

Abstract
Localizing the cellular prion protein (PrPC) in the brain is necessary for understanding the pathogenesis of prion diseases. However, the precise ultrastructural localization of PrPC still remains enigmatic. We performed the first quantitative study of the ultrastructural localization of PrPC in the mouse hippocampus using high-resolution cryoimmunogold electron microscopy. PrPC follows the standard biosynthetic trafficking pathway with a preferential localization in late endosomal compartments and on the plasma membrane of neurons and neuronal processes. PrPC is found with the same frequency within the synaptic specialization and perisynaptically, but is almost completely excluded from synaptic vesicles. Unexpectedly, PrP is also found in the cytosol in subpopulations of neurons in the hippocampus, neocortex, and thalamus but not the cerebellum. Cytosolic PrP may have altered susceptibility to aggregation, suggesting that these neurons might play a significant role in the pathogenesis of prion diseases, in particular those mammals harboring mutant PrP genes.
Keywords: prion protein, hippocampus, immunogold, localization, membrane, cytosolic
Introduction
The cellular prion protein (PrPC) is a cell-surface glycoprotein anchored by a glycosylphosphatidylinositol (GPI) moiety (Stahl et al., 1987). PrPC is expressed throughout the brain, particularly in neurons (Kretzschmar et al., 1986; Moser et al., 1995) and to a lesser extent in extraneural tissues (Bendheim et al., 1992; Ford et al., 2002). In prion diseases, PrPC is converted to an abnormal, conformationally altered isoform (PrPSc), which subsequently accumulates in the brain and results in extensive neurodegeneration with an inevitably fatal outcome (Prusiner, 1996). Therefore, localizing PrPC in the brain is an important step in understanding the biology of the normal protein and mapping changes in models of experimental prion diseases.
The precise localization of PrPC remains enigmatic because of conflicting data obtained using different techniques. Immunohistochemical studies described a somatic expression of PrPC in neurons with no signal or only a minor signal in the neuropil (DeArmond et al., 1987; Piccardo et al., 1990; Safar et al., 1990; Bendheim et al., 1992; Verghese-Nikolakaki et al., 1999; Ford et al., 2002). However, it was not determined whether PrPC was luminal [for example, in the endoplasmic reticulum (ER) or Golgi] or cytosolic. Data obtained using free-floating section immunohistochemistry (Sales et al., 1998; Haeberle et al., 2000; Moya et al., 2000) and immunoelectron microscopy (Fournier et al., 1995, 2000) indicated PrPC localization in the neuropil with a synaptic membrane prevalence. However, data obtained from synaptosomal preparations (Herms et al., 1999) and a recent ultrastructural study on the distribution of PrPC in the cerebellum (Laine et al., 2001) have favored a predominantly plasma membrane location of PrPC with no expression on synaptic vesicles or in the cytoplasm. These contradictory findings probably reflect the peculiarities inherently associated with pre-embedding techniques. Many immunoelectron microscopic procedures may result in a destruction of cellular membranes, possibly leading to an artificial redistribution of GPI-anchored proteins within the membrane (Griffiths, 1993).
Two recently published studies on the effects of proteosome inhibitors on PrPC degradation and expression of cytosolic PrP suggested that cytosolic localization of PrP is sufficient to induce neurodegeneration (Ma and Lindquist, 2002; Ma et al., 2002). It is notable that under the experimental conditions of these studies, PrP becomes insoluble and acquires partial protease resistance.
These uncertainties regarding the precise subcellular localization of PrPC therefore encouraged us to perform the first quantitative study of ultrastructural PrPC localization in the mouse brain. We used a sensitive, high-resolution detection method combining immunofluoresence and immunogold labeling of 500 and 60 nm cryosections at light and electron microscopy levels, respectively. The method uses glutaraldehyde for both optimal fixation and preventing migration of GPI-anchored proteins and circumvents the need for alcohol dehydration. Because of its potentially important role in the pathogenesis of prion diseases (DeArmond et al., 1987; Taraboulos et al., 1992a), we focused on the localization of PrPC in the hippocampus and show for the first time quantitative data of PrPC distribution at the ultrastructural level throughout the CA1 and dentate gyrus areas. We localized PrPC on all biosynthetic and endocytic transport membranous structures of hippocampal neurons, but almost no PrPC was found in synaptic vesicles. In addition, we discovered a subset of neurons in which PrP is located predominantly in the cytosol. These cells did not show any obvious signs of neurodegeneration but may have important implications in the pathogenesis of prion diseases.
Materials and Methods
Animals and preparation of tissue. Brain tissue was obtained from 22 mice at ∼12 weeks of age from four different mouse lines with the FVB background: (1) wild-type (wt) mice, (2) PrP-ablated (Prnp0/0) mice (Büeler et al., 1993), (3) transgenic 4053 mice overexpressing mouse PrPC (Telling et al., 1996), and (4) transgenic 3045 mice overexpressing hamster PrPC (Telling et al., 1996). According to PHS-NIH Guide for the Care and Use of Laboratory Animals, the mice were deeply anesthetized with Nembutal and perfused transcardially, first with PBS plus heparin (1 μl/ml) for 90 sec, then with PBS for 1 min, and finally with one of the following fixatives for 5 min: 2% paraformaldehyde (PFA) in PIPES-HEPES-EGTA-magnesium (PHEM) buffer (25 mm HEPES, 10 mm EGTA, 60 mm PIPES, 2 mm MgCl2, pH 7.2) (four B4053 mice); 2% PFA-0.2% glutaraldehyde (GA) in PHEM buffer, pH 7.2 (four B4053 mice, four wt mice, two A3045 mice, two Prnp0/0 mice); periodate-lysine-paraformaldehyde (PLP) fixative (McLean and Nakane, 1974) with a final concentration of 2% PFA (four B4053 mice); and 2% PFA-0.2% GA in PHEM buffer, pH 7.2 (two B4053 mice). After perfusion, the brains were collected, postfixed in the same fixative for 1 hr at 4°C, washed in PHEM four times, and stored at 4°C in 0.5% PFA in PHEM buffer, pH 7.2.
Reagents and antibodies. PrP-specific recombinant antibody fragments (Fabs) D13, D18, R1, R2, E123, and E149 were derived from phage libraries and have been characterized thoroughly (Peretz et al., 1997; Williamson et al., 1998; Leclerc et al., 2001; Peretz et al., 2001). SAF32 and 8H4 monoclonal antibodies were gifts from Dr. H. Axelrad (Faculty of Medicine, Pitié-Salpêtrière, Paris, France). An aliquot of Fab D18 was conjugated to UltraSmall gold particles (0.8 nm; Aurion, Wageningen, The Netherlands) to allow increased penetration into the cryosections and circumvent labeling artifacts caused by a cross-reaction with immuno-globulins in the tissue. R-GENT SE-EM and R-GENT SE-LM silver enhancement kits were purchased from Aurion. The in situ cell death detection kit [terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL)] was purchased from Roche Products (Mannheim, Germany).
Polyclonal and monoclonal antibodies against GABA, monoclonal antibodies against parvalbumin, and monoclonal 2′,3′-cyclic nucleotide 3′-phosphodiesterase (CNPase) antibodies were obtained from Sigma (St. Louis, MO). Polyclonal antibodies against parvalbumin, calretinin, and calbindin D28K were obtained from Swant (Bellinzona, Switzerland). Polyclonal antibodies against somatostatin and b-NOS were purchased from Chemicon (Temecula, CA), and monoclonal antibodies against synaptobrevin/vesicle-associated membrane protein-2 (Vamp2) were purchased from Synaptic Systems (Göttingen, Germany). Polyclonal GFAP and S100 antibodies were purchased from Dako (Glostrup, Denmark), goat polyclonal Thy-1 (GPI-anchored protein) antibodies and rabbit polyclonal antibodies against neuropeptide Y were obtained from Research Diagnostics (Flanders, NJ), and polyclonal antibodies against vasointestinal peptide were purchased from DiaSorin (Antony, France). Rabbit anti-mouse Fab and rabbit anti-mouse IgG antibodies were obtained from Pierce (Rockford, IL). Secondary anti-mouse, anti-rabbit antibodies conjugated with Alexa Fluor 488 or Texas Red were purchased from Molecular Probes (Eugene, OR). Protein-A-gold conjugates were obtained from Utrecht Medical School (The Netherlands).
Cryosectioning and immunolabeling. Cryosections were prepared as described previously (Peters, 2001). Briefly, the fixed specimens were infused with 2.3 m sucrose overnight and then frozen in liquid nitrogen. Both semithin (0.5 μm) and ultrathin sections (60 nm) were cut at -100 or -120°C with a diamond knife on a Leica (Vienna, Austria) Ultracut T equipped with a cryoattachment. Semithin sections were picked up from the diamond knife using 2.3 m sucrose and transferred to a glass slide for subsequent immunohistochemical labeling. For light microscopy, primary antibodies were detected using either secondary antibodies conjugated with Alexa Fluor 488 or Texas Red or secondary rabbit “bridging” antibodies with protein A-gold (5 nm), which was silver-enhanced with Aurion R-GENT SE-LM kit for 15 min. Ultrathin sections were picked up from the diamond knife with a 1% methyl cellulose 25CP-1.15 m sucrose solution and transferred to formvar/carbon-coated copper grids.
The immunolabeling of sections was done as described previously (Raposo et al., 1997). In brief, after blocking with 1% cold fish gelatin and 1% bovine serum albumin for 15 min, sections were incubated with primary antibody for 60 min, washed, and bridging rabbit antibodies were applied for 30 min when necessary. Sections were then incubated with protein A-gold (10 nm) for 20 min. For the double-labeling experiments, a second primary antibody followed by protein A-gold was applied consecutively after labeling with the first antibody. To rule out the possibility that noncolocalization could be attributable to interference by the primary antibody, we compared the density of immunogold labeling of each antibody in single-immunolabeling procedures or omitted one of the primary antibodies in double-labeling experiments. No significant interference was observed. Fab D18 against PrPC covalently conjugated with 0.8 nm UltraSmall gold particles was also used to label PrP. Ultra-Small gold particles were visualized using the R-GENT SE-EM silver enhancement kit, following the instructions of the manufacturer. Labeled sections were viewed with a Philips CM10 electron microscope (FEI Company, Eindhoven, The Netherlands) at 80 kV and a standard 15,000× magnification.
