

Abstract
Nicotinic acetylcholine receptors (nAChRs) expressed by dopaminergic (DA) neurons have long been considered as potential therapeutic targets for the treatment of several neuropsychiatric diseases, including nicotine and cocaine addiction or Parkinson's disease. However, DA neurons express mRNAs coding for most, if not all, neuronal nAChR subunits, and the subunit composition of functional nAChRs has been difficult to establish. Immunoprecipitation experiments performed on mouse striatal extracts allowed us to identify three main types of heteromeric nAChRs (α4β2*, α6β2*, and α4α6β2*) in DA terminal fields. The functional relevance of these subtypes was then examined by studying nicotine-induced DA release in striatal synaptosomes and recording ACh-elicited currents in DA neurons fromα4, α6, α4α6, and β2 knock-out mice. Our results establish that α6β2* nAChRs are functional and sensitive to α-conotoxin MII inhibition. These receptors are mainly located on DA terminals and consistently do not contribute to DA release induced by systemic nicotine administration, as evidenced by in vivo microdialysis. In contrast, (nonα6)α4β2* nAChRs represent the majority of functional heteromeric nAChRs on DA neuronal soma. Thus, whereas a combination of α6β2* and α4β2* nAChRs may mediate the endogenous cholinergic modulation of DA release at the terminal level, somato-dendritic (nonα6)α4β2* nAChRs most likely contribute to nicotine reinforcement.
Keywords: dopamine, knock-out mice, mesencephalon, nicotinic acetylcholine receptors, striatum, α-conotoxine MII
Introduction
The dopaminergic (DA) neurons of the ventral tegmental area (VTA) and substantia nigra pars compacta (SNc) play a key role in natural and artificial reinforcement (Berridge and Robinson, 1998), motor coordination, motor associative learning, and habit formation (Gerdeman et al., 2003). In DA neurons, nicotinic ACh receptors (nAChRs) are abundantly expressed both at the somatic (SNc, VTA) and terminal [dorsal striatum, nucleus accumbens (Nac)] levels, in which they might play different physiological roles.
nAChRs located in the VTA are critically involved in nicotine addiction. Indeed, in rats, intra-VTA infusions of nicotine increase DA concentration in the Nac (Nisell et al., 1994), an effect thought to mediate the reinforcing properties of most addictive drugs (Di Chiara and Imperato, 1988). Also, intra-VTA infusion of nicotinic antagonists blocks the effect of systemic nicotine injections on accumbal DA release (Nisell et al., 1994) and disrupt nicotine self-administration (Corrigall et al., 1994). Electrophysiological experiments demonstrate that nicotine is able to increase the firing rate of DA neurons both in vitro and in vivo (Grenhoff et al., 1986; Pidoplichko et al., 1997; Picciotto et al., 1998). Although actions on nAChRs located on GABAergic interneurons or glutamatergic terminals in the VTA, or on pedunculopontine neurons, have been proposed to contribute to these effects (Nomikos et al., 2000; Corrigall et al., 2001; Mansvelder et al., 2002), somato-dendritic nAChRs expressed by VTA DA neurons remain good candidates for the primary reinforcing action of nicotine.
The functional importance of nAChRs present on DA terminals should not be underestimated, however. In the striatum, endogenous ACh exerts a strong tonic control on action potential-dependent DA release through the activation of β2-containing (β2*) presynaptic nAChRs (Zhou et al., 2001). Furthermore, intra-Nac nicotine injections produce sensitization to the locomotor stimulant effects of systemic nicotine (Kita et al., 1992), whereas intra-striatal infusion of nicotinic antagonists blocks the induction of behavioral sensitization to amphetamine-induced stereotypies (Karler et al., 1996). These findings suggest that nAChRs on DA terminals might play a role in the control of locomotor behavior and in the development of some long-lasting adaptations associated with drug abuse.
To date, 11 neuronal nAChR subunits have been cloned in mammals, 8 of which (α3-7, β2-4) are expressed in rat DA neurons (Le Novère et al., 1996; Charpantier et al., 1998; Klink et al., 2001). In vitro, α7 is known to form homopentameric nAChRs, whereas other subunits (α2, α3, α4, α6, β2, and β4) associate to form heteropentamers with a 2α, 3β stoichiometry (for review, see Role and Berg, 1996). The structural subunits α5 and β3, so called because they lack critical residues necessary to form a ligand binding site, are incorporated into heteromeric nAChRs with at least one other α subunit and one other β subunit (Ramirez-Latorre et al., 1996; Wang et al., 1996; Groot-Kormelink et al., 1998). In vivo, studies on β2 knock-out (Ko) mice have demonstrated that functional heteromeric nAChRs expressed by DA neurons in the cell body or terminal regions contain the β2 subunit (Picciotto et al., 1998; Grady et al., 2001, 2002). Recent immunoprecipitation (IPP) experiments have examined the nature of the α subunits associating with β2 to form nAChRs on DA terminals in the rat striatum (Zoli et al., 2002).
We confirmed these results in mice and extended them using Ko mice for various nAChR subunits. Furthermore, we combined several experimental approaches ranging from electrophysiological recordings of DA neurons to in vivo microdialysis to establish the functionality and the relative abundance of the nAChR subtypes identified at the somatic and terminal level. Evidence is presented that the populations of nAChRs differ in these two compartments.
Materials and Methods
Mice. All animals were used in accordance with the Centre National de la Recherche Scientifique guidelines for care and use of laboratory animals. The generation of β2-/-, α4-/-, and α6-/- mice has been described previously (Picciotto et al., 1995; Marubio et al., 1999; Champtiaux et al., 2002). For microdialysis experiments, we used α6-/- and α6+/+ littermates obtained by heterozygous matings (N4 backcross generation with C57Bl/6J mice; Charles River, Wilmington, MA). For other experiments, Ko and matching wild-type (Wt) control colonies were bred separately. For each colony, at least five couples of homozygous breeders were produced by mating heterozygous mice, obtained after 1 (α6), 7 (α4), or 12 (β2) backcrosses with C57Bl/6J mice.
Reagents. Unless specified, all chemical reagents were purchased from Sigma (St. Louis, MO). α-Conotoxin MII (αCtxMII) was synthesized as described previously (Cartier et al., 1996).
Antibody production. Polyclonal antibodies (Abs) directed against nAChR subunits were produced in rabbit and affinity purified as described previously (Vailati et al., 1999). Peptides sequence was derived from the C-terminal (COOH) or intracytoplasmic loop (Cyt) regions of the rat (R) or human (H) subunit sequence: α2(H-Cyt), CHPLRLKLSP SYHWLE SN VDAEERE V; α3(H-Cyt), TRPTS NE GNA QK PR PLYGAELSNLNC; α4(H-Cyt), SPS DQLP PQQPLE AEKASP HPSPG P; α4(R-COOH), cgPPWLAGMI; α5(R-Cyt), DRYFTQREEAE SGAGPKSRNTLEAALDC; α6(R-Cyt), GVKDPK THTKRPAKVKFTHR KE PKLLKEC; β2(H-Cyt), RQREREGAG ALFFR EAP GAD SC; β3(R-COOH), cgPALKMWIHRFH; β4(R-Cyt), VSSHTAGLPRDARLRSSGRFR EDLQEALEGc. Lowercase letters are amino acids introduced to enable coupling to carrier protein. Underlined letters are mismatches with the mouse sequence.
Ab specificity and IPP efficiency was checked on tissue extracts from Wt and Ko mice as well as on affinity-purified nAChR subtypes (all the values reported below are the mean ± SEM of three independent determinations). Anti-α4, -α5, -α6, and -β2 Abs immunoprecipitated, respectively, 82 ± 7%, 10 ± 1%, 30 ± 2%, and 92 ± 5% of 3H-Epibatidine (Epi)-labeled nAChRs in whole brain (α4, α5, β2) or striatal (α6) extracts from Wt mice compared with 0, 0, 1.9 ± 0.4%, and 1.6 ± 0.8% in the corresponding extracts from α4, α5, α6 and β2 Ko controls. Anti-α5 and -α6 Abs immunoprecipitated, respectively, 75 ± 7% of α5* nAChRs (purified from cortex) and 75 ± 3% of α6* nAChRs (purified from retina). Anti-α3 and -β4 Abs immunoprecipitated, respectively, 2.3 ± 0.1% and 2.5 ± 1% of 3H-Epi binding sites in striatal extracts compared with 74 ± 3% and 68 ± 2% in superior cervical ganglion (known to express α3 and β4 mRNA) extracts. Anti-β3 Abs immunoprecipitated 13 ± 3% and 8 ± 3% of 3H-Epi binding sites in superior colliculus and striatal extracts, respectively (projecting regions from retina and SN/VTA, where β3 mRNA is expressed), but ≈0% of 3H-Epi binding sites in superior cervical ganglion extracts, where β3 mRNA is not detected.
