

Abstract
Synaptic plasticity requires an activity-dependent, rapid, and long-lasting modification of synaptic character, including morphology and coupling strength. Here we show that a serine protease, neuropsin, directly and specifically modifies the synaptic adhesion molecule L1, which was localized to the presynaptic site of the asymmetric synapse in the mouse hippocampus. Increased neural activity triggered the rapid, transient activation of the precursor form of neuropsin in an NMDA receptor-dependent manner. The activated neuropsin immediately cleaved L1 and released a neuropsin-specific extracellular 180 kDa fragment. This neuropsin-specific L1-cleaving system is involved in NMDA receptor-dependent synaptic plasticity, such as the Schaffer collateral long-term potentiation.
Keywords: serine protease, neuropsin, proteolysis, neural cell adhesion molecule L1, synaptic plasticity, hippocampus, Schaffer collateral long-term potentiation
Introduction
The dynamic regulation of synaptic strength by neural activity (activity-dependent synaptic plasticity) is a fundamental component of normal brain functions, such as learning and memory. Studies that have focused on this plasticity have concentrated on the postsynaptic intracellular signal transduction that leads to the regulation of neurotransmitter receptor function (Soderling and Derkach, 2000). In addition, evidence is accumulating that increased neural activity induces the rapid, synaptic morphological change that occurs during neural plasticity events (Engert and Bonhoeffer, 1999; Maletic-Savatic et al., 1999). Such morphological change is thought by many to require a rapid modification of the extracellular synaptic environment [e.g., cell adhesion and extracellular matrix (ECM) molecules] (Bailey and Kandel, 1993; Abel and Kandel, 1998; Benson et al., 2000; Colicos et al., 2001). However, the molecular mechanism underlying rapid extracellular synaptic modifications remain poorly understood. Some researchers believe that cell adhesion molecules (CAMs) are key factors in the modification of synaptic morphology. A number of studies have shown that CAMs mediate the membrane-to-membrane adhesion between presynaptic boutons and postsynapses, synapses and ECM components, and synapses and peripheral astrocytic processes (Persohn and Schachner, 1990; Miller et al., 1993; Einheber et al., 1996; Benson and Tanaka, 1998; Nishimura et al., 1998; Benson et al., 2000). In addition, CAMs play an important role in synaptic plasticity, such as long-term potentiation (LTP) and learning and memory (Luthi et al., 1994; Arami et al., 1996; Fields and Itoh, 1996; Bahr et al., 1997; Staubli et al., 1998; Tang et al., 1998; Benson et al., 2000; Bozdagi et al., 2000).
Although the involvement of CAMs in synaptic plasticity is now accepted, few studies on the modulation of CAMs during synaptic interaction have been published. Previously, we found that the extracellular serine protease neuropsin is associated with activity-dependent neural plasticity, LTP, and kindling epileptogenesis (Okabe et al., 1996; Momota et al., 1998; Yoshida and Shiosaka, 1999; Komai et al., 2000). On the basis of our findings, we hypothesized that neuropsin modifies the extracellular synaptic environment in an activity-dependent manner via the cleavage of CAMs because of the following: (1) neuropsin was specifically expressed in the pyramidal or magnocellular neurons of the hippocampus and the amygdala (Chen et al., 1995; Okabe et al., 1996), in which CAMs were involved in neural plasticity (Luthi et al., 1994; Arami et al., 1996; Fields and Itoh, 1996; Bahr et al., 1997; Staubli et al., 1998; Tang et al., 1998; Benson et al., 2000; Bozdagi et al., 2000); (2) such functions of neuropsin and CAMs overlap, and neuropsin and CAMs are both related to early-phase LTP (E-LTP) (Luthi et al., 1994; Bahr et al., 1997; Staubli et al., 1998; Tang et al., 1998; Komai et al., 2000); and (3) a deficiency in either neuropsin or CAMs has resulted in abnormalities in hippocampal neural networks in animals (Cremer et al., 1994; Demyanenko et al., 1999; Davies et al., 2001; Hirata et al., 2001). We therefore studied the sorts of CAMs, including ECM proteins, cleaved by neuropsin and how the cleavage regulates synaptic interaction. Consequently, we found that the plasticity-related CAM L1 is a specific substrate of neuropsin and that this neuropsin-L1 processing system is regulated by neural activity and is involved in hippocampal plasticity.
Materials and Methods
In vivo LTP. Male mice (10 weeks old) were deeply anesthetized with urethane (1.25 gm/kg, i.p.) and fixed in a stereotaxic frame. To record field EPSPs (fEPSPs), a tungsten recording electrode was inserted into the CA1 stratum radiatum (2.46 mm posterior, 2.0 mm lateral to bregma), and a bipolar stainless steel stimulating electrode was placed along the Schaffer collaterals (2.46 mm posterior, 2.50 mm lateral to bregma). Test stimulation (100 sec duration) was applied at intervals of 30 sec, and its intensity was adjusted to produce an fEPSP with a slope that was ∼50% of maximum. To induce LTP, theta-burst stimulation consisting of 10 burst-like trains (10 pulses at 200 Hz) at 5 Hz was applied four times every 30 sec to the Schaffer collaterals. In some experiments, the hippocampal region in which the electrode was inserted was dissected (approximately one-third of the unilateral hippocampus) at various time points before and after theta-burst stimulation and used for the measurement of the neuropsin activity.
Chemical stimulation of mouse hippocampus. According to the previous study (Li et al., 2001), intact hippocampal organs were carefully isolated from ddY (Japan, SLC, Shizuoka, Japan)- or neuropsin-deficient mice at postnatal day 14 (P14) and kept in oxygenated (95% O2 and 5% CO2) artificial CSF (ACSF) containing the following (in mm): 120 NaCl, 3 KCl, 1.2 NaH2PO4, 23 NaHCO3, 1.2 MgCl2, 2.4 CaCl2, and 11 d-glucose at room temperature for at least 1 hr before use. Chemical stimulation was provided by a bath application of 4-aminopyridine (4-AP) or NMDA at a final concentration of 100 μm for 10 min in ACSF (30°C). The application of this concentration of drugs was shown to result in no significant damage to synaptic transmission (Perreault and Avoli, 1989; Chetkovich et al., 1991). At 10 min after the application, 4-AP or NMDA was washed out with fresh ACSF of the same temperature. Glutamate receptor blockers, protein kinase inhibitors, or a rat anti-neuropsin antibody mAbB5 (2.5 μg/ml; Medical and Biological Laboratories, Nagoya, Japan) were applied in ACSF for 30 min before the chemical stimulation for control experiments. The penetration of mAbB5 into hippocampi was confirmed by an immunohistochemical method using the biotinylated anti-rat IgG (Dako, Glostrup, Denmark) recognizing mAbB5. The density of immunoreactivity in the tissue section was analyzed with the ATTO (Tokyo, Japan) densitograph system. The chemically potentiated organs were frozen in dry ice at 0, 5, 10, 15, 30, and 70 min after the application of drugs and kept at -80°C before the measurement of neuropsin proteolytic activity. Extracellular field potentials were recorded conventionally using glass micropipettes filled with 3 m NaCl positioned in the CA3 subfield and analyzed by the Chart program of the MacLab system (ADInstruments, Castle Hill, New South Wales, Australia). The healthy conditions of these hippocampal organs were checked histologically (under an electron microscopy) and electrophysiologically. The healthy synaptic response in such organs was validated by another previous study (Li et al., 2001). For Western blot analysis, the hippocampal organs were homogenized in 0.5 ml of ice-chilled buffer (20 mm Tris-HCl, 0.32 m sucrose, and 1 mm EDTA, pH 7.4). The homogenates were centrifuged for 10 min at 900 × g to remove debris, nuclei, etc. The supernatant was treated with Triton X-100 at a final concentration of 1% and was used for Western blot analysis of L1.
Measurement of proteolytic activity of endogenous neuropsin. Each hippocampal organ was homogenized separately in 1 ml of lysis buffer (1% Triton X-100, 150 mm NaCl, 5 mm EDTA, and 50 mm HEPES, pH 7.4) and then centrifuged at 15,000 rpm to remove debris. The proteolytic activity of the endogenous neuropsin in this supernatant was measured as described previously (Momota et al., 1998); briefly, the immunoprecipitate of the supernatant obtained with the monoclonal anti-NP antibody mAbF12 (Molecular and Biological Laboratories) was incubated with the synthetic substrate Pro-Phe-Arg-4-methyl-coumaryl-7-amide (MCA) at 37°C for 18 hr. All data are presented as the mean ± SEM. A statistical evaluation was done with the unpaired t test, and p values of <0.05 were considered significant.
Preparation of recombinant neuropsin, the synaptoneurosomal fraction, and the synaptic membrane fraction. The nonactive precursor form of recombinant neuropsin (r-proNP) was generated with the baculovirus-insect cell system (Shimizu et al., 1998). To obtain its active form (ractNP), activational processing by lysyl endopeptidase was performed as described previously (Shimizu et al., 1998; Kato et al., 2001). The hippocampal synaptoneurosomal (SN) fraction was prepared from adult ddY male mice (8 weeks old) and was observed under an electron microscopy according to a method reported previously (Weiler and Greenough, 1991). The purified synaptic membrane fraction was prepared from the mouse hippocampus as described previously (Lynch et al., 1982).
