

Abstract
Tolerance and physical dependence caused by chronic treatment of narcotics are good models to study basic neuronal plasticity. Activation of the NMDA subtype of the glutamate receptor has been implicated as an anti-opioid system in the development of morphine analgesic tolerance and dependence. The present study examines the specific role of the ϵ1 subunit of the NMDA receptor using mice lacking the gene encoding ϵ1 subunit of the NMDA receptor (GluRϵ1-/- mice). GluRϵ1-/- mice showed significant enhancement and prolongation of morphine anti-nociception, compared with wild-type GluRϵ1+/+ mice. GluRϵ1-/- mice also showed a marked loss of the analgesic tolerance after repeated morphine treatments. In C57BL/6J mice treated with chronic morphine after tolerance paradigm, the GluRϵ1 protein expression significantly increased in periaqueductal gray matter (PAG), ventral tegmental area (VTA) and nucleus accumbens (NAc), but not amygdala or hippocampus. The rescue of GluRϵ1 protein by electroporation into the PAG and VTA, but not NAc of GluRϵ1-/- mice significantly reversed morphine analgesic tolerance liability. Similar attempts were also performed in the naloxone-precipitated physical dependence paradigm. GluRϵ1-/- mice showed marked loss of typical withdrawal abstinence behaviors, and significant enhancement of GluRϵ1 protein expression was only observed in NAc by chronic morphine treatments after dependence paradigm. The rescue of GluRϵ1 protein by electroporation into the NAc of GluRϵ1-/- mice significantly reversed the loss of abstinence behaviors. These findings suggest that GluRϵ1 has locus-specific roles in the development of morphine analgesic tolerance and physical dependence.
Keywords: NMDA receptor, ϵ1 subunit, morphine tolerance and dependence, KO mice, locus-specific rescue, neuronal network
Introduction
The systemic administration of morphine is the most effective means of alleviating severe pain across a wide range of conditions. However, its clinical use has been limited by undesirable side effects such as tolerance. Furthermore, narcotic dependence brings serious social problems, accompanied by psychological and physical dependence. These undesirable symptoms are considered related to neuronal plasticity in the CNS. Many investigators have attempted to elucidate the mechanisms underlying the development of morphine analgesic tolerance and dependence, and some aspects of molecular and cellular mechanisms are now revealed (Ueda et al., 1995, 2001; Roth and Willins, 1999; Mamiya et al., 2001; Nestler 2001; Tsao et al., 2001; Williams et al., 2001; Bodnar and Hadjimarkou, 2002; Kieffer and Evans, 2002). However, the molecular mechanisms through the neuronal networks remain unknown. Drug-induced adaptation of neuronal networks appears a good model to study basic neuronal plasticity. Among several attempts (for review, see Crawley and Corwin, 1994; Harrison et al., 1998; Mogil and Pasternak, 2001), the idea of an anti-opioidergic system provides us with a guide for an approach to understanding the complexity of the neuronal networks. The NMDA receptor-dependent glutamatergic system is the most prominent candidate for an anti-opioid system in the development of neuronal plasticity because the administration of various kinds of NMDA receptor antagonists inhibits side effects such as morphine analgesic tolerance and dependence (for review, see Trujillo, 2000; Mao, 2002). However, a molecular-based approach has not been attempted. Native NMDA receptors comprise GluRζ (NR1) and GluRϵ (NR2) subunits (Mori and Mishina, 1995), and multiple GluRϵs [GluRϵ1-GluRϵ4 (or NR2A-NR2D)] subunits convey NMDA receptor heterogeneity to the properties of ion channels and differential functions caused by different temporal and spatial expressions (Mori and Mishina, 1995; Colwell et al., 1998). We studied the physical liability of morphine tolerance and dependence using mice lacking the gene encoding the GluRϵ1 subunit of the NMDA receptor (GluRϵ1-/-) (Sakimura et al., 1995). Furthermore, we demonstrate that the locus-specific expression by in vivo electroporation of GluRϵ1 NMDA receptors into mutant mice restores the morphine tolerance and dependence.
Materials and Methods
Mice. Male C57BL/6J mice, wild-type (GluRϵ1+/+) mice, and mutant (GluRϵ1-/-) mice lacking the NMDA receptor ϵ1 subunit gene weighing 20-22 gm were used. The GluRϵ1+/+ and GluRϵ1-/- mice with the highly homogenous genetic background of the C57BL/6J strain had been developed previously (Sakimura et al., 1995). These mice were kept in a room maintained at 22 ± 2°C with ad libitum access to a standard laboratory diet and tap water. All experiments were performed in compliance with the relevant laws and institutional guidelines. All procedures were approved by the Nagasaki University Animal Care Committee and complied with the recommendations of the International Association for the Study of Pain (Zimmermann, 1983).
Evaluation of morphine analgesic tolerance. Mice were given morphine-HCl at 10 mg/kg, subcutaneously daily for 6 d, and the nociceptive responses on the first and sixth days were compared. l-Arginine (50 mg/kg, i.p.) was injected 15 min before the administration of morphine, according to previous report (Kolesnikov et al., 1998). The Haffner tail-pinch, which is known to reflect the supraspinal nociceptive response, was performed as described previously (Ueda et al., 1997, 2000). In these experiments, an arterial clip was placed at the base of an animal's tail, and the nociceptive response in this test was evaluated by measuring the latency of biting behavior to the clip, whose strength had been adjusted for nontreated mice to show 0.3-1.0 sec latency. A cutoff time of 15 sec was used to avoid tissue damage. Morphine analgesia was expressed as the area under the curve (AUC). The tolerance index was represented as given by the following equation: tolerance index (%) = (sixth AUC/first AUC) × 100. Individual comparisons were made by the Student's t test.
Analysis of morphine withdrawal signs. Physical dependence was induced by repeated subcutaneous injections of morphine-HCl, at intervals of 8 hr, for 4 d, according to the method of Maldonado et al. (1996) with slight modification (Ueda et al., 2000). The morphine-HCl dose was progressively increased as follows: first day, 20 and 40 mg/kg; second day, 60, 80, and 100 mg/kg; third day, 100 mg/kg × 3; and fourth day, 100 mg/kg (only one injection in the morning). The control mice were treated with saline under the same conditions. Withdrawal signs were precipitated by the injection of naloxone-HCl (1 mg/kg, i.p.) 2 hr after the last morphine-HCl administration. Mice were placed individually in a clear plastic observation area (30 cm diameter, 50 cm height) with a white floor 30 min before naloxone-HCl injection. The number of withdrawal signs such as jumping, wet-dog shaking, paw tremors, and backward locomotion were counted for 30 min after the naloxone challenge. Individual comparisons were made by the Student's t test.
