

Abstract
Interleukin-1 receptor antagonist (IL-1ra) is an important anti-inflammatory cytokine that blocks all known actions of IL-1 and markedly protects against experimentally induced ischemic, excitotoxic, and traumatic brain insults. Cannabinoids (CBs) also exert potent anti-inflammatory and neuroprotective effects, but the mechanisms of their actions are unknown. Here we tested the hypothesis that the actions of CBs are mediated by endogenous IL-1ra. We report for the first time that both CB1 and CB2 receptors modulate release of endogenous IL-1ra from primary cultured glial cells. Activation of CB1 or CB2 receptors increased lipopolysaccharide-induced IL-1ra release, and specific CB1 or CB2 antagonists blocked lipopolysaccharide-induced production of IL-1ra from glial cells. Comparison of neuronal cultures from wild-type mice and mice lacking IL-1ra (knock-out) indicates that endogenous IL-1ra is essential for the neuro-protective effects of CBs against excessive activation of glutamate receptors (excitotoxicity) in response to S-AMPA or NMDA. Similarly, analysis of mixed glial cultures from IL-1ra knock-out mice indicates that endogenous IL-1ra is required for the CB-induced inhibition of nitric oxide production in response to bacterial lipopolysaccharide. These data suggest a novel neuroprotective mechanism of action for CBs in response to inflammatory or excitotoxic insults that is mediated by both CB1 and CB2 receptor-dependent pathways.
Keywords: cytokine, cannabinoid, neuroprotection, IL-1ra, excitotoxicity, nitric oxide
Introduction
Cannabinoids (CBs) exert diverse and potent actions in the brain (Howlett et al., 2002) and are under clinical evaluation as anti-inflammatory, neuroprotective, and analgesic agents (Mechoulam et al., 2002a,b). However, their mechanisms of action are primarily unknown. CBs modulate inflammatory and immune responses (Klein et al., 2000; Pertwee, 2002) and are neuroprotective in experimentally induced excitotoxicity (van der Stelt et al., 2001), ischemic (Nagayama et al., 1999), and traumatic brain injury in rodents (Panikashvili et al., 2001; Mechoulam et al., 2002b).
Exogenous and endogenous CBs exert neuroprotective and anti-inflammatory actions on glia and neurons through G-protein-coupled receptor-dependent pathways (Howlett et al., 2002). To date, two CB receptors have been identified: the CB receptor type 1 (CB1), which is expressed mainly in the brain, and the CB receptor type 2 (CB2), which is expressed primarily in the immune system, although recent data suggest that CB2 is present at low levels within the brain (Howlett et al., 2002), and additional receptors may exist. The neuroprotective actions of CBs are thought to be mediated through a variety of mechanisms, including antioxidative actions (Hampson et al., 1998), inhibition of NMDA-mediated calcium influx (Mackie and Hille, 1992; Nadler et al., 1993), and inhibition of glutamate release (Shen and Thayer, 1998). CBs act on glia and neurons to inhibit the release of proinflammatory molecules, including interleukin-1 (IL-1), tumor necrosis factor (TNF) α, and nitric oxide (NO) (Molina-Holgado et al., 1997, 2002; Shohami et al., 1997; Puffenbarger et al., 2000; Cabral et al., 2001), and enhance the release of the anti-inflammatory cytokines IL-4, IL-10 (Klein et al., 2000), and IL-6 (Molina-Holgado et al., 1998). IL-1 has been identified as an important mediator of diverse forms of experimentally induced brain damage and is expressed rapidly in response to many forms of experimental brain injury, initially by microglia and later by astrocytes (Davies et al., 1999). IL-1 receptor antagonist (IL-1ra) is a potent endogenous antagonist of all IL-1 actions in the brain (Dinarello and Thompson, 1991), protecting against ischemic, excitotoxic, and traumatic brain insults (Allan and Rothwell, 2001). Furthermore, inhibition or deletion of endogenous IL-1ra enhances ischemic brain injury (Loddick et al., 1997) and increases inflammatory responses (Josephs et al., 2000).
The aim of the present study was to test the hypothesis that the neuroprotective and anti-inflammatory effects of CBs are mediated by endogenous IL-1ra. Primary glial and neuronal cultures from C57BL/6 wild-type (WT) or IL-1ra knock-out (KO) mice (Horai et al., 1998) were exposed to inflammatory [bacterial lipopolysaccharide (LPS)] or neurotoxic (S-AMPA or NMDA) stimuli. We show here for the first time that the anti-inflammatory cytokine IL-1ra is an essential mediator of CB actions on neurons and glia and that both CB1 and CB2 receptors modulate the release of IL-1ra from primary cultured glial cells.