Although cryoultramicrotomy uses a different contrast-staining method from routine epon ultramicrotomy, cellular and subcellular profiles of cells appear similar under the electron microscope (Maunsbach and Afzelius, 1999). The only differences are the reverse contrast of membranes, which appear white, and the cytoplasm, which becomes electron dense. Cellular and subcellular profiles were thus identified and defined according to the criteria of Peters et al. (1991).
Quantification of immunogold labeling. Quantitative evaluation of hippocampal labeling was performed only on ultrathin cryosections of wt FVB mice that were fixed with 2% PFA-0.2% GA to avoid overexpression artifacts of the transgenes. The relative distributions of labeled PrPC were determined by counting gold particles over plasma and intracellular membranes of selected hippocampal cells. We estimated the membrane (gold per micrometer) and area (gold per square micrometer) labeling density on micrographs with a final 32,000× magnification by using point and intersection counting with a line and point lattice (10 mm distance) overlay as described by Weibel (1979) and Griffiths (1993). Gold particles located ≤20 nm from a visible membrane structure were assigned to that structure. The distance of 20 nm was chosen on the basis of the distance constraints of immunolabeling described below.
We used two labeling protocols on ultrathin cryosections. The first included the application of a murine Fab fragment, followed by a rabbit anti-mouse anti-Fab IgG and protein A coupled to a 10 nm gold particle. According to Griffiths (1993) and Amit et al. (1986), the length of an IgG molecule in projection is ∼8 -10 nm, and a Fab fragment is ∼5 nm. The diameter of the protein A-gold (10 nm) complex is ∼12-13 nm, but because we measure the distance to the center of the gold particle, we should consider only its radius (6 -7 nm). In summary, the complex consisting of a Fab, IgG, and protein A-gold has a projection of 19 -22 nm. Therefore, we used a ∼20 nm radius to assign gold particles to a specific structure on the sections.
The second labeling protocol involves the application of recombinant Fab fragments directly conjugated with UltraSmall gold particles (0.8 nm) and the subsequent silver enhancement for better visualization. In this case, the distance between the antigen and the center of the gold particle is ∼6 nm. The silver enhancement procedure deposits silver around the gold particle without preferential orientation, practically isotropic. Therefore, the position of the enhanced gold particle (10 -15 nm in diameter) will precisely indicate the localization of the antigen of interest.
We analyzed cells taken from a vertical strip running through CA1 from the stratum oriens to the hilus of the dentate gyrus. This included cells from the pyramidal cell layer and hilus, as well as dentate granule cells. Ten random pictures with good ultrastructural preservation were taken from each area on sections from 18 grids made from three animal samples (six grids for each sample). The gold particles were counted in the following subcellular structures: endoplasmic reticulum (including the nuclear envelope), Golgi complex, endosomes and lysosomes, tubules and vesicles without definite coat, clathrin-coated vesicles and pits, plasma membrane, mitochondria, and nucleus. Immunogold labeling on mitochondria was treated as background labeling, because PrPC has never been observed on these organelles by either biochemical or morphological methods by us or by others.
In addition, we analyzed the distribution of gold particles in the neuropil of the strata radiatum, oriens, and moleculare of the dentate gyrus, on membrane profiles in dendrites and axons, and on membranes of synaptic and perisynaptic profiles. Each class was further subdivided into the plasma membrane, internal transport vesicles, spines and endosomes (only for dendrites), mitochondria, and myelin sheaths (only for axons). Membranes of synaptic complexes were classified into synaptic vesicles, synaptic specialization (which included the two closely opposed membranes in the synapse), and the presynaptic and postsynaptic membrane (which include the membranes outside the synaptic specialization region). For our cryosections, we used the same standard criteria for subcellular structures in brain cells as those used in epon sections (Peters et al., 1991). Unidentified membrane compartments were not taken into account, because they did not show substantial labeling and represented only ∼5% of all cellular membranes.
To test that the labeling for PrP was not random, we calculated the relative labeling index (RLI) according to Mayhew et al. (2002). By superimposing a test-point lattice on the electron micrographs, we generated random points (P) on cellular compartments with the point density determined by the relative size of each compartment. The number of points was normalized to the number of observed gold particles (no), giving a value for the expected distribution of labels (ne). The RLI is calculated by dividing no by ne. Particle distributions were compared with a χ2 analysis to test whether the observed distributions differ significantly from random distributions. The partial χ2 value in each row in Table 4 was obtained using the following formula: (no - ne)2/ne. The total χ2 value was obtained by taking the sum of the partial values. If a compartment is randomly labeled, its RLI = 1 and partial χ2 = 0. If a compartment is preferentially labeled, the RLI value will be >1, and its partial χ2 value will contribute a significant portion to the total χ2 value.
Table 4.
Observed and expected distributions of gold particles in compartments of pyramidal neurons and CPrP cells, relative labeling index, and χ2 valuesa
|
Compartments |
Number of observed gold particles, no |
Number of grid points, P |
Normalized number of expected gold particles, ne = P(total no/total P) |
Relative labeling index, no/ne |
Partial χ2 values, (no - ne)2/ne |
|---|---|---|---|---|---|
| Pyramidal neurons | |||||
| Nucleus | 11 | 421 | 181 | 0.06 | 160 |
| Mitochondria | 5 | 69 | 30 | 0.24 | 20 |
| Cytosol | 13 | 799 | 344 | 0.04 | 318 |
| CPrP cells | |||||
| Nucleus | 26 | 227 | 98 | 0.27 | 51 |
| Mitochondria | 27 | 105 | 45 | 0.6 | 7.2 |
| Cytosol | 908 | 678 | 292 | 3.12b | 1308b |
| Totals
|
990
|
2299
|
990
|
1
|
1864.2c
|
The values were quantified for 10 cells in each category.
Indicates compartment is preferentially labeled.
The distribution is nonrandom, with p < 0.001.
Results
Assessment of methodology and antibodies
Four different fixatives were examined to find the optimal conditions for both ultrastructural integrity and preservation of antigenicity. We found that both PLP and 2% PFA fixatives resulted in suboptimal ultrastructure preservation for ultrathin cryosections (our unpublished data). A fixative composed of both PFA and GA gave the best results for ultrastructural integrity and antigen preservation. However, the distribution of immunolabeling was the same with all fixatives used (data not shown). Thus, we used the fixative containing 2% PFA and 0.2% GA for the immunolabeling experiments.
Raising antibodies against PrP has been difficult because of the high degree of conservation of PrP sequences between species and the inhibitory activity of anti-PrP antibodies toward lymphocytes. To circumvent this problem, we used well characterized recombinant Fabs that recognize different parts of the PrPC molecule (Peretz et al., 1997; Leclerc et al., 2001): E123 (residues 23-37), E149 (residues 72-86), D13 (residues 96 -104), D18 (residues 133-157), and R1 and R2 (residues 225-231). These Fabs were obtained from immunized Prnp0/0 mice and retrieved through phage display libraries and have been shown as reliable immunoreagents that recognize PrPC in different experimental procedures (Williamson et al., 1996, 1998; Leclerc et al., 2001; Peretz et al., 2001). Monoclonal antibodies 8H4 (which recognizes residues 158 -174) and SAF32 (which recognizes residues 52-92) were used as additional positive controls. We found no differences in labeling patterns in the murine hippocampus between these different antibodies (data not shown).
Distribution of PrPC observed by light microscopy
To determine the precise localization of PrPC, we used cryoprotected aldehyde-fixed tissue samples, which allowed us to make both semithin (0.5 μm) and ultrathin (60 nm) sections from the very same block in a serial manner. Thus, we had the ability to easily correlate immunolabeling at cellular and subcellular levels. We used two markers for semithin cryosections: (1) gold particles that were silver enhanced (Fig. 1A) and (2) a fluorescent dye marker (Fig. 1C). Both methods clearly showed that PrPC was found predominantly in the neuropil in all hippocampal layers, with a higher density in the strata oriens and radiatum, moderate immunoreactivity in the stratum lacunosum-moleculare, and weak labeling in the stratum moleculare and hilus (Fig. 1A,C). No significant differences in labeling intensity were observed among the CA1, CA2, and CA3 areas of the hippocampus (data not shown). Dendrites of pyramidal cells were mostly immunonegative inside the profile and seen as empty profiles embedded in positive surroundings (Fig. 1A,C,D). This apparently reflected the membranous localization of PrPC [see electron microscopy (EM) study below]. Specificity of the PrPC antibodies was confirmed by the absence of staining in the hippocampus with the omission of primary antibody and tissue from a Prnp0/0 mouse (Fig. 1B).
Figure 1.
