

Abstract
The node of Ranvier is a distinct domain of myelinated axons that is highly enriched in sodium channels and is critical for impulse propagation. During development, the channel subtypes expressed at the node undergo a transition from Nav1.2 to Nav1.6. Specialized junctions that form between the paranodal glial membranes and axon flank the nodes and are candidates to regulate their maturation and delineate their boundaries. To investigate these roles, we characterized node development in mice deficient in contactin-associated protein (Caspr), an integral junctional component. Paranodes in these mice lack transverse bands, a hallmark of the mature junction, and exhibit progressive disruption of axon-paranodal loop interactions in the CNS. Caspr mutant mice display significant abnormalities at central nodes; components of the nodes progressively disperse along axons, and many nodes fail to mature properly, persistently expressing Nav1.2 rather than Nav1.6. In contrast, PNS nodes are only modestly longer and, although maturation is delayed, eventually all express Nav1.6. Potassium channels are aberrantly clustered in the paranodes; these clusters are lost over time in the CNS, whereas they persist in the PNS. These findings indicate that interactions of the paranodal loops with the axon promote the transition in sodium channel subtypes at CNS nodes and provide a lateral diffusion barrier that, even in the absence of transverse bands, maintains a high concentration of components at the node and the integrity of voltage-gated channel domains.
Keywords: Caspr, myelin, sodium channels, nodes, paranodal junction, potassium channels
Introduction
The node of Ranvier is highly enriched in voltage-gated Na+ channels, reflecting its key role in the propagation of action potentials by saltatory conduction (Hille, 2001). Nodes form initially as broad complexes closely associated with processes of myelinating glia; with maturation, they become more compact and delineated (Rasband and Shrager, 2000). The Na+ channel subtype expressed at the node also undergoes a transition as nodes mature from Nav1.2, which is expressed early in development, to Nav1.6, which predominates in the adult (Caldwell et al., 2000; Boiko et al., 2001). The physiological significance of this transition and its regulation are not yet known.
Interactions with myelinating glia are essential for node formation and maintenance, although the precise mechanisms involved have remained elusive (Peles and Salzer, 2000). The paranodal junctions, which form between the spiraled, lateral edges of myelinating glia (the paranodal loops) and the axon, are potential candidates to mediate these events. They flank the node on either side and physically separate it from the juxtaparanodal axolemma, which is enriched in Kv1.1 and Kv1.2 channels. In electron micrographs, the paranodal loops closely appose and physically invaginate the axolemma. Periodic intercellular densities, the transverse bands, are present between each paranodal loop and the axon leading to the designation of the paranodal junctions as septate-like. The transverse bands develop after paranodal loops attach to the axon (Tao-Cheng and Rosenbluth, 1983; Marcus et al., 2002) and are, therefore, a hallmark of the mature junction.
The adhesion molecules contactin and Caspr (contactinassociated protein) form a cis complex on axons that is concentrated at the junctions (Einheber et al., 1997; Menegoz et al., 1997; Peles et al., 1997; Rios et al., 2000). Caspr and contactin are integral junctional components because mice deficient in these proteins exhibit severe disruptions of normal paranodal interactions (Bhat et al., 2001; Boyle et al., 2001). Defects include loss of the transverse bands and defective attachment of the glial loops to the axon, particularly in the CNS, in which glial loops are frequently everted. Similar defects are also present in mice deficient in ceramide galactosyltransferase (CGT) and galactosylceramide sulfotransferase (CST) (Dupree et al., 1998; Honke et al., 2002), enzymes required for the synthesis of the myelin glycolipids galactocerebroside and sulfatide. The precise role of these glycolipids in junction assembly has not been elucidated.
Interestingly, Na+ channels still cluster in mice with defective paranodal interactions, indicating that mature junctions (e.g., those containing transverse bands) are not required for initial clustering of sodium channels (Dupree et al., 1999; Bhat et al., 2001; Boyle et al., 2001). However, because K+ channels in these mutants are mislocalized to the paranodes, immediately adjacent to Na+ channels at the node, the transverse bands are essential for localization of K+ channels to the juxtaparanodes.
Paranodal junctions may regulate other aspects of node development, including their maturation and maintenance, and may delineate their physical boundaries. Because Caspr mutant mice have normal compact myelin but aberrant paranodes (Bhat et al., 2001), they provide an ideal system to investigate these other roles of the paranodes in node development. We now report that the distribution and density of nodal components and the transition from Nav1.2 to Nav1.6 are aberrant in the CNS of Caspr-deficient mice. These results indicate that paranodal interactions regulate the maturation of the node, including the expression of specific Na+ channel subtypes, and provide a lateral diffusion barrier that prevents dispersion of nodal components.
Materials and Methods
Antibodies. Antibodies used included a guinea pig polyclonal antibody to Caspr (1:1000) (Bhat et al., 2001); rabbit polyclonal antibodies to Nav1.6 (1:100) (Caldwell et al., 2000), to Nav1.2 (1:100; Upstate Biotechnology, Lake Placid, NY), to Kv1.1 and Kv1.2 (1:100; Alomone Labs, Ltd., Jerusalem, Israel), to βIV spectrin (1:500) (Berghs et al., 2000), and to NrCAM (1:300; a gift from M. Grumet, Rutgers University, Piscataway, NJ); and chicken antibodies to ankyrin G (1:100; a gift from S. Lambert, University of Massachusetts Medical Center, Worcester, MA) and to βIV spectrin (1:300) (Komada and Soriano, 2002). Mouse monoclonal antibodies to MBP (1:500; Sternberger Monoclonals Lutherville, MD), to neurofilament (1:2000; SM31 and SM32; Sternberger Monoclonals), to Na+ channel type II (1:100; Upstate Biotechnology), and to an epitope common to all α subunit subtypes of the voltage-gated Na+ channel (1:100; Sigma, St. Louis, MO) were also used. The secondary donkey antibodies conjugated to rhodamine, fluorescein, or Cy-5 were obtained from Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA) or Chemicon International, Inc. (Temecula, CA). Additional fluorescent secondary antibodies (goat anti-mouse Alexa488 and goat anti-rabbit Alexa568) were obtained from Molecular Probes (Eugene, OR).
Preparation of teased sciatic nerves and optic nerve sections. Sciatic nerves were removed from age-matched wild-type and mutant mice and fixed in PBS with 1 or 4% paraformaldehyde for 45 min. The nerves were then stored in PBS at 4°C until teased. Using fine needles, the individual fibers of the sciatic nerve were teased while in ice-cold PBS. Teased sciatic nerve fibers (TSNs) were then mounted on glass slides and dried overnight at room temperature and stored at -80°C until processed further. Optic nerves were dissected out and fixed in 1% paraformaldehyde in PBS for 45 min. Nerves were washed in PBS, cryoprotected with 30% sucrose, and frozen in Tissue-Tek optimal cutting temperature compound (VWR Scientific Products, New York, NY) with isopentane cooled in liquid nitrogen. Six-micrometer-thick sections were cut on a Leica CM1900 cryostat with a chamber temperature of -26°C and stage temperature of -28°C. Sections were dried and then stored at -80°C until used for immunofluorescence.
Immunofluorescence. Immunofluorescent staining of fixed tissue samples (TSNs and optic nerve sections) were performed as described previously (Rios et al., 2000). Images were captured on a Zeiss LSM 510 confocal microscope. Sciatic nerves used for the developmental analysis of Nav1.2 and Nav1.6 expression were preblocked for 30 min with a permeabilizing solution of 4% (v/v) normal goat serum, 2% (w/v) bovine γ-globulin, and 0.3% Triton X-100 in PBS. Primary antibodies were then applied overnight at room temperature. After washing in PBS, fluorescent secondary antibodies were applied in permeabilizing solution for 90 min, and the slides were washed again, dried in the dark, and cover-slipped with an antifade mounting medium (Vectashield; Vector Laboratories, Burlingame, CA). Images were acquired with a Nikon PCM-2000 laser scanning confocal microscope running SimplePCI version 4 software package (Compix, Cranberry Township, PA), attached to a Nikon Eclipse800 upright microscope.