Western blot analysis of CAMs in the SN and synaptic membrane fractions. The SN pellet was resuspended in 1 ml of lysis buffer, and then this suspension was centrifuged at 15,000 rpm for 30 min in a microfuge to clear the debris. We used this supernatant as the SN fraction. The synaptic membrane fraction was also lysed with the same buffer and microfused to remove debris. The total protein content of the SN and synaptic membrane fractions was determined using BCA Protein Assay Reagent (Pierce, Rockford, IL). We applied r-actNP at a final concentration of 250 nm to 1 mg/ml of the SN proteins prepared as described above. Then this mixture was incubated at 37°C for 5-60 min with gentle rotation. As a control, the same experiment was performed using r-proNP or its solvent only. To detect the CAMs, we performed Western blot analysis using the antibodies described below according to the conventional method. Band densities on an x-ray film were measured using a Discovery System Quantity One (Protein Databases Inc., New York, NY). Fibronectin and laminin (Invitrogen, Rockville, MD) were examined as to whether r-actNP can process them. Coomassie blue-stained band densities were analyzed with the ATTO densitograph system.
Antibodies. We used specific antibodies for Western blot analysis at adequate dilutions as follows: anti-neural cell adhesion molecule (NCAM) rabbit polyclonal antibody (Chemicon, Temecula, CA), 1:10000; anti-C-terminal L1 peptide goat polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA), 1:400; anti-pan-L1 rabbit polyclonal antibody (a gift from Dr. V. Lemmon, Case Western Reserve University, Cleveland, OH), 1:2000; anti-N-cadherin or anti-E-cadherin rabbit polyclonal antibody (Santa Cruz Biotechnology), 1:100; anti-fibronectin rabbit polyclonal antibody (Biogenesis, New Fields, UK), 1:400; anti-NMDA receptor type 1 (NMDAR1) or anti-NMDAR2B rabbit polyclonal antibody (Chemicon), 1:200; and anti-NMDAR2A, anti-glutamate receptor type 1 (GluR1), anti-GluR2/3, or anti-GluR4 rabbit polyclonal antibody (Upstate Biotechnology, Lake Placid, NY), 1 μg/ml.
L cell aggregation assay. The rat full-length L1 expression vector was provided by Dr. K. Itoh (Tokyo Metropolitan Institute of Medical Science, Tokyo, Japan). The vector was transfected into L cells using Lipofectamine reagent (Invitrogen) according to the procedure of the manufacturer. To achieve the highest efficiency transfections, transfection conditions were optimized by checking L1 protein yields using immunoblot analysis. Transfected L cells were detached from cell cultures by treatment with 0.0025% trypsin and 0.001% EDTA in PBS. Cells were resuspended in divalent cation-free HBSS supplemented with 1 mm HEPES, pH 7.3, and 0.2% BSA (HBSS-BSA) at densities of 3-4 × 106 cells/ml, and 0.6 ml was rotated at 37°C. r-actNP or r-proNP was added to HBSS-BSA at a final concentration of 250 nm. Aliquots were withdrawn after 2 hr of incubation, and particles were enumerated in a hemocytometer. Percentages of aggregated cells were represented by the index (1 - N2/N0) × 100, where N2 and N0 are the total number of particles at incubation times 2 and 0 hr, respectively.
Immunohistochemistry for L1. The preembedding and staining procedures were performed as described previously (Matsumoto et al., 1994). Sections (30 μm) were cut with a Vibratome (Dosaka, Kyoto, Japan), immunostained by using the anti-L1 C-terminal peptide polyclonal antibody at a dilution of 1:200 or the anti-pan-L1 polyclonal antibody at a dilution of 1:500, and visualized by a diaminobenzidin reaction. The Epon 812-mounted sections were observed and photographed under a light microscope. Areas of interest were cut out under the operation microscope and ultrathin-sectioned using an ultramicrotom (Leica, Wien, Austria) for electron microscopic observation (Hirata et al., 2001).
Results
Neuropsin is transiently activated in CA1 E-LTP in vivo
To reveal the involvement of the neuropsin activation in synaptic plasticity, we examined the neuropsin proteolytic activity in the hippocampus in which the Schaffer collateral LTP was induced in vivo. When theta-burst stimulation was applied to the Schaffer collateral pathway in anesthetized mice, the enhancement of fEPSP and LTP in the CA1 subfield was successfully induced (Fig. 1A). We in turn measured the immunoprecipitated endogenous neuropsin activity in hippocampi before and after theta-burst stimulation using the synthetic substrate. This assay is highly sensitive and specific for the detection of neuropsin activity for the following reasons: (1) mAbF12 specifically recognizes neuropsin in immunoprecipitation assays (Momota et al., 1998; Kato et al., 2001); and (2) we detected no proteolytic activity in neuropsin-deficient mouse hippocampi by this assay (Hirata et al., 2001). The neuropsin activity was significantly upregulated 5 and 6 min after stimulation and then decreased to the basal level at 7 min (Fig. 1B). The data strongly suggest that the rapid, transient neuropsin activation is induced as a result of physiological synaptic stimuli. In some cases, the theta-burst stimulation failed to induce synaptic potentiation (Fig. 1C,D, No LTP). We compared the neuropsin activity in the hippocampus in which the induction of synaptic potentiation was successful (Fig. 1E, LTP) or unsuccessful (Fig. 1E, No LTP) at 5 min after theta-burst stimulation. The neuropsin activity was significantly increased when the synaptic potentiation was successfully induced (Fig. 1E, p < 0.05 vs No LTP and Test Pulse). The neuropsin activity in hippocampi showing unsuccessful synaptic potentiation did not increase compared with that in control hippocampi, in which only test pulse stimulation was applied (Fig. 1E). Thus, the neuropsin activation in the CA1 E-LTP in vivo was fully dependent on the synaptic potentiation, that is, the NMDA receptor activation.
Figure 1.
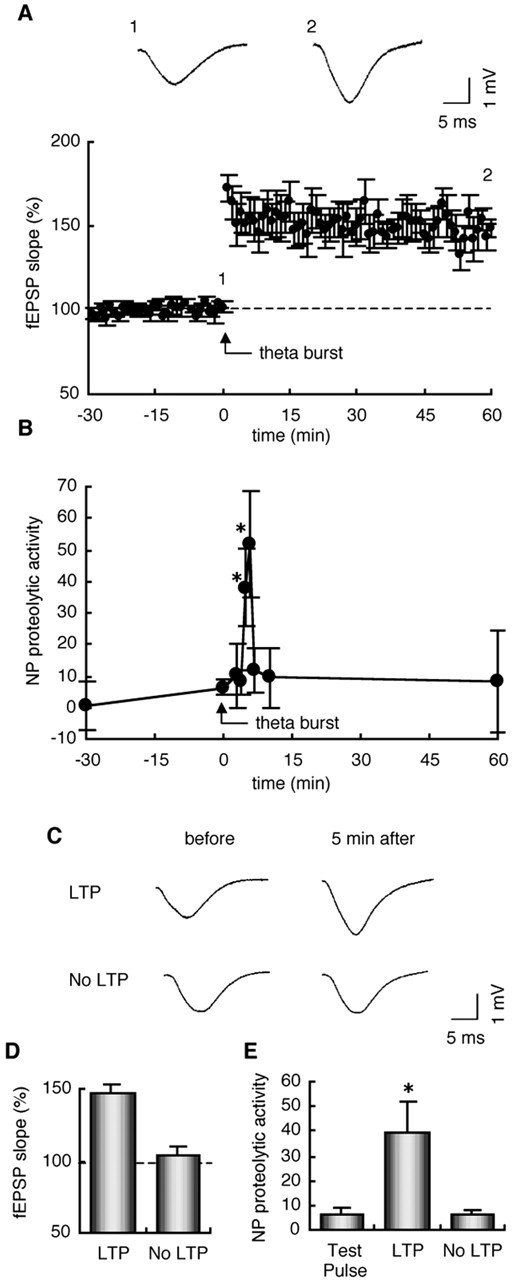
Neuropsin activation was transiently induced in the early phase of the Schaffer collateral LTP in vivo. A, Time course of changes in fEPSP slopes in anesthetized mice. The fEPSPs evoked by stimulation of the Schaffer collaterals were recorded from the CA1 stratum radiatum. The ordinate is expressed as a percentage of baseline values at time 0. Data are means ± SEM of six cases. The insets show typical field potentials immediately before (1) and 60 min after (2) theta-burst stimulation. B, Endogenous neuropsin was rapidly, transiently activated after theta-burst stimulation. The neuropsin activity at 5 and 6 min after stimulation was significantly increased[n = 5-8;*p<0.05 vs the activity before stimulation (0min)]. C, Typical field potentials immediately before and 5 min after theta-burst stimulation of the cases in which synaptic potentiation was successfully induced (LTP) and the stimulation failed to induce synaptic potentiation (No LTP). D, fEPSP slopes at 5 min after stimulation in the experiments in which the induction of synaptic potentiation was successful (LTP) or unsuccessful (No LTP). The ordinate is expressed as a percentage of baseline values at time 0. Data are means ± SEM of eight cases. E, The proteolytic activity of neuropsin in the hippocampal region in which synaptic potentiation was induced (LTP) or not (NoLTP) after the theta-burst stimulation. Neuropsin was activated only in the hippocampus in which synaptic potentiation was successfully induced (n = 8; *p < 0.05 vs Test Pulse or No LTP samples). The vertical axes of the graphs in B and E indicate the fluorescence intensity of 7-amino-4-methyl-coumarin (AMC), which was generated from the cleavage of Pro-Phe-Arg-MCA by endogenous neuropsin per 1 mg of total protein.
