

Abstract
The distribution of glutamate receptor subtypes on the surface of neurons is highly relevant for synaptic transmission and signal processing. In the present study we investigated the location and properties of functional kainate receptors (KARs) on the somatodendritic membrane of rat neocortical layer V pyramidal neurons. Infrared-guided laser stimulation was used to apply glutamate photolytically to the soma and various sites along the apical dendrite. Electrical currents, resulting from the activation of pharmacologically isolated KARs, were measured by whole-cell patch-clamp recording. In addition, KARs on somatic and dendritic outside-out patches were activated while still within the brain tissue. We found that functional KARs are located on the entire somatodendritic membrane that was examined. Fast kinetics, a linear I-V relationship, and a relatively high single-channel conductance are characteristic features of these receptors. We provide evidence that the unitary properties of somatic and dendritic KARs are identical. Regarding the subcellular distribution of KARs, our results indicate that the density of these receptors increases toward the distal dendrite. They are located mainly at extrasynaptic sites but also mediate fast synaptic signaling triggered by afferent stimulation. The differential distribution speaks in favor of a selective targeting of KARs on central neurons and may reflect a mechanism for a location-dependent regulation of synaptic efficacy. Furthermore, it is feasible to assume that extrasynaptic KARs could be activated by a “spillover” of synaptically released glutamate, ambient glutamate in the CSF, or glutamate released from adjacent astrocytes.
Keywords: AMPA receptors, brain slices, caged glutamate, dendrite, distribution, extrasynaptic, kainate receptors, layer V, neocortex, photostimulation, pyramidal neuron, rat, somatosensory, synaptic
Introduction
Ionotropic glutamate receptors are divided into three main classes: NMDA, AMPA, and kainate receptors (KARs). KARs are composed of five subunits (GluR5-GluR7, KA1-KA2), which can assemble to form functional channels. Most of our knowledge about the properties of KARs (e.g., kinetics and composition of subunits) comes primarily from studies on recombinant KARs. A general feature of these receptors is their fast kinetics, similar to those determined for AMPA receptors. KAR subunit genes are expressed throughout the brain (Wisden and Seeburg, 1993), but little is known about the properties, the subcellular distribution, and the functional roles of native postsynaptic KARs. It has been shown only recently that postsynaptic KARs contribute to the EPSC at different types of synapses (Frerking and Nicoll, 2000; Lerma et al., 2001).
Synaptic activation of postsynaptic KARs sometimes requires repetitive synaptic stimulation (Castillo et al., 1997; Vignes and Collingridge, 1997), and the KAR-mediated EPSCs have small amplitudes and show mostly slow kinetics. These observations have raised the following questions. (1) Are postsynaptic KARs located extrasynaptically? (2) If so, are KARs activated by a “spillover” of synaptically released glutamate? (3) Are the slow kinetics of native KARs an intrinsic property of heteromers, the exact subunit composition of which is unknown? (4) Do postsynaptic KARs play only a minor role in fast synaptic transmission? The studies addressing these questions in the recent past have yielded conflicting results (Min et al., 1998; Swanson and Heinemann, 1998; Bureau et al., 1999; DeVries and Schwartz, 1999; Kidd and Isaac, 1999, 2001; Cossart et al., 2002).
KAR subunits are expressed abundantly in the neocortex (Wisden and Seeburg, 1993). However, studies on neocortical KARs at the level of single neurons or synapses are scarce. KARs are located on presynaptic terminals of neocortical interneurons (Ali et al., 2001) as well as on the postsynaptic membrane of layer IV pyramidal neurons (Kidd and Isaac, 1999). Layer V pyramidal neurons also express KAR subunits (Huntley et al., 1993), but it is still unclear whether these subunits form functional receptors inserted in the somatodendritic membrane. In the present study we used a combination of infrared-guided laser stimulation and electrophysiological techniques to address this question. We found that functional KARs are located on the entire somatodendritic membrane that was examined. Furthermore, we characterized the intrinsic properties of these receptors and report that they are distributed differentially at the apical dendrite and located mainly at extrasynaptic sites.
Materials and Methods
Slice preparation. Parasagittal neocortical slices (300 μm thick) were prepared from 42 male Sprague Dawley rats (postnatal days 14-21). Before decapitation the rats were anesthetized very shortly and slightly to reduce stress response and for better handling while their heads were placed under an animal guillotine. Fluid isoflurane (0.2 ml) was dropped on a piece of fleece placed on the bottom of a vessel (volume, 3.5 l). After 3 min the animals were put into the closed vessel for 5-7 sec to make them numb. Afterward, they were taken out rapidly, and the neck was fixed under the animal guillotine. In this position the animals were kept until their initial numbness was abolished completely. In particular, it was strictly observed that the animals were breathing spontaneously and deeply the whole time to ensure that isoflurane was eliminated completely from the CNS before decapitation. This is of major importance because there is evidence that isoflurane may influence KAR function (Dildy-Mayfield et al., 1996).
After we removed the brain from the skull, a tissue block containing the somatosensory cortex was trimmed, glued to an acrylic glass cuboid (Histoacryl, Braun, Tuttlingen, Germany), and transferred to the chilled specimen holder of a vibratome (Vibracut 3, FTB, Weinheim, Germany). All steps of the preparation were performed in ice-cold artificial CSF (ACSF) oxygenated with carbogen (95% O2/5% CO2). The ACSF consisted of (in mm): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2, 25 NaHCO3, and 25 glucose, pH 7.4 (all substances were from Merck, Ismaning, Germany). The slices were transferred to glass vials containing oxygenated ACSF, incubated for 30 min at 34°C, and subsequently were stored at room temperature (22-24°C) for at least 30 min (Dodt et al., 1998).
Electrophysiology. Brain slices were placed in the recording chamber of an “infrapatch” set-up (Luigs & Neumann, Ratingen, Germany) and submerged in oxygenated ACSF (flow rate, 2-3 ml/min). Individual layer V pyramidal neurons of the somatosensory cortex were identified visually via infrared videomicroscopy and the “gradient contrast” system (Dodt et al., 2002). Whole-cell recordings (seal resistance, >1 GΩ) from somata and apical dendrites were performed in voltage-clamp mode (-60 mV holding potential), using a dSEVC (discontinuous single-electrode voltage-clamp) amplifier (SEC-10LX, npi Electronics GmbH, Tamm, Germany). Recordings were accepted only if the holding current was <100 pA. The current was low-pass filtered (1-3 kHz). The patch-clamp electrodes (open-tip resistances, 4-9 MΩ) were pulled from borosilicate glass capillaries (1.5 mm outer diameter, 0.86 mm inner diameter; Harvard Apparatus, Kent, UK) on a DMZ-Universal puller (Zeitz Instruments, Munich, Germany). The patch pipettes were filled with a solution consisting of (in mm): 105 K-gluconate (Merck), 30 KCl, 4 Mg-ATP (Sigma, Deisenhofen, Germany), 10 phosphocreatine (Sigma), 0.3 GTP (Sigma), and 10 HEPES (Biomol GmbH, Hamburg, Germany); pH 7.3-adjusted with KOH (osmolarity, 305 mOsm). In some experiments spermine (60 μm; Sigma) also was included. The cells were characterized electrophysiologically by recording the membrane potential in bridge mode. I-V relationships and spike trains were obtained by injecting hyperpolarizing and depolarizing current pulses. From 104 recorded neurons 96 cells were of the “regular spiking” and 8 cells of the “intrinsically bursting” type (Connors and Gutnick, 1990). It is feasible to assume that the properties and/or the subcellular distribution of KARs vary between these physiologically and morphologically different types of neurons. Therefore, only the regular spiking cells were included in this study. They had an average resting potential of -60 ± 0.5 mV and an input resistance of 99 ± 3 MΩ. No correction for the junction potential between the bath and pipette solutions was made. Full correction for this junction potential would make all membrane potential measurements ∼13 mV more negative than indicated. The series resistance [15 ± 0.2 MΩ for somatic (n = 81) and 23 ± 0.9 MΩ for dendritic (n = 15) whole-cell recordings] was monitored continuously during the recordings. Recordings were terminated if the series resistance changed >10%. After seal formation and patch rupture the outside-out patches were obtained by slowly withdrawing the patch pipette from the cell. Outside-out patches were accepted only if the resistance of the patch was ≥1 GΩ and the holding current <10 pA. Bipolar electrical stimulation of synapses was performed by using pipettes (∼2 μm tip diameter) pulled from borosilicate glass theta-capillaries (1.5 mm outer diameter, 1.05 mm inner diameter, 0.16 mm thickness of partition wall; Hilgenberg, Malsfeld, Germany). Both tubes of the theta-capillary were filled with ACSF and connected via Ag/AgCl wires to an isolated stimulus generator (type 2533, Devices Instruments LTD, Hertfordshire, UK). The stimulation electrode was placed near the apical dendrite (∼5 μm lateral distance). In this way the synapses, located in a defined region of the dendrite, could be activated reliably. Duration of the electrical stimuli was 0.2 msec. Data were digitized at 3-9 kHz via an ITC-16 interface (Heka Elektronik, Lambrecht/Pfalz, Germany) and stored by a Macintosh-based recording system with the standard software Pulse 8.00 (Heka Elektronik). All experiments were performed at room temperature (22-24°C).