To quantitate Na+ channel subtypes, optic nerves were triple-stained for Nav1.2 or Nav1.6, a nodal marker, and Caspr. In the case of Nav1.6, antibodies to an epitope common to Na+ channels were used; for Nav1.2, βIV spectrin was used as the nodal marker. Quantitation for each pair of markers is presented as the number of positively stained nodes averaged from six high-power fields (HPFs) (146.2 × 146.2 μm2) from two pairs of age-matched mice for each time point. To quantitate the Na+ channel subtype expression in sciatic nerve at different ages, separate images were obtained of the staining patterns in the green (Alexa488) and red (Alexa568) fluorescent channels to minimize ambiguities resulting from fluorescent bleed-through and overlap. Nodes were identified in the green channel using paranodal staining of Caspr for the wild-type mice and the pan sodium channel monoclonal for the Caspr mutants. Expression of Nav1.2 or Nav1.6 was assessed by adjusting the brightness and contrast in the red channel, so that the background staining of paranodal regions was just visible. Nodal sodium channel staining was then scored as “positive” if the intensity was discernibly brighter than the paranodal region.
To measure the intensity and length of nodal staining for ankyrin G, sodium channels, and βIV spectrin, >20 of the brightest nodes per HPF (146.2 × 146.2 μm2) that expressed all three markers were quantitated. Six HPFs were studied from age-matched littermates. To measure the length of nodes of Ranvier at the various ages in the optic nerve, all nodes of Ranvier in half of a HPF (92 × 92 μm2) were analyzed; analysis on two sets of age-matched mice (>90 nodes from six HPFs) was performed in parallel. Measurements from the 2-year-old mice were performed on one pair of mice because of limited numbers of Caspr knock-outs of this age. Quantitative analysis was performed using Zeiss LSM 510 software, statistical analysis was performed using GraphPad Prism, and graphs were generated using Microsoft Excel. To measure nodal length in the PNS, TSNs immunostained for Na+ channels or βIV spectrin were analyzed using a Zeiss Axioskop 2 microscope equipped with a Hamamatsu C-4742-95 CCD camera and Improvision OpenLab (version 2.2.5) software. Over sixty nodes of Ranvier from each animal were measured from two pairs of age-matched littermates.
Freeze fracture analysis. Mice were anesthetized with pentobarbital and fixed by transcardiac perfusion of 3% glutaraldehyde/2%formaldehyde in cacodylate buffer, pH 7.3-7.4. Spinal cord segments were dissected out and Vibratomed tangentially to obtain 50-100 μm slices through cortical fiber tracts. The slices were infiltrated with 30% glycerol, mounted between double replica plates, frozen in liquid propane, mounted in a Balzers 080 freeze-fracture unit, and fractured at -115°C. After unidirectional platinum shadowing and stabilization with evaporated carbon, replicas were cleaned in bleach, mounted on EM grids, and examined in a Philips 300 electron microscope. Micrographs enlarged to 200,000× were used for nodal particle counts, particle size measurements, and area measurements.
Electron microscopy. Two-year-old Caspr knock-out and age-matched wild-type mice were anesthetized with pentobarbitol (150 mg/kg) and perfused through the heart with 3.75% acrolein (Polysciences, Warrington, PA) and 2% paraformaldehyde in 0.1 m phosphate buffer (PB), pH 7.4. The optic nerves were removed and postfixed in 2% paraformaldehyde for 30 min and 2% osmium tetroxide for 1 hr. The nerves then were washed in PB, dehydrated in an ascending series of alcohols, and embedded in EMBed812. Ultrathin (70 nm) longitudinal sections of the nerves were collected on copper grids and counterstained with 5% uranyl acetate and Reynolds lead citrate. Thin sections were examined on a Philips CM10 electron microscope.
Results
Nodes of Ranvier are elongated in Caspr mutant mice
In previous studies, nodes of Ranvier in the Caspr mutant mice appeared to be longer and to stain more diffusely for Na+ channels than those of wild-type littermates (Bhat et al., 2001). To further characterize these changes, we analyzed the distribution of Na+ channels and that of two other nodal components, the neural cell adhesion molecule NrCAM (Bennett and Lambert, 1999) in sciatic nerves and βIV spectrin (Berghs et al., 2000) in optic nerves (Fig. 1). Sciatic nerves stained for NrCAM (red) and neurofilament (green) from wild-type and Caspr mutant mice are shown (Fig. 1a and b). Neurofilament staining appeared to be comparable in the Caspr mutant and wild-type nerves; both exhibit the significant reduction in the diameter of peripheral axons in the nodal and paranodal regions described previously (Hildebrand et al., 1994). NrCAM was concentrated at nodes of Ranvier in both cases. However, on average, the nodes are longer in the Caspr mutants than in wild types (see inset, Fig. 1a,b); similar results were also observed when other markers (Na+ channels, ankyrin G, and βIV spectrin) were used to visualize nodes (data not shown). Nodes in the optic nerve are strikingly aberrant in the Caspr mutants. The βIV spectrin staining is longer, more diffuse, and disorganized in the Caspr mutants (Fig. 1d) compared with wild-type mice (Fig. 1c); neurofilament organization does not appear to be affected.
Figure 1.
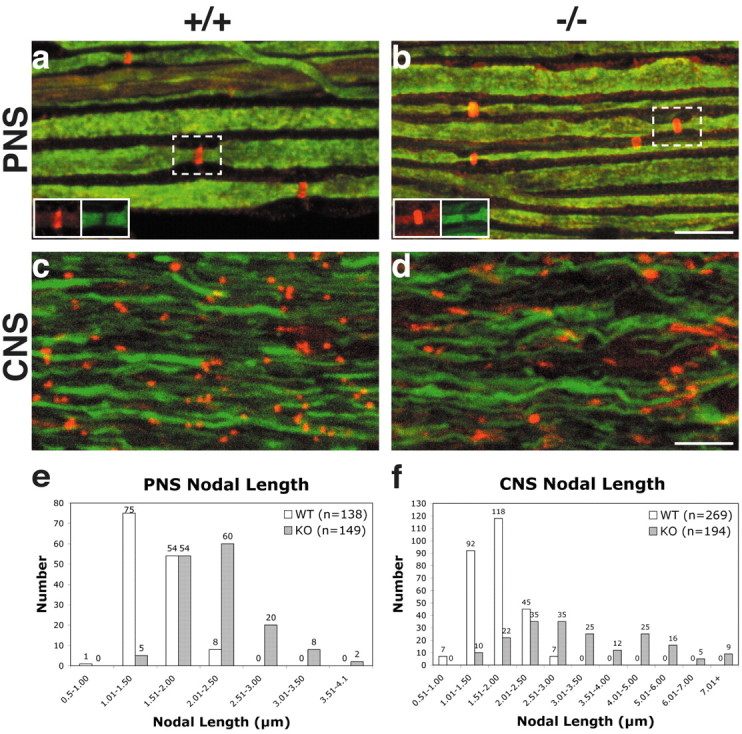
Nodes of Ranvier in the PNS and CNS are longer in Caspr mutant mice. Sciatic nerves from 4-month-old wild-type (a) and Caspr mutant (b) mice were stained for NrCAM (red) and neurofilament (green); insets show NrCAM and neurofilament immunolabeling of single nodes labeled with dashed boxes. Optic nerves from wild-type (c) and Caspr mutant (d) littermates were stained for βIV spectrin (red) and neurofilament (green). The nodes in the wild type are punctate, whereas nodes in the mutant are more variable in length and shape. Nodal lengths were quantitated in sciatic nerves (e) and optic nerves (f). The total number of nodes were binned into 0.50 μm increments and graphed for both wild-type (white) and Caspr (gray) mutants; Caspr-deficient nerves exhibit a greater range in nodal length in both the PNS and CNS. Scale bars, 10 μm.