IPPs. Extract preparation and subsequent IPP were performed as described (Vailati et al., 1999). Briefly, the dissected tissue was homogenized in 50 mm Na phosphate, pH 7.4, 1 m NaCl, 2 mm EDTA, 2 mm EGTA, and 2 mm PMSF using a potter homogenizer and centrifuged (1 hr; 60,000 × g). The pellet was collected; homogenized in 50 mm Tris-HCl, pH 7, 150 mm NaCl, 5 mm KCl, 1 mm MgCl2, 2.5 mm CaCl2, and 2 mm PMSF; centrifuged (1 hr; 40,000 × g); and then resuspended in the same buffer containing a mixture of 20 μg/ml of the leupeptin, bestatin, pepstatin A, and aprotinin protease inhibitors. Membranes were solubilized by adding 2% Triton X-100 (2 hr, 4°C). After centrifugation (1.5 hr; 60,000 × g) to remove nonsolubilized material, extracts were preincubated with 2 μm α-bungarotoxin (αBgtx), labeled with 2 nm
3H-Epi (50-66Ci/mmole; Amersham, Arlington Heights, IL), and incubated (overnight, 0°C) with a saturating concentration (20-30 μg) of each subunit-specific Ab or preimmune IgG (to measure nonspecific IPP). IPP was induced by adding beads bound with goat anti-rabbit IgG.
To determine the subunit composition of α6* nAChRs, affinity columns were prepared with anti-α6 Abs (1 mg/ml) bound to CNBr-activated Sepharose4B (Amersham). Striatal extracts from α6+/+ animals (20-40 mice) were passed three times on the affinity column. The number of α6* nAChRs in the flow-through dropped to 2.5 ± 0.8% of total nAChRs. Bound receptors were eluted by competition with the peptide (100 μm) used for α6 Ab production. IPP was then performed as on crude striatal membrane extracts. The same procedure was used to determine the subunit content of (nonα6)* nAChRs. In this case, the flow-through of the anti-α6 affinity column or crude striatal extracts from α6-/- mice were loaded on an anti-α4 affinity column.
6-Hydroxydopamine lesions. Mice were anesthetized with chloral hydrate (400 mg/kg, i.p.) and received a bilateral stereotaxic injection (4 μl/side; 4 min) of a solution containing 1× PBS, 0.02% ascorbic acid, and 6-hydroxydopamine (6-OHDA) hydrochloride (4 μg/μl) in the dorsal striatum [Bregma coordinates (in mm): anterior, 0.4; lateral, +2.0; ventral, 3.0] using a 10 μl Hamilton syringe (26 gauge). Control animals received an injection of 1× PBS and 0.02% ascorbic acid. Extracts from dorsal striatum were prepared 7 d after lesion. The extent of DA denervation was assessed by 3H-WIN35,428 (NEN, Boston, MA) binding, a ligand for DA transporter (Zoli et al., 2002).
Ligand binding on immunoimmobilized nAChRs. Subunit-specific Abs (10 μg/ml) were bound to microwells (Maxi-Sorp; Nunc, Roskilde, Denmark) by overnight incubation at 4°C. After washing to remove unbound Abs, striatal extracts prepared from α6+/+ and α4-/- mice, or from α6-/- mice, were added to the wells prepared with anti-α6 or anti-β2 Abs, respectively. After overnight incubation at 4°C, the wells were washed, and binding experiments were performed as described (Vailati et al., 1999). For saturation experiments, 125I-Epi (2200Ci/mmole; NEN) was used at concentrations ranging from 0.005 to 2 nm. For inhibition experiments, the immunoimmobilized receptors were incubated (30 min, room temperature) with various concentrations of unlabeled ligands before adding 125I-Epi at the Kd concentration. Incubation was prolonged overnight at 4°C. Nonspecific binding was measured in the presence of 100 nm unlabeled Epi. For each ligand, data from three to six experiments were analyzed using the LIGAND program (Vailati et al., 1999).
Synaptosomes. For each experiment, the striatum from one mouse was dissected on ice. Protocols for synaptosomes preparation, 3H-DA loading, and superfusion buffer (SB) composition were described previously (el-Bizri and Clarke, 1994). The superfusion apparatus comprised 20 identical channels consisting of a length of Tygon (1.14 mm inner diameter) and Teflon tubing (0.9 mm; Polylabo, Strasbourg, France) leading to and from a polypropylene superfusion chamber. Synaptosome samples immobilized on GFA/E glass fiber filters (Gelman Science, Ann Arbor, MI) were perfused with SB (37°C; 0.5 ml/min). After a 15 min washout period, samples were collected for 7 min before and 8 min after agonist application, at 1 min intervals. Antagonists, when tested, were present in SB throughout the experiment. Basal 3H-DA release was calculated as a single exponential decay from the seven fractions collected before agonist application. Peak 3H-DA release was calculated as the maximum of (3H-DA released-baseline)/baseline from the first three fractions collected after agonist application. For a particular condition (mouse genotype, agonist concentration, antagonist), peak release value was determined as the average of at least three independent triplicate experiments. For EC50 and Hill coefficient determination, peak release values were determined for different nicotine concentrations and normalized to the peak release value obtained with 3 μm nicotine (measured in parallel). Normalized dose-response curves were fitted to the Hill equation [Release = Rmax/(1+(EC50/[Nic]) n), where Rmax is maximum release, [Nic] is nicotine concentration, and n is the Hill coefficient] using SigmaPlot 4.16 (Jandel Scientific, San Rafael, CA).
Electrophysiological recordings of DA neurons. Eleven to 14-d-old mice were sacrificed by decapitation. The brain was removed rapidly and placed in ice-cold oxygenated Krebs solution containing (in mm) 126 NaCl, 2.5 KCl, 1 MgCl2, 2 CaCl2, 1.25 NaH2PO4, 26 NaHCO3, and 25 d-glucose. Coronal slices (250 μm) were obtained using a DSK-1000 slicer (Dosaka, Kyoto, Japan) and were left at least 1 hr to recover at room temperature. A single slice was transferred to the recording chamber superfused with oxygenated Krebs at 35 ± 0.5°C, at a rate of 1.8 ml/min. Atropine (1 μm) was added to the Krebs solution to inhibit muscarinic receptors. Whole-cell recordings were obtained from SNc/VTA neurons identified using infrared videomicroscopy with Nomarksy optics. Only presumptive projection neurons of large and medium size were targeted; presumptive interneurons of small size were avoided. Patch pipettes were filled with the following (in mm): 144 K-gluconate, 3 MgCl2, 0.2 EGTA, and 10 HEPES, pH 7.2, yielding a 2-4 MΩ resistance. Recordings were performed with an Axopatch-1C (Axon Instruments, Foster City, CA) amplifier operating under current-clamp or voltage-clamp mode, filtered at 1 kHz, acquired at 3.33 kHz, and analyzed using PClamp 8 (Axon Instruments). DA neurons where selected according to the long duration of their action potentials (>2.4 msec) and long after hyperpolarization (Grace and Onn, 1989; Klink et al., 2001). Fast application of ACh (1 mm) was achieved by pressure-pulse delivery (30 psi during 30 msec) to a pipette positioned at ≈30 μm from the targeted cell. One pulse was delivered every 2 min. For each neuron, a control response was determined as the average of three consecutive responses to ACh application. Only neurons exhibiting a stable response were then bath perfused with the nicotinic antagonists αCtxMII (100 nm) and/or methyllycaconitine (MLA; 1 nm). ACh pulses were administered 6, 8, and 10 min after the onset of antagonist perfusion. The antagonized response is the mean of these three responses. As observed previously in rat DA neurons (Klink et al., 2001), the effect of these nicotinic antagonists on Ach-induced currents recorded from SNc or VTA neurons were similar. Data collected from both cell types were pooled in the analysis.
In vivo microdialysis in freely moving mice. Microdialysis and DA measurements were adapted from (Malagié et al., 2001). Mice (9-12 weeks of age; 25-30 gm) were anesthetized with chloral hydrate (400 mg/kg, i.p.), and a concentric microdialysis probe (cuprophan fibers; outer diameter, 0.30 mm; active length, 2.0 mm) was implanted into the ventral striatum [Bregma coordinates (in mm): anterior, 0; lateral, +2.0; ventral, 4.5). This site was chosen after testing several rostrocaudal levels of the ventral striatum because it gave the highest and most consistent DA responses to a systemic nicotine injection in C57Bl/6J mice. After a ≈20 hr surgery recovery period, the probe was perfused continuously (1.3 μl/min) with artificial CSF (Malagié et al., 2001). DA content of dialysate samples collected every 15 min was measured using a HPLC analytical method (Malagié et al., 2001). The limit of sensitivity for DA was ∼ 0.5 fmol per sample (signal-to-noise ratio = 2). Basal DA values were measured on the five to eight fractions collected before intraperitoneal nicotine injection (0, 0.5, or 1.0 mg/kg in 0.9% NaCl; 10 μl/g). Responses to drug administration were determined over a 120 min period. At the end of the experiment, the placement of the microdialysis probes was verified histologically.