Photostimulation. Glutamate was applied to visually identified somata and apical dendrites by UV flash photolysis (wavelength, 351-364 nm) of γ-CNB (α-carboxy-2-nitrobenzyl)-caged glutamate (0.5 or 1 mm; Molecular Probes, Leiden, The Netherlands). The beam of a UV laser (Enterprise II, Coherent, Dieburg, Germany) was coupled into the epifluorescence port of the microscope (Axioskop FS, Zeiss, Jena, Germany) via a quartz fiber (Luigs & Neumann) and focused by the objective (60×, numerical aperture, 0.9; Olympus, Hamburg, Germany) onto a 1 μm spot in the specimen plane. Using a caged glutamate concentration of 0.5 mm, we determined in previous studies the site of glutamate release as a spot with a diameter of ∼10 μm (Dodt et al., 1999; Eder et al., 2002). The duration of the shuttered light pulses (Uniblitz shutter, Vincent Associates, Rochester, NY) was 3 msec. The output power of the argon ion laser (maximum, 90 mW) was adjusted by a remote control unit in increments of 1 mW. The caged glutamate used in this study has a quantum yield of 0.14, and the half-life of the major component of the photolytic reaction is in the microsecond range (Wieboldt et al., 1994). A concentration of 0.5 mm does not desensitize glutamate receptors by spontaneous hydrolysis and/or contamination with free glutamate (Eder et al., 2002). Furthermore, it also has been shown that 1 mm γ-CNB-caged glutamate neither desensitizes glutamate receptors in cultured hippocampal neurons (Wieboldt et al., 1994) nor influences the properties of layer V pyramidal neurons of 2- to 3-week-old rats (Schubert et al., 2001). To make sure that this is also valid for our experiments in which 1 mm caged glutamate was used, we performed additional control experiments. We tested whether 1 mm caged glutamate affects the amplitude of glutamate receptor-mediated EPSCs (for a detailed description of the methods used, see Eder et al., 2002). Consistent with the results from Wieboldt et al. (1994) and Schubert et al. (2001), the amplitude does not change significantly after the addition of caged glutamate [98 ± 4% as compared with control; p = 0.72 (15 EPSCs in the absence and presence of caged glutamate were averaged for 4 neurons/3 animals)]. Furthermore, exposure of the cells to the brief periods of uncaging light used in our experiments has no effect on neuronal activity (Dodt et al., 1999). During the application of caged glutamate a total amount of 4 ml of ACSF containing the caged compound was recirculated continuously and oxygenated. The following drugs were added to the ACSF: CdCl2 (Sigma), p-(3-aminopropyl)-p-diethoxymethyl-phosphinic acid (CGP35348, kind gift from Ciba-Geigy AG, Basel, Switzerland), cyclothiazide (Sigma), d-2-amino-5-phosphonopentanoate acid (d-AP-5, Sigma), GYKI 53655 (LY300168, kindly provided by Merz & Company, Frankfurt, Germany), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, Sigma), NS-102 (Sigma), picrotoxin (Sigma), and tetrodotoxin (TTX; Sigma). Depending on the substance, complete wash-in lasted 10-30 min. Data were collected as soon as complete wash-in was confirmed by a stable baseline.
Data analysis. Analysis of amplitudes, charges, kinetics, and single-channel conductance (γ) was performed by using a software written in the IgorPro 4.01 programming language (WaveMetrics, Lake Oswego, OR) by Dr. K. Becker. γ was estimated by using peak-scaled nonstationary fluctuation analysis (non-SFA). For non-SFA, sets of 50 subsequently measured glutamate responses (interstimulus interval, 45 sec) were used. From each glutamate response a segment of a fixed length was selected, ranging from the peak of the current to past the end of the decay phase (total length, 150 msec). The average current over all segments was calculated and scaled to the peak of the individual glutamate responses; then the variance of each glutamate response around the scaled average curve was computed (Traynelis et al., 1993; Silver et al., 1996). Current traces showing a variance larger than approximately three times the mean variance were excluded from the calculations. Because of the highly precise onset of the laser stimulus, the subsequently measured current traces already were aligned precisely. An additional manual alignment therefore was not necessary. The computed variances were divided into 50 equally sized bins on the amplitude scale and were plotted against the mean current value within each scaled average current bin. These data points were fit with the relationship:
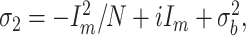 |
by a least-square algorithm. σ2 is the variance; Im is the mean current over the membrane; N is the total number of channels activated at the peak of the mean current; i is the unitary current of a single channel; and σb2 is the background variance, i.e., the average variance of the baselines (Sigworth, 1980; Traynelis et al., 1993). From the computed single-channel current (i) γ was calculated according to the relationship:
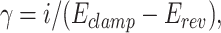 |
where Eclamp is the holding potential used in the experiments and Erev is the reversal potential derived in previous experiments (7 mV).
For spectral analysis the scaled average current was subtracted from each single current, and the variance was calculated as described above. Data segments of 1024 points in length were transformed into the frequency domain by a discrete fast-Fourier transformation (FFT), and the single-sided power spectrum was computed. To avoid frequency leakage errors, we Parzen-windowed data before transformation. The power spectrum, calculated from the binned FFT values (20 bins), was fit by the sum of n Lorentzian functions:
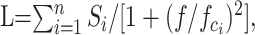 |
where L(f) is the spectral density at frequency f, Si is the power of the spectrum at f = 0, and fci is the cutoff frequency at which the spectral power is half-maximal. The time constants of the Lorentzian functions were calculated via the relationship:
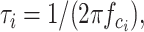 |
according to De Koninck and Mody (1997). The power of the spectrum was not corrected for the power of the mean time course as described in Sigworth (1981), because the aim of the present study was to derive only the corner frequencies. γ was derived from non-SFA analysis as described above. The necessity to introduce additional Lorentzian functions into the fits was judged visually by comparing the calculated curves with the respective data points (De Koninck and Mody, 1997).
All values are expressed as the mean ± SEM. In every brain slice only one neuron was recorded, and measurements from not more than two neurons from one animal were used for the calculation of a particular mean value. Statistical evaluation was performed with the paired or unpaired Student's t test (SigmaStat 2.03).
Results
Neocortical layer V pyramidal neurons express functional KARs on the somatodendritic membrane
To investigate whether neocortical layer V pyramidal neurons express functional KARs on the somatodendritic membrane, we applied glutamate photolytically to the soma and to sites 20 μm apart along the apical dendrite (up to 300 μm from the center of the soma). Caged glutamate (0.5 mM) was photolyzed by 3 msec light pulses of a UV laser. The electrical currents resulting from the activation of glutamate receptors were recorded from the neuronal soma. In all experiments NMDA receptors were blocked by the addition of D-AP-5 (100 μM), and TTX (1 μM) was added to isolate the neuron being investigated from synaptic inputs.
Glutamate also was applied photolytically to outside-out patches obtained from the soma or apical dendrite. It reliably evoked currents in the outside-out patch, which does not have to be transferred to an application system outside the slice, thus ensuring more physiological conditions. To evoke more prominent glutamate responses, we increased the concentration of caged glutamate to 1 mM in these experiments.
In the presence of the NMDA receptor antagonist D-AP-5, glutamate-activated currents could be elicited at all of the somatic and dendritic sites that were stimulated. At all sites the selective and noncompetitive AMPA receptor (AMPAR) antagonist GYKI 53655 (50 μM) (Vignes and Collingridge, 1997) blocked only apart of the glutamate response [49 ± 2%; p < 0.001; n = 175 stimulation sites, 12 neurons/10 animals (8 of these neurons were stimulated up to 300 μm from the center of the soma, 1 neuron up to 260 μm, and 3 neurons up to 200 μm)]. This finding suggests that this remarkably large GYKI 53655-resistant component is mediated by KARs. Further experiments confirmed this suggestion. First, an increase of the concentration of GYKI 53655 to 100 μM did not lead to a further decrease of the glutamate response amplitude (103 ± 3% as compared with 50 μM GYKI; p = 0.38; n = 7 neurons/6 animals; Fig. 1A,C). This indicates a complete block of AMPARs by GYKI 53655 (50 μM). Second, the AMPA/KAR antagonist CNQX (50 μM) (Kidd and Isaac, 1999) completely blocked the GYKI 53655-resistant component (reduction of the amplitude to 1 ± 1% as compared with 50 μM GYKI; p < 0.001; n = 6 neurons/6 animals; Fig. 1B,C). Third, in the presence of GYKI 53655 (100 μM) the selective AMPAR potentiator cyclothiazide (8 μM) had no effect on the amplitude and kinetics of the glutamate response (Fig. 1D). In contrast, in the absence of GYKI 53655, cyclothiazide caused an increase in amplitude and prolonged the decay phase by diminishing the rate of AMPAR desensitization (Fig. 1E). To detail both of these effects, we evaluated the charge transfer [98 ± 2% in the presence of GYKI 53655 (p = 0.73; n = 5 neurons/5 animals; Fig. 1G, first column); 211 ± 14% in the absence of GYKI 53655 (p < 0.01; n = 5 neurons/5 animals; Fig. 1G, second column)]. The high concentration of GYKI 53655 and the low concentration of cyclothiazide were used for the following reason. It has been shown that cyclothiazide at high concentrations reverses AMPAR antagonism of GYKI 53655 by an allosteric mechanism (Palmer and Lodge, 1993; Rammes et al., 1996). This effect might interfere with the pharmacological isolation of KAR-mediated responses by GYKI 53655, thereby causing a potentiation of the glutamate response in the presence of GYKI 53655. We observed this effect. In the presence of 50 μM GYKI 53655 cyclothiazide (100 μM) increased the amplitude and prolonged the kinetics of the glutamate response (Fig. 1 F). The increase of the charge transfer was 375 ± 25% for somatic glutamate responses (p < 0.001; n = 5 neurons/4 animals; Fig. 1G, third column) and 354 ± 24% for responses elicited at the dendrite (200 μm from the center of the soma; p = 0.001; n = 5 neurons/4 animals; Fig. 1G, fourth column). The increase of the charge transfer at the soma and the increase at the dendrite were not different (p = 0.64).
Figure 1.