To quantitate these changes, we measured the length of the nodes in Caspr mutant and wild-type nerves. Sciatic nerves from wild-type and mutant littermates, 4-6 months of age, were immunostained for Na+ channels or βIV spectrin to visualize nodes of Ranvier. In littermate controls, the average nodal length was fairly consistent (1.51 ± 0.27 μm; mean ± SD; n = 138), whereas the average node of Caspr mutant mice was significantly longer (2.19 ± 0.46 μm; n = 149; p < 0.0001 by t test). In optic nerves, the differences in nodal length were more dramatic. The mean nodal length of the Caspr mutant mice (3.41 ± 1.62 μm; n = 194) was twice that of the wild-type mice (1.67 ± 0.41 μm; n = 269; p < 0.0001 by t test) and much more variable (Fig. 2g). This increased variability in the Caspr mutants was apparent when nodes from sciatic and optic nerves were binned into groups of different lengths (Fig. 1e,f). The range of node lengths was extremely broad in the optic nerve of the Caspr mutants (1.30-8.91 μm). However, even the most elongated nodes costained for βIV spectrin, ankyrin G, and Na+ channels, indicating that the major components of the nodal complex were all present (data not shown). These findings underscore the importance of normal paranodal junctions in establishing well delineated, compact nodes of Ranvier in both the PNS and CNS.
Figure 2.

Nodes in the CNS of Caspr mutants increase in length with age. Optic nerve sections from age-matched wild-type and Caspr knock-out mice at 3 weeks, 6 months, and 2 years of age stained forβIV spectrin are shown (a-f). Scale bar, 10 μm. Node lengths were quantitated (g) and are presented as means ± SEM. In the sciatic nerve (SN), the length of the node in both wild-type and Caspr mutant mice increases slightly with age. In the optic nerve (ON), the length of the node decreases slightly in the wild-type mice but increases substantially with age in the Caspr mutant. Nodes are significantly longer in the Caspr mutants than age-matched wild-type nerves in both the CNS and PNS at all ages (*p < 0.0001).
Nodes progressively disperse in optic nerves but not sciatic nerves of Caspr mutant mice
To further address the role of Caspr and paranodal interactions in demarcating nodal boundaries, we examined the distribution of nodal components in wild-type and Caspr-deficient mice of different ages. We measured the length of nodes in optic nerves at 3 weeks, 4-6 months, and 2 years of age (Fig. 2). In wild-type mice, nodes decreased slightly in length in the optic nerves with age (Fig. 2a-c,g), consistent with reports that nodes become better delineated with age (Rasband and Shrager, 2000). Thus, the mean length of the nodes in wild types decreased from 1.88 ± 0.52 μm (mean ± SD; n = 194) at 3 weeks to 1.67 ± 0.41 μm(n = 269) at 4-6 months to 1.60 ± 0.35 μm(n = 111) at 2 years (Fig. 2g, ON). In the Caspr mutant, clusters are already significantly longer at 3 weeks (2.63 ± 0.71 μm; n = 245) and become progressively longer and more disorganized over time (Fig. 2d-f). The mean length is 3.41 ± 1.62 μm (n = 194) at 6 months and 4.21 ± 1.78 μm (n = 96) at 2 years (Fig. 2g, ON). These differences between wild-type and Caspr mutants were statistically significant at all time points (p < 0.0001).
In contrast, nodes in the sciatic nerves of Caspr mutants are slightly longer than those in wild-type mice but do not increase in size with age (Fig. 2g, SN). At 3 weeks, the mean length of nodes in wild-type mice was 1.15 ± 0.18 μm (mean ± SD; n = 123) compared with 1.53 ± 0.26 μm (n = 103) in the Caspr mutants. At 4-6 months, the mean length of nodes in wild-type mice was 1.51 ± 0.27 μm(n = 138) versus 2.19 ± 0.46 μm(n = 149) in the Caspr mutants (p < 0.0001), and at 2 years the mean length was 1.75 ± 0.16 μm(n = 58) in wild-type mice versus 2.18 ± 0.35 μm (n = 106) in Caspr mutants. In both wild-type and Caspr mutant nerves, the Schwann cell microvilli, visualized by staining for ezrin (Melendez-Vasquez et al., 2001), surround nodes of Ranvier appropriately (data not shown). These results, summarized in Figure 2g, indicate that normal paranodal junctions are required to establish nodes of appropriate size in both the CNS and PNS; a substantial increase in node size only occurs in the CNS.
The concentration of nodal components is reduced as nodes elongate
Longer nodes in the Caspr mutant nerves might form by insertion of additional sodium channels and other components to the nodal region and/or result from dispersion of existing nodal components along the axon. If new channels are added, their concentration is likely to be independent of the length of the node, whereas if existing channels disperse, the concentration should be significantly reduced in longer nodes. To distinguish between these possibilities, we analyzed the intensity of immunofluorescent labeling of Na+ channels, βIV spectrin, and ankyrin G in nodes from 4-month-old optic nerves. The intensities of individual nodal markers were measured along the long axis of each node. Nodes of similar lengths were binned into groups of 0.5 μm increments; average intensity measurements for each bin are shown in Figure 3.
Figure 3.

Concentration of nodal components is related to node length. The intensities of fluorescence staining for βIV spectrin (a,b) and Na+ channel (c,d) were measured along the length of optic nerve nodes in 4-month-old mice. Nodes are binned based on their length into 0.5 μm increments; each bin is plotted as a different color (see key). Nodes in the wild type (a,c) are of uniform size and have similar intensities. Caspr knock-outs (b,d) have many more bins because of greater variation in nodal length. The intensity of βIV spectrin (b) and Na+ channel (d) staining decreases as a function of length.
The peak intensities of βIV spectrin and Na+ channels were roughly comparable in both wild-type and Caspr mutants for nodes of normal length, e.g., 0.5-2.0 μm length (Fig. 3). However, there was a striking decrease in the intensity of immunofluorescence of nodal components in nodes >2 μm in length in the Caspr mutants (Fig. 3b,d) (data not shown for ankyrin G). The reduction in peak intensity strongly correlated to the increase in node length, except for one bin, 3.5-4 μm, which reflected measurements from only three nodes (one of which may have consisted of two smaller nodes in close register). These results indicate that longer nodes have reduced concentrations of nodal components, suggesting they have dispersed along the axons.
Ultrastructural abnormalities in the paranodal region
Previous studies of the spinal cord, cerebrum, and peripheral nerves in Caspr mutant mice demonstrated that ultrastructural abnormalities are concentrated in the paranodal and nodal regions, whereas compact myelin forms normally (Bhat et al., 2001). We examined the ultrastructure of optic nerve, the focus of the present studies, in mice up to 2 years of age. Defects were similar to those reported previously; compact myelin sheaths appeared to be normal in cross sections (data not shown), whereas various abnormalities of the paranodal region were observed on longitudinal sections (Fig. 4). No evidence of demyelination was observed. Paranodal abnormalities include everted and retracted paranodal loops (Fig. 4b) which, in some cases, resulted in extended nodal regions (data not shown). Loop eversion and retraction typically occur first in the outermost loops, e.g., those closest to the node. In some cases, multiple layers of detached paranodal loops were observed, suggesting that retraction of loops may occur in stages (Fig. 4b). Nearly all of the paranodal regions surveyed in the 2-year-old optic nerves (14 of 15) demonstrated substantial defects including everted and/or retracted loops.
Figure 4.