NMDA receptor-dependent activation of neuropsin ex vivo
To pharmacologically characterize the neuropsin activation after stimulation, we prepared unilateral intact hippocampal organs dissected from P14 mouse brain in which the hippocampal network was already established. We applied a brief chemical stimulation with the depolarizing agent 4-AP, a potassium channel blocker, to induce the depolarization and synaptic excitation (Perreault and Avoli, 1989). An extracellular recording in the CA3 subfield showed that 4-AP induced two peaks of neural activity, one after its application (Fig. 2A, a2) and the other after its washout (Fig. 2A, a4). The first peak was detected immediately after the application of 4-AP and lasted for 2 min; the second peak appeared at 10 min, immediately after the washout of 4-AP, and also lasted for 10 min (Fig. 2A). As shown in Figure 2B, proteolytic activity of neuropsin in the hippocampal organ without chemical stimulation was quite low and did not change at any point in time (Fig. 2B, NS). This result implies that neuropsin is present in a nonactive precursor form in the quiescent hippocampus. In contrast, its proteolytic activity after the 4-AP stimulation was markedly upregulated in a homologous biphasic manner (excited by both application and washout of the depolarizing agent) (Fig. 2B) to the change of neural activity (Fig. 2A). Because the NMDA receptor plays a critical role in the induction of LTP that takes place in the Schaffer collateral pathway (Malenka and Nicoll, 1993), we further investigated the effect of NMDA on the activation of neuropsin. A brief application of NMDA resulted in a biphasic activation pattern identical to that of 4-AP (Fig. 2B). To determine whether the upregulation of neuropsin activity resulted from activation of the glutamate receptor, we studied the effects of antagonists of ionotropic glutamatergic receptors on the neuropsin activity. The neuropsin activities at the first peak 5 min after stimulation (application phase) and at the second peak 15 min after stimulation (washout phase) were both completely blocked by preincubation with the NMDA receptor antagonist d-2-amino-5-phosphonovaleric acid (AP-5) (50 μm) (Fig. 2C,D). The AMPA-kainate receptor antagonist 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) (10 μm) completely blocked the neuropsin activation at 15 min (Fig. 2D, washout phase) but not at 5 min (Fig. 2C, application phase). These results showed that the neuropsin activity was critically regulated by the glutamate receptor activity, particularly by the NMDA receptor activity. The NMDA receptor activation is known to induce the rapid activation of protein kinases such as Ca2+/calmodulin-dependent protein kinase II (CaMK II), the mitogen-activated protein kinase (MAPK), and protein kinase A (PKA) and protein kinase C (PKC) (Soderling and Derkach, 2000). Therefore, we then investigated the effect of protein kinase inhibitors on the neuropsin activation. In the application phase (5 min), preincubation with the MAPK kinase (MEK) inhibitor U0126 (50 μm) partially (but not significantly) blocked the NMDA-induced neuropsin activation, but other inhibitors (CaMK inhibitor KN93, 10 μm; PKC inhibitor staurosporine, 5 μm; and PKA inhibitor H89, 20 μm) did not affect the neuropsin activation (Fig. 2C). On the other hand, in the washout phase (15 min), U0126 and H89 completely, and KN93 and staurosporine partially, inhibited the upregulation of neuropsin activity (Fig. 2D). The results showed that the upregulation of neuropsin activity depended on protein kinase signaling pathways at least in washout phase and that the signaling processes of neuropsin activation in the application and washout phases were different.
Figure 2.
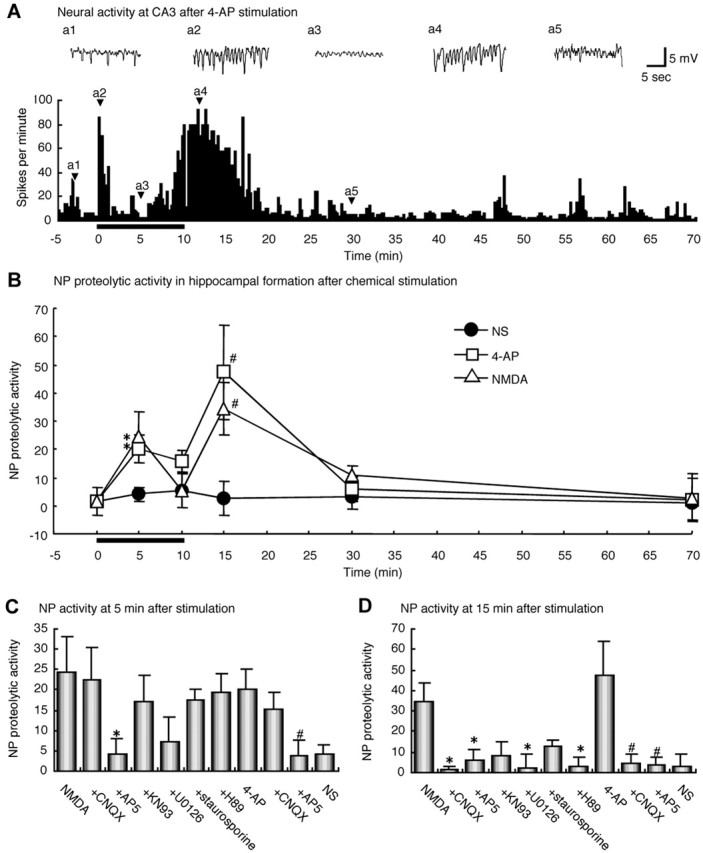
Chemical stimulation of mouse hippocampus-activated neuropsin. A, Extracellular recording in the CA3 subfield during stimulation with 4-AP. The vertical axis indicates the number of pulses beyond the threshold (2 mV) per minute. 4-AP was applied for 10 min as indicated by the bold horizontal bar below the x-axis. Insets a1-a5 show the waveforms at time points indicated by arrowheads. 4-AP induced a biphasic rise in neural activity. B, Chemical stimulation by 4-AP or NMDA also induced the neuropsin activation. 4-AP or NMDA was applied for 10 min as indicated by the bold horizontal bar. A rapid, transient activation of neuropsin was triggered by either application. The neuropsin activity increased at two time points, 5 min after the onset of stimulation by 4-AP or NMDA, and 5 min after the washout of drugs. Neuropsin activity was significantly higher at these time points than in nonstimulated controls: NS (*p < 0.05 vs NS at 5 min; #p < 0.05 vs NS at 15 min; n = 3-10 hippocampal organs per group). C, D, Glutamate receptor blockers significantly eliminated the NMDA-induced or 4-AP-induced increase in neuropsin activity at 5 min (C) and 15 min (D) after stimulation. Note that AP-5 completely blocked the increase at both time points, whereas CNQX blocked it only at 15 min. At 5 min after stimulation (C), NMDA-induced neuropsin activation was partially (not significantly) blocked by only the MEK inhibitor U0126 among protein kinase inhibitors. At 15 min (D), U0126 and the PKA inhibitor H89 significantly blocked the NMDA-induced neuropsin activation, and the inhibitory effects of other kinase inhibitors were partial (*p < 0.05 vs NMDA; #p < 0.05 vs 4-AP; n = 5-10 hippocampal organs per group). The vertical axes in B-D indicate the fluorescence intensity of AMC, which was generated from the cleavage of Pro-Phe-Arg-MCA by endogenous neuropsin in a single hippocampal organ.