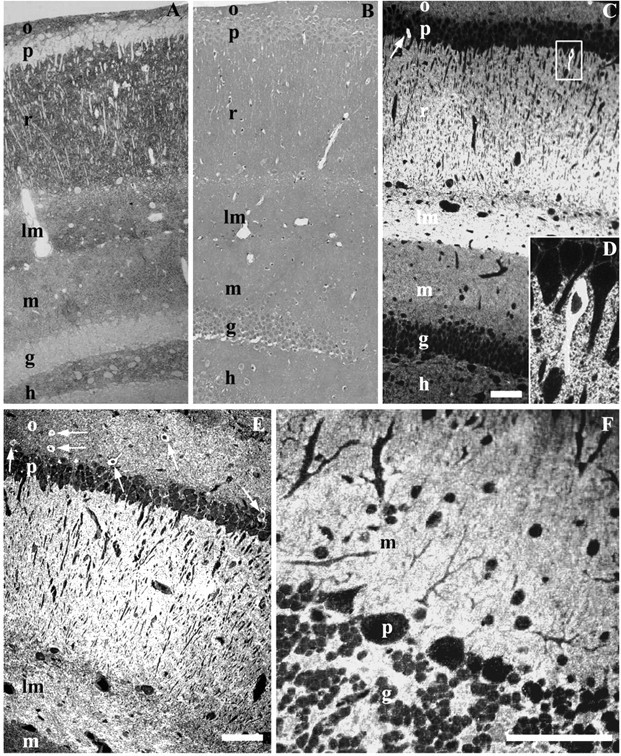
Hippocampal (CA1 and dentate gyrus) semithin cryosections labeled for PrPC with Fab D18. Labeling is concentrated in the stratum oriens (o), stratum radiatum (r), and lacunosum-moleculare (lm); less labeling is seen in the stratum moleculare (m) and hilus (h) of the dentate gyrus. Cell bodies in pyramidal (p) and granule (g) layers are free of labeling, with the exception of rare cells (D). A, Hippocampus of a wt FVB mouse. Fab D18 was used with a secondary polyclonal antibody and protein A-gold (5 nm) that was subsequently visualized by silver enhancement (Aurion). B, Hippocampus of a Prnp0/0 mouse. Labeling is the same as in A. No positive signal is visible. Sections are counterstained with Giemsa. C, Hippocampus of a wt FVB mouse. Fluorescent labeling shows a labeling pattern similar to A. The arrow points to a cell with intense PrPC labeling. The box shows the cell enlarged in D. D, A magnified view of a cell with high PrPC content. E, Hippocampus (CA1 area) of a 4053 mouse that overexpresses PrP, showing a pattern of PrPC labeling similar to A and C. Arrows point to the cells with an abundance of PrP in the cytoplasm. F, Cerebellum of a wt FVB mouse. No cells labeling positively for PrPC in the cytoplasm were found in the cerebellum. Purkinje cells (p) are free of intensive cytosolic labeling. Only faint punctate labeling is visible. Scale bars, 100 μm.
We consistently saw a small population of cell bodies that were intensely labeled by both the immunogold and immunofluorescence procedures (Fig. 1C-E). These cells with high PrPC content in cell bodies were found with similar frequency in three of the mouse lines (wt FVB, 4053, 3405) and were absent in Prnp0/0 mice. These cells were concentrated predominantly in the CA1 area in the strata pyramidale and oriens with a frequency of 1-2% of all cells. Furthermore, they were practically absent from the CA3 area and occasionally seen as small groups in the dentate gyrus. In addition, a small number of cells with high PrPC labeling in cell bodies was seen in the somatosensory neocortex (layers V and VI) and ventral lateral geniculate nucleus of the thalamus (data not shown). We refer to these cells as cytosolic PrP (CPrP) cells.
We did not observe any CPrP cells in the cerebellum of any of the murine lines that we analyzed. The immunofluorescent labeling of cerebellar PrPC was concentrated in the neuropil areas of the molecular layer and not in the cell bodies (Fig. 1F). In the neocortex and hippocampus, very weak punctate labeling in the cytoplasm of cerebellar neurons reflected PrPC localization in intracellular organelles, which was confirmed by subsequent EM analysis (data not shown).
Ultrastructural distribution of PrPC in the hippocampus
To gain insight into the precise localization of PrPC at the ultrastructural level, we used the very same blocks to produce ultrathin sections immediately after cutting semithin sections from the areas of interest. Thereby we could assess identical structures at both light and electron microscopic levels. By EM, PrPC labeling in the neuropil was predominantly found on the plasma membrane of dendrites, including spines, as well as dendritic transport vesicles, endosomes, axolemma, axonal transport vesicles, and myelin sheaths. In addition, the membranes of synaptic specializations, including presynaptic and postsynaptic membranes, and of synaptic vesicles (Figs. 2B-E, 3A) labeled positively for PrPC. However, quantitative analysis showed marked reduction of labeling on the synaptic vesicle membrane (Table 1). Prnp0/0 mice displayed no immunopositive profiles for PrPC (Fig. 2A).
Figure 2.
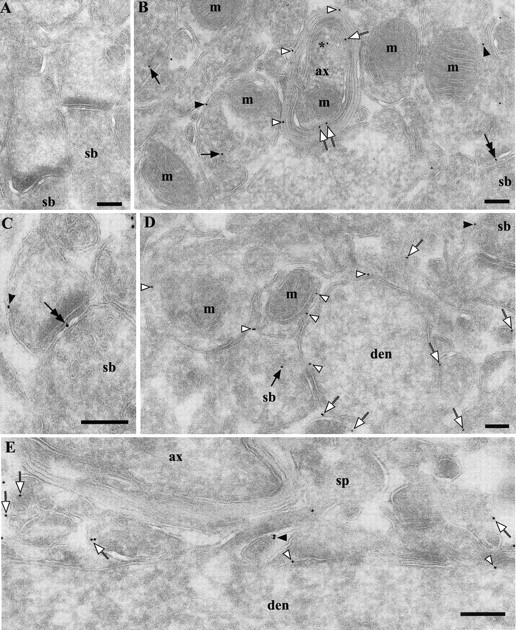
Ultrastructural localization of PrPC in the neuropil in hippocampal CA1 area. Labeling was performed using Fab D18 and protein A-gold (10 nm). A, Absence of PrPC labeling in a Prnp0/0 mouse. B, Stratum radiatum of a wt FVB mouse. Gold particles are localized on the axolemma (white arrows), myelin sheaths (white arrowheads), putative axon terminal membranes (black arrowheads), synaptic vesicles (black arrows), axonal transport vesicle (near asterisk), and synaptic specialization (double-headed arrow). Gold particles without arrows belong to processes, which are difficult to identify as being either dendritic or axonal. C, Stratum radiatum from a wt FVB mouse. As in B, gold particles can be found at the synaptic specialization (double-headed arrow) and on the membranes of the postsynaptic profile (arrowhead). D, Stratum oriens of a wt FVB mouse. Gold particles are localized on the dendritic shaft (white arrowheads), small processes (white arrows), and synaptic vesicles (black arrow). E, Stratum radiatum of a wt FVB mouse, longitudinal section of a dendrite. Gold particles are localized on the dendritic shaft (white arrowheads), membrane of a spine (black arrowhead), and small processes (white arrows). The particle inside the spine probably sits on the spinal apparatus, which is not clearly identifiable because of the tangential orientation. Relative distribution of the immunogold labels is provided in Table 1; B and D do not reflect quantitative information. ax, Myelinated axon; den, dendrite; m, mitochondria; sb, synaptic bouton; sp, spine. Scale bars, 200 nm.
Figure 3.
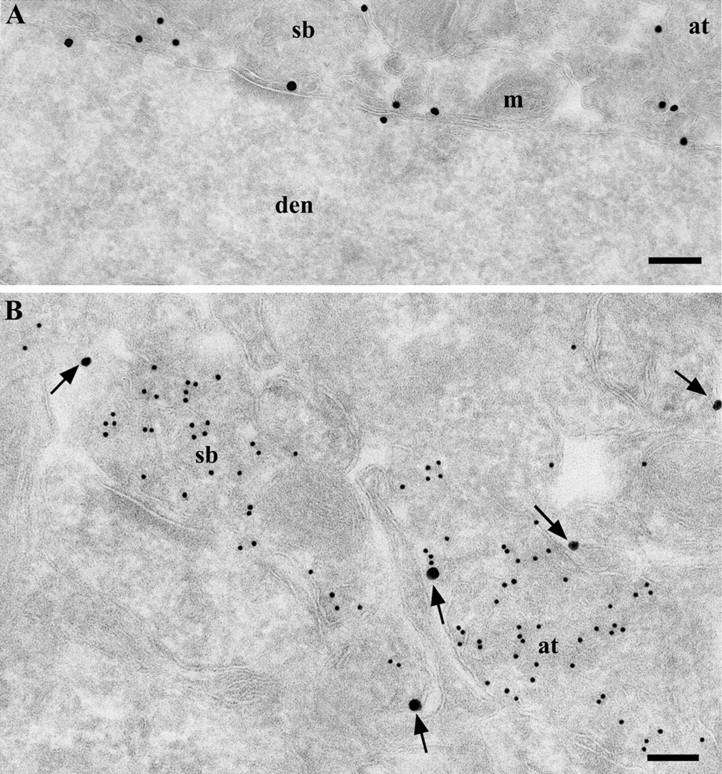
Ultrastructural localization of PrPC in the neuropil of the stratum radiatum in the hippocampal CA1 area, using Fab D18 conjugated with UltraSmall gold and enhanced with a silver enhancement kit. A, Membrane-bound localization of PrPC with dendritic cytosol free of labeling. PrPC labeling was found on the dendritic shaft, axon terminal membrane, and profiles of small processes. B, Double labeling for PrPC and VAMP2. Large particles (silver-enhanced UltraSmall gold) (arrows) represent anti-PrPC labeling on the plasma membrane of the axon terminal and dendrites; small (15 nm) gold particles indicate VAMP2 labeling of synaptic vesicles. at, Axon terminal; den, dendrite; m, mitochondria; sb, synaptic bouton. Scale bars, 200 nm.
Table 1.