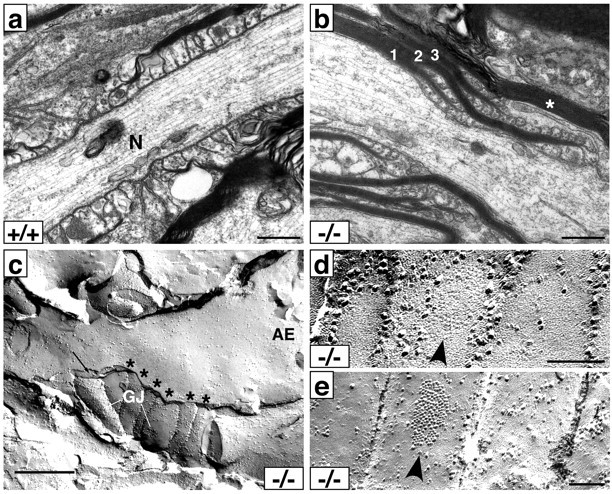
Ultrastructure and freeze fracture of the paranodal region in Caspr mutants. Electron micrographs of paranodal regions from optic nerves of 2-year-old wild-type (a) and Caspr mutant (b) mice. In the wild-type optic nerve, paranodal loops are regularly arranged and closely apposed to the axon. In contrast, in this example from the Caspr mutant optic nerve (b), a single myelin sheath separates into three distinct sets of myelin lamellae (numbered in the figure), each of which terminate with both inverted and everted loops. The outermost set of lamellae (3) has retracted from the node, which is just visible at the bottom right. A myelin sheath (*) from another cell overrides this paranodal region. c, Freeze-fracture replica showing a palisade of presumptive paranodal processes labeled above (*). Two of these display macular particle patches resembling the gap junctions (GJ) characteristic of astrocyte processes. A thin cellular process (black arrow) intervenes between these and an E-face (AE) containing node-like membrane particles in a density of ∼800/μm2 that spread well into the paranodal region. The proportion of particles 10 nm or greater is 56%, consistent with the proportion in the normal nodal axolemma. Scale bars: a-c, 0.5 μm. d,e, Details of gap junction-like patches in freeze-fracture replicas of membranes within paranodal processes. d, E-face pits; e, P-face particles. Scale bar, 0.1 μm.
Abnormalities in the distribution and density of nodal components were also apparent by freeze fracture analysis. In the replica shown (Fig. 4c), a palisade of presumptive paranodal processes (asterisks) is evident. A thin cellular process (arrow) intervenes between these loops and an E-face (AE) of a node-like membrane, which has intramembranous particles (IMPs) at a density of ∼800/μm2. Interestingly, several of the paranodal processes have macular particle patches (GJ) typical of gap junctions (Fig. 4c). Multiple examples of such patches in equivalent palisades were observed in other replicas of mutant paranodes (Fig. 4d,e, arrowheads), but none were observed in wild-type tissue. Because oligodendrocytes have been reported to form gap junctions with astrocytes but not with each other in the paranodal region (Massa and Mugnaini, 1982; Rash et al., 2001), these findings suggest extensive infiltration by astrocyte processes into the paranodes of the Caspr mutant mice, as we reported previously (Bhat et al., 2001), where they form gap junctions with adjacent oligodendrocyte loops. We cannot exclude the possibility that gap junctions may have formed between the paranodal processes themselves, as has been proposed (Sandri et al., 1977). Of particular note, the IMPs in the axonal membrane extend from the node well into the presumptive paranodal region in this replica, indicating that the barrier function of the paranode has been disrupted by the intervening cellular process. These results are consistent with the spread of nodal components along the axon observed by immunofluorescence.
Aberrant expression of Na+ channel subtypes in the Caspr mutant nerves
We next examined whether the expression of sodium channel subtypes at the nodes is perturbed. We characterized Nav1.2 and Nav1.6 expression in wild-type and Caspr mutant optic nerves from 2-year-old mice, double staining with βIV spectrin to identify all the nodes in a field (Fig. 5). In agreement with a recent report (Boiko et al., 2001), the large majority (75%; 486 of 647) of nodes in the wild type strongly expressed Nav1.6 (Fig. 5); a few nodes (7%; 44 of 647 nodes) were weakly Nav1.2 positive. In striking contrast, Nav1.2 is expressed in more than half (54%; 326 of 605) of the Caspr mutant nodes, many of which were strongly labeled, whereas the percentage of Nav1.6-positive nodes is only 26% (156 of 605) at 2 years. A few nodes did not express either Nav1.2 or Nav1.6 (Fig. 5, arrowheads), indicating that other Na+ channel subtypes are likely to be expressed. These results indicate that the paranodes play a key role in determining which sodium channel subtype is expressed at the node.
Figure 5.

Nav1.2 and Nav1.6 expression in optic nerves from wild-type and mutant mice. Optic nerve sections from 2-year-old wild-type (+/+) and Caspr mutant (-/-) littermates were stained with anti-Nav1.2 (green), anti-βIV spectrin (red), and anti-Nav 1.6 (blue) antibodies. Nav 1.2-positive nodes are not present in the wildtype (a,c), whereas the Caspr knock-out continues to express significant numbers of Nav 1.2-positive nodes (appear as yellow nodes in the merged images in d,f). Nav 1.6 is expressed at almost all the nodes of Ranvier in the wild type (magenta in the merged images in b,c) but at very few of the Caspr knock-out nodes (e,f). A few nodes (red in all panels) do not express either isoform and are indicated with arrowheads. Scale bar, 10 μm.
These differences may reflect either a failure of the Caspr mutant nodes to mature during development or reexpression of Nav1.2 and loss of Nav1.6 from nodes that had previously matured properly. To distinguish between these possibilities, we characterized the expression of Nav1.2 and Nav1.6 at postnatal day 14 (P14) and P21, a transition period between these channel subtypes in the optic nerve (Boiko et al., 2001), and again at 4-6 months. At P14, mutant and control littermates have a similar proportion of Nav1.2-positive nodes, 52% for wild types and 60% for knockouts (p > 0.05 by t test). The proportion of Nav1.2-positive nodes decreases slightly at P21, but remains comparable in both mice, corresponding to 47% for controls and 51% for knock-outs (p > 0.05). Both Na+ channel subtypes were coexpressed in ∼20% of the nodes in wild-type and Caspr mutant nerves at these early time points (data not shown). There is a significant difference by 4-6 months because only 12% of the wild-type nodes are immunoreactive for Nav1.2, and most of these are only weakly positive. In the mutant mice at this age, 44% of nodes are Nav1.2 positive (p < 0.05), and most of these are strongly positive. An inverse pattern is seen for Nav1.6 development. At P14, 45% of nodes in wild-type nerves are Nav1.6 positive; in contrast, only 21% of nodes are positive for Nav1.6 in the Caspr mutants (p < 0.05). At P21-22, roughly 60% of nodes from the wild-type optic nerves are Nav1.6 positive compared with 38% for knock-outs (p < 0.05). The percentage of Nav1.6-positive nodes appears to plateau in the mutants at this time, corresponding to 38% for the Caspr mutants at 4-6 months versus nearly 80% in the wild type. These results, which are summarized in Figure 6, indicate that nodes in the optic nerve of Caspr mutants never mature properly. Misexpression of channel subtypes in the adult is, therefore, likely to reflect persistence of this immature phenotype.
Figure 6.

Expression of Nav1.2 and Nav1.6 in wild-type and Caspr mutant mice of different ages. The percentage of nodes that are Nav1.2 or Nav1.6 positive in wild-type (+/+) or Caspr null (-/-) mice was quantitated at 14 d, 21 d, 4-6 months, and at 2 years. In the wild-type nerves, Nav1.2 is expressed in the majority of nodes but is substantially replaced by Nav1.6 at 4-6 months. In the Caspr mutants, Nav1.2 is expressed in approximately half of the nodes at all time points, and the proportion of Nav1.6-positive nodes does not increase further after P21.