Screening of synaptic neuropsin substrate(s) in hippocampus
We screened candidate substrate(s) of neuropsin from mouse hippocampus, in which neuropsin mRNA is expressed predominantly. We used three screening methods; the first one (approach 1) was comparison of two-dimensional gel electrophoresis of the hippocampal SN fraction to which was added r-actNP or nonactive r-proNP. Synaptoneurosomes, which are snowman-shaped entities consisting of a pinched-off, resealed presynaptic moiety adhered to a pinched-off, resealed postsynaptic moiety (Fig. 3A), have considerable transmission function (Weiler and Greenough, 1991). The second screening method (approach 2) was coincubation of r-actNP and the SN fraction and detection by Western blot analysis using antisera against known CAMs (L1, NCAM, N-cadherin, and E-cadherin) included in the SN fraction (Fig. 3B-E). The third screening method (approach 3) was coincubation of r-actNP and commercially available ECM proteins (fibronectin and A and B chains of laminin) (Fig. 3F,G). Using approach 1, in which we obtained at least 500 spots, no degradation of the protein spots was observed (data not shown). This may imply that the cleavage activity caused by neuropsin is restricted to macromolecules more than 100 kDa, which are not detected with this procedure. Instead, with approaches 2 and 3, we found candidate molecules that were effectively cleaved by r-actNP. Only a few macromolecules were cleaved by the incubation of r-actNP with the SN fraction or ECM proteins for a long period of time: NCAM, E-cadherin, N-cadherin, and laminin were not cleaved by r-actNP, although it was added at a considerably high concentration (Fig. 3B,D-F). Among all of the CAMs tested, L1, which belongs to the Ig superfamily, as does NCAM, was most effectively cleaved by r-actNP. Full-length L1 (200 kDa) and its C-terminal fragment (80 kDa) disappeared almost completely as a result of the incubation with r-actNP (Fig. 3C). This result indicates that either r-actNP completely degraded L1 into small pieces or this C-terminal L1 peptide-specific antibody could not detect the cleaved fragment of L1. Using another anti-pan-L1 antibody, we concluded that r-actNP cleaved L1 at only one site to generate the 180 kDa fragment (Fig. 4B) (also described below in detail). As shown in the time course of macromolecular cleavage using equimolar r-actNP, the 200 kDa band of L1 was reduced to one-half at only ∼15 min (Fig. 3H). r-actNP showed weak cleavage activity with fibronectin. The cleavage activity was characterized by the reduction of major bands and the appearance of low molecular weight bands, which appear to be fragments of fibronectin (Fig. 3G). However, even after a long incubation at a considerably high concentration of r-actNP, fibronectin was not lysed completely. After 60 min incubation with r-actNP, 61% of fibronectin remained intact (Fig. 3H). We examined whether r-actNP can cleave glutamate receptors in the SN fraction, the NMDA receptor subunits (NR1, NR2A, and NR2B), and the AMPA receptor subunits (GluR1, GluR2/3, and GluR4). Incubation of r-actNP with the SN fraction for 15 min and detection by Western blot analysis using antibodies against these subunits revealed that no degradation of these subunits was found (data not shown). Thus, the exclusively efficient cleaving activity against L1 strongly suggested that L1 was the most suitable substrate for neuropsin because endogenous neuropsin is transiently activated in a brief period (Figs. 1B, 2-3 min, 2B, 5-15 min).
Figure 3.
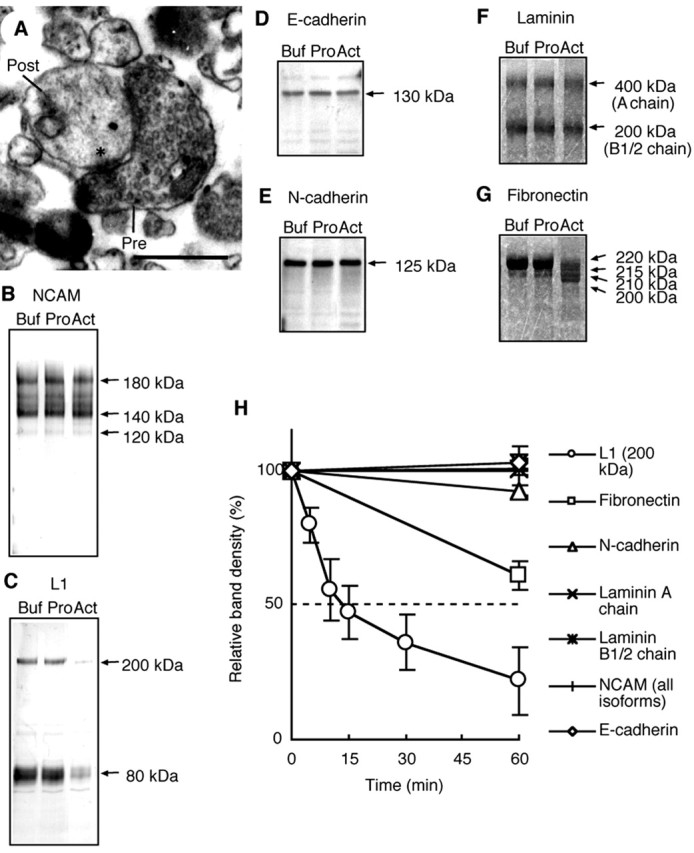
The proteolytic effects of neuropsin on CAMs and ECM proteins. A, An electron micrograph representing the prepared SN fraction. The synaptoneurosome consists of functional resealed presynaptic (Pre) plus postsynaptic (Post) entities. Scale bar, 0.5 μm. * indicates the synaptic adhesive site. B-E, Immunoblot analysis of the SN fractions treated for 60 min with a solvent (Buf), r-proNP (Pro), and r-actNP (Act) using anti-NCAM antibody (B), anti-L1 antibody (C), anti-E-cadherin antibody (D), and anti-N-cadherin antibody (E). Application of r-actNP resulted in a complete loss of intact 200 kDa L1 and a marked reduction in its 80 kDa C-terminal fragment (C). In contrast, NCAM (B), E-cadherin (D), and N-cadherin (E) were not affected by coincubation with r-actNP. F, G, Coomassie blue staining of laminin (F) and fibronectin (G) applied for 60 min with buffer only (Buf), r-proNP (Pro), and r-actNP (Act). The application of r-actNP with fibronectin resulted in a decrease in the intact 220 kDa band and the appearance of 215, 210, and 200 kDa fragments (G). In contrast, r-actNP was less effective against laminin (F). H, The time course in cleavage of CAMs and ECM molecules induced by coincubation with r-actNP. The band densities were quantified by densito-metric scanning setting densities in the SN fraction before application of r-actNP (0 min) as 100%, and the relative densities (vertical axis) were plotted at various time points after the application of r-actNP (horizontal axis). All data are presented as the mean ± SEM of two independent experiments. Of these molecules, L1 showed the highest velocity of cleavage. The 200 kDa band density of intact L1 was reduced to one-half at 15 min.
Figure 4.
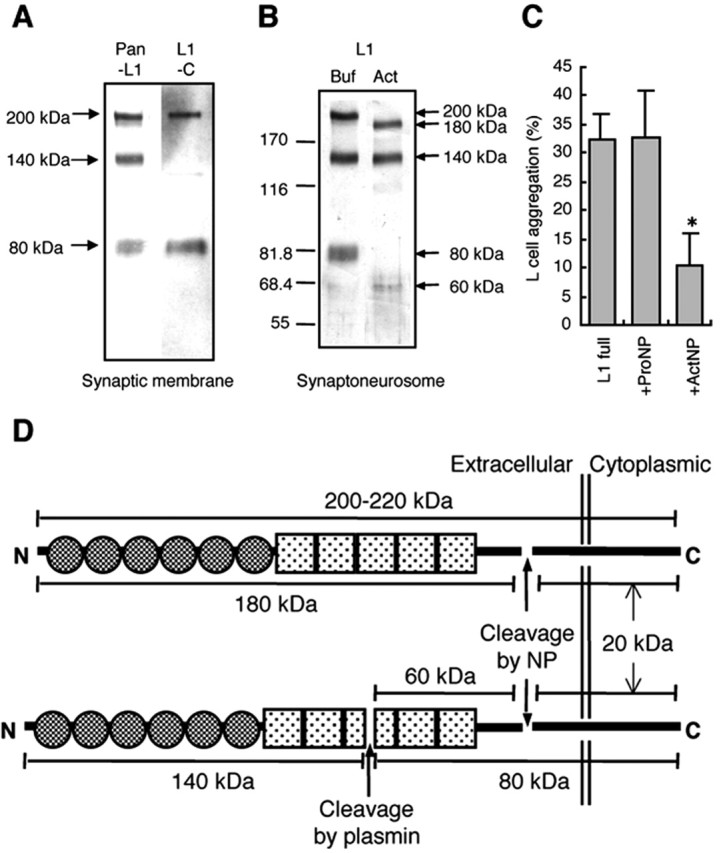
Cleavage of L1 at its extracellular membrane-proximal site by neuropsin-inhibited aggregation of L1-expressing cells. A, Western blot analysis of L1 in the synaptic membrane fraction. L1 was detected in the synaptic membrane fraction by both anti-pan-L1 antibody (Pan-L1) and C-terminal L1 peptide-specific antibody (L1-C). B, Western blot analysis of L1 cleavage by r-actNP. Full-length L1-recognizing polyclonal antibody could stain all of the degradation derivatives of L1: L1-200, L1-140, and L1--80 in the SN fraction treated with the buffer only (Buf). Coincubation of r-actNP with the SN fraction for 60 min (Act) resulted in the disappearance of L1-200 and L1-80 and, instead, the appearance of new fragments of L1-180 and L1-60. Both the band density and position of L1-140 never changed. C, The effect of extracellularly applied recombinant neuropsin on the aggregation of L1-transfected L cells. The vertical bar indicates the percentage of aggregation at 2 hr after detachment from the plastic dish. Incubation with r-actNP inhibited the aggregation of L1-transfected L cells (*p < 0.05 vs L1 full control), whereas r-proNP had no effect. Values shown are the means of five independent experiments. Error bars indicate SEM. D, Cleavage site of L1 on the basis of these experiments.