Western blot analysis. To confirm the effect of the in vivo transfection of GluRϵ1 cDNA, immunoblot analysis using 8% polyacrylamide gel, SDS-PAGE was performed as described (Yoshida and Ueda, 1999). For this experiment, 40 μg [periaqueductal gray matter (PAG) and ventral tegmental area (VTA)] or 20 μg proteins [nucleus accumbens (NAc), hippocampus, and amygdala] were used per lane. To obtain equal transfer efficiency, the saline control and morphine-treated samples for each brain region of mice (n = 6) were applied onto the same gel, and immunoblot transfers were performed using the same membrane. The membranes were incubated overnight with anti-GluRϵ1 (1:1000). After extensive washing, the membrane was incubated with peroxidase-conjugated anti-rabbit IgG (1:20,000) for 1 hr at room temperature. All visualization of immunoreactive bands was performed by the Light Capture (AE-6960/C/FC; Atto, Tokyo, Japan) with an enhanced chemiluminescent substrate for the detection of horseradish peroxidase, Super Signaling Substrate (Pierce, Rockford, IL). Because this Light Capture can repeatedly expose at intervals of 5 min and automatically superimpose these signals to obtain high contrast, we can detect enhanced signals with less background signals. Subsequently, the intensities of the immunoreactive bands were analyzed by NIH imaging for Macintosh.
Plasmid DNA. The cDNA encoding GluRϵ1 from pBKSAϵ1 (Meguro et al., 1992) was cloned into pAdTrack-cytomegalovirus (CMV). This shuttle vector is used for production of green fluorescent protein (GFP) trackable virus containing transgenes under the control of a CMV promoter (a gift from Dr. Bert Vogelstein, Howard Hughes Medical Institute and Johns Hopkins Oncology Center).
In vivo electroporation. GluRϵ1 cDNA (DGluRϵ1pAdTrack-CMV) were purified with an AccuPrep plasmid extraction kit (Bioneer Company, Daejeon, Korea) and used for gene transfer into specific brain loci. A pair of stainless steel electrodes, 0.5 mm in length and 0.3 mm in diameter, was stereotaxically inserted into both sides of the ventrolateral PAG (anterior: -3.28 mm, lateral: ±0.3 mm, height: 2.5 mm from bregma), NAc (anterior: 1.78 mm, lateral: ±0.90 mm, height: 3.75 mm from bregma), or the VTA (anterior: -3.28 mm, lateral: ±0.65 mm, height: 4.0 mm from bregma) of sodium pentobarbital-anesthetized (50 mg/kg, i.p.) mice. Electric pulses were generated with a Square Electro Porator (CUY21; TR Tech, Tokyo, Japan) at 10 pulses/sec (10 Hz). The shape of the pulse was a square wave; i.e., the voltage remained constant during the pulse duration. For the best gene transfer, cDNA at dose of 1 μg, which diluted in PBS immediately before use was electroporated into brain regions by the electric pulses (2 msec) in 40-55 V duration to get 10 mA current. There were no significant gross behavioral changes as a result of the electroporation of either the mock or recombinant cDNA into the any brain regions. Morphine was injected from day 4 after electroporation for tolerance and dependence experiments. The tolerance experiments were preformed at day 9 after electroporation, whereas the dependence experiments were performed at day 7 after electroporation. We performed histochemical confirmation of fluorescence of all mice after the behavioral experiments. We used the results with mice expressing fluorescence on both sides of the PAG, VTA, or NAc for evaluation of the effects by in vivo electroporation of GluRϵ1 or mock genes. Individual comparisons were made by the Student's t test (p < 0.05) between the mock-expressed GluRϵ1+/+ mice and the mock-expressed GluRϵ1-/- mice or the mock-expressed GluRϵ1-/- mice and the GluRϵ1-expressed GluRϵ1-/- mice.
Histological analysis. To confirm the tissue damage after the in vivo electroporation, histological analysis was performed with hematoxylineosin. Nine days after the in vivo electroporation or sham operation without electroporation, the mice were deeply anesthetized with 50 mg/kg pentobarbital intravenously and intracardially perfused with 0.1 m PBS followed by 4% paraformaldehyde fixative. Brain was dissected and cut into 10-μm-thick slices and stained with hematoxylineosin.
Statistical analysis. Statistical evaluations were performed using the Student's t test. In some experiments, statistical evaluations were also performed using the Scheffe test for multiple comparisons, after one-way or repeated measures ANOVA. Data were expressed as mean ± SEM. Significance was established at *p < 0.05; #p < 0.05.
Results
Attenuated morphine analgesic tolerance in GluRϵ1-/- mice
There were no significant differences in basal pain responses in the tail pinch test between wild-type (GluRϵ1+/+; 0.33 ± 0.02 sec) and GluRϵ1 mutant (GluRϵ1-/-; 0.31 ± 0.03 sec) mice (Fig. 1A). In this test, morphine at 10 mg/kg given subcutaneously increased the latency to show biting responses to an artery clip applied to the tail root (Fig. 1A). When the morphine analgesic activity was evaluated by AUC of time course (Fig. 1B) or using the value in the peak time for morphine analgesia (45 min after morphine administration) (Fig. 1C), an enhanced analgesia in mutant mice was observed in dose-dependent manner, compared with one in GluRϵ1+/+ mice. After daily injections of morphine for 5 d, morphine analgesia in GluRϵ1+/+ mice was markedly attenuated on day 6 (Fig. 1D), whereas the analgesic tolerance was only limited in GluRϵ1-/- mice (Fig. 1E). When the morphine analgesic activity was re-evaluated by AUC of time course morphine analgesia, the AUC value for sixth day morphine analgesia in GluRϵ1+/+ mice (158.6 ± 12.0) appears to be markedly lower than that for the first day (1086.3 ± 92.7), as shown in Figure 1F. The tolerance index calculated by the ratio was only 14.6% in GluRϵ1+/+ mice, whereas that in GluRϵ1-/- mice was significantly increased to 68.8% (Fig. 1F).
Figure 1.