In general, the Nav1.2-positive nodes in the Caspr mutants were longer than the Nav1.6-positive nodes (3.45 ± 1.82 μm vs 2.46 ± 1.24 μm at 4 months), although both were elongated compared with controls. Similarly, Nav1.2-positive nodes in developing wild-type nerves appeared to be longer than Nav1.6 nodes in the same nerves (data not shown), in agreement with previous reports (Boiko et al., 2001). Nav1.6-positive nodes also appeared to be associated with larger diameter fibers in the Caspr mutants. Finally, the total number of nodes per HPF, as detected with anti-βIV spectrin antibodies, is significantly lower (by 25% at P14 and 20% at P21) in the Caspr mutants than in their wild-type littermates (p < 0.05). Similar, modest differences were also observed at 4-6 months and at 2 years (p > 0.05), indicating that the decreased number of nodes per HPF in the Caspr mutants does not progress further.
Maturation of nodes is delayed in the PNS
Nodes undergo a similar transition from Nav1.2 to Nav1.6 during PNS development, although this switch occurs at earlier ages than in the CNS (Boiko et al., 2001). We, therefore, examined expression of Na+ channel subtypes in sciatic nerves of Caspr mutant mice during development. In contrast to optic nerves, essentially all nodes in the adult sciatic nerve of the Caspr mutant are Nav1.6 positive and Nav1.2 negative (data not shown). However, the transition in the expression of channel subtypes, in particular the downregulation of Nav1.2, was significantly delayed in the Caspr mutants. At P10 (Fig. 7), Nav1.2 persists at many more nodes in the Caspr knock-outs (47 ± 1%) than the wild-type nerves (21 ± 1.1%; p < 0.01). A small percentage (2%) of nodes in the Caspr knock-out also did not express Nav1.6 at this age, whereas all of the nodes in the wild type were Nav1.6 positive. At P21, slightly more nodes continued to express Nav1.2 in the knock-out (3 ± 1%) than in the wild type (1%); this difference was not statistically significant. These differences do not reflect delayed nerve development in the Caspr mutants because the onset of myelination and node formation began appropriately at P1-P2 and subsequent development parallels that of wild-type nerves (data not shown).
Figure 7.

The expression of Nav1.2 and Nav1.6 in P10 sciatic nerves. Wild-type (+/+) sciatic nerves from P10 mice were stained with anti-Caspr antibodies (green) and anti-Nav1.2 (red) (a-c) or anti-Nav1.6 antibodies (red) (panels g-i). Caspr mutant (-/-) sciatic nerves were labeled with anti-Na+ channel antibodies (green) and anti-Nav1.2 (red) (d-f) or anti-Nav1.6 (j-l) antibodies. Very few nodes in the wild type at this age are Nav1.2 positive (one is indicated with an arrow in a,c); in contrast, many of the Caspr mutant nodes are Nav1.2 positive (d,f). All nodes were Nav 1.6 positive in the wild-type and knock-out mice, indicating that Nav1.2 and Nav1.6 are frequently coexpressed in the P10 knock-out nerves. Scale bar, 20 μm.
Interestingly, there were significant differences (p < 0.01) in the expression of Nav1.2 at nodes between the proximal and distal segments of nerves in wild-type and knock-out mice at P10 and P21. At P10, a higher percentage of nodes in the proximal segments were Nav1.2 positive compared with the distal segments (27 ± 2% vs 18 ± 1% in the wild type; 57 ± 1.4% vs 35 ± 1.4% in the knock-out), despite the fact that myelination is thought to proceed proximally to distally in peripheral nerves (Hildebrand et al., 1994). We also noted that the mean diameter of fibers with Nav1.2-positive nodes was smaller than the diameter of fibers with Nav1.6-positive nodes. For example, at P10 the mean diameter of fibers that were Nav1.2 positive was 3.4 ± 0.6 μm in the wild type and 4.1 ± 1.1 μm in the Caspr knock-outs as compared with fiber diameters with Nav1.6-positive nodes of 4.7 ± 1.3 μm in the wild type and 5.1 ± 1.6 μm in the Caspr knock-outs. These results suggest that signals from the paranodal junctions first promote maturation of nodes, in particular the downregulation of Nav1.2, in the distal segments and larger fibers of the PNS.
Potassium channel domain abnormalities
We also analyzed the expression of Kv1.1 and Kv1.2, which are aberrantly localized to the paranodes in sciatic and optic nerves of Caspr mutant mice (Bhat et al., 2001). They colocalize with Caspr2 (data not shown), a Caspr-related protein known to be complexed indirectly with Kv1.1 and Kv1.2 (Poliak et al., 1999). Additional analysis revealed additional abnormalities (Fig. 8). Most striking is a decrease in the number of Kv1.1 clusters in the paranodes of older optic nerves (Fig. 8b,d); a similar decrease of Kv1.2 clusters was also observed (data not shown). There are fewer Kv1.1 clusters in 2-year-old than 4-month-old Caspr mutants, and both have significantly fewer clusters than the juxtaparanodal clusters in wild-type mice of the same age (Fig. 8a,c). These results indicate that the Kv1.1 clusters in the paranodes are lost over time. Indeed, the great majority of βIV spectrin clusters in the 2-year-old Caspr mutants, which are quite disorganized, are not associated with Kv1.1 clusters (Fig. 8d). Of interest, the few Kv1.1 channel clusters present in the optic nerves of 2-year-old Caspr mutants typically flank well delineated, nodal clusters of βIV spectrin (Fig. 8d, yellow arrowheads); similar results were observed for Kv1.2 (data not shown). These results suggest that the organization of the node is relatively well preserved in the older mutant nerves when Kv1.1 and Kv1.2 are present in the paranodes. This is underscored by analysis of sciatic nerves in which Kv1.1 and Kv1.2 are reliably expressed in the paranodes of Caspr mutant sciatic nerves (Fig. 8e,f), even in 2-year-old animals, and flank well delineated nodes. However, rare spectrin clusters are observed that are significantly attenuated (Fig. 8f, white arrow), and these are typically associated with aberrant paranodal Kv1.1 staining.
Figure 8.
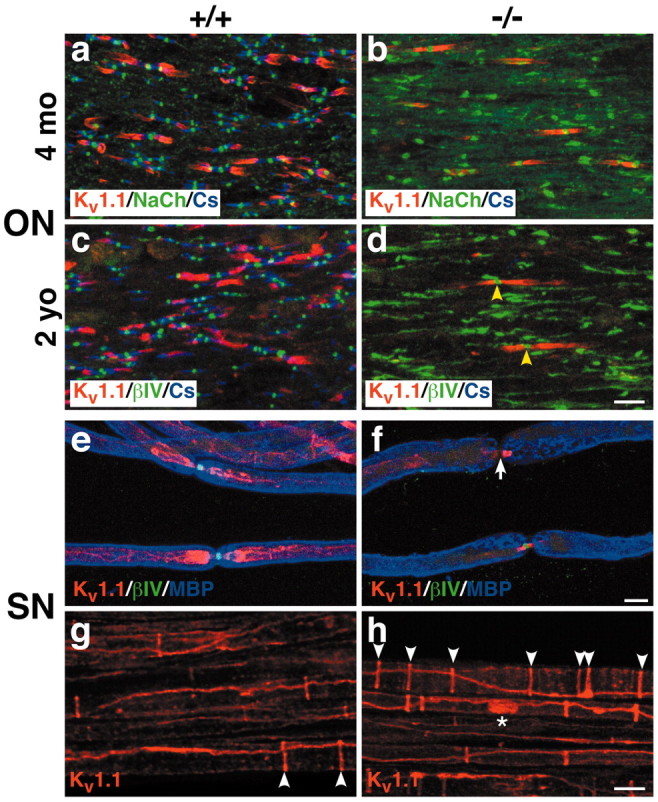
Potassium channel distribution is aberrant in Caspr-deficient mice. Optic nerve sections of 4-month-old (a,b) and 2-year-old (c,d) optic nerves from wild-type (+/+) and Caspr mutants (-/-) were triple stained for Kv1.1 (red), Caspr (blue), andβIV spectrin (green). Kv1.1 is present in many of the juxtaparanodes of the wild type (a,c) but only a few of the paranodes of the Caspr mutants (b,d), particularly at 2 years. Kv1.1 expressed in the paranodes of the mutants frequently flanks well delineated nodes; two examples are indicated (d; yellow arrowheads). Sciatic nerves from wild-type (e,g) and Caspr mutant (f,h) littermates were triple stained for Kv1.1 (red),βIV spectrin (green), and MBP (blue) (e,f) or Kv1.1 (red) alone (g,h). Kv1.1 is expressed in the juxtaparanodes of wild-type mice (e) and the paranodes of Caspr mutants (f). Deficient Kv 1.1 expression in the paranodes of Caspr mutants is associated with attenuatedβIV spectrin at the node (f, white arrow). There are more Schmidt-Lanterman clefts in the Caspr mutant (h, white arrowheads) than in the wild type (g, white arrowheads). Kv1.1 is also occasionally expressed in a band along the axon (h, asterisk). Scale bars, 10 μm.