Materials and Methods
Reagents. All tissue culture reagents were obtained from Invitrogen (Paisley, UK). The antibody for the astrocytic marker, anti-glial fibrillary acidic protein (GFAP), was from Sigma-Aldrich (Poole, UK), and antibodies for the microglial marker MAC-1 (macrophage adhesion molecule-1) and neuronal marker NeuN (neuronal-specific nuclear protein) were from Serotec (Oxford, UK). The synthetic CB agonist HU-210 was a generous gift from Prof. Raphael Mechoulam (Hebrew University, Jerusalem, Israel), and CP-55940 was from Tocris Cookson (Avon-mouth, UK). The cannabinoid antagonists SR-141617A and SR-144528 were a generous gift from Sanofi (Montpellier, France). All other re-agents were obtained from Sigma-Aldrich unless stated otherwise.
Animals. Wild-type C57BL/6 (WT) mice were obtained from Charles River (Kent, UK) and IL-1ra-deficient (KO) C57BL/6 mice were obtained from Prof. Yoichiro Iwakura (University of Tokyo, Tokyo, Japan). Animal care procedures were in accordance with the guidelines set by the European Council directives (86/609/EEC) and the Home Office, Animals (Scientific Procedures) Act 1986.
Primary mixed glial cultures. Primary, mixed glial cultures were prepared from whole brains of 1-d-old mice (WT or IL-1ra KO mice) according to published protocols (Molina-Holgado et al., 2002) and grown in T150 flasks for at least 14 d in DMEM supplemented with 10% heat-inactivated fetal bovine serum, 2 mm glutamine, and antibiotics (100 IU/ml penicillin and 100 μg/ml streptomycin solution). The medium was changed twice per week. On reaching confluence [after 13-14 d in vitro (DIV)], the cells were trypsinized and cultured (5 × 10 5 cells per well) for 2-3 d before experiments. Cultures consisted of 70% astrocytes and 30% microglia, as determined by staining with antibodies for GFAP and MAC-1, respectively.
Neuronal cell cultures. Primary cerebrocortical neurons were prepared as described previously (Moore et al., 2002). Briefly, the cortices were removed from embryonic (day 18) pups from WT or IL-1ra KO mice, chopped finely, and passed through three glass-fired pipettes of sequentially smaller bores. Cells were centrifuged (150 × g, 5 min) and resuspended in 20 ml of plating media (Neurobasal medium with B27 supplement plus antioxidants, 2 mm glutamine, 25 μm glutamate, 100 IU/ml penicillin, and 100 μg/ml streptomycin). Viable cells were counted by trypan blue exclusion and seeded (7.5 × 10 4 cells/cm2) onto plates coated with poly-d-lysine. One-half of the media was replaced after 5 and 11 DIV with maintenance medium (Neurobasal medium with B27 supplement minus antioxidants, 2 mm glutamine, 100 IU/ml penicillin, and 100 μg/ml streptomycin). After 2 DIV, cytosine arabinoside (20 μm) was added to the cultures to eliminate the majority of proliferating nonneuronal cells. These cultures consisted of >98% neurons, as assessed by NeuN immunoreactivity.
Treatment of cells. Glial cells were treated with bacterial LPS [serotype 0128:B12 (Sigma); 1 μg/ml] in DMEM for 24 hr at 37°C, in the presence or absence of the CB antagonists SR-141716A or SR-144528 (1 μm), or the synthetic CB agonists HU-210 (1 or 10 μm) or CP-55940 (0.1 or 1 μm). The culture supernatants were then analyzed for IL-1ra or nitrite as described below.
Neuronal cells were pretreated with HU-210 (1 μm) for 1 hr at 37°C and then exposed to S-AMPA (10 μm) or NMDA (20 μm) for 24 hr at 37°C, and the amount of cell death was measured as described below.
Release of IL-1ra. Immunoreactive IL-1ra in glial cell culture supernatants was assayed using a validated, mouse-specific sandwich ELISA (Safieh-Garabedian et al., 1995), supplied by Dr. Steven Poole (National Institute for Biological Standards and Control, London, UK). IL-1ra standards were assayed in triplicate and samples (100 μl) in duplicate. The assay detection limit for IL-1ra was 19 pg/ml.