Attenuation of morphine tolerance in GluRϵ1-/- mice in the tail-pinch test. A, Time course of analgesia in GluRϵ1-/- (ϵ1-/-) and GluRϵ1+/+ (ϵ1+/+) mice (n = 6) after 10 mg/kg given subcutaneously. B, Dose-dependent morphine analgesia in ϵ1-/- or ϵ1+/+ mice. A cutoff time of 15 sec was used to avoid tissue damage. The analgesic effect was represented as the AUC during 15-120 min. Maximal AUC is calculated to be 1575. C, D, Time course of morphine-induced (10 mg, s.c.) analgesia in ϵ1+/+ (C) or ϵ1-/- (D) mice with (sixth) or without (first) chronic morphine treatments (10 mg/kg, s.c.; 5 d), respectively. E, Comparison of morphine analgesia in mutant mice pretreated with or without chronic morphine treatments. Morphine (10 mg/kg, s.c.) analgesia was represented as AUC. ED50 (95% confidence limits) of morphine analgesia at first and sixth day in ϵ1+/+ mice was 8.1 (2.4-26.8) and 28.0 (15.4-50.9) mg/kg subcutaneously, respectively, whereas the one at the first and sixth day in ϵ1-/- mice was 3.0 (1.2-7.7) and 5.1 (2.8-9.4) mg/kg subcutaneously, respectively. F, Morphine tolerance index in ϵ1+/+ and ϵ1-/- mice. The tolerance index was represented as given by the following equation: tolerance index (%) = (sixth AUC/first AUC) × 100. Results were represented as the mean ± SEM of the tail-pinch latency (sec) from six separate animals. *p < 0.05 compared with ϵ1+/+ mice.
Enhancement of GluRϵ1 protein expression in specific brain regions after repeated morphine treatments
C57/BL6 mice were daily injected with morphine (10 mg/kg, s.c.) and used for the immunoblot analysis of GluRϵ1 protein expression. NAc, amygdala, the hippocampus, PAG, and VTA were chosen as pivotal brain regions in the development of morphine analgesic tolerance and neuronal plasticity after chronic morphine treatments (Mitchell et al., 2000; Williams et al., 2001; Nestler, 2001). Compared with the expression in saline-treated mice, significant increases were found in the preparations of NAc, PAG, and VTA, but not in those of amygdala and hippocampus (Fig. 2).
Figure 2.

Enhancement of GluRϵ1 protein expression in specific brain regions after repeated morphine treatments using tolerance paradigm. Mice were daily injected with saline (subcutaneously) or morphine (10 mg/kg, s.c.). The brain was isolated on the sixth day for Western blot. The proteins were separated with 8% SDS-PAGE, transferred to polyvinylidene difluoride membrane, and blotted against GluRϵ1 antibody. The results were represented as the mean ± SEM of withdrawal signs from seven separate animals. *p < 0.05, compared with saline-treated group.
Nitric oxide action in the GluRϵ1-/- mice with morphine chronic treatments
NMDA-nitric oxide (NO) cascades are reported to be involved in the morphine analgesic tolerance (Pasternak et al., 1995; Kolesnikov et al., 1998). We next assessed the effect of l-arginine, which is a NO precursor and substrate for NOS, on morphine analgesic tolerance in both GluRϵ1+/+ and GluRϵ1-/- mice. After daily administration of morphine (10 mg/kg, s.c.) for 5 d in GluRϵ1+/+ mice, morphine analgesic activity evaluated by AUC of time course analgesia significant decreased on day 4 and 5, as shown in Figure 3A. The concurrent daily administration of l-arginine (50 mg/kg, i.p.) along with morphine did not affect the acute morphine analgesia (day 1), although it accelerated the development of analgesic tolerance in GluRϵ1+/+ mice (Fig. 3A). In GluRϵ1-/- mice, on the other hand, daily morphine administration showed significant, but less marked analgesic tolerance than in GluRϵ1+/+ mice, as shown in Figure 3B. However, the concurrent daily administration of l-arginine (50 mg/kg, i.p.) along with morphine markedly accelerated the development of analgesic tolerance in GluRϵ1-/- mice. l-Arginine maximally decreased the morphine analgesia in GluRϵ1-/- mice day 4 or 5 to the level with GluRϵ1+/+ mice, which had been similarly given (Fig. 3B).
Figure 3.

l-Arginine-induced acceleration of the development of morphine tolerance in GluRϵ1+/+ and GluRϵ1-/- mice. A group of GluRϵ1+/+ (A) and GluRϵ1-/- mice (B) was administrated morphine (10 mg/kg, s.c.) in conjunction with vehicle (saline) or l-arginine (50 mg/kg, i.p.) for 5 d. Morphine (10 mg/kg, s.c.) analgesia was represented as AUC. The results were the mean ± SEM of withdrawal signs from seven separate animals. *p < 0.05 versus antinociceptive activity with day 1 morphine administration. Statistical evaluations were performed using the Scheffe test for multiple comparisons, after one-way or repeated measures ANOVA. #p < 0.05 versus vehicle-treated mice. Statistical evaluations were performed using the Student's t test.
Locus-specific reversal of morphine analgesic tolerance by gene transfer of recombinant GluRϵ1 into GluRϵ1-/- mouse brain regions
The recombinant GluRϵ1 clone with the CMV promoter was expressed in PAG, VTA, or NAc neurons using the electroporation technique. Although we electroporated 0.3-2 μg GluRϵ1 cDNA into the brain regions by the electric pulses (2 msec) in 10-20 mA current, the best gene transfer was observed when 1 μg of cDNA was electroporated into brain regions by the electric pulses (2 msec) in 40-55 V duration to get 10 mA current. As shown in Figure 4A, a marked green fluorescence used as the genetic marker was specifically observed in both sides of the ventrolateral PAG on day 9 after electroporation. The fluorescence was also observed within the limits of 0.45 mm each toward to rostrum and caudal regions from electroporation region (data not shown). On the other hand, when tissues were stained with hematoxylin-eosin, the electroporation did not damage the morphology of brain regions showing fluorescence (Fig. 4B). The expression of the GluRϵ1 protein in PAG was also confirmed with immunoblot analysis on day 4 after electroporation, and the expression lasted until at least day 9 (Fig. 4C). We started the injection of morphine into mice from day 4 after electroporation, and we performed the tolerance experiment on day 9. As shown in Figure 4D, the enhancement of acute morphine analgesia (first) and attenuation of morphine analgesic tolerance (sixth) in mice deleting GluRϵ1 gene was not affected by electroporation of mock vector into any brain regions including PAG. The electroporation of recombinant GluRϵ1 into these regions of GluRϵ1-/- mice slightly reduced the acute analgesia. However, the significant reversal of tolerance liability was observed when GluRϵ1 was electroporated into PAG and VTA, but not into NAc (Fig. 4D,E).
Figure 4.