We also observed abnormalities in the internodal distribution of Kv1.1 and Kv1.2 channels in peripheral nerves. These include an apparent increase in the number of Schmidt-Lanterman clefts (Fig. 8, compare g, h, white arrowheads), which are known to appose internodal rings of Kv1 channel staining (Arroyo et al., 1999). We have also observed that Kv1.1 is occasionally expressed in a band that stretches between two adjacent Schmidt-Lanterman clefts (Fig. 8h, asterisk); Kv1.2 staining is similarly aberrant (data not shown). These changes were not observed in wild-type controls. In the 2-year-old sciatic nerves, the number of these bands is reduced, and there is an increase in large, vesicular-like staining of Kv1.1, which may represent channel clusters that are being internalized (data not shown). These results suggest that the expression of Caspr within the internode regulates the distribution and stability of ion channel domains akin to its role in the paranodes.
Discussion
These studies further expand the role of the paranodes in the maturation and maintenance of nodes of Ranvier. They provide strong evidence that paranodal interactions delineate the lateral boundaries of the node, determine the concentration of nodal components, regulate the transition from Nav1.2 to Nav1.6, and sustain the integrity of Na+ and K+ channel domains in myelinated axons.
The density and distribution of nodal components are regulated by paranodal interactions
A major finding of this study is that nodes in the CNS of the Caspr mutants progressively increase in length; PNS nodes are modestly longer but do not continue to increase. A related finding is that the concentrations of key nodal components (ankyrin G, spectrin, and Na+ channels) decline in proportion to increases in node length in the Caspr mutants (Fig. 3). These three nodal components colocalize and are coordinately reduced in their concentration, consistent with the notion that they form a multiprotein complex of defined stoichiometry. In the longest nodes, peak intensities for spectrin were reduced by as much as 75%. Nodal IMPs also disperse along the axon and exhibit reduced density (Fig. 4). Together, these results indicate that paranodal interactions prevent the dispersion and maintain the concentration of the Na+ channel complex at the node. Functional consequences of the spreading of nodal components in the Caspr mutants may be a decrease in the conduction velocity and, potentially, conduction block at nodes with markedly reduced concentrations of Na+ channels. Such changes, together with misexpression of Na+ channel subtypes (see below), may account for the progressive neurological deficits of older Caspr mutant mice, which include spastic quadraparesis (our unpublished observations).
These findings partially agree with a recent study by Ishibashi et al. (2002). These authors reported that nodes in the CNS of CST-deficient mice, which have similar paranodal defects, are enlarged but do not increase further over time; rather, they observed a progressive loss of up to 90% of Na+ and K+ channel clusters by 4-5 months. In striking contrast, whereas K+ channel clusters are lost in older Caspr mice (Fig. 8), the number of nodes per HPF in the optic nerve is only modestly reduced (∼25%), even at 2 years. There was no obvious change in the number of PNS nodes. The slight reduction in the number of optic nerve nodes per HPF in the Caspr mutants seems to reflect a change in the organization of optic nerve fibers, which are more widely spaced in these mice, rather than an actual loss of nodes per fiber (S. Einheber and J. Salzer, unpublished observations). Whether discrepancies between these two studies reflect methodological differences in how nodes were identified and quantitated or, alternatively, real differences between these murine mutants will require further study.
Paranodal interactions provide a lateral diffusion barrier independent of the transverse bands
Our findings indicate that interactions of paranodal loops with the axon provide a barrier to the lateral diffusion of nodal components even in the absence of transverse bands. Schwann cell paranodal loops are appropriately oriented toward the axon in the Caspr mutants, although they tend to be more widely separated from the axon than normal (Bhat et al., 2001). Because PNS nodes are only modestly larger in the Caspr mutants, these paranodal interactions appear to be sufficient to prevent dispersion of the node. Contact of the nodal complex with putative receptors on the Schwann cell microvilli (Melendez-Vasquez et al., 2001) may also provide a signal in trans that immobilizes the nodal complex and prevents its diffusion into the paranode. Ezrin staining demonstrated that the microvilli remain associated with PNS nodes in the Caspr mutants (data not shown). These microvillar interactions, together with Schwann cell attachment to the basal lamina, may also account for the relative preservation of the nodal and paranodal organization in the PNS.
Results in the CNS also suggest that the paranodal loops act as diffusion barriers (Rosenbluth, 1976). Paranodal abnormalities in the CNS are complex and variable. Some glial loops remain attached to the axon in the CNS (Fig. 4) and may even display intervening densities with the axolemma (Bhat et al., 2001), although such EM dense material is never organized into transverse bands. These incomplete paranodal loop-axon interactions resemble those of early, immature junctions in which paranodal loops closely appose the axon but transverse bands have not yet formed; such immature junctions, nevertheless, demarcate the boundary of early nodes (Tao-Cheng and Rosenbluth, 1983). Paranodal loop interactions in the CGT knock-out, which also lack transverse bands, were recently reported to demarcate nodal IMP clusters (Rosenbluth et al., 2003).
Attachment of paranodal loops to the axon in the absence of the Caspr-contactin complex suggests the existence of residual adhesive interactions. Other adhesion molecules that may promote axon-loop interactions include a presumptive complex of TAG-1 on the glial cell (Traka et al., 2002) and Caspr2 and K+ channels on the axon (Poliak et al., 1999). This complex is consistently displaced into the paranodes in the PNS of Caspr mutants but is variably present in paranodes of the CNS of the Caspr mutants (Fig. 8) and CGT mutants (Poliak et al., 2001; Ishibashi et al., 2002), presumably reflecting detachment of central loops. Interestingly, when K+ channels are expressed in central paranodes, nodes are well delineated, likely indicating persistent paranodal interactions (Fig. 8).
In most cases, however, paranodal loops detach and retract in the CNS of the Caspr mutants. Loops that are everted, or are separated by intervening glial processes (Fig. 4b,c), appear not to function as diffusion barriers (Fig. 4c). These findings also indicate that the transverse bands are required for the stable attachment of the central paranodal loops to the axon. Paranodal loops appear to detach over months, in agreement with recent analysis of the CGT mutant, which demonstrated that paranodal loops first appose and later detach from the axon (Marcus et al., 2002). In the Caspr mutant, 40-50% of the paranodes in the spinal cord and cerebrum had everted loops at 6 weeks, whereas nearly all paranodes examined in the optic nerve at 2 years had substantial abnormalities (data not shown). Paranodal loops adjacent to the node detach first, followed by those closer to the juxtaparanodes (Fig. 4). In some cases, processes of other cells may infiltrate between the paranodal loops and the axon, potentially contributing to their detachment (data not shown). Evolution of these paranodal defects seems likely to account for the progressive dispersion of the nodes over time.