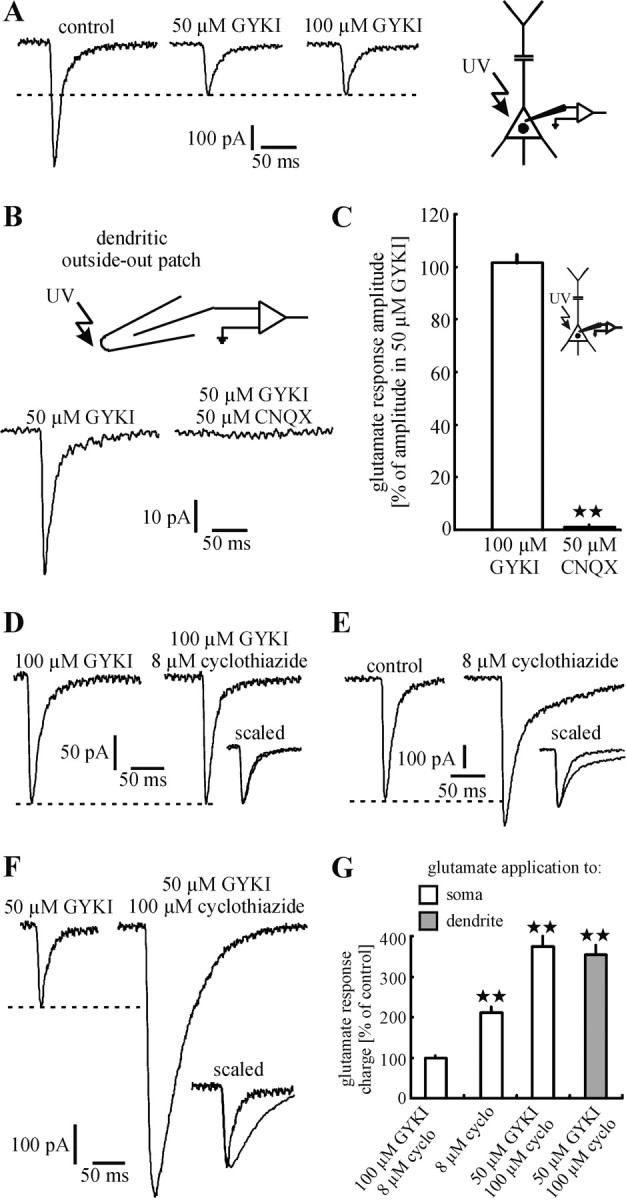
Neocortical layer V pyramidal neurons express functional KARs on the somatodendritic membrane. A, In the presence of d-AP-5 (control) photolytic application of glutamate to the somatodendritic membrane elicited inward currents, which were blocked only partially by GYKI 53655 (50 μm). The remaining glutamate responses, mediated by KARs because they were not affected by an increase in the concentration of GYKI 53655 to 100 μm, were blocked completely by CNQX (50 μm; B, C) and not potentiated by cyclothiazide (8 μm; D). In contrast, cyclothiazide (8 μm) potentiated mixed AMPA/KAR-mediated responses (in the absence of GYKI 53655; E). Because of an allosteric reversion of AMPAR antagonism of GYKI 53655 by cyclothiazide at a high concentration (100 μm), the glutamate responses were also potentiated in the presence of GYKI 53655 (50 μm; F). A, B, D-F show single representative experiments. C, G, Statistical evaluation of the experiments shown in A, B, D-F. A significant difference (p < 0.01) is indicated by two asterisks.
Although presented in more detail in the next sections, it should be noted at this point that photolytic application of glutamate to somatic or dendritic outside-out patches also evoked reliably KAR-mediated currents (Fig. 1B).
Properties of KARs activated by photolytically applied glutamate
Kinetics
KARs in cultured cells and heterologous expression systems show fast kinetics similar to those determined for AMPARs (Lerma et al., 2001). However, KAR-mediated EPSCs mostly show markedly slower kinetics [rise time, 10-20 msec; decay time constant (τ), >100 msec]. To investigate whether KARs of layer V pyramidal neurons share the characteristic features of fast activation and desensitization, we determined the rise time (10-90%) and τ of glutamate responses that were evoked and recorded at the soma.
For the interpretation of the following results, it has to be noted that the kinetics of photolytically evoked glutamate responses are two to three times as long as those of synaptically evoked glutamate responses (Dodt et al., 1999; Eder et al., 2002). Because the rise time of a photolytically evoked current is equal to the length of the uncaging pulse plus the time during which any agonist that is produced can diffuse to additional receptors, we can make a rough estimate of this kinetic parameter but probably have not the necessary resolution to detect small differences among the rise times of responses elicited at different stimulation sites. Thus τ of photolytically evoked currents is the more significant parameter when comparing the kinetics of ion channels located at different sites of the neuronal membrane.
Mixed AMPA/KAR-mediated currents had, on average, a rise time of 4.0 ± 0.1 msec and τ of 18.9 ± 1.4 msec (n = 15 sweeps, 15 neurons/13 animals; Fig. 2A,D). The respective values determined for pure KAR-mediated currents were 3.9 ± 0.2 and 18.5 ± 1.2 msec (n = 15 sweeps, 15 neurons/13 animals; Fig. 2A,D). Similar values were obtained from KAR-mediated responses, which were elicited at somatic outside-out patches (rise time, 3.8 ± 0.6 msec; τ, 17.2 ± 1.4 msec; n = 7 sweeps, 7 patches/7 animals; Fig. 2C,D).
Figure 2.
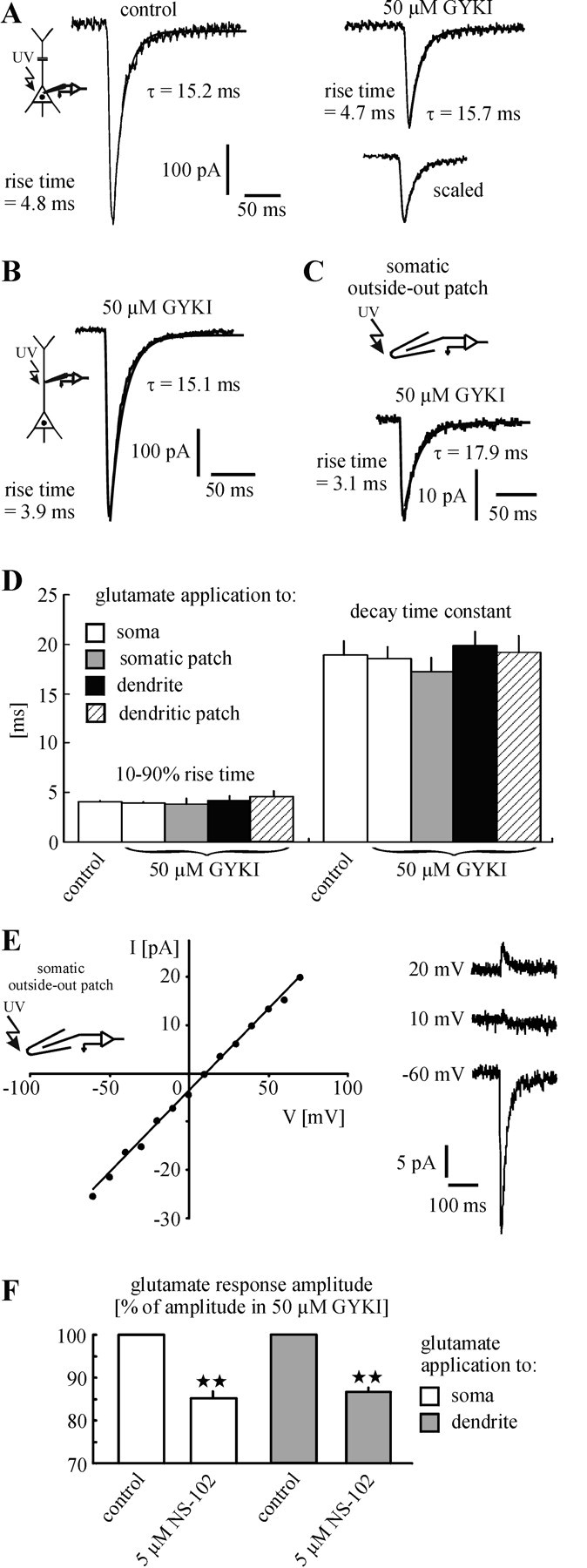
Properties of KARs. A-D, KARs on layer V pyramidal neurons show fast kinetics similar to those of AMPARs. Statistical evaluation revealed no difference between the rise time values of mixed AMPA/KAR-mediated responses (control) and pure KAR-mediated currents that were evoked at the soma (A). This is also the case for the decay time constant (τ). Comparison of the kinetics of KAR-mediated responses, which were elicited at somata (A), somatic outside-out patches (C), apical dendrites (B), and dendritic outside-out patches (no example shown; see Figs. 1B, 4C) revealed no significant differences (D). This result refers to identical unitary properties of somatic and dendritic KARs. A-C show single traces. The current decay was best fit monoexponentially (smooth lines). E, KAR-mediated responses, which were evoked at somatic outside-out patches, showed a linear I-V relationship and had a mean reversal potential of 7 ± 2 mV (n = 6 patches). A single experiment is shown. F, The GluR5/GluR6 antagonist NS-102 decreased the amplitude of KAR-mediated currents that were elicited at the soma and apical dendrite. This result refers to an involvement of GluR5 and/or GluR6 in the formation of KARs on layer V pyramidal neurons.
In additional experiments we examined the kinetics of dendritic KARs. To avoid electrotonic slowing of the glutamate response kinetics by a likely loss of spatial control of the dendritic membrane potential during somatic whole-cell recording, we recorded the cells at the dendrite (at a distance of ∼200 μm from the center of the soma) and photolytically applied glutamate close to the patch pipette. The responses mediated by dendritic KARs showed fast kinetics similar to those mediated by somatic KARs (rise time, 4.2 ± 0.4 msec; τ, 19.8 ± 1.5 msec; n = 8 sweeps, 8 neurons/7 animals; Fig. 2B,D). Similar values were obtained from KAR-mediated responses, which were elicited at dendritic outside-out patches (rise time, 4.5 ± 0.7 msec; τ, 19.1 ± 1.9 msec; n = 7 sweeps, 7 patches/7 animals; Fig. 2D). The current decay of all sweeps was best fit monoexponentially. Statistical evaluation revealed no significant differences among all mean rise time values (p = 0.24-0.83). This was also the case for τ (p = 0.31-0.85).
The results described above indicate that KARs on layer V pyramidal neurons have fast kinetic properties resembling those of AMPARs. Furthermore, the kinetics of somatic and dendritic KARs are identical.