Quantification of PrPc labeling density on the membranes of and within dendritic, axonal, and synaptic profiles from the neuropil of the CA1 area and dentate gyrusa
|
|
Neuropil area |
||||
|---|---|---|---|---|---|
| Membranous profiles
|
Stratum oriens (n = 17)
|
Stratum radiatum (n = 20)
|
Stratum moleculare (n = 17)
|
||
| Dendrites | |||||
| Dendritic shaft | 1.54 ± 0.04 | 1.63 ± 0.06 | 0.62 ± 0.04 | ||
| Spines | 1.58 ± 0.1 | 1.52 ± 0.09 | 0.58 ± 0.1 | ||
| Transport vesicles/tubules | 1.08 ± 0.06 | 1.12 ± 0.09 | 0.48 ± 0.1 | ||
| Endosomes/lysosomes | 1.46 ± 0.2 | 1.38 ± 0.08 | 0.58 ± 0.05 | ||
| Axons | |||||
| Axolemma | 0.94 ± 0.09 | 0.84 ± 0.07 | 0.55 ± 0.06 | ||
| Transport vesicles/tubules | 0.44 ± 0.04 | 0.37 ± 0.03 | 0.35 ± 0.03 | ||
| Myelin sheaths | 0.38 ± 0.05 | 0.35 ± 0.03 | 0.36 ± 0.04 | ||
| Synaptic complexes | |||||
| Presynaptic bouton | 1.6 ± 0.08 | 1.65 ± 0.02 | 0.82 ± 0.11 | ||
| Synaptic specialization | 1.51 ± 0.15 | 1.48 ± 0.2 | 0.79 ± 0.12 | ||
| Postsynaptic bouton | 1.64 ± 0.1 | 1.63 ± 0.12 | 0.71 ± 0.1 | ||
| Synaptic vesicles | 0.12 ± 0.012 | 0.14 ± 0.013 | 0.096 ± 0.015 | ||
| Mitochondria
|
0.056 ± 0.014
|
0.073 ± 0.016
|
0.04 ± 0.008
|
||
Values represent the number of gold particles per 1 μm of membrane (gold per micrometer). Mitochondria were used to assess background labeling. Results are presented as mean ± SEM.
Light microscopy of immunolabeled sections suggested no preferential accumulation of PrPC in the profiles of the neuropil. Being aware that PrPC has been described previously as being enriched in synapses (Sales et al., 1998; Fournier et al., 2000; Haeberle et al., 2000; Moya et al., 2000), we checked the accessibility of other synaptic proteins for immunolabeling on ultrathin cryosections. In performing double-labeling experiments, we encountered the problem of false colocalization resulting from an interaction between secondary antibodies and protein A-gold (data not shown). We circumvented this artifact by using anti-PrPC Fab fragments directly conjugated with UltraSmall Aurion gold particles (0.8 nm) that were subsequently enlarged by silver enhancement (Aurion). Despite a slight decrease in contrast and section quality, this method excluded artifactual effects such as erroneous colocalization. Direct labeling with gold-conjugated Fabs demonstrated a similar labeling pattern (Fig. 3A) to that observed with indirect labeling using Fabs in combination with secondary antibodies.
Sections were colabeled for the synaptic vesicle-specific protein synaptobrevin (VAMP2) and PrPC. As expected, VAMP2 was enriched in synaptic vesicles, whereas PrPC was primarily seen on the plasma membrane (Fig. 3B). The levels of PrPC labeling in different layers of the hippocampus and in different profiles were assessed quantitatively, as described below.
In both neuronal and glial cells, PrP was detected on the ER, Golgi complex, endosomes, uncoated transport vesicles, and plasma membrane (Fig. 4A,B), which are important parts of the biosynthetic and endocytic pathways. No labeling was detected in the coated rims of Golgi cisternae (Fig. 4A), clathrin-coated pits of the plasma membrane, or clathrin-coated vesicles (Fig. 4C). Generally, immunolabeling was negligible or absent from mitochondrial and nuclear structures. Although the distribution of PrPC in glial cells (confirmed by GFAP labeling) was not quantified, EM analysis suggests that they do not express PrPC at levels comparable with neurons, because surrounding neuropil structures often had more gold particles than glial cells and their processes (Fig. 5A,B, respectively). Electron micrographs from ultrathin sections, which were cut directly after semithin sections, revealed the subcellular location of PrP in those neurons with cell bodies that were immunopositive for PrPC (i.e., CPrP cells) by light microscopy. Strikingly, the majority of the immunogold particles was not associated with surrounding membranous structures (Fig. 6A,D) but were located in the cytosol. Therefore, we designate this form of PrP as “cytPrP.”
Figure 4.
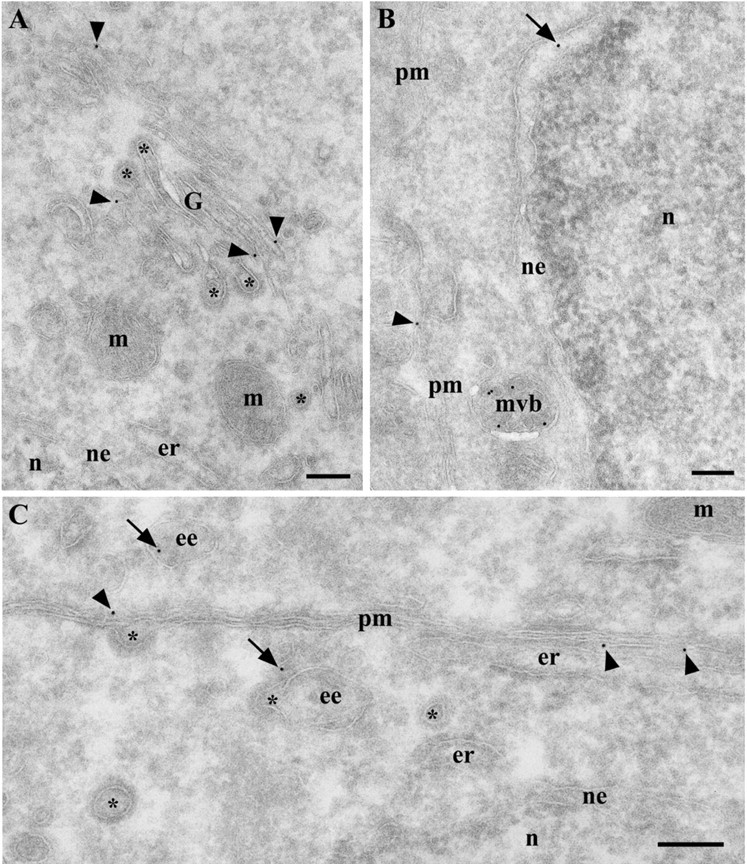
PrPC labeling in neuronal cell bodies in the hippocampus of a wt FVB mouse. A, Pyramidal neuron. Gold particles were found in the Golgi complex (arrowheads) and were absent from coated rims (asterisks) of Golgi cisternae. B, Granule neuron from the dentate gyrus. Gold particles are present in the nuclear envelope (arrow), which is a part of the ER, on late endosomes-multivesicular bodies (mvb), and on the plasma membrane (arrowhead). C, Pyramidal neuron. Gold particles are found on the plasma membrane (arrowheads) and early endosomes (arrows) but not in clathrin-coated pits or clathrin-coated vesicles (asterisks). ee, Early endosome; er, endoplasmic reticulum; G, Golgi complex; m, mitochondria; mvb, late endosomal multivesicular body; n, nucleus; ne, nuclear envelope; pm, plasma membrane; asterisks, lumen of coated vesicles, pits, and rims. Scale bars, 200 nm.
Figure 5.
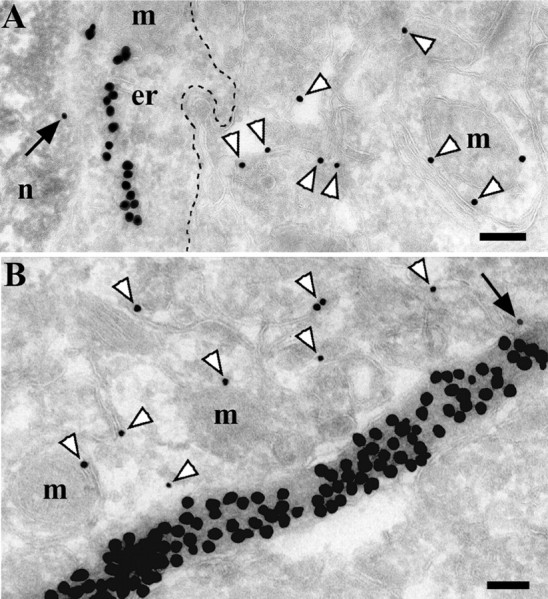
PrPC is not enriched in astrocytes in the hippocampus. A, B, A strocyte and astrocytic process. Double labeling for PrPC (small particles) and GFAP (large particles) is shown. Both labels were enhanced with silver. PrPC is more concentrated on neuropil membranes (white arrowheads) than on astrocytic membranes (black arrows). The dashed line in A delineates the astrocytic plasma membrane. er, Endoplasmic reticulum; m, mitochondria; n, nucleus. Scale bars, 200 nm.
Figure 6.
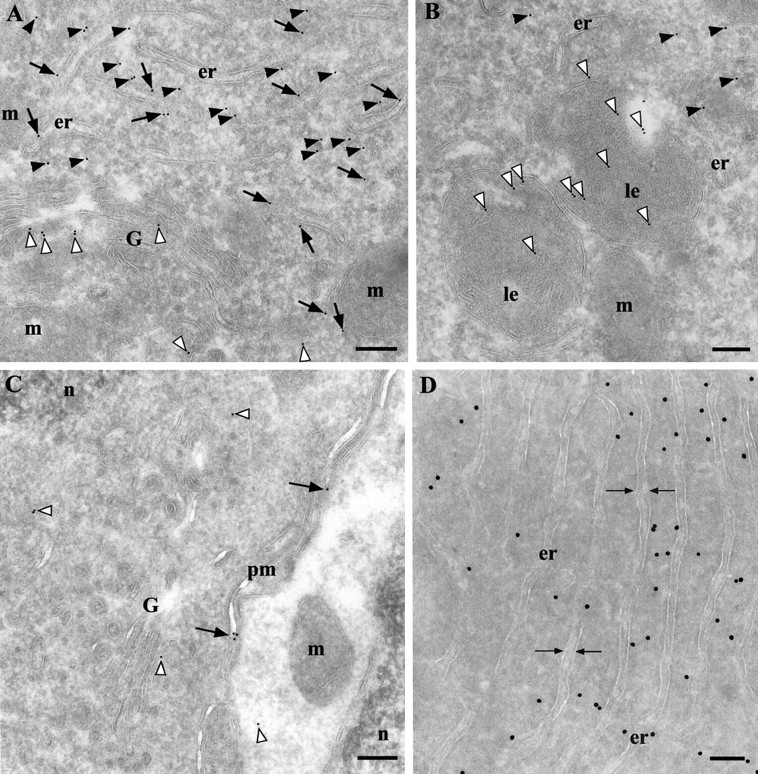
PrP is concentrated in the cytosol (black arrowheads) in a population of neurons. A, Gold particles also label PrP in the ER lumen (black arrows), Golgi cisternae and vesicles (white arrowheads), and in late endosomes (B, white arrowheads). C, Although cells that are positive for cytPrP have a denser cytosol than surrounding neurons, this is not a distinctive feature because there are cells with a dense cytosol displaying only membrane-bound PrPC labeling on the plasma membrane (black arrows) and intracellular organelles (white arrowheads). D, Direct immunolabeling with Fab D18 demonstrates the same cytPrP abundance as seen with indirect labeling methods (compare with A and B). The space indicated by the opposing arrows shows the lumen of the ER. er, Endoplasmic reticulum; G, Golgi complex; le, late endosome; m, mitochondria; n, nucleus; pm, plasma membrane. Scale bars, 200 nm.