Locus-specific rescue of morphine tolerance by gene transfer of recombinant GluRϵ1 into GluRϵ1-/- mouse brain regions. A, GFP fluorescence to confirm the accurate expression of the recombinant gene on day 9 after electroporation. A marked green fluorescence used as the genetic marker was specifically observed in both sides of the ventrolateral PAG. B, Lack of morphological change in the ventrolateral PAG region by electroporation on day 9. The hematoxylin-eosin staining of ventrolateral PAG region expressing GFP fluorescence in A (+). The staining of corresponding PAG region without electroporation (-). C, Western blot of GluRϵ1 (ϵ1) in the PAG from mock-transferred ϵ1+/+ or ϵ1-/- or recombinant ϵ1-transferred ϵ1-/-[ϵ1+/+(m), ϵ1-/- (m) or ϵ1-/- (ϵ1), respectively] mice at days 4 and 9. D, Comparison of morphine analgesia in ϵ1+/+ (m), ϵ1-/- (m), or ϵ1-/- (ϵ1) mice with or without chronic morphine treatments. Morphine (10 mg/kg, s.c.) analgesia was represented as AUC. E, Morphine tolerance index in ϵ1 gene mutant mice with gene transfer into several brain regions. Results were represented as the mean ± SEM of the tail-pinch latency (in seconds) from six separate animals. The details are described in the legend of Figure 1.
Attenuated morphine dependence in GluRϵ1-/- mice
We proceeded with the study of the physical dependence liability of morphine in both GluRϵ1-/- and GluRϵ1+/+ mice, because we confirmed the classical signs of high-dose (20-100 mg/kg, s.c.) morphine-induced behaviors, such as Straub's tail symptom and increased locomotive behaviors in these mice. The intraperitoneal injection of naloxone (1 mg/kg) precipitated somatic symptoms of withdrawal abstinence behaviors, such as jumping, wet-dog shaking, paw tremors, and backward locomotion in the GluRϵ1+/+ mice chronically treated with increasing doses of morphine, according to the morphine dependence paradigms. All of these abstinence scores during 30 min after the naloxone challenge were significantly attenuated in the GluRϵ1-/- mice, but the withdrawal abstinence was relatively less marked (Fig. 5).
Figure 5.
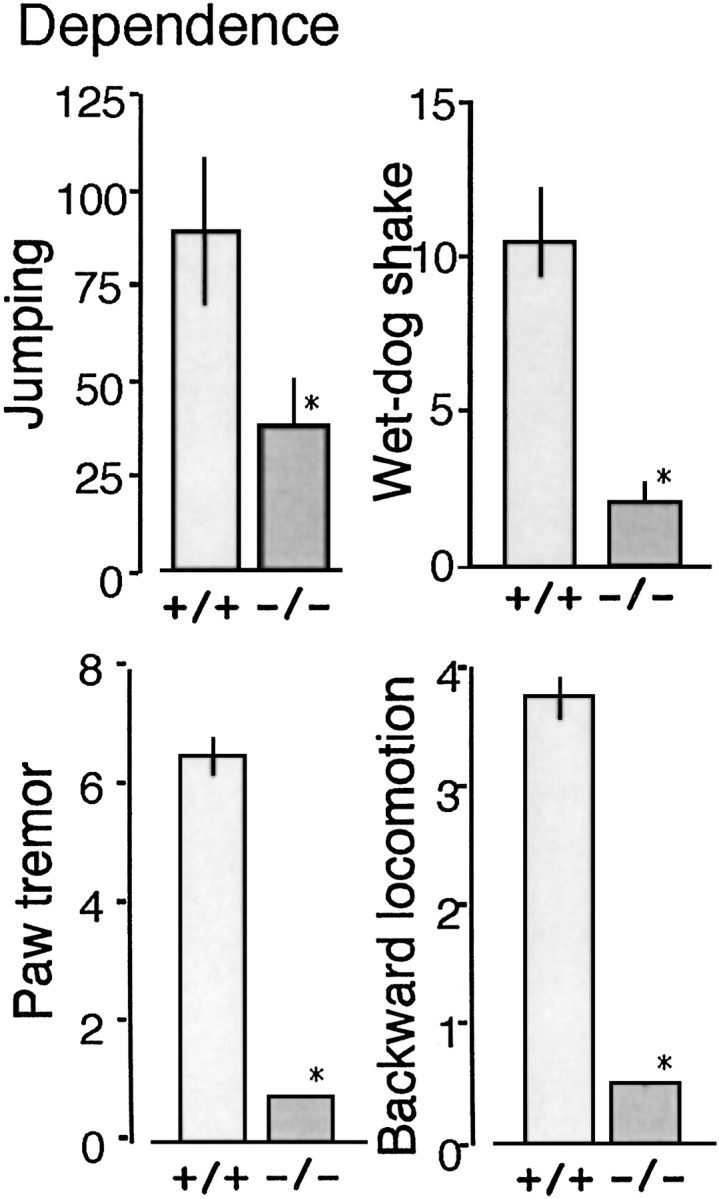
Attenuated morphine dependence in GluRϵ1-/- mice. Mice were pretreated with morphine-HCl every 8 hr as follows: first day, 20 and 40 mg/kg; second day, 60, 80, and 100 mg/kg; third day, 100 mg/kg × 3; and fourth day, 100 mg/kg. The number of jumps, wet-dog shakes, paw tremors, and backward locomotions as withdrawal signs were counted for 30 min after the naloxone (1 mg/kg, i.p.) challenge 2 hr after the final morphine-HCl injection. The results were represented as the mean ± SEM of withdrawal signs from seven separate animals. *p < 0.05, compared with ϵ1+/+ mice.
Enhancement of GluRϵ1 protein expression in NAc after chronic morphine treatments using dependence paradigm
Compared with the GluRϵ1 protein expression in saline-treated C57BL/6J mice, a significant increase was found only in NAc, but not in PAG, VTA, amygdala, or hippocampus, whereas there was a slight but significant decrease in the expression in amygdala (Fig. 6).
Figure 6.

Enhanced GluRϵ1 protein expression in NAc after repeated morphine treatments using dependence paradigm. Mice were pretreated with morphine-HCl following dependence paradigm. The brain was isolated 2 hr after the final morphine injection for Western blot. The results were represented as the mean ± SEM of withdrawal signs from seven separate animals. The details are described in the legend of Figure 2.
Reversal of attenuated morphine dependence in GluRϵ1-/-
mice by the in vivo transfection of recombinant GluRϵ1 into NAc As shown in Figure 7A, when recombinant GluRϵ1 was transfected in NAc, a marked green fluorescence was specifically observed in NAc (core and shell) on day 7 after electroporation. The in vivo electroporation of mock cDNA into NAc of GluRϵ1+/+ mice slightly affected some of naloxone-precipitated withdrawal abstinence scores, compared with those in background C57BL/6J mice, as shown in Figures 5 and 7. However, the relative attenuation of these scores in GluRϵ1-/- mice compared with those in GluRϵ1+/+ mice was not significantly affected by mock cDNA electroporation. The electroporation of GluRϵ1 cDNA into NAc of GluRϵ1-/- mice recovered the decreased withdrawal abstinence behaviors. Significant recovery was observed in wet-dog shaking, paw tremors, and backward locomotion, but the recovery of jumping was only partial.