Results
α4, α6, and β2 are the most abundant nAChR subunits in mice striatum
To determine the subunit composition of heteromeric nAChRs in DA terminal fields, we performed IPP on mouse striatal membrane extracts. For each subunit, we raised a polyclonal Ab against a peptide chosen to minimize intersubunit cross-reactivity. Ab specificity and IPP efficiency were tested on tissue extracts from Wt and α4, α5, α6, and β2 Ko mice (see Materials and Methods). Before IPP, incubation of the solubilized extracts with 2 nM 3H-Epi, a highly specific nicotinic ligand, ensured that only nAChRs were quantified by radioactive counting of the immunoprecipitated material. A preincubation step with 2 μm unlabeled αBgtx prevented the binding of 3H-Epi to homomeric α7 nAChRs.
In α6+/+ mice (Fig. 1A), the vast majority (97%) of striatal Epi binding sites could be immunoprecipitated using the anti-β2 Ab. High levels of α4* (67%) and α6* (30%) nAChRs were also detected. In contrast, the contribution of α2 (3%), α3 (2%), and β4 (2%) subunits to striatal Epi binding sites was minimal. Finally, a small fraction of heteromeric nAChRs contained the structural subunits α5 (12%) and β3 (8%). Similar results have been obtained using striatal extracts from α4+/+ mice (Fig. 1B) and from rat (Zoli et al., 2002).
Figure 1.

Subunit composition of striatal nAChRs. A, B, Solubilized striatal membrane extracts from α6-/-, α4-/-, α6+/+, and α4+/+ mice, preincubated with 2 nm3H-Epi, were immunoprecipitated with subunit-specific Abs. Results are expressed in femtomoles of immunoprecipitated 3H-Epi/mg of protein (n = 3-5; *p < 0.05 compared with matching Wt control; Student's t test). Total 3H-Epi and 125I-αBgtX binding to striatal extracts is indicated. C, IPP experiments were conducted on extracts from the dorsal striatum of α6+/+ mice after 6-OHDA lesions. Each value is the mean of two to three independent determinations on different groups of animals (*p < 0.05 compared with saline-lesioned groups; Student's t test). The extent of DA denervation was assessed by 3H-WIN35,428 binding, a ligand for DA transporter (204 ± 26 and 41 ± 3 fmol/mg protein in the saline- and 6-OHDA-lesioned groups, respectively). D, α6* and α4* nAChRs were purified from α6+/+ striatal extracts using an anti-α6 and an anti-α4 affinity column. (nonα6)α4* nAChRs were purified on an anti-α4 affinity column using striatal extracts from α6-/- mice or from α6+/+ mice after α6 depletion. The subunit content of purified nAChRs was determined by IPP. The amount of 3H-Epi immunoprecipitated is expressed as percentage of total 3H-Epi bound to the purified material before IPP (n = 3).
Because only a portion of striatal nAChRs are located on DA terminals (Clarke and Pert, 1985), we performed a selective denervation of DA terminals using 6-OHDA. In DA denervated dorsal striatum of α6+/+ mice (Fig. 1C), we observed a 90% reduction of α6* and β3* nAChRs. The number of α4* and β2* nAChRs was only reduced by 50%, whereas the number of 125I- αBgtx binding sites was not affected by the lesion. These results indicate that striatal α6* and β3* nAChRs are located on DA terminals (exclusively), whereas α7* nAChRs are not. Striatal α4* and β2* nAChRs are both expressed on DA terminals and non-DA cells or terminals.
IPP experiments were also conducted in α4-/- and α6-/- animals to check for the consequences of single subunit gene inactivation on the composition of striatal nAChRs. As reported previously (Champtiaux et al., 2002), we could not detect any significant decrease in total Epi binding in striatal extracts from α6-/- compared with α6+/+ mice (83.3 ± 5.1 and 89.4 ± 4.6 fmol/mg of protein, respectively). This finding can be explained by the upregulation of α4* nAChRs seen in α6-/- animals (Fig. 1A). In contrast, in α4-/- animals, the total number of Epi binding sites was decreased by 86%. Among the remaining Epi binding sites, 68% contained the α6 subunit and 81% the β2 subunit (Fig. 1B). Interestingly, in α4-/- animals, the number of α6* nAChRs was reduced by 57% compared with α4+/+ mice, suggesting the existence of α4α6* nAChRs in Wt mice.
Subunit composition of purified nAChRs. Existence of
α4α6β2* oligomers.
To test this hypothesis, we looked at the subunit composition of α6* nAChRs purified from α6+/+ striatal extracts on an affinity column bearing anti-α6 antibody. The proportion of α6* nAChRs in the flow-through of the affinity column dropped to 2.5 ± 0.8% of total nAChRs, demonstrating the efficiency of the depletion. Purified nAChRs were then eluted from the column by an excess of the α6 peptide, and their subunit content was determined by IPP. Using this procedure, we found that a significant fraction of α6* nAChRs also contain the α4 (35%) and/or the β3 (13%) subunits (Fig. 1D). The specificity of the purification was confirmed on α6-/- striatal extracts (yielding only 2% of the number of Epi binding sites obtained using a similar amount of α6+/+ striatal tissue).
A similar procedure, using an anti-α4 affinity column, allowed us to purify α4* nAChRs from α6+/+ striatal extracts and confirmed the presence of the α6 subunit in 14% of α4* purified nAChRs. The structural subunits α5 and β3 were also found in 9% and 11% of α4-purified receptors, respectively.
At last, purification of α4* nAChRs from α6+/+ striatal extracts after α6 depletion or from α6-/- striatal extracts revealed that (nonα6)α4* nAChRs were essentially composed of the α4 and β2 subunits [most likely (α4)2(β2)3].
Pharmacological characterization of immunoimmobilized receptors.
Having determined the composition of the major nAChR subtypes expressed in the mouse striatum, we studied their pharmacology after immunoimmobilization using anti-α6 and anti-β2 Abs. β2-Immunoimmobilized nAChRs from α6-/- animals (mainly α4β2, according to our IPP experiments and, thus, referred to as “α4β2” nAChRs for clarity), α6-immunoimmobilized nAChRs from α4-/- animals (mainly α6β2, referred to as “α6β2”), and α6-immunoimmobilized nAChRs from α6+/+ animals (containing a mix of α6β2 and α4α6β2 nAChRs) were studied. Affinity for 125I-Epi was determined by saturation experiments, whereas displacement of 125I-Epi binding to immunoimmobilized receptors was used to determine Ki values for nicotinic agonists(cytisine, Ach, and nicotine) or antagonists (dtubocurarine, dihydro-β-erythroidine, αCtxMII, MLA) (Fig. 2, Table 1).
Figure 2.
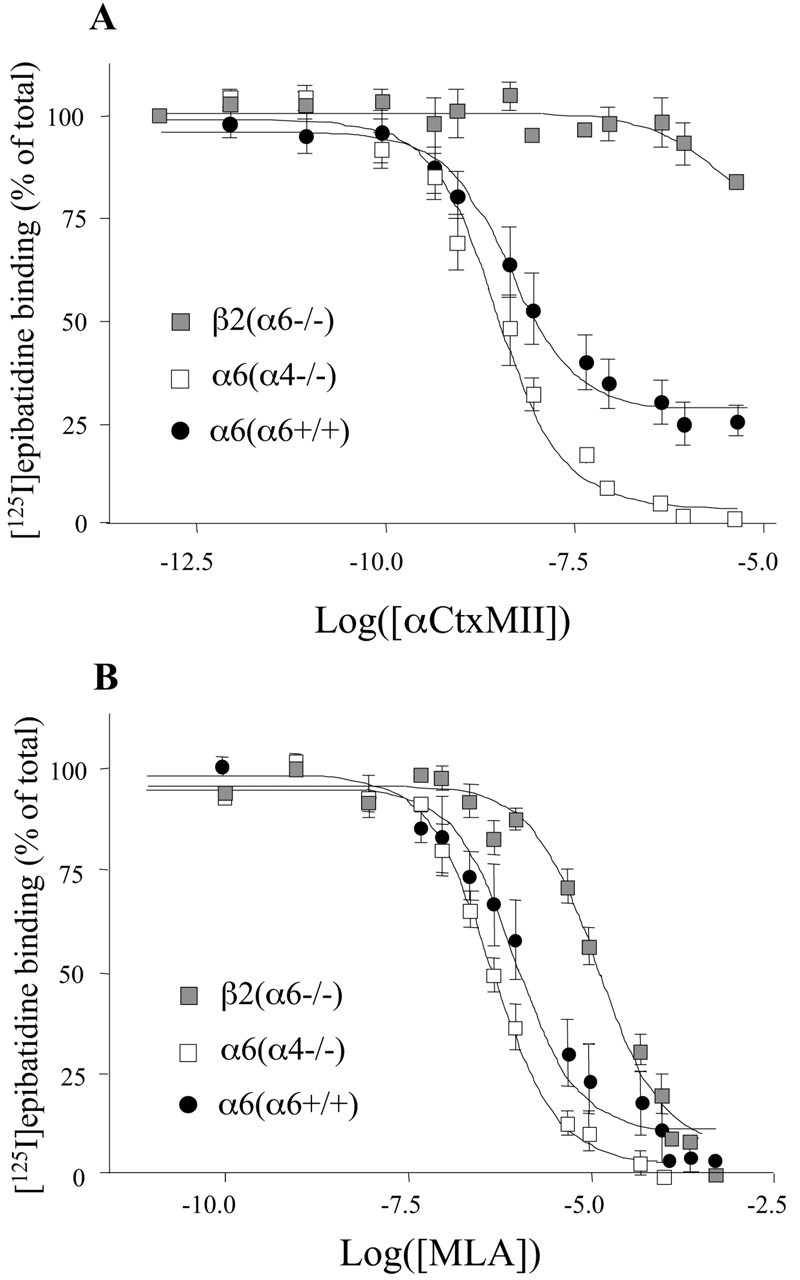
Displacement of [125I]epibatidine binding to immunoimmobilized nAChRs. αCtxMII (A) and MLA (B) were used to displace 125I-Epi binding to immunoimmobilized striatal nAChRs. Three types of nAChR preparation have been tested in these experiments: β2 immunoimmobilized nAChRs from α6-/- animals [β2(α6-/-)] and α6-immunoimmobilized nAChRs from α4-/- [α6 (α4-/-)] or α6+/+ [α6 (α6+/+)] animals. According to our IPP experiments, these three preparations are likely to contain primarily α4β2, α6β2, and a mix of α4α6β2 and α6β2 nAChRs, respectively. Data points were fitted to a one- or two-site model using the LIGAND program (see Table 1 for fitting parameter values). For both ligands, this analysis revealed the existence of a heterogeneous population of binding sites only in α6-immunoimmobilized nAChRs from Wt mice. This result is in agreement with the existence of α4α6β2 nAChRs. Each point is the mean ± SEM of at least four separate determinations.