Morphologically, CPrP cells appear to have abundant dense cytoplasms, long cisternae of the ER, well developed Golgi complexes, endosomal and lysosomal structures, well structured mitochondria, and often a nucleus with an irregular shape. The cells show an irregular distribution of cytPrP, shifted to the periphery of the cell and mostly excluded from the pericentrosomal region. The cytosolic labeling can be classified as specific because it exceeded >100× (see below) the background labeling in pyramidal neurons, which were immunonegative by light microscopy. A small number of gold particles was found in the lumen of biosynthetic pathway organelles, such as the ER, Golgi complex, and endosomes (Fig. 6A,B). From these data, we conclude that there could be two isoforms of PrPC in these cells, one that is membrane-bound and another that is cytosolic.
We detected cytPrP with Fabs recognizing the central region (D18 and D13) (Fig. 6), N-terminal region (Est123 and Est149), and C-terminal region (R1 and R2) of PrPC. Fab D18 conjugated with UltraSmall gold gave the same pattern of labeling (Fig. 6D). Furthermore, SAF32 and 8H4 monoclonal antibodies were equally able to detect cytPrP (data not shown). This argues that full-length PrP molecules that were present in the cytosol probably bound to some factor or aggregated into multimeric complexes, which prevent diffusion into the nucleus. Theoretically, a proteosome could cut PrP molecules into fragments, which are recognizable by all applied antibodies. However, this scenario seems much less likely, because either fragments of degraded proteins are destroyed very quickly by various peptidases present in the cytosol or the peptides should be detectable in the nucleus where they are unavailable for degradation (Reits et al., 2003).
The soma and dendritic processes of cells with cytPrP receive synaptic input from other neurons (Fig. 7A). Occasionally, axonal terminals with unusually high labeling for PrPC were also found in the neuropil (Fig. 7B). We assume that these axonal terminals are derived from CPrP cells, but it remains to be proven because of the low resolution of the immunogold method in structures densely packed with membranes. Furthermore, CPrP cells were negative for GFAP, CNPase, and S100 glial cells markers (data not shown), and their morphology was different to that of glia but more closely resembled that of interneurons.
Figure 7.
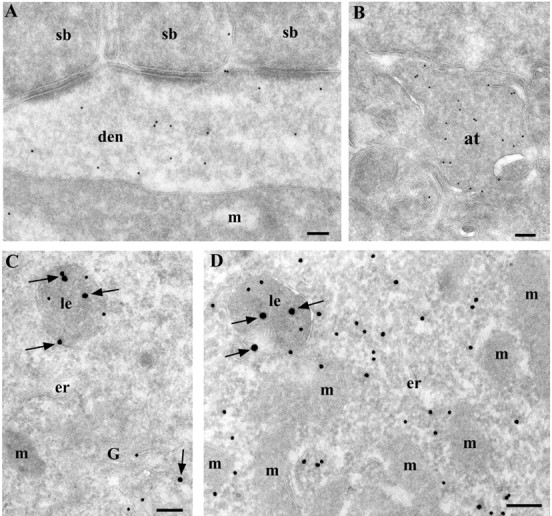
Cells with cytPrP are probably of a neuronal nature and have a functional GPI-biosynthetic machinery. A, Cells with cytPrP receive synaptic input from other neurons on their cell body and dendrites. B, An axonal terminal shows high labeling for PrPC and is most likely derived from a CPrP cell. Because of the high abundance of synaptic vesicles, the exact localization of PrPC is unclear. C, Normal CA1 pyramidal neurons have no PrP (small particles) or Thy-1 (big particles; arrows) in cytosol. PrPC and Thy-1 are concentrated in the Golgi complex and late endosomes. D, CPrP cells demonstrate both cytosolic and membrane-bound PrP (small particles) and only membrane-bound Thy-1, another GPI-anchored protein (big particles; arrows). at, Axon terminal; den, dendrite; er, endoplasmic reticulum; G, Golgi complex; m, mitochondria; le, late endosome; sb, synaptic bouton. Scale bars, 200 nm.
Previous studies have shown that inhibitory interneurons may play a prominent role in the pathogenesis of prion diseases (Belichenko et al., 1999; Bouzamondo et al., 2000). Thus, we attempted to determine whether these cells are indeed of the interneuronal GABAergic type using several different markers for particular cell populations. None of the CPrP cells were immunopositive for calbindin, calreticulin, parvalbumin, vasointestinal peptide, neuropeptide Y, somatostatin, and b-NOS (data not shown). Therefore, the specific type of neurons to which CPrP cells belong remains to be determined.
The cytosolic localization of PrP could reflect a particular state of the neuron, which may cause or result from a dysfunction of the machinery that generates GPI-anchored proteins. To check for the latter possibility, we labeled these neurons for both PrPC and another abundant GPI-anchored protein, Thy-1. In pyramidal neurons as well as CPrP cells, the labeling against Thy-1 was concentrated on late endosomes and the plasma membrane (Fig. 7C,D) without any indication of cytosolic distribution. This argues that the GPI-adding machinery functions normally in these neurons, and another factor must account for the presence of both cytosolic and membrane-bound PrP.
CPrP cells are neither necrotic nor apoptotic
Whether PrP accumulation in the cytosol reflects stress or damage to the cells is unknown. However, morphological examination of these neurons did not reveal organelle swelling (mitochondria, ER, Golgi apparatus), disaggregation of polyribosomes, or cell and nuclear membrane breaks that are clearly indicative of neuronal necrosis. Furthermore, we did not observe any apoptotic signs such as chromatin clumping, condensation of cytoplasmic content, or accumulation of autophagic lysosomes. Mitochondria displayed organized structure with well preserved cristae, inner and outer membranes. Some of these neurons had a denser cytosol than pyramidal neurons, but it was not a feature distinguishing these neurons from others. We saw a number of “dense cells” without signs of PrP accumulation in the cytosol (Fig. 6C). Moreover, many neurons with cytPrP appeared to have a normal cytosolic density, similar to other neurons. Although morphological analysis remains the “gold standard” for assessment and quantification of apoptosis (Hall, 1999), we nevertheless checked CPrP cells on semithin sections using an apoptosis detection kit (on the basis of TUNEL methodology). None of the CPrP cells appeared to be apoptotic (Fig. 8A-C).
Figure 8.

Correlated light (A-C) and electron (D,E) microscopic pictures of the same CPrP cell. A-C, Serial semithin hippocampal sections labeled against PrPC (red) and TUNEL staining (green). A, Section treated with 1 U per 1 ml of DNaseI as the positive control. Nuclei from all cells are highly fluorescent, indicating broken DNA strands. B, C, Serial sections not treated with DNaseI. CPrP and pyramidal neurons show no signal for broken DNA strands, indicating that these cells are not apoptotic. The asterisk in C indicates a cell imaged by EM in D. D, Low-magnification EM image of the cell marked with an asterisk in C. The blue line delineates the border of the CPrP cell. Note the absence of chromatine clumping. E, Higher magnification of the rectanglular area in D with cytosolic immunogold (15 nm) labeling against PrP. The majority of the immunogold is localized in the cytosol (arrowheads). er, Endoplasmic reticulum; m, mitochondria; n, nucleus; pm, plasma membrane. Scale bars: (in C) A--C, 50 μm; D, 2 μm; E, 200 nm.
Quantification of PrPC in the hippocampus
The use of gold particles and the good ultrastructural preservation produced by ultracryomicrotomy gave us the opportunity to quantify the distribution of PrPC in the hippocampus. All segments of dendritic membranes (dendritic shaft, spines, transport vesicles, and endocytic structures) showed approximately the same density of gold particles per unit of membrane (Table 1). The same structures in the molecular layer of the dentate gyrus had less labeling than those in the strata oriens and radiatum, matching precisely the labeling pattern observed by LM via immunofluorescence and silver enhancement. Axonal membranes had a lower gold particle density than dendritic membranes. Myelin sheaths were labeled with fewer gold particles per length of membrane than axolemma (Table 1). Interestingly, we found similar PrPC concentrations on the membrane of presynaptic and postsynaptic profiles and on those within the synapse. No preferential labeling was observed within the synaptic specialization. Synaptic vesicles were labeled just above the background level determined for mitochondria, suggesting an exclusion of PrPC from this structure.