Figure 7.
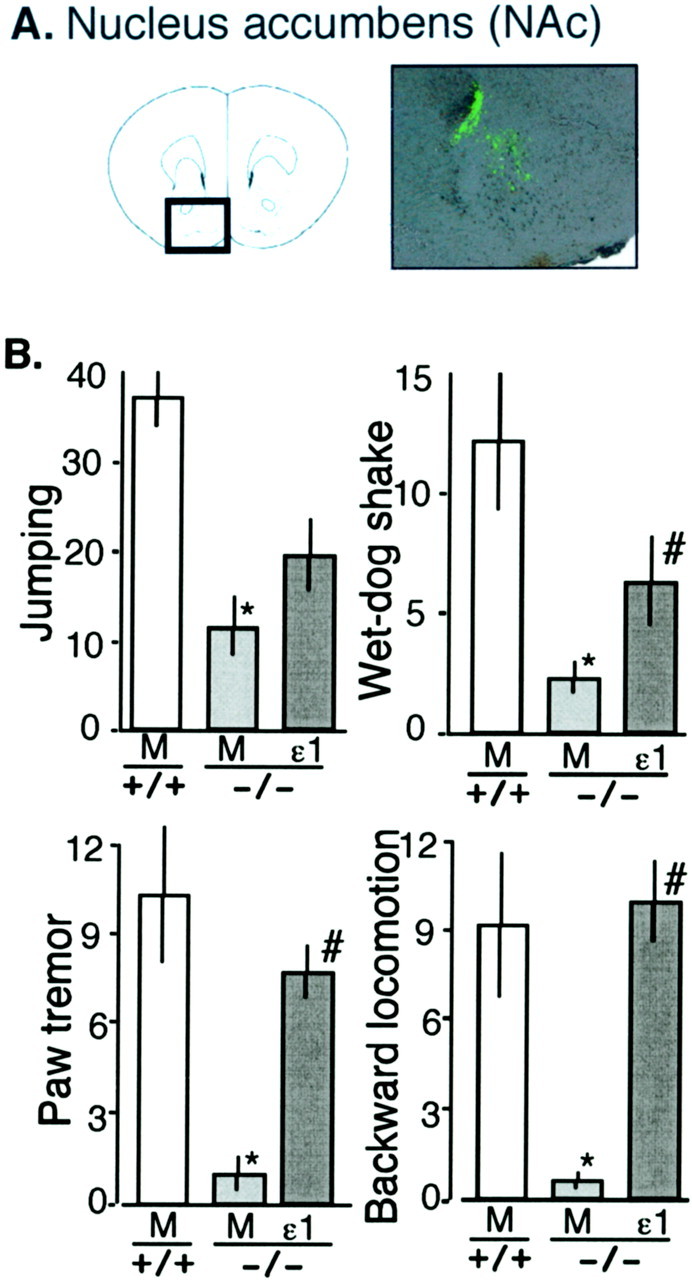
Reversal of attenuated morphine dependence by gene transfer of recombinant GluRϵ1 into NAc of GluRϵ1-/- mice. A, GFP fluorescence to confirm the accurate expression of recombinant gene on day 7 after electroporation into NAc. B, Reversal of attenuated morphine dependence. Withdrawal signs were precipitated by naloxone-HCl (1 mg/kg, i.p.) 2 hr after the final morphine-HCl administration. The results were represented as the mean ± SEM of withdrawal signs from six separate animals.
Discussion
Here, we used GluRϵ1-/- mice and in vivo electroporation technique to rescue the GluRϵ1 protein expression to see the involvement of this receptor subunit in the neuronal plasticity observed in morphine tolerance and dependence. For the study of morphine tolerance, we focused the analgesic activity, a major pharmacological action of morphine.
The lack of alteration in pain threshold in GluRϵ1-/- mice (Fig. 1A) is consistent with other reports, which used different nociception tests (Minami et al., 1998; Kiyama et al., 1998; Kishimoto et al., 2001). Similar lack of alteration in pain has been also reported with the GluRϵ4-/- mice (Minami et al., 1998), although heterozygous mutant mice for the GluRϵ2 subunit showed decrease in the pain threshold (Wainai et al., 2001). Therefore, at least GluRϵ1 unlikely plays a major role in the determination of basal pain threshold. However, because NMDA receptors mediate a wide range of brain processes, including central sensitization during persistent pain (Yaksh, 1993; Woolf and Costigan, 1999), the role of GluRϵ1 subunit in the NMDA receptor-mediated central sensitization during persistent pain remains to be determined.
The acute morphine analgesia was slightly potent in GluRϵ1-/- mice, compared with that in GluRϵ1+/+ mice (Fig. 1A-C). These findings suggest that opioidergic neuron could be connected to the glutamatergic neurons activating postsynaptic GluRϵ1 NMDA receptor subunit through unknown inhibitory neuron. Thus, the inhibitory opioid actions on nociception are partially counteracted by these glutamatergic neurons. This view could be supported by the reports that NMDA receptor antagonists potentiate the morphine analgesia (Plesan et al., 1998; Lutfy et al., 1999).
Since the first discovery that MK-801, an NMDA receptor antagonist inhibits the tolerance (Trujillo and Akil, 1991), a number of reports have supported the involvement of NMDA receptor in the development of morphine analgesic tolerance (for review, see Trujillo, 2000; Mao and Mayer, 2001; Mao, 2002). In the present study we first observed that GluRϵ1 NMDA receptor subunit is involved in the morphine analgesic tolerance by use of GluRϵ1-/- mice (Fig. 1C). As a result of several attempts to examine biochemical changes (gene and protein expression and protein phosphorylation) in the brain regions of mice with chronic morphine treatments, we succeeded in the detection of significant change in GluRϵ1 protein expression. Among brain regions related to morphine analgesia and tolerance (including associated tolerance) (Mitchell et al., 2000; Nestler, 2001; Williams et al., 2001), significant increase in the GluRϵ1 protein expression after chronic morphine treatments was observed in PAG, VTA, and NAc (Fig. 2). These findings suggest that GluRϵ1 NMDA receptor subunit is specifically involved in the development of morphine analgesic tolerance as an anti-opioid system to counteract inhibitory morphine actions.