Table 1.
Pharmacological profile of immunoimmobilized nAChRs
|
Main subtypes |
α6 (α6+/+) α6β2, α4α6β2 |
β2 (α6−/−) α4β2 |
α6 (α4−/−) α6β2 |
|---|---|---|---|
| Ligand | |||
| Kd, pM | |||
| [125I]Epibatidine | 46 (20) | 75 (34) | 34 (24) |
| Ki, nM | |||
| Cytisine | 0.8 (19) | 0.2 (14) | 1.3 (21) |
| Nicotine | 2.8 (40) | 1.7 (19) | 3.8 (31) |
| ACh | 16.6 (23) | 9.6 (23) | ND |
| Dihydro-β-erythroidine | 611 (21) | 341 (20) | ND |
| d-Tubocurarine | 5072 (20) | 20980 (31) | ND |
| αCtxMII (high affinity) | 2.14 (41) | 1.07 (34) | |
| αCtxMII (low affinity) | >10000 | >10000 | |
| MLA (high affinity) | 123 (87) | 231 (23) | |
| MLA (low affinity)
|
21900 (43)
|
14300 (27)
|
|
The Kd and Ki values were derived from saturation and competition binding curves obtained on α6-immunoimmobilized striatal nAChRs from α6+/+ and α4−/− animals [columns α6(α6+/+) and α6(α4−/−), respectively] and on the β2-immunoimmobilized nAChRs from α6−/− animals [β2(α6−/−)]. According to our IPP experiments, the main immunoimmobilized subtypes are indicated. Curves obtained from three to six separate experiments were fitted using a nonlinear least-squares analysis program and the F test. For αCtxMII and MLA, data analysis revealed the existence of a low-and high-affinity binding site in α6* nAChRs of Wt animals. Numbers in parentheses represent the coefficient of variation as a percentage.
For the agonists, cytisine exhibited a higher affinity for α4β2 nAChRs than for α6β2 nAChRs. For the antagonists, αCtxMII and MLA both showed a high affinity for α6β2 nAChRs (1.07 nM and 231 nM, respectively) and a low affinity for α4β2 nAChRs (>10 μm and 14.3 μm, respectively). In α6-immunoimmobilized nAChRs from α6+/+ mice, 125I-Epi displacement by αCtxMII and MLA was biphasic (p < 0.01 compared with a monophasic model), suggesting the existence of a heterogenous population of sites. The estimated proportion of low-affinity sites was 28 ± 4% for αCtxMII and 40 ± 11% for MLA. For both ligands, the estimated Ki value for the low- and high-affinity binding sites were in good agreement with those obtained on α4β2 and α6β2 receptors, respectively (Table 1) (Mogg et al., 2002). Our interpretation of these results is that, in a fraction of α6* nAChRs, one of the two Epi binding sites (located at the interface between the α and β subunits) is made up of an α4β2 interface with low affinity for αCtxMII and MLA. Control experiments on α6-/- animals rule out the possibility of a nonselective immunoimmobilization of (nonα6)* nAChRs by our anti-α6 Ab (data not shown). These findings correlate well with IPP results showing that α4α6β2* nAChRs are present in the striatum of Wt mice.
In striatal synaptosomes, α6β2* and α4(nonα6)β2* nAChRs each contribute 50% of the effect of nicotine on DA release
The three main heteromeric nAChR subtypes found in striatum are α4β2, α6β2, and α4α6β2. However, whereas α6* nAChRs are mainly located on DA terminals, a significant proportion of striatal α4β2* nAChRs is not present on DA terminals (Fig. 1C). To more directly evaluate the relative contributions of α4* and α6* nAChRs to DA function at the terminal level, we studied nicotine-stimulated DA release in striatal synaptosomes.
Synaptosomes loaded with 3H-DA and immobilized on glass fiber filters were perfused with a saline buffer (unless specified, synaptosomes were obtained from adult mice whole striatum). When applied on synaptosomal preparations from α4+/+ or α6+/+ mice, nicotine (3 μm for maximal effect) stimulated DA release by ∼ 100% above baseline level (peak value) (Fig. 3A). This effect was entirely blocked by mecamylamine (mean inhibition, 94 ± 3%; n = 6) or by removing Ca2+ from perfusion buffer (mean inhibition, 92 ± 4%; n = 3). In addition, a mix of NMDA (AP-5, 100 μm) and non-NMDA (CNQX, 10 μm) ionotropic glutamate receptor antagonists did not alter DA response to nicotine (104 ± 5% of control response determined in parallel using an antagonist-free buffer; n = 4), ruling out the possibility of an indirect action of nicotine via stimulation of glutamate release. The effect of 3 μm nicotine on DA release was reduced by half in both α4-/- and α6-/- animals (compared with α4+/+ and α6+/+ animals, respectively) and completely abolished in α4-/-α6-/- or β2-/- animals (Fig. 3A). The effect of KCl (23 mM) application on DA release was not altered by inactivation of any nAChR subunit (data not shown). These results demonstrate that all nAChRs mediating the effects of nicotine on synaptosomal DA release contain the β2 subunit in association with either α4or α6 or both subunits. As will be detailed in Discussion, a specific contribution of α4α6β2* nAChRs to the effect of nicotine in Wt mice is suggested by the fact that nicotine potency is decreased by eightfold as a consequence of α4 inactivation (Fig. 3C) but is unaffected by gene targeting of the α6 subunit (Fig. 3B).
Figure 3.
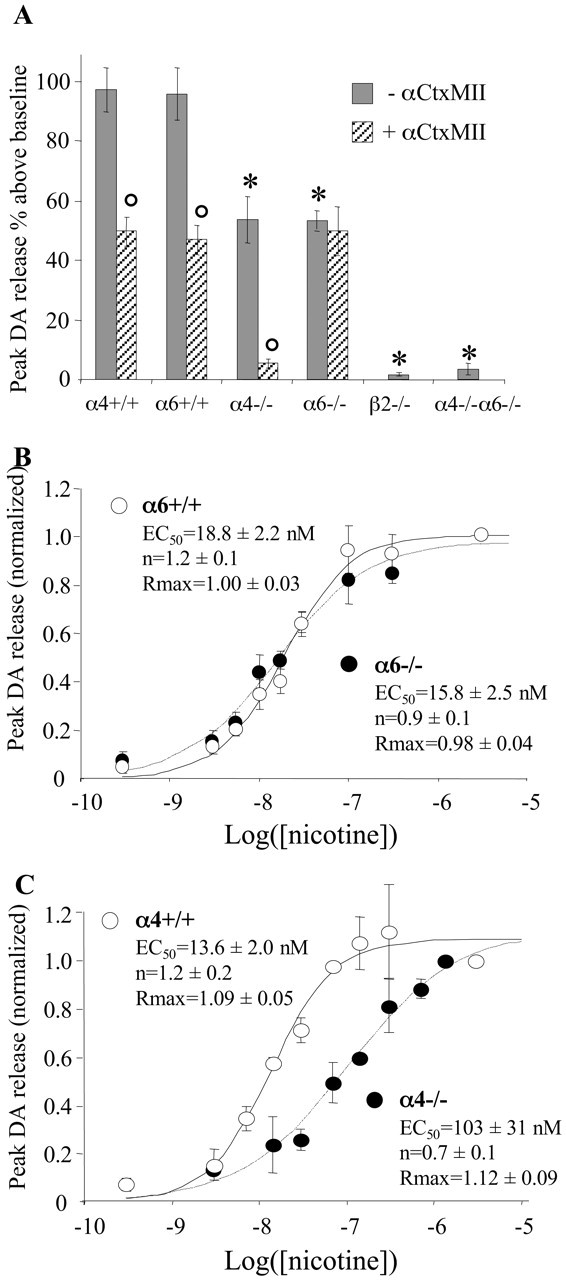
Nicotine-induced DA release in striatal synaptosomes. A, Striatal synaptosomes from α4-/-, α6-/-, β2-/-, and α4-/-α6-/- animals and their respective Wt controls were prepared and loaded with 3H-DA. The effect of 3 μm l-nicotine on DA release was determined in the presence or absence of 100 nm αCtxMII. Peak 3H-DA release values (expressed as percentage of release above baseline levels) represent the mean ± SEM of at least three determinations. *p < 0.01 compared with matching Wt control (Student's t test); °p < 0.01 compared with αCtxMII-free condition (paired Student's t test). B, C, Dose-response curves for nicotine-induced DA release. For each synaptosome preparation, data were normalized to the response obtained with 3 μm nicotine. Dose-response curves were fitted to the Hill equation: Release = Rmax/(1+(EC50/[Nic])n).