Proteins linked to the plasma membrane by either a GPI anchor or a transmembrane domain travel along the secretory pathway. This begins with the synthesis of the protein, then translocation into the ER, and finally movement through the Golgi complex toward the final destination, the plasma membrane. At various stages along the pathway, the proteins may become concentrated. We therefore analyzed the distribution of PrPC on various organelles and membranes that are involved with the trafficking pathway from four hippocampal cell populations: pyramidal neurons (CA1), granule neurons (dentate gyrus), hilar neurons, and CPrP cells. We quantified the distribution in the same manner as for the neuropil, counting the number of gold particles per micrometer of organelle membrane (Table 2). The data are grouped according to cell type (pyramidal neurons, granule neurons, hilar neurons, and CPrP cells) and organelles (ER, Golgi, endosomes, transport vesicles, and plasma membrane). The overall distribution of gold particles was similar in all types of cells analyzed. The lowest concentration of PrPC was detected in the ER and Golgi complex. An increase in PrPC concentration occurs on the plasma membrane and in late endosomes-multivesicular bodies. Our findings correlate nicely with previously published results about the folate receptor, a GPI-anchored protein in cultured cells (Mayor et al., 1998; Chatterjee et al., 2001). Labeling for cytPrP was negligible in all neurons except CPrP cells, in which the number of gold particles exceeded >200 times the usual background level from the three other types of neurons (Table 3).
Table 2.
Quantification of PrPc labeling on the membrane in different neuronal populations in the CA1 area and dentate gyrusa
|
Neurons |
Endoplasmic reticulum |
Golgi complex |
Endosomes/lysosomes |
Plasma membrane |
Mitochondria |
|---|---|---|---|---|---|
| Pyramidal (n = 18) | 0.44 ± 0.03 | 0.71 ± 0.03 | 1.4 ± 0.2 | 1.37 ± 0.11 | 0.037 ± 0.002 |
| Hilar (n = 18) | 0.23 ± 0.01 | 0.4 ± 0.01 | 0.59 ± 0.06 | 0.58 ± 0.04 | 0.03 ± 0.01 |
| Granule (n = 21) | 0.25 ± 0.01 | 0.36 ± 0.03 | 0.58 ± 0.07 | 0.64 ± 0.03 | 0.03 ± 0.01 |
| CPrP cells (n = 20)
|
0.38 ± 0.02
|
0.64 ± 0.05
|
1.51 ± 0.12
|
1.53 ± 0.08
|
0.06 ± 0.008
|
Values are number of gold particles per 1 μm of membrane (gold per micrometer) in 77 cells from three different animals, given as mean ± SEM. Mitochondrial membranes were used to assess background labeling.
Table 3.
Quantification of gold-labeled PrP in the cytosol in different populations of neuronsa
|
Neurons |
Nucleus |
Mitochondria |
Cytosol |
|---|---|---|---|
| Pyramidal (n = 18) | 0.12 ± 0.04 | 0.87 ± 0.6 | 0.09 ± 0.04 |
| Hilar (n = 18) | 0.14 ± 0.08 | 1.09 ± 0.73 | 0.09 ± 0.04 |
| Granule (n = 21) | 0.11 ± 0.04 | 0.54 ± 0.26 | 0 |
| CPrP cells (n = 20)
|
0.845 ± 0.16
|
2.6 ± 0.53
|
23.4 ± 3.6
|
Gold particles were quantified over an area of the cytosol that was free of organelles. To assess background labeling, gold particles were quantified over the area of mitochondria and nucleus (gold per square micrometer). Results are presented as mean ± SEM.
The statistical significance of labeling for cytPrP was assessed by the RLI (Mayhew et al., 2002) on the basis of a comparison between the expected and observed distributions of the gold particles. The expected gold particle distribution is derived from a lattice of test points used for morphometric counting. The null hypothesis corresponds to a random distribution (RLI = 1) of the marker between compartments such as the nucleus, mitochondria, and cytosol. The RLI for PrP in the cytosol of CPrP cells attained a value of 3.12 (Table 4), which indicates preferential nonrandom labeling. The total χ2 value (the sum of partial values) is 1864.2, indicating that the null hypothesis must be rejected (p < 0.001). The partial χ2 values show that the most important contributor to the total χ2 value is cytPrP.
Discussion
We determined the ultrastructural distribution of PrPC in murine hippocampus using a number of recombinant Fabs generated against different parts of PrPC, the N terminus, central region, and C terminus. In addition, we also present the first quantitative data on the localization of PrPC in the CA1 region and dentate gyrus of the hippocampus of several lines of wt in-bred mice.
We found the following: (1) PrPC generally follows the standard biosynthetic trafficking pathway in brain neurons with prominent presence in endosomes and the plasma membrane; (2) PrPC has a ubiquitous distribution on the neuronal plasma membrane and cellular processes without preferential accumulation at synaptic specializations; (3) PrPC is found with the same frequency on presynaptic as well as postsynaptic membranes and within the synapse; (4) PrPC is almost excluded from the membrane of synaptic vesicles; and (5) PrP is expressed in the cytosol in a small population of neurons in the hippocampus, thalamus, and somatosensory neocortex but not in the cerebellum.
Localization and quantification of PrPC in the hippocampus
At the light microscopic level, we saw preferential PrPC labeling of the strata oriens, radiatum, and moleculare of the dentate gyrus, which closely resembles previous results (Sales et al., 1998; Moya et al., 2000). Because of an abundance of synapses in these areas, it was concluded that the pattern of staining is “very close to synaptic in nature” and similar to the rab3 labeling (Moya et al., 2000). The punctate appearance of labeling could reflect the local concentrations of GPI-anchored proteins on the plasma membrane of neurites, as shown previously (Madore et al., 1999). Furthermore, double labeling for PrPC and either synaptophysin (Fournier et al., 2000) or rab3 (Moya et al., 2000) was not resolved well enough to claim unambiguously the colocalization of these proteins on the same subcellular structure. We demonstrated clearly that synaptobrevin and PrPC are localized on separate synaptic compartments. Therefore, we argue against a significant biological role of PrPC in synaptic vesicles. However, it does not preclude an indirect role.
The very low level of labeling of synaptic vesicles in our study does not deny the potential involvement of the synapse in the pathogenesis of prion diseases, as proposed by Fournier et al. (2000). The exclusion of PrPC from synaptic vesicles in our studies correlates with the morphological and biochemical data by Laine et al. (2001) and Herms et al. (1999). The presence of PrPC in all parts of the plasma membrane, whether somatic, axonal, synaptic, or dendritic membranes (with the highest concentration on the last), argues for a more general physiological function than merely a synaptic one. Various potential functions have been proposed by several authors, including oxidative stress protection (Brown et al., 2001), copper metabolism (Pauly and Harris, 1998), signal transduction (Mouillet-Richard et al., 2000), and mediator of intercellular contacts (Rieger et al., 1999; Schmitt-Ulms et al., 2001).
The relative concentration of PrPC in organelles along the trafficking pathway observed in our study agrees well with the general theory that proteins are concentrated along this pathway en route to the plasma membrane. Here, we present quantitative morphological results on the localization and concentration of PrPC in subcellular organelles of the trafficking pathway in neurons in situ. These results are in agreement with the intracellular retention of GPI-anchored proteins in endosomal compartments (Mayor et al., 1998) and with data concerning the recycling of PrPC from plasma membranes via endosomes (Shyng et al., 1993). Reports on the concentration of PrPSc in endosomal and lysosomal structures (Caughey et al., 1991; McKinley et al., 1991; Borchelt et al., 1992; Taraboulos et al., 1992b; Arnold et al., 1995) underscore the importance of these sites for PrPC degradation and possibly PrPC-to-PrPSc conversion.
The absence of PrP labeling in clathrin-coated structures contradicts the suggestions by Laine et al. (2001). However, their data could be attributed to either the possible diffusion of the peroxidase reaction product to nearby locations, giving false-positive results, or too low concentrations of PrPC in clathrin-coated structures, which were undetectable by our immunogold procedure. Because it is known that neurons do not have calveoli, we can speculate that PrPC recycles through endosomes via a nonclathrin, noncalveoli pathway (Peters et al., 2003).
The almost ubiquitous distribution of PrPC on the neuronal plasma membrane and cellular processes without a preferential accumulation at synaptic specializations suggests the absence of active retention mechanisms, allowing unhampered diffusion of PrPC along cellular membranes. This diffusion could play a major role in PrPSc propagation, because it was shown that a defective fast axonal transport did not interfere with prion neuroinvasion (Kunzi et al., 2002). It is entirely plausible that PrPSc, which retains the GPI anchor, could physically contact PrPC on adjacent cells or even be physically translocated to the membranes from neighboring cells (Liu et al., 2002) at sites of very close apposition to cellular membranes, including at the synapse. Additionally, the finding of significant PrPC labeling in myelin sheaths points to the possible involvement of oligodendrocytes in the propagation of prion diseases.
Neurons containing cytPrP
Our studies revealed the existence of neurons containing cytPrP. These cells showed a very different morphology from glial cells and are negative for GFAP (an astroglial marker) and CNPase and S100 (oligodendrocytic markers). We observed synapses on cell bodies and processes as well as axonal terminals with a high PrPC content, which probably belong to CPrP cells. On the basis of our LM and EM observations, we assumed CPrP cells to be of neuronal nature. However, we are still focusing our efforts on identifying the neuronal subtype. An antibody against GABA (the most commonly used marker for interneurons) did not give any positive labeling in cryosections. We believe that this can be explained by the fact that a very small molecule, such as GABA, is not adequately retained in cryosections, despite the use of glutaraldehyde. The additional cross-linking through an embedding medium, such as resin, may be necessary to preserve GABA localization in ultrathin sections (Bouzamondo et al., 2000), but it inevitably destroys the antigen sites for PrPC antibodies (our unpublished observations).
PrP accumulation in the cytosol might depend on the circadian cycle of the cells or reflect damage and stress. However, morphological examination of these neurons did not show any organelle swelling, disaggregating of polyribosomes, or breaks in the cell and nuclear membranes that are indicative of neuronal necrosis. Furthermore, we did not find any morphological or immunocytochemical apoptotic indicators. The absence of TUNEL labeling revealed that CPrP cells do not possess breaks in nuclear DNA that are characteristic of apoptotic cells. Therefore, we conclude that these cells did not suffer any observable damage that could be responsible for the unusual localization of PrP.