Reversal potential
To characterize KARs on layer V pyramidal neurons further under the best possible control of voltage, we applied glutamate in the presence of D-AP-5 and GYKI 53655 to somatic outside-out patches at different holding potentials. Measured at the peak, the KAR-mediated responses showed a linear I-V relationship and had a reversal potential close to 0 mV (7 ± 2 mV; n = 6 patches/6 animals; Fig. 2E). This finding indicates that the KARs involved in these responses belong predominantly to the Ca2+-impermeable form, because Ca2+-permeable KARs show strong inward rectification caused by a voltage-dependent block by polyamines (Burnashev et al., 1996). To exclude the possibility that the linear I-V relationship results from washout of polyamines, we performed control experiments in which spermine (60 μM) was added to the internal solution. These experiments also revealed a linear I-V relationship (n = 3 patches/3 animals; data not shown).
The expression of GluR5 and GluR6 seems to be characteristic for neocortical pyramidal neurons (Huntley et al., 1993; Kidd and Isaac, 1999). To test whether GluR5 and/or GluR6 subunits also are involved in the formation of functional KARs on layer V pyramidal neurons, we applied NS-102 (5 μM), an antagonist of GluR5 and GluR6 (Verdoorn et al., 1994; Wilding and Huettner, 1996), to the superfusion medium. NS-102 indeed decreased the amplitude of KAR-mediated currents, which were evoked either at the somata (15 ± 2% decrease; p = 0.002; n = 5 neurons/5 animals) or at the apical dendrites (200 μm from the center of the soma; 14 ± 1% decrease; p < 0.001; n = 5 neurons/5 animals; Fig. 2F). Statistical evaluation revealed no significant difference between the decrease at the soma and the decrease at the dendrite (p = 0.54). KARs containing edited GluR5 and/or GluR6 subunits are impermeable for Ca2+ ions and show a linear I-V relationship (Lerma et al., 2001). Thus our data strongly suggest that edited GluR5 and/or GluR6 subunits are involved in the formation of functional postsynaptic KARs on layer V pyramidal neurons.
Infrared-guided laser stimulation: a new method to examine single-channel properties of ligand-gated ion channels
To reveal γ of somatic and dendritic KARs, we performed whole-cell recording and glutamate application either at the soma or at the dendrite (at a distance of ∼200 μm from the center of the soma), respectively. The recorded glutamate responses were analyzed by using non-SFA. Both somatic and dendritic KARs showed nearly identical values of γ [7.7 ± 0.7 pS for somatic KARs; n = 6 neurons/5 animals (Fig. 3A,C); 8.1 ± 0.9 pS for dendritic KARs; n = 6 neurons/6 animals (Fig. 3B,C); p = 0.78].
Figure 3.
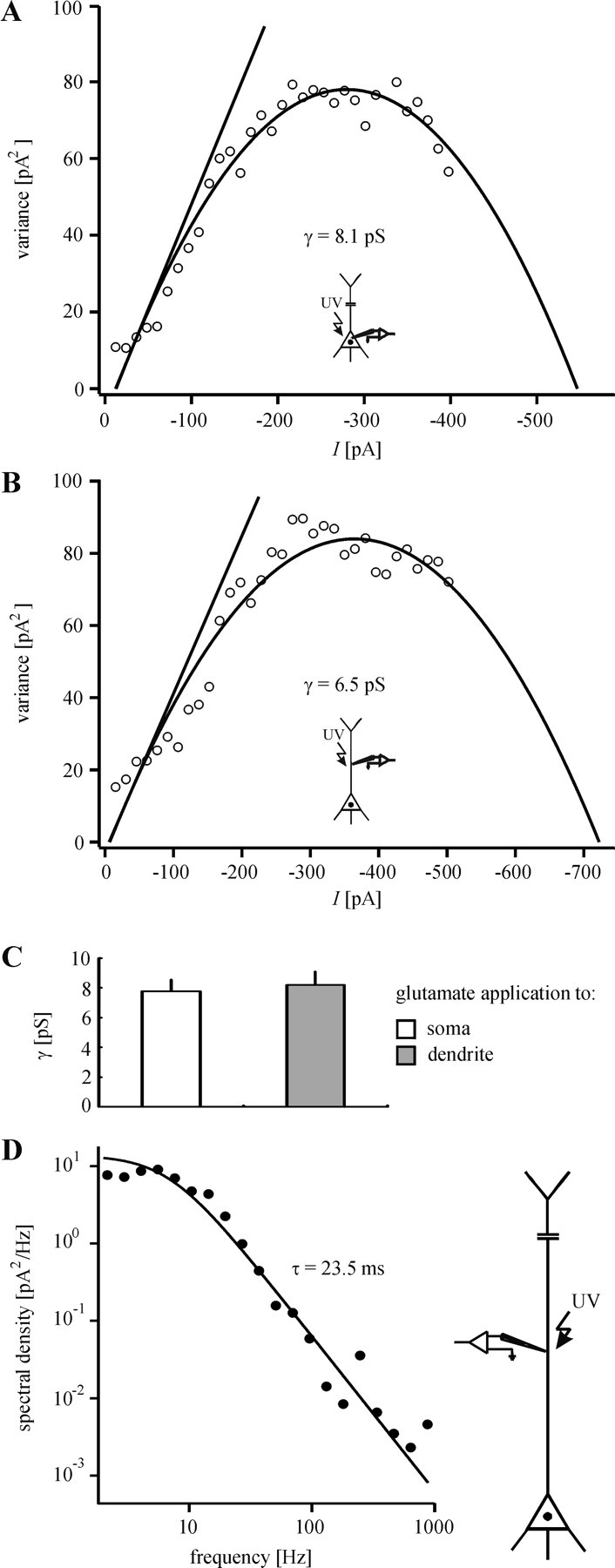
Unitary properties of KARs. The single-channel conductance (γ) of somatic (A) and dendritic (B) KARs was estimated by nonstationary fluctuation analysis. Consistent with the identical unitary properties of somatic and dendritic KARs, statistical evaluation revealed no significant difference between γ of somatic and γ of dendritic KARs (C). A,B show single experiments. D, The kinetics of KAR-mediated responses were examined by spectral analysis. Fits of sums of Lorentzian functions to the variance power spectra showed that the data can be described best by a single Lorentzian function. The presence of only a single corner frequency agrees well with the observation that, in the time domain, the decay phase of the evoked currents can be best fit monoexponentially. The mean time constants obtained by both ways are nearly identical.
We also investigated the kinetics of the KAR-mediated currents by spectral analysis. Fitting sums of Lorentzian functions to the power spectra of the measured current noise showed that the course of these spectra can be described best by a single Lorentzian function (Fig. 3D). This is consistent with our observation that the decay phase of the evoked currents can be well fit by a single exponential function (Fig. 2A-C). Our results, therefore, provide evidence that the kinetics of KARs on neocortical layer V pyramidal neurons can be described by a single time constant. As shown in Figure 2D, the kinetics of somatic and dendritic KARs do not differ significantly. We obtained the same result by spectral analysis. For this reason the time constants for all data sets were pooled. The mean time constant is 22.7 ± 2.1 msec (n = 12 neurons/11 animals). This agrees with the time constant of the exponential curves fit to the evoked currents (∼20 msec; Fig. 2D).
Subcellular distribution of KARs and AMPARs
Location-dependent ratio of the number of KARs to AMPARs
To examine the distribution of KARs and AMPARs on the somatodendritic membrane of layer V pyramidal neurons, we applied glutamate photolytically to sites 20 μm apart along the apical dendrite (up to 300 μm from the center of the soma). Glutamate application to the soma reaches a much larger membrane area than glutamate application to the dendrite. To avoid this influence of the geometry of the neuron on the profile of the dendritic glutamate sensitivity, we started dendritic stimulation at a distance of 40 μm from the center of the soma. The tapering of the soma into the dendrite ends approximately at this distance (Eder et al., 2001). In the presence of D-AP-5 the glutamate sensitivity of each site stimulated was measured before and after the addition of GYKI 53655 (50 μM). The amplitudes of the inward currents were expressed in percentage of the response evoked 40 μm from the center of the soma. Interestingly, the amplitudes of the mixed AMPA/KAR-mediated responses showed nearly the same profile as the amplitudes of the pure KAR-mediated responses (p ≥ 0.18 for all stimulation sites; n = 12 neurons/9 animals; Fig. 4A). This result suggests that the relative ratio of the number of KARs to AMPARs does not change toward the distal dendrite. This conclusion presupposes that the glutamate sensitivity of the stimulated membrane area is directly proportional to the number of activated receptors. Such a correlation can be assumed for the following reasons. (1) The kinetics of AMPARs and KARs are nearly identical (Fig. 2A,D). Thus the peak current of mixed AMPA/KAR-mediated responses represents for both glutamate receptor subtypes the time point of maximum channel opening. Furthermore, we can exclude a differential dendritic low-pass filtering of AMPAR- and KAR-mediated currents that could arise from imperfect spatial control of membrane potential during our voltage-clamp measurements. (2) Loss of voltage control with distance could result in the activation of dendritic voltage-dependent conductances that can affect the amplitude and shape of neuronal responses to glutamate (Johnston et al., 1996). Such effects on the photolytically evoked glutamate responses can be excluded because voltage-dependent Na+ channels were blocked in all experiments by the addition of TTX. Furthermore, blocking of Ca2+ channels by the addition of CdCl2 (200 μM) does not affect the amplitude of mixed AMPA/KAR-mediated responses, which were elicited at the most distal part of the dendrite that was examined (300 μm from the center of the soma; 102 ± 4% as compared with control; p = 0.99; n = 5 neurons/4 animals; Fig. 4B). This observation can be explained by the fairly uniform distribution of voltage-dependent Ca2+ conductances across the somatodendritic axis of layer V pyramidal neurons (Markram et al., 1995; Oakley et al., 2001; Migliore and Shepherd, 2002). Such a uniform distribution also has been found for KA channels (Migliore and Shepherd, 2002). (3) It is possible that local differences in the glutamate affinity of the receptors, rather than in the absolute number of these receptors, are responsible for the profiles of glutamate sensitivity observed. However, the nearly identical kinetics and values of γ of somatic and dendritic KARs (Figs. 2A,D, 3) are very indicative that these receptors have similar intrinsic properties.