Because αCtxMII appears highly selective for α6β2* nAChRs in binding experiments (Table 1), we examined the effect of this toxin on nicotine-elicited DA release to determine the relative contribution of α6β2* and (nonα6)α4β2* nAChRs. As described previously (Kulak et al., 1997; Kaiser et al., 1998; Sharples et al., 2000), αCtxMII (100 nM) reduced the effect of 3 μm nicotine on DA release by 50% in α4+/+ or α6+/+ mice whole striatum (Fig. 3A). The inhibitory effect of αCtxMII was identical on synaptosomal preparations from the dorsal or ventral part of the striatum (mean inhibition: 49 ± 5% and 47 ± 7%, respectively; n = 3). Finally, αCtxMII completely blocked the effect of nicotine in α4-/- animals but had no inhibitory effect in α6-/- mice. Taken together, these data demonstrate that the αCtxMII-sensitive fraction of nicotine-induced DA release is mediated by α6* nAChRs and, thus, that α6* nAChRs and α4(nonα6)* nAChRs each contribute to 50% of the effect of nicotine in Wt mice striatum, regardless of the anatomical region examined (dorsal or ventral part).
To more easily compare results obtained in synaptosomal experiments and those derived from the study of ACh-induced currents in DA neurons (see below), we also studied ACh-elicited DA release in striatal synaptosomes from young (postnatal days 11-14) α6+/+ animals. In these conditions, in the presence of atropine (1 μm), αCtxMII blocked 53 ± 4% of peak DA release elicited by ACh (10 μm for maximal effect) application (n = 4).
Slow ACh-elicited currents in SNc/VTA DA neurons are mediated by α4β2* and α6β2* nAChRs
To expand the results on DA terminals to somato-dendritic nAChRs, we studied the electrophysiological response of DA neurons to ACh application. DA neurons in the SNc and VTA were identified according to their characteristic electrophysiological signature (see Materials and Methods). In Wt mice, in the presence of atropine to block muscarinic currents, fast ACh (1 mM) application elicited inward currents (Fig. 4) entirely blocked by mecamylamine (10 μm; n = 2; data not shown). Previous experiments on α7-/- and β2-/- animals have demonstrated that both α7 homomeric and β2 heteromeric nAChRs differentially contribute to this response (Klink et al., 2001). Whereas α7-mediated currents display fast activation and decay kinetics, β2* nAChRs-mediated currents exhibit a slow activation and a long duration. The α partners of the β2 subunit were investigated using α4-/-, α6-/-, and α4-/-α6-/- mice (Table 2, Fig. 4).
Figure 4.

ACh-elicited currents in SNc/VTA DA neurons. Effect of αCtxMII. Representative traces of ACh-elicited currents in DA neurons from α6+/+, α4-/-, α6-/-, and α4-/-α6-/- mice. Mean αCtxMII (100 nm) inhibition values are shown in Table 2. In α4-/-α6-/- mice, or inα4-/- animals after αCtxMII application, residual current exhibited the kinetic (fast rise and decay) and pharmacological characteristics (complete inhibition by 1 nm MLA) of α7 nAChR-mediated currents.
Table 2.
ACh-elicited current in DA neurons, waveform parameters, and effect of αCtxMII
|
|
Wt (n = 15) |
α6−/− (n = 7) |
α4−/− (n = 7) |
α4α6−/− (n = 14) |
α4−/− after αCtxMII (n = 4) |
|---|---|---|---|---|---|
| Amplitude (pA) | 202 ± 24 | 160 ± 26 | 107 ± 18* | 147 ± 43 | |
| Rise time (msec) | 225 ± 32 | 189 ± 39 | 29 ± 6** | 25 ± 7** | 15 ± 1 |
| Decay time (msec) | >1000 | >1000 | 496 ± 148° | 179 ± 43 | 162 ± 43 |
| Half width (msec) | 647 ± 97 | 670 ± 135 | 87 ± 28**° | 21 ± 3** | 27 ± 11 |
| αCtxMII inhibition (%)
|
19 ± 3†
|
6 ± 5
|
64 ± 5†
|
|
|
Rise and decay time were measured between 10 and 90% of the peak value. Half-width is the time between 50% of the peak value on the rising phase and 50% of the peak value on the falling phase. In Wt and α6−/− animals, decay time was not accurately measured because, in many cases, current did not come back to baseline levels before the end of the registering period (1.5 sec). In α4−/− animals after αCtxMII inhibition, kinetic parameters were calculated only when the amplitude of residual current was sufficient to make a precise measure (>40pA). *p<0.05, **p<0.01 compared with Wt. °p<0.05 compared with α4−/−α6−/−; Student's t test. αCtxMII inhibition is expressed as a percentage of basal response before antagonist application. Maximal amplitude and current kinetics were measured on three successive responses before and after (6 min) αCtxMII (100 nM) bath perfusion. Because αCtxMII inhibitory effect was small and irreversible (even after 10 min washout period), only neurons exhibiting a stable response to ACh application were kept in the analysis. †p<0.001 compared with αCtxMII-free condition; paired Student's t test.
The amplitude of ACh-elicited response is markedly decreased in α4-/- animals. Residual currents in α4-/- animals display faster activation kinetics than in Wt mice, but still exhibit a slow decay. This current waveform was never seen in β2-/- mice, suggesting that α4β2* nAChRs are not the only β2* nAChRs subtype contributing to the slow ACh-elicited current in Wt mice. To investigate whether α6β2* nAChRs could be involved, we studied the effect of αCtxMII (100 nM). In Wt mice, this toxin inhibited only a small fraction of ACh-gated currents (mean inhibition, 19 ± 3%; n = 15; p < 0.001; paired Student's t test), in agreement with the observation of a small (20%), non-statistically significant decrease of mean current amplitude in α6-/- animals compared to their Wt controls. The specificity of αCtxMII for α6* nAChRs was checked in α6-/- mice where it had no significant effect on ACh-elicited currents (mean inhibition, 6 ± 5%; n = 7). Two lines of evidence indicate that the modest inhibitory effect of αCtxMII seen in Wt mice is not because of partial efficacy or inadequate concentration of the antagonist. First, in α4-/- animals, αCtxMII inhibited 64% of the ACh-elicited response. Second, the residual αCtxMII-resistant current exhibited all the kinetic (Table 2) and pharmacological characteristics (complete inhibition by 1 nM MLA; n = 4) of α7* homomeric nAChRs. In α4-/-α6-/- animals in the absence of αCtxMII, the α7* homomeric current was the only residual response observed (n = 14) and was entirely blocked by MLA (n = 3).
Altogether, these results demonstrate that both α4 and α6 subunits, in association with the β2 subunit, form functional nAChRs that mediate the slow current component of ACh-elicited response in DA neurons from Wt animals.
In vivo, α6* nAChRs do not contribute to the effect of systemic nicotine injections on DA release
As mentioned previously, the reinforcing properties of nicotine have been attributed to its ability to increase accumbal DA levels (for review, see Di Chiara, 2000). To examine the involvement of α6* nAChRs in this effect, we studied the consequences of systemic nicotine injection on DA levels in the ventral striatum of α6-/- and α6+/+ mice. DA levels were monitored by in vivo microdialysis in freely moving mice. Basal extracellular DA levels in the ventral striatum did not differ between the two genotypes [12.6 ± 0.6 and 13.4 ± 0.5 fmol/20 μl of dialysate in α6+/+ (n = 11) and α6-/- animals (n = 10), respectively]. After nicotine (0.5 or 1 mg/kg; free base), but not after saline injection, an increase in striatal DA levels was observed in α6+/+ and α6-/- animals (Fig. 5A-C). When plotting DA outflow (in percentage of basal levels) as a function of time, one can measure the area under the curve (AUC), which is an estimate of DA release over the 120 min post-treatment period (Fig. 5D). A two-way ANOVA on AUC values was performed, with drug treatment (saline, 0.5 or 1 mg/kg nicotine) and mouse genotype (α6+/+ or α6-/-)as main factors. This statistical analysis revealed a significant treatment factor (F(2,30) = 8.35; p < 0.001), no significant genotype factor (F(1,30) = 0.016; p = 0.872), and no significant genotype-treatment interaction (F(2,30) = 0.311; p = 0.73).