To confirm the synaptic localization of L1 as shown above, the purified synaptic membrane fraction was examined by Western blot analysis using two different anti-L1 antibodies. We could detect L1 in the synaptic membrane fraction prepared from mouse hippocampi using anti-pan L1 and anti-C-terminal L1 peptide antibodies (Fig. 4A). The experiment could also determine its site of cleavage by neuropsin. Using an anti-pan-L1 polyclonal antibody that recognizes the whole sequence of L1, the 200 kDa full-length L1 (L1-200), the 140 kDa N-terminal fragment (L1-140), and the 80 kDa C-terminal fragment (L1-80) were detected (Fig. 4A,B). The L1-140 and L1-80 fragments are known to be posttranslationally processed end products cleaved by plasmin in the third fibronectin-like domain of L1 (Fig. 4D) (Nayeem et al., 1999). We found that neuropsin cleaved an alternative site of L1 that is unidentified so far. When the SN fraction was preincubated with r-actNP before gel electrophoresis, the L1-200 and L1-80 bands completely disappeared in the blot, and instead, 180 kDa (L1-180) and 60 kDa (L1-60) bands appeared (Fig. 4B). However, the size and density of L1-140 were never affected. These results indicated that neuropsin cleaves the extracellular domain of L1 20 kDa from its C-terminal at only one site (Fig. 4D). However, additional study such as amino acid sequence analysis of cleaved stump of L1 by neuropsin might be necessary to identify the accurate cleavage site.
To examine whether extracellularly applied r-actNP can induce proteolysis-dependent segregation, we analyzed the effect of r-actNP on L1-transfected L cells, which themselves do not express CAMs significantly. L1-transfected L cells readily aggregated as observed previously (Hillenbrand et al., 1999) (Fig. 4C). The application of r-actNP markedly inhibited the aggregation of L1-transfected L cells, whereas treatment with r-proNP had no effect on their aggregation (Fig. 4C). The application of r-actNP to the human embryonic kidney cell line 293, which expresses CAMs such as integrin αVβ1 (Bodary and McLean, 1990) and Ig superfamily LAR (Aicher et al., 1997), did not affect cellular aggregation (data not shown).
The results show significant effects of neuropsin on L1: (1) L1 is the most suitable substrate for neuropsin; (2) neuropsin cleaves the extracellular domain of L1; (3) L1 is localized to synaptic membrane; and (4) the cleavage caused segregation of L1-expressing cultured cells.
NMDA-dependent cleavage of L1 by neuropsin ex vivo
Because it was found that L1 is the most suitable substrate for neuropsin, it is important to determine whether the cleavage of L1 depends on endogenous neuropsin activated by potentiated neural activity as observed in Figure 2B. We therefore analyzed the time course of neuropsin-specific cleavage of L1 by 4-AP and NMDA in hippocampus. We detected L1 protein in the stimulated hippocampal organ by immunoblotting with anti-pan-L1 polyclonal antibody recognizing full-length L1. The new 180 kDa band of L1, which was exclusively produced by neuropsin (Fig. 4B), appeared in the hippocampal organ at 15 and 30 min after stimulation by 4-AP or NMDA (Fig. 5A). On the other hand, the intensity of the L1-180 band was faint when the hippocampus was not stimulated by these agents (Fig. 5A,B, NS). The temporal change in L1-180 band density (Fig. 5B, top) and the density ratio of L1-180/L1-200 (Fig. 5B, bottom) revealed a rapid increase in the chemically stimulated hippocampus. The band density increased markedly from 10 to 70 min after 4-AP and NMDA treatment (Fig. 5B, 4-AP, NMDA), whereas it did not show any increase when the organ was nonstimulated (Fig. 5B, NS). The application of 4-AP or NMDA resulted in a rapid increase in the band density of L1-180 at as early as 5 min (NMDA) or 10 min (4-AP). Such an early response was consistent with the time course of the increase in the enzymatic activity of neuropsin (Fig. 2B). The increase in L1-180 density continued between 15 and 70 min after the stimulation (Fig. 5B). The increase was completely blocked after pretreatment with the NMDA receptor antagonist AP-5 (Fig. 5A,B, 4-AP + AP5), showing that this increase was dependent on the activation of the NMDA receptor. To clarify whether the appearance of L1-180 was mediated by endogenous neuropsin, we performed the same experiment using NMDA-stimulated hippocampus from neuropsin-deficient mice. The L1-180 band density was found to be negligible [Fig. 5A,B, NMDA (NP-/-)]. To further confirm the dependence on the proteolytic action of endogenous neuropsin, we analyzed whether the activity-neutralizing anti-neuropsin antibody mAbB5 blocked the NMDA-induced increase in L1-180. The penetration of mAbB5 into hippocampus was checked by an immunohistological analysis using biotinylated anti-rat IgG recognizing mAbB5. Strong immunoreactivity for bath-applied mAbB5 was found in the stratum oriens, pyramidale, and radiatum in the hippocampus. The density of immunoreactivity in the core region of CA1 subfield of treated hippocampus (5.08 ± 2.01 × 106/1.44 mm2) was significantly higher than the control without mAbB5 (1.16 ± 0.78 × 106/1.44 mm2; p < 0.05). No increase of L1-180 was found in NMDA-stimulated hippocampal organs treated with neutralizing antibody (Fig. 5A,B, NMDA + mAbB5). This neutralizing antibody also impaired the hippocampal Schaffer collateral E-LTP (H. Tamura, K. Matsumoto-Miyai, and S. Shiosaka, unpublished observations) (Komai et al., 2000), suggesting that neuropsin-dependent L1 cleavage is significant for the Schaffer collateral synaptic plasticity. We further examined whether the endogenous neuropsin activated by increased neural activity can cleave N-cadherin and fibronectin because the involvement of N-cadherin in synaptic plasticity was well characterized (Bozdagi et al., 2000) and r-actNP showed a weak proteolytic effect on fibronectin (Fig. 3G,H). Unlike in the SN fraction, anti-N-cadherin antibody recognized three bands (125, 105, and 90 kDa) in the hippocampal homogenate, but none of the immunoreactive bands showed any change on chemical stimulation (Fig. 5C). As shown in Figure 5D, fibronectin also exhibited no change in band density. These results showed that neither N-cadherin nor fibronectin was cleaved by endogenous neuropsin in such a short period. Altogether, increased neural activity triggered the cleavage of L1 via endogenous neuropsin ex vivo.
Figure 5.
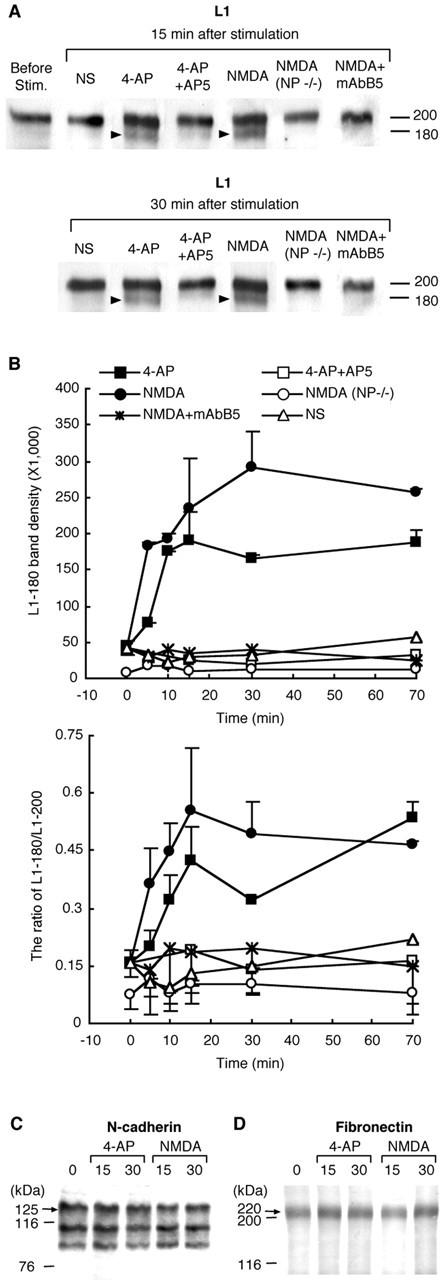
Chemical stimulation induced neuropsin-dependent L1 cleavage. A, Immunoblot analysis showed that L1-180 (arrowheads) increased in the hippocampal organs stimulated by 4-AP or NMDA but not in the absence of stimulation (NS) or in the neuropsin-deficient hippocampus stimulated with NMDA [NMDA (NP-/-)] at both 15 and 30 min after treatment. The 4-AP-induced or NMDA-induced increase of L1-180 was blocked by preincubation with AP-5 (4-AP + AP5) or anti-neuropsin antibody mAbB5 (NMDA + mAbB5). B, Temporal change in the L1-180 band density (top) and the band density ratio of L1-180/L1-200 (bottom graph) after chemical stimulation. Error bars indicate SEM. A rapid neuropsin-dependent increase in L1-180 was induced by chemical stimulation with 4-AP and NMDA. C, D, Immunoblot analyses using anti-N-cadherin (C) and anti-fibronectin (D) antibodies revealed that no change in N-cadherin and fibronectin in the hippocampal organs was induced by chemical stimulation (4-AP or NMDA).