Figure 4.
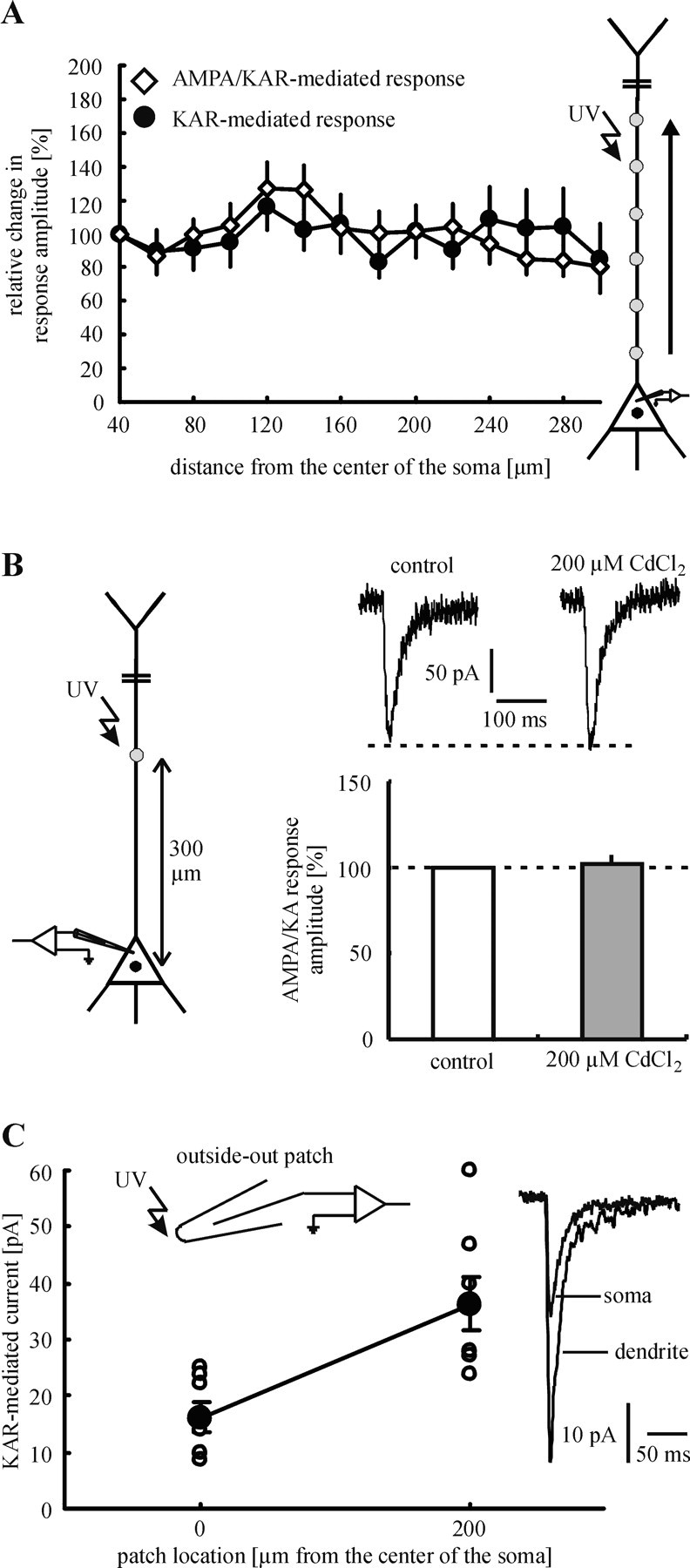
Subcellular distribution of KARs and AMPARs. A, Glutamate was applied photolytically to sites 20 μm apart along the apical dendrite. The profile of the dendritic glutamate sensitivity does not change after the addition of GYKI 53655. This result strongly suggests that the relative ratio of the number of KARs to AMPARs does not change toward the distal dendrite. B, Voltage-dependent Ca2+ channels do not affect the amplitude of mixed AMPA/KAR-mediated currents in our experiments. In the presence of d-AP-5 (control) mixed AMPA/KAR-mediated responses were evoked by glutamate application to the most distal part of the dendrite that was examined (300 μm from the center of the soma). To block voltage-dependent Ca2+ channels, we added CdCl2 (200 μm) to the superfusion medium. This pharmacological treatment changed neither the amplitude nor the size of the glutamate responses. C, Glutamate responses, which were evoked at somatic outside-out patches, showed smaller amplitudes on average than glutamate responses that were elicited at dendritic outside-out patches. Because of the evidence for identical unitary properties of somatic and dendritic KARs, this finding indicates an increasing density of KARs toward the distal dendrite.
The results described above strongly suggest that the relative ratio of the number of KARs to AMPARs does not change toward the distal dendrite. This suggestion is strengthened further by the observation that cyclothiazide exponentiates mixed AMPA/KAR-mediated responses at the soma and the dendrite to the same degree (Fig. 1G, third and fourth column).
Estimation of the density of KARs
In addition to γ, peak-scaled non-SFA also reveals the maximal number (N) of activated channels at the peak of the mean current (see Materials and Methods). In our experiments the mean values of N were 718 ± 79 for somatic stimulation (n = 6 neurons/5 animals) and 627 ± 90 for dendritic stimulation (at a distance of ∼200 μm from the center of the soma; n = 6 neurons/6 animals). To obtain the minimal density of KARs, we have to divide these values by the membrane surface that was exposed to glutamate. In previous studies we estimated the site of glutamate release as a spot of ∼10 μm diameter (Dodt et al., 1999; Eder et al., 2002). Thus the stimulated dendritic membrane area was assumed to be the surface of a 10-μm-long cylinder of 1 μm radius (= 62.8 μm2). The diameter of the somata of layer V pyramidal neurons was estimated as 10-15 μm. To consider the upper and lower side of the neuron, we calculated the stimulated somatic membrane surface as two times the area of a circle of 10 μm in diameter (= 157 μm2). Using these values, we estimated the minimal density of KARs at the soma as 4.5 receptors/μm2. The respective value for the dendrite (at a distance of ∼200 μm from the center of the soma) is 9.9 receptors/μm2. These results indicate that the density of KARs is higher at the more distal dendrite (at a distance of 200 μm from the soma ∼2× the density at the soma).
Additional experiments provided more direct evidence for an increased density of KARs at the more distal dendrite. In these experiments the glutamate was applied photolytically to somatic and dendritic outside-out patches. To achieve constant experimental conditions, we held constant the following parameters: open-tip resistance of patch pipettes (8 MΩ), concentration of caged glutamate (1 mM), intensity of laser light (5 mW measured beyond the objective), stimulation duration (3 msec), and the depth of the stimulation site within the brain slice (∼30 μm). KAR-mediated responses elicited at dendritic outside-out patches showed larger amplitudes than those evoked at somatic outside-out patches (36.3 ± 4.6 pA for dendritic outside-out patches, n = 7 patches/7 animals; 16.2 ± 2.5 pA for somatic outside-out patches, n = 7 patches/7 animals; p < 0.01; Fig. 4C). Because of our evidence for identical unitary properties of somatic and dendritic KARs, these results are consistent with an increased density of KARs at the dendrite (also ∼2× the density at the soma).
The results described in the previous section strongly suggest that the relative ratio of the number of KARs to AMPARs does not change toward the distal dendrite. As a consequence, an increased density of KARs at the more distal dendrite implies that the density of AMPARs also is higher at dendritic sites.
Evidence for a mainly extrasynaptic location of KARs
The slow kinetic properties of most KAR synapses described up to now and the need for repetitive stimulation to open the KAR channels (Castillo et al., 1997; Vignes and Collingridge, 1997) have been interpreted by some groups as evidence for their putative extrasynaptic location (Benveniste et al., 1984; Lerma, 1997; Mayer, 1997). To investigate whether this holds true for KARs on neocortical layer V pyramidal neurons, we performed the following experiment. Infrared-guided laser stimulation was used to apply glutamate to the apical dendrite at a distance of 200 μm from the center of the soma. In this region (layer IV) many afferents form glutamatergic synapses with the apical dendrite of layer V pyramidal neurons (Peters, 1987). In the same experiment, glutamatergic synapses, located in the region of photolytic glutamate release, were stimulated electrically. For this purpose a small theta-capillary (∼2 μm tip diameter) was placed near the dendrite (also 200 μm from the center of the soma, ∼5 μm lateral distance). This kind of stimulation allowed us to activate selectively those synapses located in a defined region of the dendrite. Control experiments confirmed the high spatial resolution of this stimulation technique (Fig. 5D). Because synaptic currents were blocked completely by TTX (1 μM), they had to be the result of action potential generation in afferents (data not shown). In all experiments with synaptic stimulation, potential GABAergic inputs were blocked by specific antagonists: picrotoxin (75 μM) for GABAA receptors and CGP35348 (200 μM) for GABAB receptors.
Figure 5.