Figure 5.
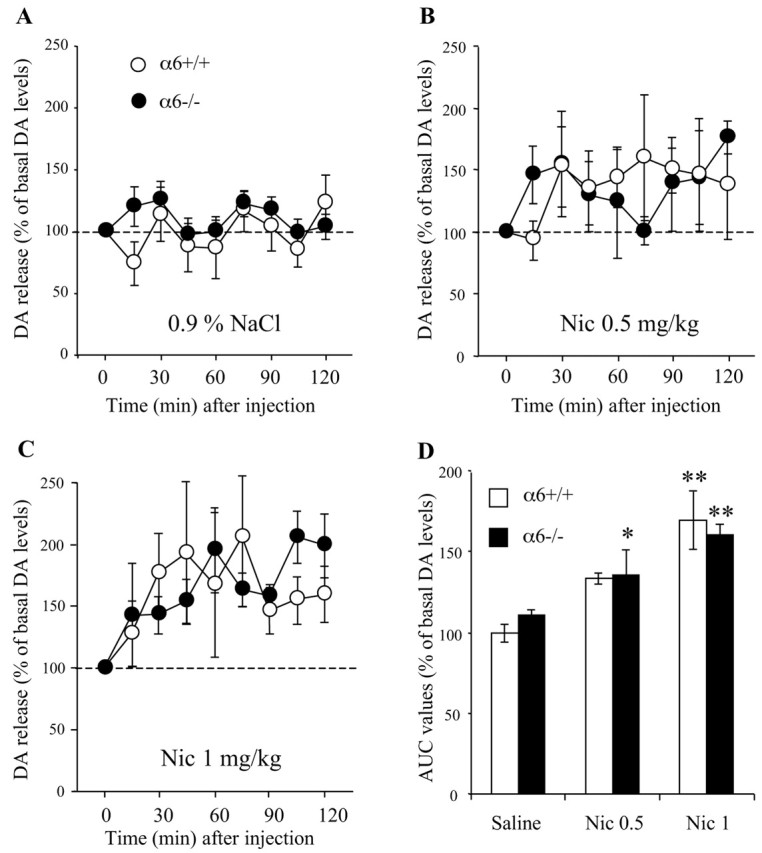
Nicotine-elicited DA release in the ventral striatum of α6-/- mice. A-C, Mice (n = 4-7 animals per group) were injected intraperitoneally with a solution of 0.5 or 1 mg kg-1 nicotine (free base), and extracellular DA levels were measured by microdialysis in the ventral striatum. Results are expressed as percentage of basal DA levels (mean of 5-8 samples collected immediately before treatment). Basal DA levels in perfusate did not differ between the two genotypes. D, AUC values, representing the amount of DA outflow collected during the 0-120 min post-treatment period, are expressed as percentage of basal values. (*p < 0.05 and **p < 0.01 relative to the corresponding control NaCl-treated group; two-way ANOVA). No statistically significant difference between α6+/+ and α6-/- mice is observed.
These results demonstrate that in the range of doses tested, the effect of systemic nicotine injections on DA release in the ventral striatum is not mediated by α6* nAChRs. However, in view of the upregulation of α4* Epi binding sites in the striatum of α6-/- mice, one could argue that the lack of influence of the α6 mutation on nicotine-induced DA release is because of functional compensation. Although we cannot rule out this hypothesis, such compensation was not seen in efflux experiments. Indeed, in striatal synaptosomes from α6-/- mice, the effect of nicotine on DA release was reduced by 50% compared with α6+/+ mice, a decrease closely corresponding to the αCtxMII-sensitive fraction of nicotine effect in Wt mice (Fig. 3A).
Discussion
In this article, we have established that the main heteromeric nAChR subtypes expressed by DA neurons contain the α4 and/or the α6 subunits in association with the β2 subunit. IPP experiments on striatal extracts demonstrate that the most abundant subunits accounting for the majority of high-affinity 3H-Epi binding sites in DA terminal fields are the α4, α6, and β2 subunits. Studying nicotine-elicited DA release in striatal synaptosomes or recording ACh-elicited currents on DA neurons further establishes the existence of two types of functional heteromeric nAChRs in DA neurons. In both types of experiments, the response to nicotinic agonists includes an αCtxMII-sensitive fraction, mediated by α6β2* nAChRs and an αCtxMII-resistant fraction attributed to α4(nonα6)β2* nAChRs. The partial inhibitory effect of αCtxMII on nicotine-induced DA release had already been observed but was initially attributed to the presence of α3β2* nAChRs on DA terminals (Kulak et al., 1997; Kaiser et al., 1998; Sharples et al., 2000). Indeed, in Xenopus oocytes, αCtxMII was reported as selective for α3β2* nAChRs (Cartier et al., 1996). We found little evidence for the presence of α3* nAChRs in the striatum, confirming previous in situ hybridization results showing a very low concentration of α3 mRNA in DA neurons (Le Novère et al., 1996). IPP experiments showed that at the terminal level only 2% of 3H-Epi binding sites contain the α3 subunit. Moreover, in α6-/- animals, the inhibitory effect of αCtxMII on nicotine-induced DA release or on ACh-elicited currents is entirely abolished. These results are in good agreement with the recent observations that high-affinity 125I-αCtxMII binding sites are preserved in the striatum of α3-/- animals (Whiteaker et al., 2002) but completely disappear in α6-/- animals (Champtiaux et al., 2002). Ultimately, experiments on α4-/-α6-/- double mutant mice establish that the α4 and α6 subunits are necessary constituents of all functional heteromeric nAChRs in DA neurons.
In addition to the simple α6β2 and α4β2 combinations, our experiments reveal the existence of an α4α6β2 subtype in DA neurons. This was initially suggested by the decrease of α6* Epi binding sites in the striatum of α4-/- animals (Fig. 1B) and was further confirmed by IPP performed on purified α6* nAChRs. With this approach, we found that 35% of α6* nAChRs also contain the α4 subunit. We also found that 28% of 125I-Epi binding sites on α6 immunoimmobilized receptors (from Wt animals) were resistant to αCtxMII displacement, most likely reflecting the existence of an α4β2 interface in α6* nAChRs. Functionally, α4α6β2* nAChRs were difficult to distinguish from α6β2* nAChRs, because αCtxMII is likely to block both subtypes. Nonetheless, a clue as to the role of α4α6β2* nAChRs is given by the study of DA release in synaptosomes. In this experiment, we found that the potency of nicotine is lower on α6β2* nAChRs, the only remaining nAChR subtype in the striatum of α4-/- mice, than on α4β2* nAChRs (Fig. 3, compare EC50 values in α4-/- and α6-/- animals). Accordingly, we found an eightfold increase in the EC50 value in α4-/- animals compared with α4+/+ mice, revealing low potency α6β2* nAChRs. However, we did not observe the expected corresponding decrease in the EC50 value in α6-/- mice. Such a decrease would have been expected if α6β2* and α4β2* nAChRs each contributed to 50% of nicotine effect in Wt mice. These findings, in contrast, are compatible with a preponderant role of α4α6β2* nAChRs in nicotine-mediated DA release in Wt mice because α4α6β2* and α4β2* nAChRs were shown to have a similar affinity for nicotine when expressed in Xenopus oocytes (Kuryatov et al., 2000). According to this hypothesis, the contribution of α6β2 nAChRs to nicotine-induced DA release, minimal in Wt mice, is only revealed in α4-/- mice. Altogether, these results demonstrate the existence of α4α6β2* nAChRs in DA neurons and suggest a preponderant role of this subtype in mediating the αCtxMII-sensitive fraction of nicotine-elicited DA release.
Whereas the main heteromeric nAChRs subtypes identified in mouse DA neurons are constituted of the α4β2, α6β2, and α4α6β2 combinations, a contribution of the structural subunits β3 and α5 was also revealed. Interestingly, the α5 subunit was found preferentially associated with α4* nAChRs (α5 was present in 9% of purified α4* nAChRs compared with only 1% of α6* nAChRs; Fig. 1D), whereas the β3 subunit was detected at similar levels in both α6* and α4* nAChRs (11% of purified α4* and 13% of purified α6* nAChRs also contained β3). These results are in partial agreement with previous observations made in the rat striatum showing a preferential association of the α5 and β3 subunits with α4* and α6* nAChRs, respectively (Zoli et al., 2002).