Assessment of cell death. Cytotoxicity was measured by the release of cytosolic lactate dehydrogenase (LDH) by dead and dying cells using the CytoTox-96 LDH assay (Promega, Southampton, UK) according to the instructions of the manufacturer. Total LDH release was calculated by incubating untreated cells with 9% Triton X-100 for 45 min (37°C) to induce maximal cell lysis. Values for treated cells were then expressed as a percentage of the total LDH release. Background LDH release induced by media alone was subtracted from the experimental values.
Assay of nitric oxide synthase activity. NO synthase activity was assessed by measurement of nitrite (a breakdown product of NO) accumulation. Aliquots (100 μl) of culture supernatants were mixed with 100 μl of Griess reagent, a colorimetric indicator of the presence of nitrite (Green et al., 1982), and incubated in the dark at room temperature for 10 min. The absorbance was measured at 570 nm in a microplate reader. Fresh culture medium served as controls in all experiments.
Statistical analysis. Results are presented as the mean ± SEM of at least three different experiments performed on separate cell preparations, and duplicate or triplicate determinations were performed in each experiment. To determine differences between groups, one-way ANOVA was followed by posteriori Newman-Keuls multiple comparison. Statistical significance was established at p < 0.05.
Results
Cannabinoid agonists and antagonists modulate LPS-induced IL-1ra release from glial cells
Wild-type, primary, mixed glial cultures were used to test whether LPS-induced IL-1ra release is regulated by CB receptors, using specific and potent antagonists for the CB1 (SR-141617A) and CB2 (SR-144528) receptors (Howlett et al., 2002). Basal release of IL-1ra was not affected by incubation with CB1 or CB2 antagonists alone (data not shown). LPS increased release of IL-1ra by 15-fold (Fig. 1a), and this was almost completely abolished by cotreatment with either CB receptor antagonist (SR-141716A or SR-144528, by -12-fold and -13.6-fold, respectively). Thus, CB receptors regulate LPS-induced IL-1ra release. Moreover, the synthetic CB agonists HU-210 and CP-55940 had no effect on basal IL-1ra release (data not shown), but they enhanced LPS-induced IL-1ra release (by 28 and 15%, respectively) in WT mixed glial cultures (Fig. 1b,c).
Figure 1.
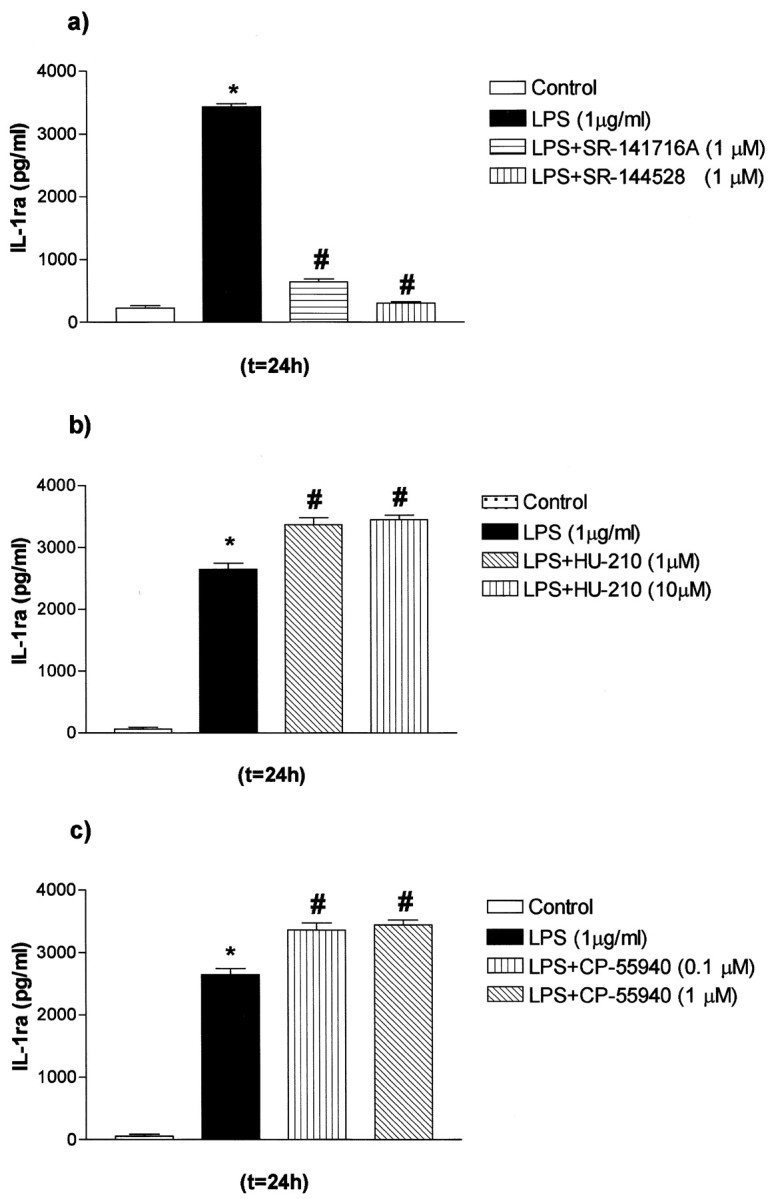
