction of excessive grooming. Br J Pharmacol 123: 1503-1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson SJ, Akil H, Richard III CW, Barchas JD ( 1978) Evidence for two separate opiate peptide neuronal systems. Nature 275: 226-228. [DOI] [PubMed] [Google Scholar]
- Wilson BD, Bagnol D, Kaelin CB, Ollmann MM, Gantz I, Watson SJ, Barsh GS ( 1999) Physiological and anatomical circuitry between Agouti-related protein and leptin signaling. Endocrinology 140: 2387-2397. [DOI] [PubMed] [Google Scholar]
- Wirth MM, Giraudo SQ ( 2001) Effect of Agouti-related protein delivered to the dorsomedial nucleus of the hypothalamus on intake of a preferred versus a non-preferred diet. Brain Res 897: 169-174. [DOI] [PubMed] [Google Scholar]
- Wirth MM, Olszewski PK, Yu C, Levine AS, Giraudo SQ ( 2001) Paraventricular hypothalamic alpha-melanocyte-stimulating hormone and MTII reduce feeding without causing aversive effects. Peptides 22: 129-134. [DOI] [PubMed] [Google Scholar]
- Yaswen L, Diehl N, Brennan MB, Hochgeschwender U ( 1999) Obesity in the mouse model of pro-opiomelanocortin deficiency responds to peripheral melanocortin. Nat Med 5: 1066-1070. [DOI] [PubMed] [Google Scholar]