To identify the loci in the brain that are involved in the tolerance liability of morphine, we attempted to rescue the protein expression in specific brain regions of GluRϵ1-/- mice. First we confirmed whether the pathways downstream to NMDA receptor are intact in the GluRϵ1-/- mice, because the question that always comes up with a mutant mice is whether there are general organizational changes in the animals caused by the absence of the protein of interest during development. The role of NMDA receptors in opioid tolerance is often placed in the context of an NMDA-NO cascade (Pasternak et al., 1995; Kolesnikov et al., 1998). As l-arginine, the substrate for NOS, accelerated the morphine analgesic tolerance in GluRϵ1-/- mice as well as in GluRϵ1+/+ mice (Fig. 3), the pathways downstream from NMDA receptor were proven to be intact in GluRϵ1-/- mice.
We performed the in vivo electroporation of GluRϵ1 cDNA to rescue the protein expression in specific brain regions of GluRϵ1-/- mice. As immunoblot analyses revealed that the protein expression lasted for >9 d after the electroporation into PAG (Fig. 4A), we planned the experimental designs to complete chronic morphine treatments and behavioral tests within 9 d after the start of electroporation. Under these conditions, we performed direct gene transfection into mutant mice to elucidate the brain locus-specific rescue of morphine analgesia and tolerance. As shown in Figure 4D, the electroporation of recombinant GluRϵ1 cDNA into PAG, VTA, or NAc partially reversed the enhanced acute morphine analgesia in GluRϵ1-/- mice, suggesting that the anti-opioid system through GluRϵ1 activation regulates morphine analgesia in all three regions. However, morphine analgesic tolerance in GluRϵ1-/- mice was significantly reversed by the electroporation into PAG and VTA, but not NAc. Thus, it is evident that the GluRϵ1 system in PAG and VTA plays a critical role in the development of morphine analgesic tolerance. There are reports that PAG neurons are related to the descending mechanisms of the pain-inhibitory roles of opioids, whereas dopaminergic neurons in VTA are related to ascending ones (Franklin, 1998; Millan, 2002).
There is a view that anti-reward systems involving anti-opioidergic systems, called allostasis, can contribute to motivational changes associated with chronic administration of drugs of abuse (Koob and Le Moal, 2001). The glutamate or NMDA receptor system and some anti-opioid peptidergic systems are known to be representative of anti-opioid systems (Harrison et al., 1998; Trujillo, 2000; Ueda et al., 2000; Mao and Mayer, 2001). In the present study, we observed that naloxone-precipitated withdrawal abstinence behaviors were significantly attenuated in the GluRϵ1-/- mice (Fig. 5). These findings suggest that GluRϵ1 system is also involved in the development of morphine dependence. To examine the validity of the speculation that the enhanced GluRϵ1 anti-opioid system is also related to the plasticity in morphine dependence, C57BL/6J mice were treated with morphine by dependence paradigm and used for the analysis of GluRϵ1 protein expression. Among these five brain loci, only NAc showed a significant increase in GluRϵ1 protein levels, whereas amygdala showed a slight decrease (Fig. 6). It should be noticed that no significant change was observed in either PAG or VTA, although a significant increase in GluRϵ1 protein levels was observed in both brain loci after chronic morphine treatments by tolerance paradigm (Fig. 2). Such difference may be explained by the shorter period of morphine pretreatments for dependence paradigm (4 d), compared with that for tolerance one (6 d). But this possibility unlikely explains the selective enhancement of GluRϵ1 protein expression in NAc. Alternatively another feedback mechanism to reset the gene expression of GluRϵ1 may work through unidentified plasticity in neuronal networks. Significant, but weak decrease in GluRϵ1 protein expression in amygdala might be another target related to different behavioral changes such as reverse tolerance (sensitization) caused by chronic morphine treatments.
It is interesting to examine whether highly integrated behaviors observed in morphine withdrawal abstinence could be recovered by locus-specific expression of GluRϵ1 in the NAc of GluRϵ1-/- mice. In GluRϵ1-/- mice transfected with GluRϵ1 cDNA, withdrawal behaviors such as wet-dog shake, paw tremors, and backward locomotion were significantly recovered, whereas withdrawal jumping behavior was only partial (Fig. 7B). These findings suggest that some of withdrawal symptoms are derived from plastic changes in relatively narrow brain loci. Among these four withdrawal behaviors, only jumping behavior shows some resistance to the locus-specific rescue of GluRϵ1 cDNA into NAc (Fig. 7B). This finding may be related to our previous finding that the intrathecal injection of naloxone more effectively induces withdrawal jumping in chronic morphine-treated mice than the supraspinal injection (Ueda et al., 1987).
Because GluRϵ1-/- mice still have some liability to morphine analgesic tolerance and physical dependence, it is possible that other anti-opioid systems are also involved in this plasticity. We have previously proposed that nociceptin/orphanin FQ receptor (NOP) system plays an important role in the development of morphine analgesic tolerance and physical dependence by use of specific NOP antagonist and NOP-/- mice (Ueda et al., 1997, 2000). In this study, the inhibition of tolerance development is more clearly observed by the intrathecal injection of NOP antagonist than by the supraspinal injection. These findings suggest that spinal anti-opioid mechanisms through N/OFQ system are more likely involved in the plasticity in morphine analgesic tolerance. Other anti-opioids may be also involved in the development of morphine tolerance and dependence. Specific cholecystokinin 2 (CCK2) receptor antagonist blocked the expression of associative morphine analgesic tolerance (Mitchell et al., 2000), whereas CCK2 receptor-deficient mice showed greater naloxone-precipitated withdrawal responses than in wild-type mice (Pommier et al., 2002). On the other hand, specific neuropeptide FF antagonist or its antibody increased morphine analgesia, reversed morphine analgesic tolerance, and attenuated naloxone-precipitated morphine withdrawal syndromes (Malin et al., 1990; Lake et al., 1992). In addition to these mechanisms, there are reports that some opioid systems regulate the development of morphine analgesic tolerance in experiments using mutant mice for delta opioid receptor or preproenkephalin genes (Zhu et al., 1999; Nitsche et al., 2002). Thus, other anti-opioid systems or opioid systems as well as NMDA system may be also related to the residual mechanisms for the development of morphine tolerance or dependence.