Modulation of IL-1ra release by LPS and CB antagonists and agonists in primary mixed glial cultures from C57BL/6 (WT) mice. Mixed glial cultures were treated with medium (Control) or LPS in the presence or absence of HU-210, CP-55940, SR-141716A, or SR-144528. a, Effect of LPS and SR-141716A or SR-144528 on IL-1ra release. b, Effect of LPS and HU-210 on IL-1ra release. c, Effect of LPS and CP-55940 on IL-1ra release. Data are presented as mean ± SEM of four independent experiments on separate cultures, each performed in triplicate. Statistical differences (one-way ANOVA and Newman-Keuls multiple test): *p < 0.001 versus control; #p < 0.001 versus LPS.
Endogenous IL-1ra mediates the neuroprotective actions of cannabinoids on excitotoxicity in primary neuronal cultures
We next investigated whether endogenous IL-1ra contributes to the neuroprotective actions of CBs against excessive activation of glutamate receptors (excitotoxicity), induced by exposure of primary cerebrocortical neurons from WT and IL-1ra KO mice to S-AMPA or NMDA. In WT neuronal cells, both glutamatergic agonists increased cell death significantly (Fig. 2a,b), and the CB agonist HU-210 significantly inhibited the cell death induced by S-AMPA (-38%) or NMDA (-51%) (Fig. 2a,b). S-AMPA and NMDA were also cytotoxic in cells from IL-1ra KO mice, but the neuroprotective effects of HU-210 were completely abolished (Fig. 2c,d).
Figure 2.

Neuroprotective effects of HU-210 on excitotoxicity in primary neuronal cultures. Neuronal cultures from WT (a, b) or IL-1ra KO (c, d) mice were pretreated with HU-210 before addition of S-AMPA (a, c) or NMDA (b, d). Data are presented as mean ± SEM of four independent experiments on separate cultures, each performed in triplicate. Statistical differences (one-way ANOVA and Newman-Keuls multiple test): *p < 0.001 versus control or HU210 alone; #p < 0.001 versus S-AMPA or NMDA.
Endogenous IL-1ra mediates the anti-inflammatory actions of cannabinoids in glial cells
The contribution of endogenous IL-1ra to the actions of CBs was further analyzed by testing the effects of the synthetic cannabinoid agonists HU-210 and CP-55940 on LPS-induced NO synthase activity, because NO is an important mediator of neuroinflammation and neurodegeneration (Hobbs et al., 1999). HU-210 or CP-55940 significantly inhibited the release of NO induced by LPS in WT glia (-55 and -25%, respectively) (Fig. 3a,b). In contrast, HU-210 and CP-55940 failed to inhibit the LPS-induced release of NO in mixed glial cultures from IL-1ra KO mice (Fig. 3c,d). Similar effects were observed when mixed glial cultures from KO mice were coincubated with LPS and the synthetic CB methanandamide or 11-OH-CBN (data not shown).
Figure 3.