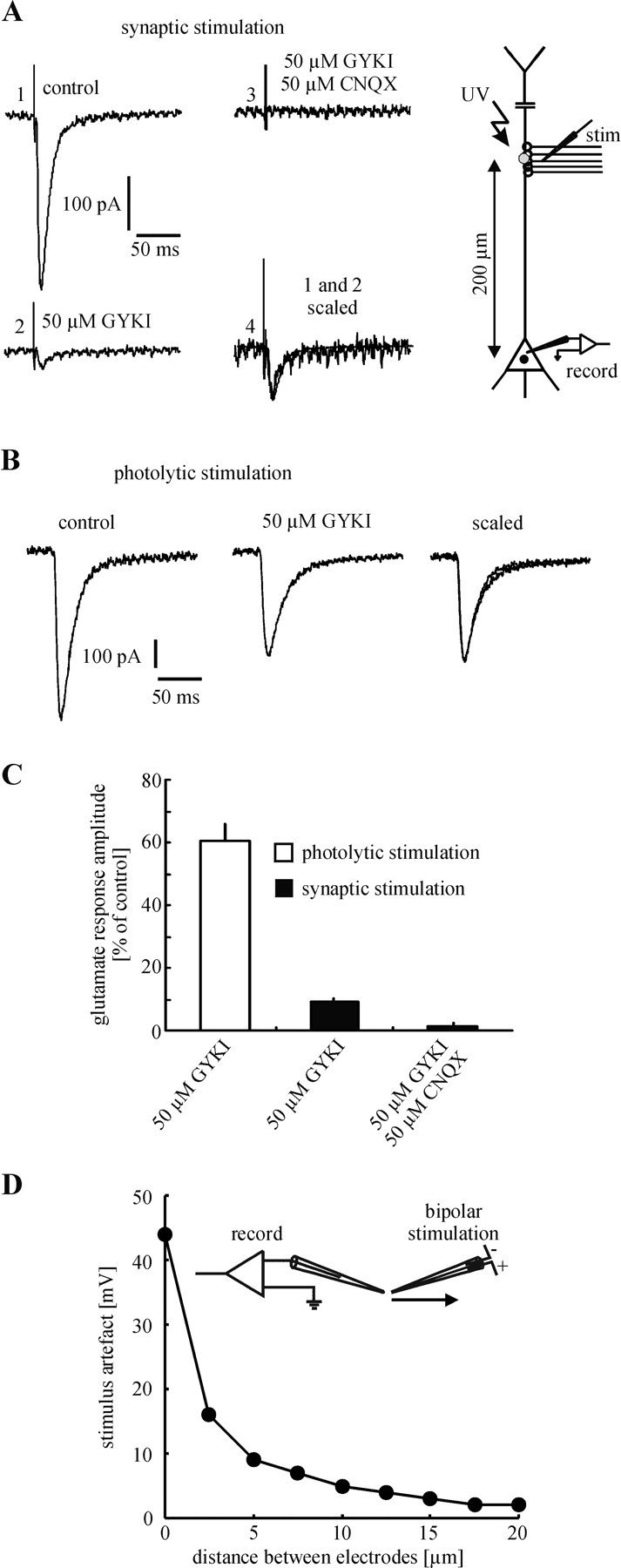
Evidence for a mainly extrasynaptic location of KARs. A, Glutamatergic synapses were activated in a defined region of the apical dendrite by local electrical stimulation. In the presence of d-AP-5 (control) (1) single stimuli reliably evoked EPSCs. GYKI 53655 blocked ∼90% of the EPSC amplitude (2). Because the remaining current was blocked completely by CNQX (3), it has to be mediated by KARs. Interestingly, the mixed AMPA/KAR-mediated EPSC and the isolated KAR-mediated EPSC showed nearly identical fast kinetics (4). In the same experiment glutamate was applied photolytically to the region of synaptic stimulation (B). Only ∼40% of the glutamate response amplitude was blocked by GYKI 53655. Because photolytic application of glutamate also activates extrasynaptic receptors, these results strongly suggest that KARs are located mainly at extrasynaptic sites. A, B, Single experiments are shown. C, Statistical evaluation of the experiments shown in A, B. D, Spatial resolution of electrical stimulation (single experiment).
In the presence of D-AP-5 single electrical stimuli reliably evoked an EPSC (n = 12 neurons/10 animals). The amplitude of this current was strongly diminished by the addition of GYKI 53655 (50 μM; 9 ± 1% as compared with control; Fig. 5A,C). Because the remaining component was blocked completely by CNQX (50 μM; 2 ± 1% as compared with control), it obviously was mediated by KARs. Interestingly, the mixed AMPA/KAR-mediated EPSC (in the absence of GYKI 53655) and the isolated KAR-mediated EPSC showed nearly identical fast kinetics (Fig. 5A). In contrast to the strong antagonistic effect of GYKI 53655 on the EPSC, the application of GYKI 53655 led to a much smaller decrease of the amplitude of the photolytically evoked glutamate response (61 ± 6% as compared with control; p < 0.001; Fig. 5B,C). Because photolytically released glutamate has access to synaptic as well as to extrasynaptic receptors (Pettit and Augustine, 2000; Frick et al., 2001; Matsuzaki et al., 2001) and a single electrical stimulus should not cause an intense spillover of synaptically released glutamate to extrasynaptic sites, the most likely explanation for this observation is a mainly extrasynaptic location of KARs. Such a conclusion presupposes that the activated AMPARs and KARs have a similar affinity to glutamate. To test this hypothesis, we elicited mixed AMPA/KAR-mediated responses at different concentrations of caged glutamate (0.5 and 1 mM) and compared the KAR components of these responses after the addition of GYKI 53655. Figure 6, A and B, shows that GYKI 53655 decreased the amplitude of the mixed AMPA/KAR-mediated response by ∼60%, independently of the concentration of caged glutamate [57 ± 2% for 0.5 mM caged glutamate (n = 7 neurons/5 animals) and 58 ± 3% for 1 mM caged glutamate (n = 7 neurons/6 animals)]. Statistical evaluation revealed no significant difference between these values (p = 0.69). This result indicates that AMPARs and KARs of layer V pyramidal neurons have similar affinities to glutamate and strongly supports the assumption of a mainly extrasynaptic location of KARs.
Figure 6.

KARs and AMPARs of layer V pyramidal neurons have similar affinities to glutamate. A, Using two different concentrations of caged glutamate (0.5 and 1 mm), we applied glutamate photolytically to the neuronal soma. Independently from the concentration of caged glutamate, GYKI 53655 decreased the glutamate response amplitude by the same amount (∼60%). This result indicates that the KARs and AMPARs mediating the glutamate responses have similar affinities to glutamate. B, Statistical evaluation of the experiments shown in A.
Discussion
The present study describes the existence, properties, and spatial distribution of functional postsynaptic KARs on neocortical layer V pyramidal neurons. We show that KARs are located on the entire somatodendritic membrane that was examined. We also show that laser photolysis of caged substances is well suited to apply neurotransmitters to outside-out patches within a brain slice and to examine single-channel properties of ligand-gated ion channels.
Properties and subcellular distribution of postsynaptic KARs
Our results indicate that somatic and dendritic KARs have similar intrinsic properties. This is consistent with studies showing that the unitary properties of AMPA and NMDA receptors on the somata and dendrites of hippocampal pyramidal neurons and cerebellar Purkinje cells are identical (Spruston et al., 1995; Häusser and Roth, 1997; Andrásfalvy and Magee, 2001).
The mostly slow kinetic properties of KAR synapses and the need for repetitive stimulation to open the KAR channels have been interpreted by some groups as evidence for an extrasynaptic location of KARs, which can be activated by a spillover of synaptically released glutamate (Lerma et al., 2001). However, there are also arguments against the spillover hypothesis (Min et al., 1998; Kidd and Isaac, 2001; Cossart et al., 2002). Two additional models compete as an explanation, at least for the slow time course of KAR-mediated EPSCs. First, intracellular signals and/or accessory proteins of the SAP90/PSD-95 family have been suggested as modifying the kinetic properties of KARs (Garcia et al., 1998; Swanson and Heinemann, 1998; Kidd and Isaac, 2001). Second, native KARs might be composed of heteromers with different intrinsic properties from recombinant homomeric channels (Frerking and Nicoll, 2000). Our data shed new light on the kinetic properties and subcellular distribution of postsynaptic KARs. We provide strong evidence for a mainly extrasynaptic and minor synaptic location of KARs, which can be activated without repetitive stimulation. Furthermore, the fast kinetics seem to be a general feature of extrasynaptic as well as synaptic KARs. Fast KAR-mediated EPSCs also were observed in the retina (DeVries and Schwartz, 1999) and recently were recorded from CA1 interneurons (Cossart et al., 2002).
In summary, the discussion about the synaptic and/or extrasynaptic localization, as well as about the reasons for fast/slow kinetics of KARs, remains puzzling and raises the question whether general rules exist regarding these issues. Therefore, it seems likely that the specific targeting of KARs on the postsynaptic membrane of central neurons, as well as the intrinsic properties of these receptors, depends on the brain region, type of neuron, type of synapse, and age. This point of view is strengthened further by the following observations. (1) KAR subunits are expressed in the brain in a complex mosaic (Wisden and Seeburg, 1993). (2) Quantal release of glutamate, which should not cause a spillover of glutamate to extrasynaptic sites, evokes slow KAR-mediated EPSCs in CA3 pyramidal neurons, fast KAR-mediated EPSCs in CA1 interneurons, but no synaptic response in CA1 pyramidal cells (Cossart et al., 2002), which also express functional KARs (Bureau et al., 1999). (3) Nonrepetitive stimulation of the thalamus evokes KAR-mediated EPSCs in layer IV pyramidal cells of 3- to 8-d-old rats (Kidd and Isaac, 1999). However, these EPSCs show a slow time course and are mediated by receptors, which probably contain unedited GluR5 and/or GluR6 subunits. In contrast, our data strongly suggest that layer V pyramidal neurons of 14- to 21-d-old rats express postsynaptic KARs, which contain edited GluR5 and/or GluR6 subunits.
The single-channel conductance of ligand-gated ion channels frequently is estimated by non-SFA. Non-SFA usually is applied to synaptically evoked currents or to currents that are elicited by application of the agonist to outside-out patches. However, these two methods have the following problems. First, synaptic currents can be evoked at the intact neuron in a physiological environment but also reflect presynaptic influences that can distort the estimated value of γ (Robinson and Kawai, 1993; Silver et al., 1996). Second, agonist application to outside-out patches prevents presynaptic influences but, with the use of a standard external application system, cannot be performed within the slice. In the present study we show that infrared-guided laser stimulation is a well suited and simple method to estimate γ of ligand-gated ion channels, circumventing these problems.
Our estimation of ∼8 pS agrees well with other studies, which determined the range of KAR single-channel conductances from very small values (<1 pS) to values comparable to AMPARs (∼10 pS) (Frerking and Nicoll, 2000). However, there is evidence that high-conductance KARs contain unedited GluR5 and/or GluR6 subunits (Lerma et al., 2001). Therefore, our relatively high value of ∼8 pS, at first glance, does not seem to be consistent with our evidence for edited GluR5 and/or GluR6 subunits. We cannot explain this discrepancy conclusively. However, the subunit compositions and interactions of native KARs are nearly unknown. Thus it would not be surprising if some features of native KARs diverge from those of artificially composed KARs.