Presynaptic localization of L1 in hippocampus
Because the present study revealed the involvement of the neuropsin-L1 processing system in synaptic potentiation, and presumably Schaffer collateral plasticity, it is important to examine whether L1 protein is associated with the synaptic machinery. Strong dot-like L1 immunoreactivity was found to be scattered in the stratum radiatum of the CA1 subfield by a light microscopic observation of L1 immunohistochemistry using C-terminal L1 peptide-specific antibody (Fig. 6A,B). Axon bundles projecting from the CA3 cell bodies, which were Schaffer collateral pathways, were immunostained (Fig. 6A,B, arrowheads). The L1-immunoreactive fiber bands projected toward the stratum radiatum of the CA1 subfield. Epon-embedded immunostained tissue sections were sectioned further after bright-field micrographs had been taken and observed under an electron microscope. The immunoreactive nerve terminals apparently formed asymmetrical synapses on the dendrites and dendritic spines of the CA1 pyramidal neurons (Fig. 6C). L1 immunoreactivity in the CA1 subfield was confined to the presynapses that apposed nonimmunoreactive dendritic spines protruding from apical and secondary apical dendrites (Fig. 6C-E). These observations imply that L1 protein exhibits heterophilic binding with unidentified CAMs localized in postsynapses. We also confirmed the L1 localization by immunohistochemical analysis using another anti-pan-L1 antibody, which was applied in former immunohistochemical studies (Miller et al., 1993). This antibody stained the Schaffer collateral axons (Fig. 6F) slightly stronger than the C-terminal L1 peptide-specific antibody. The immunoreactivity showed completely the same pattern in its presynaptic boutons under both light and electron microscopes (Fig. 6G). Thus, our biochemical (Fig. 4A) and immunohistochemical (Fig. 6) data strongly showed the presynaptic localization of L1 in the hippocampal CA1 subfield.
Figure 6.
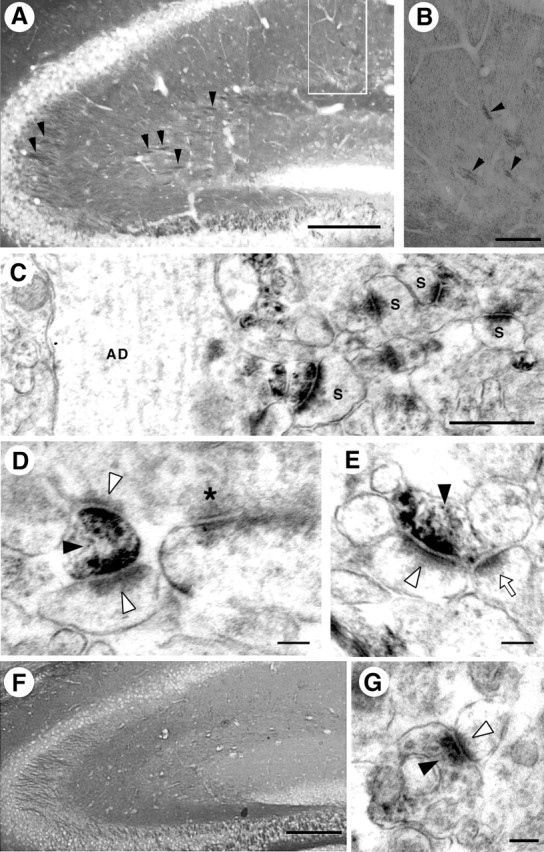
L1 immunoreactivity in the mouse hippocampus. A-E, L1 immunohistochemistry using C-terminal L1 peptide-specific antibody. A, B, Light microscopic observation of L1 immunoreactivity in Epon-embedded mouse hippocampal tissue. Schaffer collateral axons projecting from CA3 to CA1 showed intense L1-immunoreactive bundles (A, B, arrowheads). B shows a higher-magnification view of the stratum radiatum of the CA1 subfield indicated by the white rectangle in A. L1-immunoreactivity showed a fine granular pattern. C, Electron microscopic observation of the immunoreactivity in the stratum radiatum of the CA1 subfield. L1 was exclusively localized to the presynaptic membrane in the asymmetrical synapses. AD, Apical dendrite; S, dendritic spine. D, E, L1 localization in the multisynaptic boutons (black arrowheads). L1 immunoreactivity was observed in both presynaptic sites in some multisynaptic boutons (D, white arrowheads). In another multisynaptic bouton, only one presynaptic site was positive (E, left synapse, white arrowhead) and the other was negative (E, right synapse, white arrow). A nonimmunoreactive presynaptic bouton is also seen in D (asterisk). F, G, L1 immunohistochemistry using anti-pan-L1 antibody. F, Light microscopic observation of the immunoreactivity with anti-pan-L1 antibody revealed a similar pattern to that with C-terminal L1 peptide-specific antibody (A). G, Anti-pan-L1 antibody also labeled the presynaptic bouton (black arrowhead) in the CA1 stratum radiatum. The postsynaptic site is indicated by a white arrowhead. Scale bars: A, F, 200 μm; B, 50 μm; C, 500 nm; D, E, G, 100 nm.
Not all of the presynaptic boutons forming asymmetric synapses were immunoreactive for L1. Non-L1-immunoreactive synaptic boutons were also frequently seen (Fig. 6D, asterisk). The immunoreactive boutons are generally small compared with nonimmunoreactive axodendritic asymmetry synapses (Fig. 6D,E). In some cases, there were L1-associated and nonassociated synapses at the presynaptic membrane in one multisynapse bouton (Fig. 6E), suggesting that two kinds of synaptic interactions exist, L1-dependent and L1-independent, in a single bouton.
Overall, the present study clearly showed that a neuropsin-dependent presynaptic L1-cleaving system that is activated in an NMDA receptor-dependent manner is present in the specific subpopulation of hippocampal CA1 synapses.
Discussion
The present study showed that increased neural activity triggered a rapid, transient increase in the proteolytic activity of a limbic-specific serine protease, neuropsin (Chen et al., 1995; Momota et al., 1998; Komai et al., 2000). Neuropsin is secreted into the extracellular space in a nonactive precursor form (proNP) and activated by the processing of four N-terminal amino acids after its release (Shimizu et al., 1998). Most of the neuropsin in brain is detected in the soluble fraction as a nonactive proNP by subfractionation assay (Shimizu et al., 1998; Kato et al., 2001). Most neuropsin is released via a constitutive secretory pathway in neuropsin-transfected cells (Oka et al., 2002). Therefore, our previous study, as well as the present study, suggests that most neuropsin is stored in the extracellular space as a nonactive proNP and that its enzymatic activation is regulated by neural activity. In this study, we observed that the NMDA receptor blocker AP-5 could effectively block both the induction of neuropsin activity and the proteolytic modification of L1 by neuropsin. This observation implies that postsynaptic signals transmitted through the NMDA receptor activate proNP stored in the extracellular space via a still unidentified pathway. The activation of neuropsin may occur downstream of the MAPK, PKA, PKC, or CaMK II signaling pathway. In the present study, the neuropsin activation in the washout phase was blocked by the inhibitors against MEK and PKA, whereas that in the application phase was only partially blocked by the MEK inhibitor. The difference of these two phases is still unknown, although it might represent the existence of several complex signaling pathways regulating the neuropsin activation and inactivation. Postsynaptic signaling events are known to be responsible for E-LTP (Soderling and Derkach, 2000), and we reported previously that neuropsin exhibited a regulatory effect on the Schaffer collateral E-LTP (Komai et al., 2000). The application of various concentrations of recombinant neuropsin resulted in a bell-shaped dose-response curve for the amplitude of the tetanic-stimulation-induced E-LTP, suggesting that neuropsin acts as a regulatory molecule in E-LTP via its proteolytic action. Endogenous neuropsin was transiently activated after chemical stimulations ex vivo (Fig. 2B,5-15 min) and the theta-burst stimulation-inducing CA1 LTP in vivo (Fig. 1B, 2-3 min). Because neuropsin rapidly cleaved L1 in vitro (Fig. 3C,H) and ex vivo (Fig. 5A,B), L1 was thought to be the most suitable substrate for neuropsin in the synaptic plasticity event. Because L1 is also known to contribute to the earliest phase of LTP (Luthi et al., 1994), our finding is consistent with theirs.