The transition in channel subtypes is regulated by paranodal interactions
Previous studies of a dysmyelinating mutant suggested that compact myelin and/or paranodal junctions regulate the expression of Nav1.6 at mature nodes (Boiko et al., 2001). Because compact myelin is essentially normal in the Caspr mutant (Bhat et al., 2001), our results indicate that paranodal interactions play a key role in promoting the transition of Na+ channel subtypes. In the CNS, this transition is substantially aberrant, affecting both the downregulation of Nav1.2 and the increase in Nav1.6. Some optic nerve nodes do mature properly, consistent with the variable nature of the paranodal perturbation. Our results contrast with a study of the jimpy mouse that suggested that this transition can occur in the absence of Caspr-positive paranodal interactions (Jenkins and Bennett, 2002). However, oligodendrocytes in the jimpy mutant undergo programmed cell death (Knapp et al., 1986), and nodes analyzed in this study may have formed in association with myelin sheaths, and paranodes, that were later lost as shown in a similar myelin mutant (Arroyo et al., 2002). In addition, the absence of Caspr does not preclude significant paranodal interactions as discussed previously.
Nothing is known about how the paranodes regulate this transition. Normally, adult retinal ganglion neurons express both channel subtypes: Nav1.2 is specifically localized to the proximal, unmyelinated segment of optic nerve axons whereas Nav1.6 is expressed at mature nodes in the distal, myelinated portion, replacing Nav1.2 (Boiko et al., 2001). Deficient expression of Nav1.6 at Caspr mutant nodes in the optic nerve could, therefore, result from limited transcription of this channel subtype and subsequent persistence of Nav1.2. In potential agreement, mutant mice that express Nav1.6 at reduced levels continue to express Nav1.2 at nodes quite late in development (Kearney et al., 2002). Myelin is known to downregulate Nav1.2 in the CNS (Westenbroek et al., 1992) possibly, on the basis of the results reported here, via paranodal signals. Studies of the rat mutant taiep (Black et al., 1999) suggest that myelin may regulate the relative abundance of sodium channel subtypes by transcriptional mechanisms. Whether transcription of Na+ channel subtypes is aberrant in the Caspr mutants is not yet known.
Alternatively, Nav1.6 may be expressed but not targeted to nodes in the Caspr mutant optic nerves, similar to its absence from the unmyelinated, proximal segment of the optic nerve. Recently, Nav1.6 was shown to be expressed in the initial segments, but not the nodes, of the hypomyelinated optic nerves of the shiverer mouse (Boiko et al., 2003), strongly suggesting that myelination is required for its targeting, but not necessarily its expression. Our results suggest that targeting of Nav1.6 is directed by paranodal interactions. Potentially, the density of nodal components or the molecular composition of the flanking paranodes, both of which are abnormal in the Caspr mutants, may affect Nav1.6 targeting. Because all PNS nodes in the Caspr mutant eventually express Nav1.6 after a delay (Fig. 7), the Caspr-contactin complex cannot itself be an essential targeting signal; it may contribute to this transition, however, by enhancing paranodal interactions. Cytoplasmic regions of Na+ channels (Garrido et al., 2001) and/or differential association of channel subtypes with ancillary β subunits (Kaplan et al., 2001; Ratcliffe et al., 2001) may direct their targeting to the node in combination with appropriate paranodal signals.
In summary, these results indicate that interactions of the paranodal loops with the axon, independent of the transverse bands, prevent dispersion of sodium channels, sustaining their density at concentrations critical for normal saltatory conduction. Paranodal interactions are also required for the appropriate transition of sodium channel subtypes at the node. Characterization of the molecular nature of the paranodal diffusion barrier and identification of the paranodal signals that regulate the transition of channel subtypes at the node are key questions for future investigation.
Footnotes
This work was supported by National Institutes of Health Grants NS38208 and 43474 (J.L.S.), NS34375 (S.R.L.), NS37475 (J.R.), and GM63074 (M.B.); by Grant KO1-CA 78437 from the National Cancer Institute (M.B.); and by National Multiple Sclerosis Society Grants RG 3439 (J.L.S.) and RG 2539 (J.R.). M.B. is a recipient of the Howard Temin Career Development Award and Hirschl Foundation. We thank Teresa Milner for assistance with EM, Ori Peles for antibodies to Caspr2, Masa Komada and Michele Solimena for anti-βIV spectrin antibodies, Marty Grumet for antibodies to NrCAM, Steve Lambert for antibodies to ankyrin G, and Carmen Melendez-Vasquez for comments on this manuscript.
Correspondence should be addressed to Dr. James L. Salzer, Department of Cell Biology and Neurology, New York University School of Medicine, 550 First Avenue, New York, NY 10016. E-mail: Jim.Salzer@med.nyu.edu.
M. Bhat's present address: Department of Cell and Molecular Physiology, University of North Carolina School of Medicine, Chapel Hill, NC 27599.
Copyright © 2003 Society for Neuroscience 0270-6474/03/237001-11$15.00/0
References
- Arroyo EJ, Xu YT, Zhou L, Messing A, Peles E, Chiu SY, Scherer SS ( 1999) Myelinating Schwann cells determine the internodal localization of Kv1.1, Kv1.2, Kvbeta2, and Caspr. J Neurocytol 28: 333-347. [DOI] [PubMed] [Google Scholar]
- Arroyo EJ, Xu T, Grinspan J, Lambert S, Levinson SR, Brophy PJ, Peles E, Scherer SS ( 2002) Genetic dysmyelination alters the molecular architecture of the nodal region. J Neurosci 22: 1726-1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett V, Lambert S ( 1999) Physiological roles of axonal ankyrins in survival of premyelinated axons and localization of voltage-gated sodium channels. J Neurocytol 28: 303-318. [DOI] [PubMed] [Google Scholar]
- Berghs S, Aggujaro D, Dirkx Jr R, Maksimova E, Stabach P, Hermel JM, Zhang JP, Philbrick W, Slepnev V, Ort T, Solimena M ( 2000) BetaIV spectrin, a new spectrin localized at axon initial segments and nodes of Ranvier in the central and peripheral nervous system. J Cell Biol 151: 985-1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat MA, Rios JC, Lu Y, Garcia-Fresco GP, Ching W, Martin MS, Li J, Einheber S, Chesler M, Rosenbluth J, Salzer JL, Bellen HJ ( 2001) Axon-glia interactions and the domain organization of myelinated axons requires Neurexin IV/Caspr/Paranodin. Neuron 30: 369-383. [DOI] [PubMed] [Google Scholar]
- Black JA, Fjell J, Dib-Hajj S, Duncan ID, O'Connor LT, Fried K, Gladwell Z, Tate S, Waxman SG ( 1999) Abnormal expression of SNS/PN3 sodium channel in cerebellar Purkinje cells following loss of myelin in the taiep rat. NeuroReport 10: 913-918. [DOI] [PubMed] [Google Scholar]
- Boiko T, Rasband MN, Levinson SR, Caldwell JH, Mandel G, Trimmer JS, Matthews G ( 2001) Compact myelin dictates the differential targeting of two sodium channel isoforms in the same axon. Neuron 30: 91-104. [DOI] [PubMed] [Google Scholar]
- Boiko T, Van Wart A, Caldwell JH, Levinson SR, Trimmer JS, Matthews G ( 2003) Functional specialization of the axon initial segment by isoform-specific sodium channel targeting. J Neurosci 23: 2306-2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle MET, Berglund EO, Murai KK, Weber L, Peles E, Ranscht B ( 2001) Contactin orchestrates assembly of the septate-like junctions at the paranode in myelinated peripheral nerve. Neuron 30: 385-397. [DOI] [PubMed] [Google Scholar]
- Caldwell JH, Schaller KL, Lasher RS, Peles E, Levinson SR ( 2000) Sodium channel Na(v)1.6 is localized at nodes of ranvier, dendrites, and synapses. Proc Natl Acad Sci USA 97: 5616-5620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupree JL, Coetzee T, Blight A, Suzuki K, Popko B ( 1998) Myelin galactolipids are essential for proper node of Ranvier formation in the CNS. J Neurosci 18: 1642-1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupree JL, Girault JA, Popko B ( 1999) Axo-glial interactions regulate the localization of axonal paranodal proteins. J Cell Biol 147: 1145-1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einheber S, Zanazzi G, Ching W, Scherer S, Milner TA, Peles E, Salzer JL ( 1997) The axonal membrane protein Caspr, a homologue of neurexin IV, is a component of the septate-like paranodal junctions that assemble during myelination. J Cell Biol 139: 1495-1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrido JJ, Fernandes F, Giraud P, Mouret I, Pasqualini E, Fache MP, Jullien F, Dargent B ( 2001) Identification of an axonal determinant in the C-terminus of the sodium channel Na(v)1.2. EMBO J 20: 5950-5961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand C, Bowe CM, Remahl IN ( 1994) Myelination and myelin sheath remodelling in normal and pathological PNS nerve fibres. Prog Neurobiol 43: 85-141. [DOI] [PubMed] [Google Scholar]
- Hille B ( 2001) Ion channels of excitatory membranes. Sunderland, MA: Sinauer Associates.