We provide strong evidence for a distance-dependent increase in the density of KARs and AMPARs in the apical dendrites of layer V pyramidal neurons. Such an increase also has been described for AMPARs on hippocampal CA1 pyramidal cells (Andrásfalvy and Magee, 2001) and seems to be responsible for counterbalancing some of the filtering effects of dendrites (Stricker et al., 1996; Magee and Cook, 2000). Interestingly, evidence for an increasing density toward the distal apical dendrite also was provided for GABAA receptors (Eder et al., 2001). Thus our results support the idea that an increasing density toward the distal dendrite is a general principle for ligand-gated ion channels with fast kinetics.
Functional roles of postsynaptic KARs
The generally small amplitude and mostly slow kinetics of the KAR component of EPSCs raise the question of whether these receptors play only a minor role in fast synaptic transmission. But what is the physiological role for postsynaptic KARs? An attractive hypothesis was postulated by Frerking and Nicoll (2000). In their model the KARs are ideally suited for temporal summation of synaptic inputs at low frequencies. In this way the information about the global afferent firing rate may be transmitted.
Regarding extrasynaptic KARs, these receptors may be activated by a spillover of synaptically released glutamate (Scanziani, 2002), glutamate released from the somatodendritic membrane itself (Ali et al., 2001), or ambient glutamate present in the extra-cellular fluid at persistent micromolar concentrations (Lerma et al., 1986; Timmerman and Westerink, 1997). Such concentrations are sufficient to activate NMDA receptors tonically on pyramidal neurons (Sah et al., 1989). Also KARs might be activated in this way. Such a mechanism would be attractive for a fine tuning of the neuronal excitability. This hypothesis is strengthened by the high sensitivity of KARs to resting levels of glutamate (∼100 μM), which are high enough to open the channels and, at the same time, low enough to produce only an incomplete desensitization (Paternain et al., 1998; Lerma et al., 2001).
Furthermore, extrasynaptic KARs may mediate glutamatergic communication between glial cells and neurons. Several observations support this hypothesis. It has been shown that hippocampal and cortical astrocytes release glutamate in a Ca2+-dependent process (Parpura et al., 1994; Bezzi et al., 1998). Complementing this finding, spontaneous astrocytic Ca2+ oscillations propagate as waves to neighboring astrocytes and trigger glutamate receptor-mediated excitation of neurons located along the wave path (Parri et al., 2001).
The epileptogenic action of kainate is a well known phenomenon. A direct causation between the appearance of seizures and the activation of KARs has been shown by Mulle et al. (1998). Although there is evidence for a decrease of inhibition via presynaptic KARs located on GABAergic terminals (Frerking and Nicoll, 2000), the exact mechanisms underlying the epileptogenic action of kainate are not yet clear. Besides an indirect excitation by a decreased GABAergic inhibition, it is also conceivable that glial glutamate directly excites the postsynaptic neuronal membrane by activating extrasynaptic KARs. Several observations support such a glia-neuron interaction. It has been shown that transients of Ca2+ oscillations in neocortical astrocytes generalize under epileptiform conditions (Tashiro et al., 2002). Such changes in the intracellular Ca2+ concentration can cause glutamate release in cortical astrocytes. Interestingly, Bezzi et al. (2001) observed that prostaglandins, which are often the product of altered CNS functions, stimulate this kind of glial glutamate release. Probably in this way the prostaglandins increase the concentration of extracellular glutamate (Malmberg et al., 1995). Such enhanced levels of extracellular glutamate may be involved in the generation of seizures and epileptiform activity (Engstrom et al., 2001).
Recent studies report physiological functions of extrasynaptic NMDA and extrasynaptic metabotropic glutamate receptors (Brasnjo and Otis, 2001; Li et al., 2002). It is likely also that the extrasynaptic KARs we found play a physiological role. It seems worth clarifying whether indeed they represent an important target for extrasynaptic glutamate and/or whether they provide a pool of glutamate receptors that may be recruited to the synapse during activity-dependent or developmentally regulated changes in synaptic strength.
Footnotes
This study was supported by Sonderforschungsbereich Grant 391. S.C.A. is grateful to Dr. H. C. K. Peter and the Claussen-Simon Stiftung des Stifterverbandes der Deutschen Wissenschaft for supporting her scientific work.
Correspondence should be addressed to Dr. Matthias Eder, Max-Planck-Institute of Psychiatry, Kraepelinstrasse 2, 80804 Munich, Germany. E-mail: Eder@mpipsykl.mpg.de.
Copyright © 2003 Society for Neuroscience 0270-6474/03/236660-11$15.00/0
References
- Ali AB, Rossier J, Staiger JF, Audinat E ( 2001) Kainate receptors regulate unitary IPSCs elicited in pyramidal cells by fast-spiking interneurons in the neocortex. J Neurosci 21: 2992-2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrásfalvy BK, Magee JC ( 2001) Distance-dependent increase in AMPA receptor number in the dendrites of adult hippocampal CA1 pyramidal neurons. J Neurosci 21: 9151-9159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benveniste H, Drejer J, Schousboe A, Diemer NH ( 1984) Elevation of the extracellular concentrations of glutamate and aspartate in rat hippocampus during transient cerebral-ischemia monitored by intracerebral microdialysis. J Neurochem 43: 1369-1374. [DOI] [PubMed] [Google Scholar]
- Bezzi P, Carmignoto G, Pasti L, Vesce S, Rossi D, Rizzini BL, Pozzan T, Volterra A ( 1998) Prostaglandins stimulate calcium-dependent glutamate release in astrocytes. Nature 391: 281-285. [DOI] [PubMed] [Google Scholar]
- Bezzi P, Domercq M, Brambilla L, Galli R, Schols D, De Clercq E, Vescovi A, Bagetta G, Kollias G, Meldolesi J, Volterra A ( 2001) CXCR4-activated astrocyte glutamate release via TNFα: amplification by microglia triggers neurotoxicity. Nat Neurosci 4: 702-710. [DOI] [PubMed] [Google Scholar]
- Brasnjo G, Otis TS ( 2001) Neuronal glutamate transporters control activation of postsynaptic metabotropic glutamate receptors and influence cerebellar long-term depression. Neuron 31: 607-616. [DOI] [PubMed] [Google Scholar]
- Bureau I, Bischoff S, Heinemann SF, Mulle C ( 1999) Kainate receptor-mediated responses in the CA1 field of wild-type and GluR6-deficient mice. J Neurosci 19: 653-663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnashev N, Villarroel A, Sakmann B ( 1996) Dimensions and ion selectivity of recombinant AMPA and kainate receptor channels and their dependence on Q/R site residues. J Physiol (Lond) 496: 165-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo PE, Malenka RC, Nicoll RA ( 1997) Kainate receptors mediate a slow postsynaptic current in hippocampal CA3 neurons. Nature 388: 182-186. [DOI] [PubMed] [Google Scholar]
- Connors BW, Gutnick MJ ( 1990) Intrinsic firing patterns of diverse neocortical neurons. Trends Neurosci 13: 99-104. [DOI] [PubMed] [Google Scholar]
- Cossart R, Epsztein J, Tyzio R, Becq H, Hirsch J, Ben Ari Y, Crepel V ( 2002) Quantal release of glutamate generates pure kainate and mixed AMPA/kainate EPSCs in hippocampal neurons. Neuron 35: 147-159. [DOI] [PubMed] [Google Scholar]
- De Koninck Y, Mody I ( 1997) Endogenous GABA activates small-conductance K+ channels underlying slow IPSCs in rat hippocampal neurons. J Neurophysiol 77: 2202-2208. [DOI] [PubMed] [Google Scholar]
- DeVries SH, Schwartz EA ( 1999) Kainate receptors mediate synaptic transmission between cones and “Off” bipolar cells in a mammalian retina. Nature 397: 157-160. [DOI] [PubMed] [Google Scholar]
- Dildy-Mayfield JE, Eger 2nd EI, Harris RA ( 1996) Anesthetics produce subunit-selective actions on glutamate receptors. J Pharmacol Exp Ther 276: 1058-1065. [PubMed] [Google Scholar]
- Dodt HU, Frick A, Kampe K, Zieglgänsberger W ( 1998) NMDA and AMPA receptors on neocortical neurons are differentially distributed. Eur J Neurosci 10: 3351-3357. [DOI] [PubMed] [Google Scholar]
- Dodt HU, Eder M, Frick A, Zieglgänsberger W ( 1999) Precisely localized LTD in the neocortex revealed by infrared-guided laser stimulation. Science 286: 110-113. [DOI] [PubMed] [Google Scholar]
- Dodt HU, Eder M, Schierloh A, Zieglgänsberger W ( 2002) Infrared-guided laser stimulation of neurons in brain slices. Sci STKE 120: PL2. [DOI] [PubMed] [Google Scholar]
- Eder M, Rammes G, Zieglgänsberger W, Dodt HU ( 2001) GABAA and GABAB receptors on neocortical neurons are differentially distributed. Eur J Neurosci 13: 1065-1069. [DOI] [PubMed] [Google Scholar]
- Eder M, Zieglgänsberger W, Dodt HU ( 2002) Neocortical long-term potentiation and long-term depression: site of expression investigated by infrared-guided laser stimulation. J Neurosci 22: 7558-7568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engstrom ER, Hillered L, Flink R, Kihlstrom L, Lindquist C, Nie JX, Olsson Y, Silander HC ( 2001) Extracellular amino acid levels measured with intra-cerebral microdialysis in the model of post-traumatic epilepsy induced by intracortical iron injection. Epilepsy Res 43: 135-144. [DOI] [PubMed] [Google Scholar]