In this study, we found that L1 is a Schaffer collateral presynaptic component. The localization of L1 in the presynaptic components was confirmed with two different analyses in the present study: (1) Western blot analysis of the synaptic membrane fraction; and (2) light microscopic and electron microscopic observations of immunohistochemistry using two kinds of anti-L1 antibodies. Thus, we concluded that L1 was present in synapses of the hippocampal CA1 subfield. The present study is consistent with a light microscopic immunohistochemical study demonstrating that L1 is localized to the unmyelinated Schaffer collateral fiber system (Miller et al., 1993). In addition, we first showed the localization of L1 in presynaptic membranes of the Schaffer collateral pathway in the CA1 striatum radiatum by an electron microscopic observation. The present study is also consistent with a recent proteomic analysis (Husi et al., 2000), which demonstrated that the NMDA receptor multiprotein complex contains L1 protein and may function synergistically in the physiological context of NMDA receptor-dependent synaptic plasticity. Electrophysiological studies have revealed that various antibodies against NCAM and L1 reduced the amplitude of the Schaffer collateral LTP elicited by theta-burst stimulation (Luthi et al., 1994). On the other hand, E-cadherin and N-cadherin are localized between the presynaptic and postsynaptic membranes in the hippocampal CA3 subfield as a primary adhesion moiety, although they localize outside of synaptic active sites (Fannon and Colman, 1996). Thus, it appears that the extracellular macromolecules, which are localized in the synapses, play an important role in synaptic transmission and neural plasticity via modifications of cell-to-cell binding. However, what happens in the cleft space and the binding machinery between presynapses and postsynapses is not clear. We found that the neuropsin-dependent cleavage of presynaptic L1 is triggered by neural activity, which induces synaptic excitation, and that the blocking of neuropsin by the neutralizing antibody resulted in a concomitant reduction in NMDA-induced L1 processing (Fig. 5) and the amplitude of Schaffer collateral LTP (Komai et al., 2000), suggesting that the neuropsin-dependent L1 processing might be essential for the expression of synaptic plasticity. This hypothesis is strongly supported by the observation that the neuropsin activation was induced in the early phase of LTP in vivo in a synaptic potentiation-dependent manner (Fig. 1). Such an activity-dependent cleavage of L1 by neuropsin is a first model of the protease CAMs regulatory system, and presumably, this system induces the rapid morphological change in presynapses and postsynapses demonstrated by other investigators (Bailey and Kandel, 1993; Engert and Bonhoeffer, 1999; Maletic-Savatic et al., 1999; Toni et al., 1999; Geinisman et al., 2001; Toni et al., 2001) because such a cleavage may result in a decrease in synaptic adhesion and subsequent increase in the flexibility of synaptic structures.
Several studies showed that ankyrins and syntenin-1 bind to the cytoplasmic region of the L1 subfamily (Brummendorf and Lemmon, 2001). Ankyrins are multifunctional membrane adapter proteins that have the potential to couple diverse membrane proteins such as voltage-gated sodium channels to the spectrin-based membrane skeleton (Srinivasan et al., 1988). Syntenin-1 contains two PDZ (postsynaptic density 95/Discs large/zona occludens 1) domains and interacts with other neural receptors (Koroll et al., 2001). Because we observed the cleavage of L1 after chemical stimulation at synapses, this modification may induce a dynamic rearrangement of the submembrane skeleton and membrane protein clustering, as well as a decrease in synaptic adhesion. Because L1 is localized only in presynapses, the neuropsin-L1 modification system may be a kind of retrograde transmission of postsynaptic signals for the rearrangement of presynaptic structures. Such a model provides a novel view of the molecular mechanism underlying synaptic structural plasticity. In conclusion, we identified here the activity-dependent, neuropsin-specific proteolytic modification of presynaptic L1 and showed that this system is significantly related to the early phase of LTP in the Schaffer collateral pathway.
Footnotes
This work was supported by a grant-in-aid for Scientific Research on Priority Areas (A) from the Ministry of Science, Sport, Culture, and Technology, and the Sasakawa Scientific Research Grant from the Japan Science Society. We thank Dr. S. F. Traynelis for reading this manuscript and making helpful suggestions, Dr. M. Tohyama for supportive encouragement, Drs. T. Tsumoto, Y. Hata, F. Kimura, and S. Komai for discussion and technical advice on electrophysiology, Dr. K. Itoh for advice on the L1 experiment, Drs. N. Matsuki, Y. Ikegaya, and K. Nakao for technical advice on the in vivo LTP experiment, and Y. Sato and M. Sano for technical assistance.
Correspondence should be addressed to Dr. Kazumasa Matsumoto-Miyai, Division of Structural Cell Biology, Nara Institute of Science and Technology, 8916-5 Takayama, Ikoma, Nara 630-0192, Japan. E-mail: kmatsumo@bs.aist-nara.ac.jp.
Copyright © 2003 Society for Neuroscience 0270-6474/03/237727-10$15.00/0
References
- Abel T, Kandel E ( 1998) Positive and negative regulatory mechanisms that mediate long-term memory storage. Brain Res Brain Res Rev 26: 360-378. [DOI] [PubMed] [Google Scholar]
- Aicher B, Lerch MM, Muller T, Schilling J, Ullrich A ( 1997) Cellular redistribution of protein tyrosine phosphatases LAR and PTPsigma by inducible proteolytic processing. J Cell Biol 138: 681-696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arami S, Jucker M, Schachner M, Welzl H ( 1996) The effect of continuous intraventricular infusion of L1 and NCAM antibodies on spatial learning in rats. Behav Brain Res 81: 81-87. [DOI] [PubMed] [Google Scholar]
- Bahr BA, Staubli U, Xiao P, Chun D, Ji ZX, Esteban ET, Lynch G ( 1997) Arg-Gly-Asp-Ser-selective adhesion and the stabilization of long-term potentiation: pharmacological studies and the characterization of a candidate matrix receptor. J Neurosci 17: 1320-1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CH, Kandel ER ( 1993) Structural changes accompanying memory storage. Annu Rev Physiol 55: 397-426. [DOI] [PubMed] [Google Scholar]
- Benson DL, Tanaka H ( 1998) N-cadherin redistribution during synaptogenesis in hippocampal neurons. J Neurosci 18: 6892-6904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson DL, Schnapp LM, Shapiro M, Huntley GW ( 2000) Making memories stick: cell-adhesion molecules in synaptic plasticity. Trends Cell Biol 10: 473-482. [DOI] [PubMed] [Google Scholar]
- Bodary SC, McLean JW ( 1990) The integrin beta 1 subunit associates with the vitronectin receptor alpha v subunit to form a novel vitronectin receptor in a human embryonic kidney cell line. J Biol Chem 265: 5938-5941. [PubMed] [Google Scholar]
- Bozdagi O, Shan W, Tanaka H, Benson DL, Huntley GW ( 2000) Increasing numbers of synaptic puncta during late-phase LTP: N-cadherin is synthesized, recruited to synaptic sites, and required for potentiation. Neuron 28: 245-259. [DOI] [PubMed] [Google Scholar]
- Brummendorf T, Lemmon V ( 2001) Immunoglobulin superfamily receptors: cis-interactions, intracellular adapters and alternative splicing regulate adhesion. Curr Opin Cell Biol 13: 611-618. [DOI] [PubMed] [Google Scholar]
- Chen ZL, Yoshida S, Kato K, Momota Y, Suzuki J, Tanaka T, Ito J, Nishino H, Aimoto S, Kiyama H, Shiosaka S ( 1995) Expression and activity-dependent changes of a novel limbic-serine protease gene in the hippocampus. J Neurosci 15: 5088-5097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chetkovich DM, Gray R, Johnston D, Sweatt JD ( 1991) N-methyl-d-aspartate receptor activation increases cAMP levels and voltage-gated Ca2+ channel activity in area CA1 of hippocampus. Proc Natl Acad Sci USA 88: 6467-6471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colicos MA, Collins BE, Sailor MJ, Goda Y ( 2001) Remodeling of synaptic actin induced by photoconductive stimulation. Cell 107: 605-616. [DOI] [PubMed] [Google Scholar]