A main issue arising from the evidence of the diversity of nAChRs in DA neurons is whether they are differentially expressed in different parts of DA pathways. So far, most studies have only revealed subtle differences in the subunit composition or pharmacology of nAChRs expressed in the mesolimbic or nigrostriatal DA pathways (Klink et al., 2001; Grady et al., 2002; Wooltorton et al., 2003). In the present study, αCtxMII was shown to inhibit nicotine-induced DA release to the same extent in synaptosomal preparations from the dorsal and ventral parts of the striatum, suggesting a similar proportion of α6* versus α4(nonα6)* nAChRs in both regions. Our data, however, reveal a heterogeneity in the nature of nAChRs mediating nicotinic agonists effects on DA neuron soma or terminals. Whereas αCtxMII can block 50% of the effect of nicotine on DA release in striatal synaptosomes, it has only a modest effect (<20% inhibition) on ACh-elicited currents in SN/VTA DA cell bodies. Moreover, ligand binding autoradiography experiments on mouse brain sections demonstrate that the ratio of 125I-αCtxMII binding sites versus total 3H-Epi binding sites, taken as an estimate of the proportion of α6* nAChRs within the total nAChR population, is more than three times higher in the Nac than in the VTA (M. Zoli, L. Marubio, N. Champtiaux, and J. P. Changeux, unpublished observations). Finally, IPP experiments show that α6* nAChRs account for 30% of 3H-Epi binding sites in the striatum (Fig. 1A,B) but only 5% in SN/VTA (data not shown). Thus, α6* nAChRs appear to be preferentially addressed to DA nerve terminal compartment. In contrast, α7* nAChRs appear to be exclusively present on DA neuronal cell bodies. Indeed, although α7-mediated nicotinic currents could be detected at the somatic level, the lack of residual effect of nicotine on DA release in striatal synaptosomes from α4-/-α6-/- or β2-/- mice, as well as the lack of effect of 6-OHDA lesions on 125I-αBgtx binding to striatal extracts, demonstrate the absence of α7* nAChRs on DA terminals (Kaiser and Wonnacott, 2000).
The mechanisms responsible for the differential targeting of the α6* and α7* nAChRs are unknown, but the paucity of α6* nAChRs on DA neuron soma has important physiological implications. Indeed, it is known that activation of nAChRs located in the VTA, but not in the Nac, is critical for mediating DA release in the ventral striatum induced by systemic nicotine injections and maintaining nicotine self-administration (Corrigall et al., 1994; Nisell et al., 1994). In agreement with these observations, whereas the α6 subunit gene inactivation did not modify the effect of systemic nicotine on DA release in the ventral striatum, in α4-/- animals, systemic nicotine injections (0.5 and 1 mg/kg) failed to stimulate striatal DA outflow (Marubio et al., 2003). Thus, α4* rather than α6* nAChRs, presumably located in the VTA, are critical for nicotine reinforcement. In contrast, both α4* and α6* nAChRs are likely to contribute to the endogenous cholinergic modulation of DA release at the terminal level. In vitro, it was recently demonstrated that action potential-dependent DA release is under control of β2* nAChRs located on DA terminals and tonically activated by endogenous ACh (Zhou et al., 2001). The disruption of this control in β2-/- animals, leading to 80% decrease in electrically-evoked DA release, might explain the reduced sensitivity to the reinforcing effects of cocaine seen in β2-/- mice (Zachariou et al., 2001). In addition, the induction of behavioral sensitization to amphetamine-induced stereotypes can be blocked by intrastriatal infusions of nicotinic antagonists (Karler et al., 1996). By tonically controlling DA release in DA terminal fields, endogenous activation of terminal α4β2* and α6β2* nAChRs is, thus, likely to modulate important aspects of natural and artificial reinforcement.
In summary (Fig. 6), in mouse DA neurons, heteromeric nAChRs contain the β2 subunit associated with the α4 and/or the α6 subunits. On DA terminals, α6β2* and (nonα6)α4β2* nAChRs each contribute to 50% of nicotine effect on synaptosomal DA release. In contrast, (nonα6)α4β2* nAChRs mediate most of the slow ACh-elicited currents in the somato-dendritic compartment. Other evidence suggests that although α6* nAChRs are mainly located on DA nerve terminals, α7* nAChRs are only present on DA neuronal soma. Whereas a critical contribution of somato-dendritic α4β2* nAChRs to nicotine reinforcement is likely, the physiological role of α4β2* and α6β2* nAChRs expressed on striatal DA terminals remains to be investigated further.
Figure 6.

Subunit composition of functional nAChRs expressed by DA neurons. The relative proportion of the different nAChR subtypes mediating nicotine-induced DA release in striatal synaptosomes or ACh-elicited currents in SN/VTA are indicated (gray insets). The contribution of subunits in brackets to functional α6* nAChRs remains to be fully established. In addition to its direct actions on nAChRs expressed by DA neurons, nicotine might increase DA concentration in the Nac by at least two other mechanisms. By acting on α7 nAChRs located on cortical glutamatergic terminals, nicotine stimulates intra-VTA glutamate release and alters the firing pattern of DA neurons via NMDA receptor activation (Nomikos et al., 2000). At concentrations close to those found in smokers' blood, nicotine desensitizes α4β2 nAChRs on GABAergic interneurons, thus relieving the inhibitory control they exert on DA neurons (Mansvelder et al., 2002). Both direct and indirect mechanisms are likely to play a role in nicotine reinforcement.
Footnotes
This work was supported by the Collège de France, the Association pour la Recherche sur le Cancer, the Association Française contre les Myopathies, European Economic Community contracts 097038 and 097038, and European Community (EC) project HPRNCT2002OO258. F.C. is supported by grants from the Italian Ministero dell'Istruziona, l'Universita e la Ricerca (2002052378), the Progetto Strategico Neuroscienze, the Italian Ministry of Health (ICS 030.3/RA 0048), and EC contract HPRN-CT-2002-00258. J.M.M. is supported by National Institutes of Health Grants MH53631 and GM48677. We thank Drs. M. Zoli, L. M. Marubio, and N. Mechawar for critical reading of this manuscript and Drs. M. De Biasi, A. L. Beaudet, and A. Orr-Urtreger for providing α5-/- mice.
Correspondence should be addressed to Dr. Jean-Pierre Changeux, Laboratoire de Neurobiologie Moléculaire, Institut Pasteur, 25-28 rue du Dr Roux, 75015 Paris, France. E-mail: changeux@pasteur.fr.
Copyright © 2003 Society for Neuroscience 0270-6474/03/237820-10$15.00/0
References
- Berridge KC, Robinson TE ( 1998) What is the role of dopamine in reward: hedonic impact, reward learning, or incentive salience? Brain Res Rev 28: 309-369. [DOI] [PubMed] [Google Scholar]
- Cartier GE, Yoshikami D, Gray WR, Luo S, Olivera BM, McIntosh JM ( 1996) A new α-conotoxin which targets α3β2 nicotinic acetylcholine receptors. J Biol Chem 271: 7522-7528. [DOI] [PubMed] [Google Scholar]
- Champtiaux N, Han ZY, Bessis A, Rossi FM, Zoli M, Marubio L, McIntosh JM, Changeux JP ( 2002) Distribution and pharmacology of α6-containing nicotinic acetylcholine receptors analyzed with mutant mice. J Neurosci 22: 1208-1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charpantier E, Barneoud P, Moser P, Besnard F, Sgard F ( 1998) Nicotinic acetylcholine subunit mRNA expression in dopaminergic neurons of the rat substantia nigra and ventral tegmental area. NeuroReport 9: 3097-3101. [DOI] [PubMed] [Google Scholar]
- Clarke PB, Pert A ( 1985) Autoradiographic evidence for nicotine receptors on nigrostriatal and mesolimbic dopaminergic neurons. Brain Res 348: 355-358. [DOI] [PubMed] [Google Scholar]
- Corrigall WA, Coen KM, Adamson KL ( 1994) Self-administered nicotine activates the mesolimbic dopamine system through the ventral tegmental area. Brain Res 653: 278-284. [DOI] [PubMed] [Google Scholar]
- Corrigall WA, Coen KM, Zhang J, Adamson KL ( 2001) GABA mechanisms in the pedunculopontine tegmental nucleus influence particular aspects of nicotine self-administration selectively in the rat. Psychopharmacology (Berl) 158: 190-197. [DOI] [PubMed] [Google Scholar]
- Di Chiara G ( 2000) Role of dopamine in the behavioural actions of nicotine related to addiction. Eur J Pharmacol 393: 295-314. [DOI] [PubMed] [Google Scholar]