- Frerking M, Nicoll RA ( 2000) Synaptic kainate receptors. Curr Opin Neurobiol 10: 342-351. [DOI] [PubMed] [Google Scholar]
- Frick A, Zieglgänsberger W, Dodt HU ( 2001) Glutamate receptors form hot spots on apical dendrites of neocortical pyramidal neurons. J Neurophysiol 86: 1412-1421. [DOI] [PubMed] [Google Scholar]
- Garcia EP, Mehta S, Blair LA, Wells DG, Shang J, Fukushima T, Fallon JR, Garner CC, Marshall J ( 1998) SAP90 binds and clusters kainate receptors causing incomplete desensitization. Neuron 21: 727-739. [DOI] [PubMed] [Google Scholar]
- Häusser M, Roth A ( 1997) Dendritic and somatic glutamate receptor channels in rat cerebellar Purkinje cells. J Physiol (Lond) 501: 77-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntley GW, Rogers SW, Moran T, Janssen W, Archin N, Vickers JC, Cauley K, Heinemann SF, Morrison JH ( 1993) Selective distribution of kainate receptor subunit immunoreactivity in monkey neocortex revealed by a monoclonal antibody that recognizes glutamate receptor subunits GluR5/6/7. J Neurosci 13: 2965-2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston D, Magee JC, Colbert CM, Christie BR ( 1996) Active properties of neuronal dendrites. Annu Rev Neurosci 19: 165-186. [DOI] [PubMed] [Google Scholar]
- Kidd FL, Isaac JT ( 1999) Developmental and activity-dependent regulation of kainate receptors at thalamocortical synapses. Nature 400: 569-573. [DOI] [PubMed] [Google Scholar]
- Kidd FL, Isaac JT ( 2001) Kinetics and activation of postsynaptic kainate receptors at thalamocortical synapses: role of glutamate clearance. J Neurophysiol 86: 1139-1148. [DOI] [PubMed] [Google Scholar]
- Lerma J ( 1997) Kainate reveals its targets. Neuron 19: 1155-1158. [DOI] [PubMed] [Google Scholar]
- Lerma J, Herranz AS, Herreras O, Abraira V, Martin DR ( 1986) In vivo determination of extracellular concentration of amino acids in the rat hippocampus. A method based on brain dialysis and computerized analysis. Brain Res 384: 145-155. [DOI] [PubMed] [Google Scholar]
- Lerma J, Paternain AV, Rodriguez-Moreno A, Lopez-Garcia JC ( 2001) Molecular physiology of kainate receptors. Physiol Rev 81: 971-998. [DOI] [PubMed] [Google Scholar]
- Li B, Chen N, Luo T, Otsu Y, Murphy TH, Raymond LA ( 2002) Differential regulation of synaptic and extra-synaptic NMDA receptors. Nat Neurosci 5: 833-834. [DOI] [PubMed] [Google Scholar]
- Magee JC, Cook EP ( 2000) Somatic EPSP amplitude is independent of synapse location in hippocampal pyramidal neurons. Nat Neurosci 3: 895-903. [DOI] [PubMed] [Google Scholar]
- Malmberg AB, Hamberger A, Hedner T ( 1995) Effects of prostaglandin E2 and capsaicin on behavior and cerebrospinal fluid amino acid concentrations of unanesthetized rats: a microdialysis study. J Neurochem 65: 2185-2193. [DOI] [PubMed] [Google Scholar]
- Markram H, Helm PJ, Sakmann B ( 1995) Dendritic calcium transients evoked by single back-propagating action potentials in rat neocortical pyramidal neurons. J Physiol (Lond) 485: 1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, Ellis-Davies GCR, Nemoto T, Miyashita Y, Iino M, Kasai H ( 2001) Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat Neurosci 4: 1086-1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer M ( 1997) Kainate receptors—finding homes at synapses. Nature 389: 542-543. [DOI] [PubMed] [Google Scholar]
- Migliore M, Shepherd GM ( 2002) Emerging rules for the distributions of active dendritic conductances. Nat Rev Neurosci 3: 362-370. [DOI] [PubMed] [Google Scholar]
- Min MY, Rusakov DA, Kullmann DM ( 1998) Activation of AMPA, kainate, and metabotropic receptors at hippocampal mossy fiber synapses: role of glutamate diffusion. Neuron 21: 561-570. [DOI] [PubMed] [Google Scholar]
- Mulle C, Sailer A, Perez-Otano I, Dickinson-Anson H, Castillo PE, Bureau I, Maron C, Gage FH, Mann JR, Bettler B, Heinemann SF ( 1998) Altered synaptic physiology and reduced susceptibility to kainate-induced seizures in GluR6-deficient mice. Nature 392: 601-605. [DOI] [PubMed] [Google Scholar]
- Oakley JC, Schwindt PC, Crill WE ( 2001) Initiation and propagation of regenerative Ca2+-dependent potentials in dendrites of layer 5 pyramidal neurons. J Neurophysiol 86: 503-513. [DOI] [PubMed] [Google Scholar]
- Palmer AJ, Lodge D ( 1993) Cyclothiazide reverses AMPA receptor antagonism of the 2,3-benzodiazepine, GYKI 53655. Eur J Pharmacol 244: 193-194. [DOI] [PubMed] [Google Scholar]
- Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S, Haydon PG ( 1994) Glutamate-mediated astrocyte-neuron signaling. Nature 369: 744-747. [DOI] [PubMed] [Google Scholar]
- Parri HR, Gould TM, Crunelli V ( 2001) Spontaneous astrocytic Ca2+ oscillations in situ drive NMDAR-mediated neuronal excitation. Nat Neurosci 4: 803-812. [DOI] [PubMed] [Google Scholar]
- Paternain AV, Rodriguez-Moreno A, Villarroel A, Lerma J ( 1998) Activation and desensitization properties of native and recombinant kainate receptors. Neuropharmacology 37: 1249-1259. [DOI] [PubMed] [Google Scholar]
- Peters A ( 1987) Number of neurons and synapses in primary visual cortex. In: Cerebral cortex: further aspects of cortical function, including hippocampus (Peters A, Jones EG, eds), pp 267-294. New York: Plenum.
- Pettit DL, Augustine GJ ( 2000) Distribution of functional glutamate and GABA receptors on hippocampal pyramidal cells and interneurons. J Neurophysiol 84: 28-38. [DOI] [PubMed] [Google Scholar]
- Rammes G, Swandulla D, Collingridge GL, Hartmann S, Parsons CG ( 1996) Interactions of 2,3-benzodiazepines and cyclothiazide at AMPA receptors: patch clamp recordings in cultured neurones and area CA1 in hippocampal slices. Br J Pharmacol 117: 1209-1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson HP, Kawai N ( 1993) Single channel properties at the synaptic site. EXS 63: 250-265. [DOI] [PubMed] [Google Scholar]
- Sah P, Hestrin S, Nicoll RA ( 1989) Tonic activation of NMDA receptors by ambient glutamate enhances excitability of neurons. Science 246: 815-818. [DOI] [PubMed] [Google Scholar]
- Scanziani M ( 2002) Competing on the edge. Trends Neurosci 25: 282-283. [DOI] [PubMed] [Google Scholar]
- Schubert D, Staiger JF, Cho N, Kotter R, Zilles K, Luhmann HJ ( 2001) Layer-specific intracolumnar and transcolumnar functional connectivity of layer V pyramidal cells in rat barrel cortex. J Neurosci 21: 3580-3592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigworth FJ ( 1980) The variance of sodium current fluctuations at the node of Ranvier. J Physiol (Lond) 307: 97-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigworth FJ ( 1981) Interpreting power spectra from nonstationary membrane current fluctuations. Biophys J 35: 289-300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver RA, Cull-Candy SG, Takahashi T ( 1996) Non-NMDA glutamate receptor occupancy and open probability at a rat cerebellar synapse with single and multiple release sites. J Physiol (Lond) 494: 231-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruston N, Jonas P, Sakmann B ( 1995) Dendritic glutamate receptor channels in rat hippocampal CA3 and CA1 pyramidal neurons. J Physiol (Lond) 482: 325-352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stricker C, Field AC, Redman SJ ( 1996) Statistical analysis of amplitude fluctuations in EPSCs evoked in rat CA1 pyramidal neurones in vitro. J Physiol (Lond) 490: 419-441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson GT, Heinemann SF ( 1998) Heterogeneity of homomeric GluR5 kainate receptor desensitization expressed in HEK293 cells. J Physiol (Lond) 513: 639-646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tashiro A, Goldberg J, Yuste R ( 2002) Calcium oscillations in neocortical astrocytes under epileptiform conditions. J Neurobiol 50: 45-55. [DOI] [PubMed] [Google Scholar]
- Timmerman W, Westerink BH ( 1997) Brain microdialysis of GABA and glutamate: what does it signify? Synapse 27: 242-261. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Silver RA, Cull-Candy SG ( 1993) Estimated conductance of glutamate receptor channels activated during EPSCs at the cerebellar mossy fiber-granule cell synapse. Neuron 11: 279-289. [DOI] [PubMed] [Google Scholar]
- Verdoorn TA, Johansen TH, Drejer J, Nielsen EO ( 1994) Selective block of recombinant GluR6 receptors by NS-102, a novel non-NMDA receptor antagonist. Eur J Pharmacol 269: 43-49. [DOI] [PubMed] [Google Scholar]
- Vignes M, Collingridge GL ( 1997) The synaptic activation of kainate receptors. Nature 388: 179-182. [DOI] [PubMed] [Google Scholar]
- Wieboldt R, Gee KR, Niu L, Ramesh D, Carpenter BK, Hess GP ( 1994) Photolabile precursors of glutamate: synthesis, photochemical properties, and activation of glutamate receptors on a microsecond time scale. Proc Natl Acad Sci USA 91: 8752-8756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilding TJ, Huettner JE ( 1996) Antagonist pharmacology of kainate- and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid-preferring receptors. Mol Pharmacol 49: 540-546. [PubMed] [Google Scholar]
- Wisden W, Seeburg PH ( 1993) A complex mosaic of high-affinity kainate receptors in rat brain. J Neurosci 13: 3582-3598. [DOI] [PMC free article] [PubMed] [Google Scholar]