Modulation of NO release by LPS and CB agonists in primary mixed glial cultures from WT or IL-1ra KO mice. Primary mixed glial cultures were treated with medium (Control) or LPS in the presence or absence of HU-210 or CP-55940. a, b, Effect of HU-210 or CP-55940 on LPS-induced NO2[minus] production in mixed glial cultures from WT mice. c, d, Effect of HU-210 or CP-55940 on LPS-induced NO2[minus] production in mixed glial cultures from IL-1ra KO mice. Data are presented as mean ± SEM of four independent experiments on separate cultures, each performed in triplicate. Statistical differences (one-way ANOVA and Newman-Keuls multiple test): *p < 0.001 versus control; #p < 0.001 versus LPS
Discussion
Cannabinoids have potent anti-inflammatory and neuroprotective effects in the brain, but the underlying mechanisms are unclear. We and others have shown previously that CBs can modulate production of inflammatory mediators, including the cytokines TNFα and IL-6 (Molina-Holgado et al., 1998; Klein et al., 2000; Puffenbarger et al., 2000) and generation of Th1 and Th2 phenotypes (Yuan et al., 2002). The present report extends these studies and suggests for the first time that the neuro-protective and anti-inflammatory effects of CBs are mediated by CB-induced release of endogenous IL-1ra in primary neurons or glial cells in vitro. The synthetic CB agonists HU-210 and CP-55940, which have similar affinities for CB1 and CB2 receptors (Howlett et al., 2002), increased LPS-induced IL-1ra release from primary glial cells. Similar results were obtained with other cannabinoids, including 11-OH-CBN, and the endogenous cannabinoid anandamide (data not shown). The potent and specific antagonists for CB1 and CB2 receptors, SR-141716A and SR-144528, respectively, significantly abrogated the LPS-induced IL-1ra release, implicating both CB receptors in this effect. Because neither the CB agonists nor antagonists altered basal release, these results indicate that cannabinoids enhance release of IL-1ra specifically in response to proinflammatory stimuli. The exact participation of CB1, CB2, or new CB receptors in these neuro-protective actions remains to be elucidated.
HU-210 and CP-55940 may promote LPS-induced IL-1ra expression and release directly, or they could modulate IL-1ra expression via an intermediate, such as IL-6. In support of the latter hypothesis, it has been reported that neutralization of endogenous IL-6 suppresses induction of IL-1ra (Jordan et al., 1995), and CB agonists modulate IL-6 production in glial cells by a CB receptor-mediated pathway (Molina-Holgado et al., 1998).
CBs are neuroprotective in a variety of in vitro and in vivo models of neuronal excitotoxicity. In the present study, low doses of HU-210 reduced cell death of WT neurons in response to S-AMPA or NMDA receptor activation. In contrast, HU-210 failed to protect against S-AMPA- or NMDA-induced death of neurons from IL-1ra KO mice. These results are consistent with previous observations that endogenous IL-1ra is neuroprotective (Loddick et al., 1997) and suggest that CB-induced neuroprotection is mediated, at least in part, by endogenous IL-1ra.
Many cell types are susceptible to excessive NO generation, and reduction of NO production markedly improves cell survival and is potentially beneficial in the treatment of a range of pathologies, including septic shock, neurodegeneration, and inflammation (Hobbs et al., 1999). CBs inhibit generation of NO by glial cells (Cabral et al., 2001; Molina-Holgado et al., 2002), and the present results demonstrate that reduction of LPS-induced NO generation by CBs in glial cells is dependent on endogenous IL-1ra.
In summary, the results presented here support our hypothesis that endogenous IL-1ra mediates the neuroprotective and anti-inflammatory actions of CBs in primary neurons and glia. These effects appear to be mediated by both CB1 and CB2 receptors. CB-induced IL-1ra release may negatively regulate IL-1β actions in the brain, via IL-1ra blocking the IL-1 receptor (IL-1RI), after inflammatory or excitotoxic insults. It is tempting to speculate therefore that the neuroprotective and anti-inflammatory actions of CBs depend in part on modification of the balance between proinflammatory and anti-inflammatory cytokines. These findings have important implications for our understanding of the mechanisms of action of CBs in diverse CNS disorders and for the development of new neuroprotective therapies.
Footnotes
This work was supported by a Wellcome Trust International Travelling Postdoctoral Fellowship (F.M.-H.), Medical Research Council (United Kingdom) Recognizing Our Potential Awards Grant G9819368, Comisíon Interministerial de Ciencia y Tecnología (Ministerio de Educación y Cultura, Spain), and European Union BioMed-2 Programme Contract BMH4-CT-97-2942. The IL-1ra knock-out mice were a generous gift from Prof. Yoichiro Iwakura (University of Tokyo, Tokyo, Japan). The cannabinoid HU-210 was a generous gift from Prof. Raphael Mechoulam (Hebrew University, Jerusalem, Israel).