- Di Chiara G, Imperato A ( 1988) Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci USA 85: 5274-5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- el-Bizri H, Clarke PB ( 1994) Blockade of nicotinic receptor-mediated release of dopamine from striatal synaptosomes by chlorisondamine and other nicotinic antagonists administered in vitro. Br J Pharmacol 111: 406-413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdeman GL, Partridge JG, Lupica CR, Lovinger DM ( 2003) It could be habit forming: drugs of abuse and striatal synaptic plasticity. Trends Neurosci 26: 184-192. [DOI] [PubMed] [Google Scholar]
- Grace AA, Onn SP ( 1989) Morphology and electrophysiological properties of immunocytochemically identified rat dopamine neurons recorded in vitro. J Neurosci 9: 3463-3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady SR, Meinerz NM, Cao J, Reynolds AM, Picciotto MR, Changeux JP, McIntosh JM, Marks MJ, Collins AC ( 2001) Nicotinic agonists stimulate acetylcholine release from mouse interpeduncular nucleus: a function mediated by a different nAChR than dopamine release from striatum. J Neurochem 76: 258-268. [DOI] [PubMed] [Google Scholar]
- Grady SR, Murphy KL, Cao J, Marks MJ, McIntosh JM, Collins AC ( 2002) Characterization of nicotinic agonist-induced [(3)H]dopamine release from synaptosomes prepared from four mouse brain regions. J Pharmacol Exp Ther 301: 651-660. [DOI] [PubMed] [Google Scholar]
- Grenhoff J, Aston-Jones G, Svensson TH ( 1986) Nicotinic effects on the firing pattern of midbrain dopamine neurons. Acta Physiol Scand 128: 351-358. [DOI] [PubMed] [Google Scholar]
- Groot-Kormelink PJ, Luyten WH, Colquhoun D, Sivilotti LG ( 1998) A reporter mutation approach shows incorporation of the “orphan” subunit beta3 into a functional nicotinic receptor. J Biol Chem 273: 15317-15320. [DOI] [PubMed] [Google Scholar]
- Kaiser S, Wonnacott S ( 2000) α-Bungarotoxin-sensitive nicotinic receptors indirectly modulate [(3)H]dopamine release in rat striatal slices via glutamate release. Mol Pharmacol 58: 312-318. [DOI] [PubMed] [Google Scholar]
- Kaiser SA, Soliakov L, Harvey SC, Luetje CW, Wonnacott S ( 1998) Differential inhibition by α-conotoxin-MII of the nicotinic stimulation of [3H]dopamine release from rat striatal synaptosomes and slices. J Neurochem 70: 1069-1076. [DOI] [PubMed] [Google Scholar]
- Karler R, Calder LD, Bedingfield JB ( 1996) A novel nicotinic-cholinergic role in behavioral sensitization to amphetamine-induced stereotypy in mice. Brain Res 725: 192-198. [DOI] [PubMed] [Google Scholar]
- Kita T, Okamoto M, Nakashima T ( 1992) Nicotine-induced sensitization to ambulatory stimulant effect produced by daily administration into the ventral tegmental area and the nucleus accumbens in rats. Life Sci 50: 583-590. [DOI] [PubMed] [Google Scholar]
- Klink R, de Kerchove d'Exaerde A, Zoli M, Changeux JP ( 2001) Molecular and physiological diversity of nicotinic acetylcholine receptors in the midbrain dopaminergic nuclei. J Neurosci 21: 1452-1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulak JM, Nguyen TA, Olivera BM, McIntosh JM ( 1997) Alpha-conotoxin MII blocks nicotine-stimulated dopamine release in rat striatal synaptosomes. J Neurosci 17: 5263-5270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuryatov A, Olale F, Cooper J, Choi C, Lindstrom J ( 2000) Human α6 AChR subtypes: subunit composition, assembly, and pharmacological responses. Neuropharmacology 39: 2570-2590. [DOI] [PubMed] [Google Scholar]
- Le Novère N, Zoli M, Changeux JP ( 1996) Neuronal nicotinic receptor α6 subunit mRNA is selectively concentrated in catecholaminergic nuclei of the rat brain. Eur J Neurosci 8: 2428-2439. [DOI] [PubMed] [Google Scholar]
- Malagié I, Trillat AC, Bourin M, Jacquot C, Hen R, Gardier AM ( 2001) 5-HT1B autoreceptors limit the effects of selective serotonin re-uptake inhibitors in mouse hippocampus and frontal cortex. J Neurochem 76: 865-871. [DOI] [PubMed] [Google Scholar]
- Mansvelder HD, Keath JR, McGehee DS ( 2002) Synaptic mechanisms underlie nicotine-induced excitability of brain reward areas. Neuron 33: 905-919. [DOI] [PubMed] [Google Scholar]
- Marubio LM, del Mar Arroyo-Jimenez M, Cordero-Erausquin M, Lena C, Le Novere N, de Kerchove d'Exaerde A, Huchet M, Damaj MI, Changeux JP ( 1999) Reduced antinociception in mice lacking neuronal nicotinic receptor subunits. Nature 398: 805-810. [DOI] [PubMed] [Google Scholar]
- Marubio LM, Gardier AM, Durier S, David D, Klink R, del Mar Arroyo-Jimenez M, McIntosh JM, Rossi FM, Champtiaux N, Zoli M, Changeux JP ( 2003) Effects of nicotine in the dopaminergic system of mice lacking the α4 subunit of neuronal nicotinic acetylcholine receptors. Eur J Neurosci 17: 1329-1337. [DOI] [PubMed] [Google Scholar]
- Mogg AJ, Whiteaker P, McIntosh JM, Marks M, Collins AC, Wonnacott S ( 2002) Methyllycaconitine is a potent antagonist of α-conotoxin-MII-sensitive presynaptic nicotinic acetylcholine receptors in rat striatum. J Pharmacol Exp Ther 302: 197-204. [DOI] [PubMed] [Google Scholar]
- Nisell M, Nomikos GG, Svensson TH ( 1994) Systemic nicotine-induced dopamine release in the rat nucleus accumbens is regulated by nicotinic receptors in the ventral tegmental area. Synapse 16: 36-44. [DOI] [PubMed] [Google Scholar]
- Nomikos GG, Schilstrom B, Hildebrand BE, Panagis G, Grenhoff J, Svensson TH ( 2000) Role of α7 nicotinic receptors in nicotine dependence and implications for psychiatric illness. Behav Brain Res 113: 97-103. [DOI] [PubMed] [Google Scholar]
- Picciotto MR, Zoli M, Lena C, Bessis A, Lallemand Y, LeNovere N, Vincent P, Pich EM, Brulet P, Changeux JP ( 1995) Abnormal avoidance learning in mice lacking functional high-affinity nicotine receptor in the brain. Nature 374: 65-67. [DOI] [PubMed] [Google Scholar]
- Picciotto MR, Zoli M, Rimondini R, Lena C, Marubio LM, Pich EM, Fuxe K, Changeux JP ( 1998) Acetylcholine receptors containing the β2 subunit are involved in the reinforcing properties of nicotine. Nature 391: 173-177. [DOI] [PubMed] [Google Scholar]
- Pidoplichko VI, DeBiasi M, Williams JT, Dani JA ( 1997) Nicotine activates and desensitizes midbrain dopamine neurons. Nature 390: 401-404. [DOI] [PubMed] [Google Scholar]
- Ramirez-Latorre J, Yu CR, Qu X, Perin F, Karlin A, Role L ( 1996) Functional contributions of α5 subunit to neuronal acetylcholine receptor channels. Nature 380: 347-351. [DOI] [PubMed] [Google Scholar]
- Role LW, Berg DK ( 1996) Nicotinic receptors in the development and modulation of CNS synapses. Neuron 16: 1077-1085. [DOI] [PubMed] [Google Scholar]
- Sharples CG, Kaiser S, Soliakov L, Marks MJ, Collins AC, Washburn M, Wright E, Spencer JA, Gallagher T, Whiteaker P, Wonnacott S ( 2000) UB-165: a novel nicotinic agonist with subtype selectivity implicates the α4β2* subtype in the modulation of dopamine release from rat striatal synaptosomes. J Neurosci 20: 2783-2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vailati S, Hanke W, Bejan A, Barabino B, Longhi R, Balestra B, Moretti M, Clementi F, Gotti C ( 1999) Functional alpha6-containing nicotinic receptors are present in chick retina. Mol Pharmacol 56: 11-19. [DOI] [PubMed] [Google Scholar]
- Wang F, Gerzanich V, Wells GB, Anand R, Peng X, Keyser K, Lindstrom J ( 1996) Assembly of human neuronal nicotinic receptor α5 subunits with α3, β2, and β4 subunits. J Biol Chem 271: 17656-17665. [DOI] [PubMed] [Google Scholar]
- Whiteaker P, Peterson CG, Xu W, McIntosh JM, Paylor R, Beaudet AL, Collins AC, Marks MJ ( 2002) Involvement of the alpha3 subunit in central nicotinic binding populations. J Neurosci 22: 2522-2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooltorton JR, Pidoplichko VI, Broide RS, Dani JA ( 2003) Differential desensitization and distribution of nicotinic acetylcholine receptor subtypes in midbrain dopamine areas. J Neurosci 23: 3176-3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachariou V, Caldarone BJ, Weathers-Lowin A, George TP, Elsworth JD, Roth RH, Changeux JP, Picciotto MR ( 2001) Nicotine receptor inactivation decreases sensitivity to cocaine. Neuropsychopharmacology 24: 576-589. [DOI] [PubMed] [Google Scholar]
- Zhou FM, Liang Y, Dani JA ( 2001) Endogenous nicotinic cholinergic activity regulates dopamine release in the striatum. Nat Neurosci 4: 1224-1229. [DOI] [PubMed] [Google Scholar]
- Zoli M, Moretti M, Zanardi A, McIntosh JM, Clementi F, Gotti C ( 2002) Identification of the nicotinic receptor subtypes expressed on dopaminergic terminals in the rat striatum. J Neurosci 22: 8785-8789. [DOI] [PMC free article] [PubMed] [Google Scholar]