- Honke K, Hirahara Y, Dupree J, Suzuki K, Popko B, Fukushima K, Fukushima J, Nagasawa T, Yoshida N, Wada Y, Taniguchi N ( 2002) Paranodal junction formation and spermatogenesis require sulfoglycolipids. Proc Natl Acad Sci USA 99: 4227-4232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi T, Dupree JL, Ikenaka K, Hirahara Y, Honke K, Peles E, Popko B, Suzuki K, Nishino H, Baba H ( 2002) A myelin galactolipid, sulfatide, is essential for maintenance of ion channels on myelinated axon but not essential for initial cluster formation. J Neurosci 22: 6507-6514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins SM, Bennett V ( 2002) Developing nodes of Ranvier are defined by ankyrin-G clustering and are independent of paranodal axoglial adhesion. Proc Natl Acad Sci USA 99: 2303-2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan MR, Cho M, Ullian EM, Isom LL, Levinson SR, Barres BA ( 2001) Differential control of clustering of the sodium channels Na(v)1.2 and Na(v)1.6 at developing CNS nodes of Ranvier. Neuron 30: 105-119. [DOI] [PubMed] [Google Scholar]
- Kearney JA, Buchner DA, De Haan G, Adamska M, Levin SI, Furay AR, Albin RL, Jones JM, Montal M, Stevens MJ, Sprunger LK, Meisler MH ( 2002) Molecular and pathological effects of a modifier gene on deficiency of the sodium channel Scn8a (Na(v)1.6). Hum Mol Genet 11: 2765-2775. [DOI] [PubMed] [Google Scholar]
- Knapp PE, Skoff RP, Redstone DW ( 1986) Oligodendroglial cell death in jimpy mice: an explanation for the myelin deficit. J Neurosci 6: 2813-2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komada M, Soriano P ( 2002) βIV-Spectrin regulates sodium channel clustering through ankyrin-G at axon initial segments and nodes of Ranvier. J Cell Biol 156: 337-348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus J, Dupree JL, Popko B ( 2002) Myelin-associated glycoprotein and myelin galactolipids stabilize developing axo-glial interactions. J Cell Biol 156: 567-577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massa PT, Mugnaini E ( 1982) Cell junctions and intramembrane particles of astrocytes and oligodendrocytes: a freeze-fracture study. Neuroscience 7: 523-538. [DOI] [PubMed] [Google Scholar]
- Melendez-Vasquez CV, Rios JC, Zanazzi G, Lambert S, Bretscher A, Salzer JL ( 2001) Nodes of Ranvier form in association with ezrin-radixin-moesin (ERM)-positive Schwann cell processes. Proc Natl Acad Sci USA 98: 1235-1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menegoz M, Gaspar P, Bert ML, Galvez T, Burgaya F, Palfrey C, Ezan P, Arnos F, Girault J-A ( 1997) Paranodin, a glycoprotein of neuronal paranodal membranes. Neuron 19: 319-331. [DOI] [PubMed] [Google Scholar]
- Peles E, Salzer JL ( 2000) Molecular domains of myelinated axons. Curr Opin Neurobiol 10: 558-565. [DOI] [PubMed] [Google Scholar]
- Peles E, Nativ M, Lustig M, Grumet M, Schilling J, Martinez R, Plowman GD, Schlessinger J ( 1997) Identification of a novel contactin-associated transmembrane receptor with multiple domains implicated in protein-protein interactions. EMBO J 16: 978-988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poliak S, Gollan L, Martinez R, Custer A, Einheber S, Salzer JL, Trimmer JS, Shrager P, Peles E ( 1999) Caspr2, a new member of the neurexin super-family, is localized at the juxtaparanodes of myelinated axons and associates with K+ channels. Neuron 24: 1037-1047. [DOI] [PubMed] [Google Scholar]
- Poliak S, Gollan L, Salomon D, Berglund EO, Ohara R, Ranscht B, Peles E ( 2001) Localization of Caspr2 in myelinated nerves depends on axon-glia interactions and the generation of barriers along the axon. J Neurosci 21: 7568-7575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband MN, Shrager P ( 2000) Ion channel sequestration in central nervous system axons. J Physiol (Lond) 525: 63-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rash JE, Yasumura T, Dudek FE, Nagy JI ( 2001) Cell-specific expression of connexins and evidence of restricted gap junctional coupling between glial cells and between neurons. J Neurosci 21: 1983-2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratcliffe CF, Westenbroek RE, Curtis R, Catterall WA ( 2001) Sodium channel β1 and β3 subunits associate with neurofascin through their extracellular immunoglobulin-like domain. J Cell Biol 154: 427-434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios JC, Melendez-Vasquez CV, Einheber S, Lustig M, Grumet M, Hemperly J, Peles E, Salzer JL ( 2000) Contactin-associated protein (Caspr) and contactin form a complex that is targeted to the paranodal junctions during myelination. J Neurosci 20: 8354-8364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbluth J ( 1976) Intramembranous particle distribution at the node of Ranvier and adjacent axolemma in myelinated axons of the frog brain. J Neurocytol 5: 731-745. [DOI] [PubMed] [Google Scholar]
- Rosenbluth J, Dupree JL, Popko B ( 2003) Nodal sodium channel domain integrity depends on the conformation of the paranodal junction, not on the presence of transverse bands. Glia 41: 318-325. [DOI] [PubMed] [Google Scholar]
- Sandri C, Van Buren JM, Akert K ( 1977) Membrane morphology of the vertebrate nervous system. Prog Brain Res 46: 201-265. [PubMed] [Google Scholar]
- Tao-Cheng JH, Rosenbluth J ( 1983) Axolemmal differentiation in myelinated fibers of rat peripheral nerves. Brain Res 285: 251-263. [DOI] [PubMed] [Google Scholar]
- Traka M, Dupree JL, Popko B, Karagogeos D ( 2002) The neuronal adhesion protein TAG-1 is expressed by Schwann cells and oligodendrocytes and is localized to the juxtaparanodal region of myelinated fibers. J Neurosci 22: 3016-3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westenbroek RE, Noebels JL, Catterall WA ( 1992) Elevated expression of type II Na+ channels in hypomyelinated axons of shiverer mouse brain. J Neurosci 12: 2259-2267. [DOI] [PMC free article] [PubMed] [Google Scholar]