- Cremer H, Lange R, Christoph A, Polmann M, Vopper G, Roes J, Brown R, Baldwin S, Kraemer P, Scheff S ( 1994) Inactivation of the N-CAM gene in mice results in size reduction of the olfactory bulb and deficits in spatial learning. Nature 367: 455-459. [DOI] [PubMed] [Google Scholar]
- Davies B, Kearns IR, Ure J, Davies CH, Lathe R ( 2001) Loss of hippocampal serine protease BSP1/neuropsin predisposes to global seizure activity. J Neurosci 21: 6993-7000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demyanenko GP, Tsai AY, Maness PF ( 1999) Abnormalities in neuronal process extension, hippocampal development, and the ventricular system of L1 knockout mice. J Neurosci 19: 4907-4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einheber S, Schnapp LM, Salzer JL, Cappiello ZB, Milner TA ( 1996) Regional and ultrastructural distribution of the alpha 8 integrin subunit in developing and adult rat brain suggests a role in synaptic function. J Comp Neurol 370: 105-134. [DOI] [PubMed] [Google Scholar]
- Engert F, Bonhoeffer T ( 1999) Dendritic spine changes associated with hippocampal long-term plasticity. Nature 399: 66-70. [DOI] [PubMed] [Google Scholar]
- Fannon AM, Colman DR ( 1996) A model for central synaptic junctional complex formation based on the differential adhesive specificities of the cadherins. Neuron 17: 423-434. [DOI] [PubMed] [Google Scholar]
- Fields RD, Itoh K ( 1996) Neural cell adhesion molecules in activity-dependent development and synaptic plasticity. Trends Neurosci 19: 473-480. [DOI] [PubMed] [Google Scholar]
- Geinisman Y, Berry RW, Disterhoft JF, Power JM, Van der Zee EA ( 2001) Associative learning elicits the formation of multiple-synapse boutons. J Neurosci 21: 5568-5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillenbrand R, Molthagen M, Montag D, Schachner M ( 1999) The close homologue of the neural cell adhesion molecule L1 (CHL1): patterns of expression and promotion of neurite outgrowth by heterophilic interactions. Eur J Neurosci 11: 813-826. [DOI] [PubMed] [Google Scholar]
- Hirata A, Yoshida S, Inoue N, Matsumoto-Miyai K, Ninomiya A, Taniguchi M, Matsuyama T, Kato K, Iizasa H, Kataoka Y, Yoshida N, Shiosaka S ( 2001) Abnormalities of synapses and neurons in the hippocampus of neuropsin-deficient mice. Mol Cell Neurosci 17: 600-610. [DOI] [PubMed] [Google Scholar]
- Husi H, Ward MA, Choudhary JS, Blackstock WP, Grant SGN ( 2000) Proteomic analysis of NMDA receptor-adhesion protein signaling complexes. Nat Neurosci 3: 661-669. [DOI] [PubMed] [Google Scholar]
- Kato K, Kishi T, Kamachi T, Akisada M, Oka T, Midorikawa R, Takio K, Dohmae N, Bird PI, Sun J, Scott F, Miyake Y, Yamamoto K, Machida A, Tanaka T, Matsumoto K, Shibata M, Shiosaka S ( 2001) Serine proteinase inhibitor 3 and murinoglobulin I are potent inhibitors of neuropsin in adult mouse brain. J Biol Chem 276: 14562-14571. [DOI] [PubMed] [Google Scholar]
- Komai S, Matsuyama T, Matsumoto K, Kato K, Kobayashi M, Imamura K, Yoshida S, Ugawa S, Shiosaka S ( 2000) Neuropsin regulates an early phase of Schaffer-collateral long-term potentiation in the murine hippocampus. Eur J Neurosci 12: 1479-1486. [DOI] [PubMed] [Google Scholar]
- Koroll M, Rathjen FG, Volkmer H ( 2001) The neural cell recognition molecule neurofascin interacts with syntenin-1 but not syntenin-2, both of which reveal self-associating activity. J Biol Chem 276: 10646-10654. [DOI] [PubMed] [Google Scholar]
- Li J, Shen H, Naus CCG, Zhang L, Carlen PL ( 2001) Upregulation of gap junction connexin 32 with epileptoform activity in the isolated mouse hippocampus. Neuroscience 105: 589-598. [DOI] [PubMed] [Google Scholar]
- Luthl A, Laurent JP, Figurov A, Muller D, Schachner M ( 1994) Hippocampal long-term potentiation and neural cell adhesion molecules L1 and NCAM. Nature 372: 777-779. [DOI] [PubMed] [Google Scholar]
- Lynch G, Halpain S, Baudry M ( 1982) Effects of high-frequency synaptic stimulation on glutamate receptor binding studied with a modified in vitro hippocampal slice preparation. Brain Res 244: 101-111. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA ( 1993) NMDA-receptor-dependent synaptic plasticity: multiple forms and mechanisms. Trends Neurosci 16: 521-527. [DOI] [PubMed] [Google Scholar]
- Maletic-Savatic M, Malinow R, Svoboda K ( 1999) Rapid dendritic morphogenesis in CA1 hippocampal dendrites induced by synaptic activity. Science 283: 1923-1927. [DOI] [PubMed] [Google Scholar]
- Matsumoto K, Wanaka A, Takatsuji K, Muramatsu H, Muramatsu T, Tohyama M ( 1994) A novel family of heparin-binding growth factors, pleiotrophin and midkine, is expressed in the developing rat cerebral cortex. Brain Res Dev Brain Res 79: 229-241. [DOI] [PubMed] [Google Scholar]
- Miller PD, Chung WW, Lagenaur CF, DeKoskey ST ( 1993) Regional distribution of neural cell adhesion molecule (NCAM) and L1 in human and rodent hippocampus. J Comp Neurol 327: 341-349. [DOI] [PubMed] [Google Scholar]
- Momota Y, Yoshida S, Ito J, Shibata M, Kato K, Sakurai K, Matsumoto K, Shiosaka S ( 1998) Blockade of neuropsin, a serine protease, ameliorates kindling epilepsy. Eur J Neurosci 10: 760-764. [DOI] [PubMed] [Google Scholar]
- Nayeem N, Silletti S, Yang X, Lemmon VP, Reisfeld RA, Stallcup WB, Montgomery AM ( 1999) A potential role for the plasmin(ogen) system in the posttranslational cleavage of the neural cell adhesion molecule L1. J Cell Sci 112: 4739-4749. [DOI] [PubMed] [Google Scholar]
- Nishimura SL, Boylen KP, Einheber S, Milner TA, Ramos DM, Pytela R ( 1998) Synaptic and glial localization of the integrin alphavbeta8 in mouse and rat brain. Brain Res 791: 271-282. [DOI] [PubMed] [Google Scholar]
- Oka T, Hakoshima T, Itakura M, Yamamori S, Takahashi M, Hashimoto Y, Shiosaka S, Kato K ( 2002) Role of loop structures of neuropsin in the activity of serine protease and regulated secretion. J Biol Chem 277: 14724-14730. [DOI] [PubMed] [Google Scholar]
- Okabe A, Momota Y, Yoshida S, Hirata A, Ito J, Nishino H, Shiosaka S ( 1996) Kindling induces neuropsin mRNA in the mouse brain. Brain Res 728: 116-120. [PubMed] [Google Scholar]
- Perreault P, Avoli M ( 1989) Effects of low concentration of 4-aminopyridine on CA1 pyramidal cells of the hippocampus. J Neurophysiol 61: 953-970. [DOI] [PubMed] [Google Scholar]
- Persohn E, Schachner M ( 1990) Immunohistological localization of the neural cell adhesion molecules L1 and NCAM in the developing hippocampus of the mouse. J Neurocytol 19: 807-819. [DOI] [PubMed] [Google Scholar]
- Shimizu C, Yoshida S, Shibata M, Kato K, Momota Y, Matsumoto K, Shiosaka T, Midorikawa R, Kamachi T, Kawabe A, Shiosaka S ( 1998) Characterization of recombinant and brain neuropsin, a plasticity-related serine protease. J Biol Chem 273: 11189-11196. [DOI] [PubMed] [Google Scholar]
- Soderling TR, Derkach VA ( 2000) Postsynaptic protein phosphorylation and LTP. Trends Neurosci 23: 75-80. [DOI] [PubMed] [Google Scholar]
- Srinivasan Y, Elmer L, Davis J, Bennett V, Angelides K ( 1988) Ankyrin and spectrin associate with voltage-dependent sodium channels in brain. Nature 333: 177-180. [DOI] [PubMed] [Google Scholar]
- Staubli U, Chun D, Lynch G ( 1998) Time-dependent reversal of long-term potentiation by an integrin antagonist. J Neurosci 18: 3460-3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang L, Hung CP, Schuman EM ( 1998) A role for the cadherin family of cell adhesion molecules in hippocampal long-term potentiation. Neuron 20: 1165-1175. [DOI] [PubMed] [Google Scholar]
- Toni N, Buchs PA, Nikonenko I, Bron CR, Muller D ( 1999) LTP promotes formation of multiple spine synapses between a single axon terminal and a dendrite. Nature 402: 421-425. [DOI] [PubMed] [Google Scholar]
- Toni N, Buchs PA, Nikonenko I, Povilaitite P, Parisi L, Muller D ( 2001) Remodeling of synaptic membranes after induction of long-term potentiation. J Neurosci 21: 6245-6251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiler IJ, Greenough WT ( 1991) Potassium ion stimulation triggers protein translation in synaptoneurosomal polyribosomes. Mol Cell Neurosci 2: 305-314. [DOI] [PubMed] [Google Scholar]
- Yoshida S, Shiosaka S ( 1999) Plasticity-related serine proteases in the brain [review]. Int J Mol Med 3: 405-409. [DOI] [PubMed] [Google Scholar]