Although it seems unusual for a cell to have a protein in two such distinct locations (the membranes of the trafficking pathway and the cytosol), it is entirely plausible that a protein such as PrPC could have roles in more than one compartment of a cell (Hegde and Lingappa, 1999). Such a diversity of function from a single gene has been observed for the protein calreticulin, which was found to function in the ER, cytosol, and nucleus (Smith and Koch, 1989; Burns et al., 1994; Coppolino et al., 1997). Similarly, dual localization of other proteins or protein domains has also been described for the plasminogen activator protein (Belin et al., 1996) and the hepatitis B virus envelope protein (Swameye and Schaller, 1997). It has been shown that PrPC has a rather complex signal sequence, which directs it to the lumen of the ER (Zhang and Ling, 1995; Hegde et al., 1998; Holscher et al., 2001; Kim et al., 2001). Therefore, it is possible that the synthesis of the different topological forms of PrPC varies between different cell types, according to the expression of different cytoplasmic components of the translocation machinery (Hegde and Lingappa, 1999).
The dislocation of PrP from the ER to the cytosol has been demonstrated previously in cell culture systems under certain conditions, such as in a reducing environment and glycosylation (Ma and Lindquist, 2001) or proteosomal (Yedidia et al., 2001) inhibition. This could explain the presence of cytPrP in situ. However, the absence of GPI-anchored Thy-1 in the cytosol of CPrP cells argues that other GPI-anchored proteins preserve their usual localization. Because proteosome inhibition should affect many proteins, an unknown specific mechanism for PrP accumulation in the cytosol may be present.
It was recently shown that transfected cytPrP appears to be toxic in both cell culture and transgenic animals, in a cell type-dependent manner. Only cerebellar cells appeared to be affected in mice that expressed PrPC without an ER translocation signal (Ma et al., 2002). Accumulation of cytPrP in “susceptible” neurons might be responsible for some of the variants of prion diseases in which cytPrP aggregates kill the cell and cause release of infectious prions. These prions might then initiate the vicious circle of prion propagation. However, our studies show that cytPrP is present in normal rodent brains in a population of neurons that appears healthy and shows no cellular degeneration. Thus, we can infer that cytPrP is not toxic in some neurons but highly toxic when overexpressed in specific cell populations. More work is needed to elucidate the causes of such striking differences resulting from cytPrP.
Footnotes
We thank Hang Nguyen for outstanding editorial work, Erik Bos for excellent technical assistance, and E. van Donselaar for extensive preliminary testing of many anti-PrPC antibodies. We are grateful to M. van der Valk for generous advice, K. de Goeij, J. Bulthuis, and M. Tjin-A-Koeng for technical assistance in pilot experiments, and N. Ong for help with the photographs.
Correspondence should be addressed to Dr. Peter J. Peters, The Netherlands Cancer Institute, Antoni van Leeuwenhoek Hospital, Plesmanlaan 121-H4, 1066 CX Amsterdam, The Netherlands. E-mail: p.peters@nki.nl.
Copyright © 2003 Society for Neuroscience 0270-6474/03/237183-11$15.00/0
References
- Amit AG, Mariuzza RA, Phillips SE, Poljak RJ ( 1986) Three-dimensional structure of an antigen-antibody complex at 2.8 Å resolution. Science 233: 747-753. [DOI] [PubMed] [Google Scholar]
- Arnold JE, Tipler C, Laszlo L, Hope J, Landon M, Mayer RJ ( 1995) The abnormal isoform of the prion protein accumulates in late-endosome-like organelles in scrapie-infected mouse brain. J Pathol 176: 403-411. [DOI] [PubMed] [Google Scholar]
- Belichenko PV, Miklossy J, Belser B, Budka H, Celio MR ( 1999) Early destruction of the extracellular matrix around parvalbumin-immunoreactive interneurons in Creutzfeldt-Jakob disease. Neurobiol Dis 6: 269-279. [DOI] [PubMed] [Google Scholar]
- Belin D, Bost S, Vassalli JD, Strub K ( 1996) A two-step recognition of signal sequences determines the translocation efficiency of proteins. EMBO J 15: 468-478. [PMC free article] [PubMed] [Google Scholar]
- Bendheim PE, Brown HR, Rudelli RD, Scala LJ, Goller NL, Wen GY, Kascsak RJ, Cashman NR, Bolton DC ( 1992) Nearly ubiquitous tissue distribution of the scrapie agent precursor protein. Neurology 42: 149-156. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Taraboulos A, Prusiner SB ( 1992) Evidence for synthesis of scrapie prion proteins in the endocytic pathway. J Biol Chem 267: 16188-16199. [PubMed] [Google Scholar]
- Bouzamondo E, DeArmond SJ, Ralston HJ, Prusiner SB, Milroy AM ( 2000) Selective neuronal vulnerability during experimental scrapie infection: insights from an ultrastructural investigation. Brain Res 874: 210-215. [DOI] [PubMed] [Google Scholar]
- Brown DR, Clive C, Haswell SJ ( 2001) Antioxidant activity related to copper binding of native prion protein. J Neurochem 76: 69-76. [DOI] [PubMed] [Google Scholar]
- Büeler H, Aguzzi A, Sailer A, Greiner R-A, Autenried P, Aguet M, Weissmann C ( 1993) Mice devoid of PrP are resistant to scrapie. Cell 73: 1339-1347. [DOI] [PubMed] [Google Scholar]
- Burns K, Duggan B, Atkinson EA, Famulsk KS, Nemer M, Bleackley RC, Michalak M ( 1994) Modulation of gene expression by calreticulin binding to the glucocorticoid receptor. Nature 367: 476-480. [DOI] [PubMed] [Google Scholar]
- Caughey B, Raymond GJ, Ernst D, Race RE ( 1991) N-terminal truncation of the scrapie-associated form of PrP by lysosomal protease(s): implications regarding the site of conversion of PrP to the protease-resistant state. J Virol 65: 6597-6603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S, Smith ER, Hanada K, Stevens VL, Mayor S ( 2001) GPI anchoring leads to sphingolipid-dependent retention of endocytosed proteins in the recycling endosomal compartment. EMBO J 20: 1583-1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppolino MG, Woodside MJ, Demaurex N, Grinstein S, St. Arnaud R, Dedhar S ( 1997) Calreticulin is essential for integrin-mediated calcium signalling and cell adhesion. Nature 386: 843-847. [DOI] [PubMed] [Google Scholar]
- DeArmond SJ, Mobley WC, DeMott DL, Barry RA, Beckstead JH, Prusiner SB ( 1987) Changes in the localization of brain prion proteins during scrapie infection. Neurology 37: 1271-1280. [DOI] [PubMed] [Google Scholar]
- Ford MJ, Burton LJ, Li H, Graham CH, Frobert Y, Grassi J, Hall SM, Morris RJ ( 2002) A marked disparity between the expression of prion protein and its message by neurones of the CNS. Neuroscience 111: 533-551. [DOI] [PubMed] [Google Scholar]
- Fournier J-G, Escaig-Haye F, De Villemeur TB, Robain O ( 1995) Ultrastructural localization of cellular prion protein (PrPC) in synaptic boutons of normal hamster hippocampus. C R Acad Sci III 318: 339-344. [PubMed] [Google Scholar]
- Fournier JG, Escaig-Haye F, Grigoriev V ( 2000) Ultrastructural localization of prion proteins: physiological and pathological implications. Microsc Res Tech 50: 76-88. [DOI] [PubMed] [Google Scholar]
- Griffiths G ( 1993) Fine structure immunocytochemistry. New York: Springer.
- Haeberle AM, Ribaut-Barassin C, Bombarde G, Mariani J, Hunsmann G, Grassi J, Bailly Y ( 2000) Synaptic prion protein immuno-reactivity in the rodent cerebellum. Microsc Res Tech 50: 66-75. [DOI] [PubMed] [Google Scholar]
- Hall PA ( 1999) Assessing apoptosis: a critical survey. Endocr Relat Cancer 6: 3-8. [DOI] [PubMed] [Google Scholar]
- Hegde RS, Lingappa VR ( 1999) Regulation of protein biogenesis at the endoplasmic reticulum membrane. Trends Cell Biol 9: 132-137. [DOI] [PubMed] [Google Scholar]
- Hegde RS, Mastrianni JA, Scott MR, DeFea KA, Tremblay P, Torchia M, DeArmond SJ, Prusiner SB, Lingappa VR ( 1998) A transmembrane form of the prion protein in neurodegenerative disease. Science 279: 827-834. [DOI] [PubMed] [Google Scholar]
- Herms JW, Tings T, Gall S, Madlung A, Giese A, Siebert H, Schurmann P, Windl O, Brose N, Kretzschmar H ( 1999) Evidence of presynaptic location and function of the prion protein. J Neurosci 19: 8866-8875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holscher C, Bach UC, Dobberstein B ( 2001) Prion protein contains a second endoplasmic reticulum targeting signal sequence located at its C terminus. J Biol Chem 276: 13388-13394. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Rahbar R, Hegde RS ( 2001) Combinatorial control of prion protein biogenesis by the signal sequence and transmembrane domain. J Biol Chem 276: 26132-26140. [DOI] [PubMed] [Google Scholar]
- Kunzi V, Glatzel M, Nakano MY, Greber UF, Van Leuven F, Aguzzi A ( 2002) Unhampered prion neuroinvasion despite impaired fast axonal transport in transgenic mice overexpressing four-repeat tau. J Neurosci 22: 7471-7477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretzschmar HA, Prusiner SB, Stowring LE, DeArmond SJ ( 1986) Scrapie prion proteins are synthesized in neurons. Am J Pathol 122: 1-5. [PMC free article] [PubMed] [Google Scholar]
- Laine J, Marc ME, Sy MS, Axelrad H ( 2001) Cellular and subcellular morphological localization of normal prion protein in rodent cerebellum. Eur J Neurosci 14: 47-56. [DOI] [PubMed] [Google Scholar]
- Leclerc E, Peretz D, Ball H, Sakurai H, Legname G, Serban A, Prusiner SB, Burton DR, Williamson RA ( 2001) Immobilized prion protein undergoes spontaneous rearrangement to a conformation having features in common with the infectious form. EMBO J 20: 1547-1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Li R, Pan T, Liu D, Petersen RB, Wong BS, Gambetti P, Sy MS ( 2002) Intercellular transfer of the cellular prion protein. J Biol Chem 277: 47671-47678. [DOI] [PubMed] [Google Scholar]
- Ma J, Lindquist S ( 2001) Wild-type PrP and a mutant associated with prion disease are subject to retrograde transport and proteasome degradation. Proc Natl Acad Sci USA 98: 14955-14960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Lindquist S ( 2002) Conversion of PrP to a self-perpetuating PrPSc-like conformation in the cytosol. Science 298: 1785-1788. [DOI] [PubMed] [Google Scholar]
- Ma J, Wollmann R, Lindquist S ( 2002) Neurotoxicity and neurodegeneration when PrP accumulates in the cytosol. Science 298: 1781-1785. [DOI] [PubMed] [Google Scholar]
- Madore N, Smith KL, Graham CH, Jen A, Brady K, Hall S, Morris R ( 1999) Functionally different GPI proteins are organized in different domains on the neuronal surface. EMBO J 18: 6917-6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maunsbach AB, Afzelius BA ( 1999) Biomedical electron microscopy. Illustrated methods and interpretations, pp 196-206. San Diego: Academic.