Correspondence should be addressed to Nancy J. Rothwell, School of Biological Sciences, 1.124 Stopford Building, Oxford Road, Manchester, M13 9PT, UK. E-mail: nancy.rothwell@man.ac.uk.
F. Molina-Holgado's present address: Neurology Unit, Department of Clinical Veterinary Medicine, University of Cambridge, Cambridge, CB3 OES, UK.
J. D. Moore's present address: Department of Pathology, University of Cambridge, Cambridge, CB2 1QP, UK.
Copyright © 2003 Society for Neuroscience 0270-6474/03/236470-05$15.00/0
References
- Allan SM, Rothwell NJ ( 2001) Cytokines and acute neurodegeneration. Nat Rev Neurosci 2: 734-744. [DOI] [PubMed] [Google Scholar]
- Cabral GA, Harmon KN, Carlisle SJ ( 2001) Cannabinoid-mediated inhibition of inducible nitric oxide production by rat microglial cells: evidence for CB1 receptor participation. Adv Exp Med Biol 493: 207-214. [DOI] [PubMed] [Google Scholar]
- Davies CA, Loddick SA, Toulmond S, Stroemer RP, Hunt J, Rothwell NJ ( 1999) The progression and topographic distribution of interleukin-1beta expression after permanent middle cerebral artery occlusion in the rat. J Cereb Blood Flow Metab 19: 87-98. [DOI] [PubMed] [Google Scholar]
- Dinarello CA, Thompson RC ( 1991) Blocking IL-1: interleukin 1 receptor antagonist in vivo and in vitro. Immunol Today 12: 404-410. [DOI] [PubMed] [Google Scholar]
- Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR ( 1982) Analysis of nitrate, nitrite, and [15N]nitrate in biological fluids. Anal Biochem 126: 131-138. [DOI] [PubMed] [Google Scholar]
- Hampson AJ, Grimaldi M, Axelrod J, Wink D ( 1998) Cannabidiol and (-)Delta9-tetrahydrocannabinol are neuroprotective antioxidants. Proc Natl Acad Sci USA 95: 8268-8273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs AJ, Higgs A, Moncada S ( 1999) Inhibition of nitric oxide synthase as a potential therapeutic target. Annu Rev Pharmacol Toxicol 39: 191-220. [DOI] [PubMed] [Google Scholar]
- Horai R, Asano M, Sudo K, Kanuka H, Suzuki M, Nishihara M, Takahashi M, Iwakura Y ( 1998) Production of mice deficient in genes for interleukin (IL)-1alpha, IL-1beta, IL-1alpha/beta, and IL-1 receptor antagonist shows that IL-1beta is crucial in turpentine-induced fever development and glucocorticoid secretion. J Exp Med 187: 1463-1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, Devane WA, Felder C, Herkenham M, Mackie K, Martin BR, Mechoulam R, Pertwee RG ( 2002) International Union of Pharmacology XXVII classification of cannabinoid receptors. Pharmacol Rev 54: 161-202. [DOI] [PubMed] [Google Scholar]
- Jordan M, Otterness IG, Ng R, Gessner A, Rollinghoff M, Beuscher HU ( 1995) Neutralization of endogenous IL-6 suppresses induction of IL-1 receptor antagonist. J Immunol 154: 4081-4090. [PubMed] [Google Scholar]
- Josephs MD, Solorzano CC, Taylor M, Rosenberg JJ, Topping D, Abouhamze A, Mackay SL, Hirsch E, Hirsh D, Labow M, Moldawer LL ( 2000) Modulation of the acute phase response by altered expression of the IL-1 type 1 receptor or IL-1ra. Am J Physiol Regul Integr Comp Physiol 278: R824-R830. [DOI] [PubMed] [Google Scholar]
- Klein TW, Lane B, Newton CA, Friedman H ( 2000) The cannabinoid system and cytokine network. Proc Soc Exp Biol Med 225: 1-8. [DOI] [PubMed] [Google Scholar]
- Loddick SA, Wong M-L, Bongiorno PB, Gold PW, Licinio J, Rothwell NJ ( 1997) Endogenous interleukin-1 receptor antagonist is neuroprotective. Biochem Biophys Res Commun 234: 211-215. [DOI] [PubMed] [Google Scholar]