The present study using in vivo electroporation of specific gene into mice deleting the gene provides a new strategy to identify the brain loci involved in drug-induced behaviors of interest. This strategy seems to have more advantages in the case with pharmacological actions after chronic drug treatments. To study the brain loci of interest, the microinjection of antagonists could be another approach. However, in the case with the present study, long-term infusion of NMDA receptor antagonists should be applied, but it might be difficult to avoid unexpected side effects of such compounds. In addition, specific antagonist for GluRϵ1 is not available. Currently the use of spatial and temporal conditional mutant mice might be a good strategy. However, available promoters still cover broad brain regions. Therefore, the strategy of rescue of the receptor by gene transfer into specific loci in mutant mice looks promising. Adenovirus infection used for this purpose has some limitations in its efficiency of gene transfer, and higher titers of adenovirus may cause toxic effects (Nestler, 2000). Additionally, the use of lentiviruses highly efficient for transfection into neurons has limitations in experimental conditions to avoid infection in humans. Furthermore, the size of the DNA needs to be appropriately small for construction into such viral vectors. Thus, the direct gene transfection into mutant mice by in vivo electroporation would be convenient and useful strategy to elucidate the brain locus-specific recovery of morphine tolerance and dependence.
In summary, we have found, by use of mutant mice deleting specific gene and in vivo electroporation technique that GluRϵ1 NMDA receptor in PAG and VTA plays a critical role in the development of analgesic tolerance, and the receptor in NAc plays a specific role in the development of physical dependence to chronic morphine. This approach using the locus-specific rescue of gene expression may further clarify the neuronal plasticity, which modifies psychological dependence and sensitization after chronic morphine treatments.
Footnotes
Parts of this study were supported by Special Coordination Funds of the Science and Technology Agency of the Japanese Government, Human Frontier Science Program and Grants-in-Aid from the Ministry of Education, Science, Culture, and Sports of Japan. We thank Akira Yoshida, Toshiko Kawashima, and Md. Harunor Rashid for technical help. We also thank Dr. Bert Vogelstein (Howard Hughes Medical Institute and Johns Hopkins Oncology Center) for the generous gift of pAdTrack-CMV vector.
Correspondence should be addressed to Dr. Hiroshi Ueda, Division of Molecular Pharmacology and Neuroscience, Nagasaki University Graduate School of Biomedical Sciences, 1-14 Bunkyo-machi, Nagasaki 852-8521, Japan. E-mail: ueda@net.nagasaki-u.ac.jp.
Copyright © 2003 Society for Neuroscience 0270-6474/03/236529-08$15.00/0
References
- Bodnar RJ, Hadjimarkou MM ( 2002) Endogenous opiates and behavior: 2001. Peptides 23: 2307-2365. [DOI] [PubMed] [Google Scholar]
- Colwell CS, Cepeda C, Crawford C, Levine MS ( 1998) Postnatal development of glutamate receptor-mediated responses in the neostriatum. Dev Neurosci 20: 154-163. [DOI] [PubMed] [Google Scholar]
- Crawley JN, Corwin RL ( 1994) Biological actions of cholecystokinin. Peptides 15: 731-755. [DOI] [PubMed] [Google Scholar]
- Franklin KB ( 1998) Analgesia and abuse potential: an accidental association or a common substrate? Pharmacol Biochem Behav 59: 993-1002. [DOI] [PubMed] [Google Scholar]
- Harrison LM, Kastin AJ, Zadina JE ( 1998) Opiate tolerance and dependence: receptors, G-proteins, and antiopiates. Peptides 19: 1603-1630. [DOI] [PubMed] [Google Scholar]
- Kieffer BL, Evans CJ ( 2002) Opioid tolerance-in search of the holy grail. Cell 108: 587-590. [DOI] [PubMed] [Google Scholar]
- Kishimoto Y, Kawahara S, Mori H, Mishina M, Kirino Y ( 2001) Long-trace interval eyeblink conditioning is impaired in mutant mice lacking the NMDA receptor subunit epsilon 1. Eur J Neurosci 13: 1221-1227. [DOI] [PubMed] [Google Scholar]
- Kiyama Y, Manabe T, Sakimura K, Kawakami F, Mori H, Mishina M ( 1998) Increased thresholds for long-term potentiation and contextual learning in mice lacking the NMDA-type glutamate receptor ϵ1 subunit. J Neurosci 18: 6704-6712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolesnikov Y, Jain S, Wilson R, Pasternak GW ( 1998) Lack of morphine and enkephalin tolerance in 129/SvEv mice: evidence for a NMDA receptor defect. J Pharmacol Exp Ther 284: 455-459. [PubMed] [Google Scholar]
- Koob GF, Le Moal M ( 2001) Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology 24: 97-129. [DOI] [PubMed] [Google Scholar]
- Lake JR, Hebert KM, Payza K, Deshotel KD, Hausam DD, Witherspoon WE, Arcangeli KA, Malin DH ( 1992) Analog of neuropeptide FF attenuates morphine tolerance. Neurosci Lett 146: 203-206. [DOI] [PubMed] [Google Scholar]
- Lutfy K, Doan P, Weber E ( 1999) ACEA-1328, a NMDA receptor/glycine site antagonist, acutely potentiates antinociception and chronically attenuates tolerance induced by morphine. Pharmacol Res 40: 435-442. [DOI] [PubMed] [Google Scholar]
- Maldonado R, Blendy JA, Tzavara E, Gass P, Roques BP, Hanoune J, Schutz G ( 1996) Reduction of morphine abstinence in mice with a mutation in the gene encoding CREB. Science 273: 657-659. [DOI] [PubMed] [Google Scholar]
- Malin DH, Lake RJ, Hammond MV, Fowler DE, Rogillio RB, Brown SL, Sims JL, Leecraft BM, Yang HYT ( 1990) FMRF-NH2-like mammalian octapeptide: possible role in opiate dependence and abstinence. Peptides 11: 969-972. [DOI] [PubMed] [Google Scholar]
- Mamiya T, Noda Y, Ren X, Hamdy M, Furukawa S, Kameyama T, Yamada K, Nabeshima T ( 2001) Involvement of cyclic AMP systems in morphine physical dependence in mice: prevention of development of morphine dependence by rolipram, a phosphodiesterase 4 inhibitor. Br J Pharmacol 132: 1111-1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao J ( 2002) Opioid-induced abnormal pain sensitivity: implications in clinical opioid therapy. Pain 100: 213-217. [DOI] [PubMed] [Google Scholar]