- Mayhew TM, Lucocq JM, Griffiths ( 2002) Relative labelling index: a novel stereological approach to test for non-random immunogold labelling of organelles and membranes on transmission electron microscopy thin sections. J Microsc 205: 153-164. [DOI] [PubMed] [Google Scholar]
- Mayor S, Sabharanjak S, Maxfield FR ( 1998) Cholesterol-dependent retention of GPI-anchored proteins in endosomes. EMBO J 17: 4626-4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinley MP, Taraboulos A, Kenaga L, Serban D, Stieber A, DeArmond SJ, Prusiner SB, Gonatas N ( 1991) Ultrastructural localization of scrapie prion proteins in cytoplasmic vesicles of infected cultured cells. Lab Invest 65: 622-630. [PubMed] [Google Scholar]
- McLean IW, Nakane PK ( 1974) Periodate-lysine-paraformaldehyde fixative. A new fixation for immunoelectron microscopy. J Histochem Cytochem 22: 1077-1083. [DOI] [PubMed] [Google Scholar]
- Moser M, Colello RJ, Pott U, Oesch B ( 1995) Developmental expression of the prion protein gene in glial cells. Neuron 14: 509-517. [DOI] [PubMed] [Google Scholar]
- Mouillet-Richard S, Ermonval M, Chebassier C, Laplanche JL, Lehmann S, Launay JM, Kellermann O ( 2000) Signal transduction through prion protein. Science 289: 1925-1928. [DOI] [PubMed] [Google Scholar]
- Moya KL, Sales N, Hassig R, Creminon C, Grassi J, Di Giamberardino L ( 2000) Immunolocalization of the cellular prion protein in normal brain. Microsc Res Tech 50: 58-65. [DOI] [PubMed] [Google Scholar]
- Pauly PC, Harris DA ( 1998) Copper stimulates endocytosis of the prion protein. J Biol Chem 273: 33107-33110. [DOI] [PubMed] [Google Scholar]
- Peretz D, Williamson RA, Matsunaga Y, Serban H, Pinilla C, Bastidas RB, Rozenshteyn R, James TL, Houghten RA, Cohen FE, Prusiner SB, Burton DR ( 1997) A conformational transition at the N terminus of the prion protein features in formation of the scrapie isoform. J Mol Biol 273: 614-622. [DOI] [PubMed] [Google Scholar]
- Peretz D, Williamson RA, Kaneko K, Vergara J, Leclerc E, Schmitt-Ulms G, Mehlhorn IR, Legname G, Wormald MR, Rudd PM, Dwek RA, Burton DR, Prusiner SB ( 2001) Antibodies inhibit prion propagation and clear cell cultures of prion infectivity. Nature 412: 739-743. [DOI] [PubMed] [Google Scholar]
- Peters A, Palay SL, Webster HF ( 1991) The fine structure of the nervous system. New York: Oxford UP.
- Peters PJ ( 2001) Cryo-immunogold electron microscopy. In: Current protocols in cell biology (Bonifacino JS, ed), pp 4.7.1-4.7.12. New York: Wiley. [DOI] [PubMed]
- Peters PJ, Mironov A, Vey M, Peretz D, van Donselaar E, Leclerc E, Erpel S, DeArmond SJ, Burton DR, Williamson RA, Prusine SB ( 2003) Trafficking of prion proteins through a caveolae-mediated endosomal pathway. J Cell Biol, in press. [DOI] [PMC free article] [PubMed]
- Piccardo P, Safar J, Ceroni M, Gajdusek DC, Gibbs Jr CJ ( 1990) Immunohistochemical localization of prion protein in spongiform encephalopathies and normal brain tissue. Neurology 40: 518-522. [DOI] [PubMed] [Google Scholar]
- Prusiner SB ( 1996) Molecular biology and pathogenesis of prion diseases. Trends Biochem Sci 252: 482-487. [DOI] [PubMed] [Google Scholar]
- Raposo G, Tenza D, Mecheri S, Peronet R, Bonnerot C, Desaymard C ( 1997) Accumulation of major histocompatibility complex class II molecules in mast cell secretory granules and their release upon degranulation. Mol Biol Cell 8: 2631-2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reits E, Griekspoor A, Neijssen J, Groothuis T, Jalink K, van Veelen P, Janssen H, Calafat J, Drijfhout JW, Neefjes J ( 2003) Peptide diffusion, protection, and degradation in nuclear and cytoplasmic compartments before antigen presentation by MHC class I. Immunity 18: 97-108. [DOI] [PubMed] [Google Scholar]
- Rieger R, Lasmezas CI, Weiss S ( 1999) Role of the 37 kDa laminin receptor precursor in the life cycle of prions. Transfus Clin Biol 6: 7-16. [DOI] [PubMed] [Google Scholar]
- Safar J, Ceroni M, Piccardo P, Liberski PP, Miyazaki M, Gajdusek DC, Gibbs Jr CJ ( 1990) Subcellular distribution and physicochemical properties of scrapie associated precursor protein and relationship with scrapie agent. Neurology 40: 503-508. [DOI] [PubMed] [Google Scholar]
- Sales N, Rodolfo K, Hassig R, Faucheux B, Di Giamberardino L, Moya KL ( 1998) Cellular prion protein localization in rodent and primate brain. Eur J Neurosci 10: 2464-2471. [DOI] [PubMed] [Google Scholar]
- Schmitt-Ulms G, Legname G, Baldwin MA, Ball HL, Bradon N, Bosque PJ, Crossin KL, Edelman GM, DeArmond SJ, Cohen FE, Prusiner SB ( 2001) Binding of neural cell adhesion molecules (N-CAMs) to the cellular prion protein. J Mol Biol 314: 1209-1225. [DOI] [PubMed] [Google Scholar]
- Shyng SL, Huber MT, Harris DA ( 1993) A prion protein cycles between the cell surface and an endocytic compartment in cultured neuroblastoma cells. J Biol Chem 21: 15922-15928. [PubMed] [Google Scholar]
- Smith MJ, Koch GL ( 1989) Multiple zones in the sequence of calreticulin (CRP55, calregulin, HACBP), a major calcium binding ER/SR protein. EMBO J 8: 3581-3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl N, Borchelt DR, Hsiao K, Prusiner SB ( 1987) Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 51: 229-240. [DOI] [PubMed] [Google Scholar]
- Swameye I, Schaller H ( 1997) Dual topology of the large envelope protein of duck hepatitis B virus: determinants preventing pre-S translocation and glycosylation. J Virol 71: 9434-9441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taraboulos A, Jendroska K, Serban D, Yang SL, DeArmond SJ, Prusiner SB ( 1992a) Regional mapping of prion proteins in brains. Proc Natl Acad Sci USA 89: 7620-7624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taraboulos A, Raeber AJ, Borchelt DR, Serban D, Prusiner SB ( 1992b) Synthesis and trafficking of prion proteins in cultured cells. Mol Biol Cell 3: 851-863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telling GC, Haga T, Torchia M, Tremblay P, DeArmond SJ, Prusiner SB ( 1996) Interactions between wild-type and mutant prion proteins modulate neurodegeneration in transgenic mice. Genes Dev 10: 1736-1750. [DOI] [PubMed] [Google Scholar]
- Verghese-Nikolakaki S, Michaloudi H, Polymenidou M, Groschup MH, Papadopoulos GC, Sklaviadis T ( 1999) Expression of the prion protein in the rat forebrain-an immunohistochemical study. Neurosci Lett 272: 9-12. [DOI] [PubMed] [Google Scholar]
- Weibel ER ( 1979) Morphometry of the human lung: the state of the art after two decades. Bull Physiopathol Respir (Nancy) 15: 999-1013. [PubMed] [Google Scholar]
- Williamson RA, Peretz D, Smorodinsky N, Bastidas R, Serban H, Mehlhorn I, DeArmond SJ, Prusiner SB, Burton DR ( 1996) Circumventing tolerance to generate autologous monoclonal antibodies to the prion protein. Proc Natl Acad Sci USA 93: 7279-7282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson RA, Peretz D, Pinilla C, Ball H, Bastidas RB, Rozenshteyn R, Houghten RA, Prusiner SB, Burton DR ( 1998) Mapping the prion protein using recombinant antibodies. J Virol 72: 9413-9418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yedidia Y, Horonchik L, Tzaban S, Yanai A, Taraboulos A ( 2001) Proteasomes and ubiquitin are involved in the turnover of the wild-type prion protein. EMBO J 20: 5383-5391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JT, Ling V ( 1995) Involvement of cytoplasmic factors regulating the membrane orientation of P-glycoprotein sequences. Biochemistry 34: 9159-9165. [DOI] [PubMed] [Google Scholar]