- Mackie K, Hille B ( 1992) Cannabinoids inhibit N-type calcium channels in neuroblastoma-glioma cells. Proc Natl Acad Sci USA 89: 3825-3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechoulam R, Panikashvili D, Shohami E ( 2002a) Cannabinoids and brain injury: therapeutic implications. Trends Mol Med 8: 58-61. [DOI] [PubMed] [Google Scholar]
- Mechoulam R, Spatz M, Shohami E ( 2002b) Endocannabinoids and neuro-protection. Science STKE 129: 1-6. [DOI] [PubMed] [Google Scholar]
- Molina-Holgado F, Lledo A, Guaza C ( 1997) Anandamide suppresses nitric oxide and TNF-alpha responses to Theiler's virus or endotoxin in astrocytes. NeuroReport 8: 1929-1933. [DOI] [PubMed] [Google Scholar]
- Molina-Holgado F, Molina-Holgado E, Guaza C ( 1998) The endogenous cannabinoid anandamide potentiates interleukin-6 production by astrocytes infected with Theiler's murine encephalomyelitis virus by a receptor-mediated pathway. FEBS Lett 433: 139-142. [DOI] [PubMed] [Google Scholar]
- Molina-Holgado F, Molina-Holgado E, Guaza C, Rothwell NJ ( 2002) Role of CB1 and CB2 receptors in the inhibitory effects of cannabinoids on lipopolysaccharide induced nitric oxide release in astrocyte cultures. J Neurosci Res 67: 829-836. [DOI] [PubMed] [Google Scholar]
- Moore JD, Rothwell NJ, Gibson RM ( 2002) Involvement of caspases and calpains in cerebrocortical neuronal cell death is stimulus-dependent. Br J Pharmacol 135: 1069-1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadler V, Mechoulam R, Sokolovsky M ( 1993) Blockade of 45Ca2+ influx through the N-methyl-d-aspartate receptor ion channel by the nonpsychoactive cannabinoid HU-211. Brain Res 622: 79-85. [DOI] [PubMed] [Google Scholar]
- Nagayama T, Sinor AD, Simon RP, Chen J, Graham SH, Jin K, Greenberg DA ( 1999) Cannabinoids and neuroprotection in global and focal cerebral ischemia and in neuronal cultures. J Neurosci 19: 2987-2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panikashvili D, Simeonidou C, Ben-Shabat S, Hanus L, Breuer A, Mechoulam R, Shohami E ( 2001) An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature 413: 527-531. [DOI] [PubMed] [Google Scholar]
- Pertwee RG ( 2002) Cannabinoids and multiple sclerosis. Pharmacol Ther 95: 165-174. [DOI] [PubMed] [Google Scholar]
- Puffenbarger RA, Boothe AC, Cabral GA ( 2000) Cannabinoids inhibit LPS-inducible cytokine mRNA expression in rat microglial cells. Glia 29: 58-69. [PubMed] [Google Scholar]
- Safieh-Garabedian B, Poole S, Allchorne A, Winter J, Woolf CJ ( 1995) Contribution of interleukin-1 to the inflammation-induced increase in nerve growth factor levels and inflammatory hyperalgesia. Br J Pharmacol 115: 1265-1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen M, Thayer SA ( 1998) Cannabinoid receptor agonists protect cultured rat hippocampal neurons from excitotoxicity. Mol Pharmacol 54: 459-462. [DOI] [PubMed] [Google Scholar]
- Shohami E, Gallily R, Mechoulam R, Bass R, Ben-Hur T ( 1997) Cytokine production in the brain following closed head injury: dexanabinol (HU-211) is a novel TNF-alpha inhibitor and an effective neuroprotectant. J Neuroimmunol 72: 169-177. [DOI] [PubMed] [Google Scholar]
- van der Stelt M, Veldhuis WB, van Haaften GW, Fezza F, Bisogno T, Bär PR, Veldink GA, Vliegenthart JFG, Di Marzo V, Nicolay K ( 2001) Exogenous anandamide protects rat brain against acute neuronal injury in vivo. J Neurosci 21: 8765-8771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan M, Kiertscher SM, Cheng Q, Zoumalan R, Tashkin DP, Roth MD ( 2002) Delta 9-Tetrahydrocannabinol regulates Th1/Th2 cytokine balance in activated human T cells. J Neuroimmunol 133: 124-131. [DOI] [PubMed] [Google Scholar]