- Mao J, Mayer DJ ( 2001) Spinal cord neuroplasticity following repeated opioid exposure and its relation to pathological pain. Ann NY Acad Sci 933: 175-184. [DOI] [PubMed] [Google Scholar]
- Meguro H, Mori H, Araki K, Kushiya E, Kutsuwada T, Yamazaki M, Kumanishi T, Arakawa M, Sakimura K, Mishina M ( 1992) Functional characterization of a heteromeric NMDA receptor channel expressed from cloned cDNAs. Nature 357: 70-74. [DOI] [PubMed] [Google Scholar]
- Millan MJ ( 2002) Descending control of pain. Prog Neurobiol 66: 355-474. [DOI] [PubMed] [Google Scholar]
- Minami T, Sugatani J, Sakimura K, Abe M, Mishina M, Ito S ( 1998) Absence of prostaglandin E2-induced hyperalgesia in NMDA receptor epsilon subunit knockout mice. Br J Pharmacol 120: 1522-1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell JM, Basbaum AI, Fields HL ( 2000) A locus and mechanism of action for associative morphine tolerance. Nat Neurosci 3: 47-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogil JS, Pasternak GW ( 2001) The molecular and behavioral pharmacology of the orphanin FQ/nociceptin peptide and receptor family. Pharmacol Rev 53: 381-415. [PubMed] [Google Scholar]
- Mori H, Mishina M ( 1995) Structure and function of the NMDA receptor channel. Neuropharmacology 34: 1219-1237. [DOI] [PubMed] [Google Scholar]
- Nestler EJ ( 2000) Genes and addiction. Nat Genet 26: 277-281. [DOI] [PubMed] [Google Scholar]
- Nestler EJ ( 2001) Molecular neurobiology of addiction. Am J Addict 10: 201-217. [DOI] [PubMed] [Google Scholar]
- Nitsche JF, Schuller AG, King MA, Zengh M, Pasternak GW, Pintar JE ( 2002) Genetic dissociation of opiate tolerance and physical dependence in delta-opioid receptor-1 and preproenkephalin knock-out mice. J Neurosci 22: 10906-10913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternak GW, Kolesnikov YA, Babey AM ( 1995) Perspectives on the N-methyl-d-aspartate/nitric oxide cascade and opioid tolerance. Neuropsychopharmacology 13: 309-313. [DOI] [PubMed] [Google Scholar]
- Plesan A, Hedman U, Xu XJ, Wiesenfeld-Hallin Z ( 1998) Comparison of ketamine and dextromethorphan in potentiating the antinociceptive effect of morphine in rats. Anesth Analg 86: 825-829. [DOI] [PubMed] [Google Scholar]
- Pommier B, Beslot F, Simon A, Pophillat M, Matsui T, Dauge V, Roques BP, Noble F ( 2002) Deletion of CCK2 receptor in mice results in an upregulation of the endogenous opioid system. J Neurosci 22: 2005-2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth BL, Willins DL ( 1999) What's all the RAVE about receptor internalization? Neuron 23: 629-631. [DOI] [PubMed] [Google Scholar]
- Sakimura K, Kutsuwada T, Ito I, Manabe T, Takayama C, Kushiya E, Yagi T, Aizawa S, Inoue Y, Sugiyama H, Mishina M ( 1995) Reduced hippocampal LTP and spatial learning in mice lacking NMDA receptor epsilon 1 subunit. Nature 373: 151-155. [DOI] [PubMed] [Google Scholar]
- Trujillo KA ( 2000) Are NMDA receptors involved in opiate-induced neural and behavioral plasticity? A review of preclinical studies. Psychopharmacology (Berl) 151: 121-141. [DOI] [PubMed] [Google Scholar]
- Trujillo KA, Akil H ( 1991) Inhibition of morphine tolerance and dependence by the NMDA receptor antagonist MK-801. Science 251: 85-87. [DOI] [PubMed] [Google Scholar]
- Tsao P, Cao T, von Zastrow M ( 2001) Role of endocytosis in mediating downregulation of G-protein-coupled receptors. Trends Pharmacol Sci 22: 91-96. [DOI] [PubMed] [Google Scholar]
- Ueda H, Tamura S, Satoh M, Takagi H ( 1987) Excess release of substance P from the spinal cord of mice during morphine withdrawal and involvement of the enhancement of presynaptic Ca2+ entry. Brain Res 425: 101-105. [DOI] [PubMed] [Google Scholar]
- Ueda H, Miyamae T, Hayashi C, Watanabe S, Fukushima N, Sasaki Y, Iwamura T, Misu Y ( 1995) Protein kinase C involvement in homologous desensitization of delta-opioid receptor coupled to Gi1-phospholipase C activation in Xenopus oocytes. J Neurosci 15: 7485-7499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda H, Yamaguchi T, Tokuyama S, Inoue M, Nishi M, Takeshima H ( 1997) Partial loss of tolerance liability to morphine analgesia in mice lacking the nociceptin receptor gene. Neurosci Lett 237: 136-138. [DOI] [PubMed] [Google Scholar]
- Ueda H, Inoue M, Takeshima H, Iwasawa Y ( 2000) Enhanced spinal nociceptin receptor expression develops morphine tolerance and dependence. J Neurosci 20: 7640-7647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda H, Inoue M, Matsumoto T ( 2001) Protein kinase C-mediated inhibition of mu-opioid receptor internalization and its involvement in the development of acute tolerance to peripheral mu-agonist analgesia. J Neurosci 21: 2967-2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wainai T, Takeuchi T, Seo N, Mishina M ( 2001) Regulation of acute nociceptive responses by the NMDA receptor GluRepsilon2 subunit. NeuroReport 12: 3169-3172. [DOI] [PubMed] [Google Scholar]
- Williams JT, Christie MJ, Manzoni O ( 2001) Cellular and synaptic adaptations mediating opioid dependence. Physiol Rev 81: 299-343. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Costigan M ( 1999) Transcriptional and posttranslational plasticity and the generation of inflammatory pain. Proc Natl Acad Sci USA 96: 7723-7730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaksh TL ( 1993) The spinal pharmacology of facilitation of afferent processing evoked by high-threshold afferent input of the postinjury pain state. Curr Opin Neurol Neurosurg 6: 250-256. [PubMed] [Google Scholar]
- Yoshida A, Ueda H ( 1999) Activation of Gi1 by lysophosphatidic acid receptor without ligand in the baculovirus expression system. Biochem Biophys Res Commun 259: 78-84. [DOI] [PubMed] [Google Scholar]
- Zhu Y, King MA, Schuller AG, Nitsche JF, Reidl M, Elde RP, Unterwald E, Pasternak GW, Pintar JE ( 1999) Retention of supraspinal delta-like analgesia and loss of morphine tolerance in delta opioid receptor knockout mice. Neuron 24: 243-252. [DOI] [PubMed] [Google Scholar]
- Zimmermann M ( 1983) Ethical guidelines for investigations of experimental pain in conscious animals. Pain 16: 109-110. [DOI] [PubMed] [Google Scholar]