

Abstract
Insulin resistance and diabetes might promote neurodegenerative disease, but a molecular link between these disorders is unknown. Many factors are responsible for brain growth, patterning, and survival, including the insulin-insulin-like growth factor (IGF)-signaling cascades that are mediated by tyrosine phosphorylation of insulin receptor substrate (IRS) proteins. Irs2 signaling mediates peripheral insulin action and pancreatic β-cell function, and its failure causes diabetes in mice. In this study, we reveal two important roles for Irs2 signaling in the mouse brain. First, disruption of the Irs2 gene reduced neuronal proliferation during development by 50%, which dissociated brain growth from Irs1-dependent body growth. Second, neurofibrillary tangles containing phosphorylated tau accumulated in the hippocampus of old Irs2 knock-out mice, suggesting that Irs2 signaling is neuroprotective. Thus, dysregulation of the Irs2 branch of the insulin-Igf-signaling cascade reveals a molecular link between diabetes and neurodegenerative disease.
Keywords: diabetes, insulin receptor substrates, Irs2, growth factors, tau phosphorylation, brain size, neuron survival, Alzheimer's disease
Introduction
Among mammals including mice, the size of the brain and its components are related to body size (Finlay and Darlington, 1995). Patterning and size of the brain are determined by the coordinated proliferation, death, and migration of neuronal precursors in the cerebellum, hippocampus, retinal ganglion, and other regions (Strom and Williams, 1998; Airey et al., 2001; Lu et al., 2001; Chenn and Walsh, 2002). Various factors regulate these processes, including extracellular matrix proteins, immunoglobins, peptide growth factors, inflammatory cytokines, and neurotransmitters (Horner and Gage, 2000). Developmental signals are especially important, including the sonic hedgehog pathway that balances proliferation or differentiation of neuronal precursors, and the wnt→β-catenin pathway that controls cell cycle exit (Goldowitz and Hamre, 1998; Chenn and Walsh, 2002). At least 50% of brain and body growth is mediated by the insulin-Igf-signaling system (Liu et al., 1993). Various heterologous signals integrate the effect of the insulin-like growth factors (IGFs) with other systems. Cytoskeletal signals might modulate insulin-IGF signaling through the activity of ρ GTPases (Sordella et al., 2002), and growth hormone upregulates peripheral but not central IGF1 levels, so the body can grow faster than the brain (Lupu et al., 2001). These relationships provide a molecular basis for understanding species-specific expansion of the brain and body size during development and neonatal growth (Ludwig et al., 1996; Goldowitz and Hamre, 1998).
The receptors for insulin and IGF1 are tyrosine kinases that mediate phosphorylation of the insulin receptor substrates (IRSs), including Irs1, Irs2, Irs3, and Irs4 (Yamada et al., 1997; Yenush and White, 1997). Irs1 and Irs2 are widely expressed and mediate insulin action in most tissues (Sun et al., 1992, 1995; Bernal et al., 1998), Irs3 is restricted to rodent adipose tissue but might be missing in people (Lavan et al., 1997; Bjornholm et al., 2002), and Irs4 is primarily detected in the brain, including the thymus, pituitary, and hypothalamus (Fantin et al., 1999; Uchida et al., 2000). Irs1 mediates the effects of Igf1 on somatic growth, because Irs1-/- mice are ∼50% smaller than normal. In contrast, Irs2-/- mice reach nearly normal size (Withers et al., 1999). Although both Irs1-/- and Irs2-/- mice are insulin resistant, only Irs2-/- mice develop diabetes resulting from impaired pancreatic β-cell survival and function (Bruning et al., 1998; Withers et al., 1998, 1999; Kulkarni et al., 1999; Michael et al., 2000; Previs et al., 2000). Female Irs2 knock-out mice are infertile and display increased food intake and mild obesity, suggesting that Irs2 signaling also promotes hypothalamic control of appetite and reproduction (Burks et al., 2000).
Phosphorylated IRS proteins activate multiple signaling pathways, including the phosphatidylinositol 3 (PI3)-kinase and extracellular signal-regulated kinase (ERK) cascades that directly regulate various physiological processes (Saltiel, 2001). Activation of the PI3-kinase→ Akt cascade promotes neuronal growth and survival by phosphorylating Foxo-transcription factors to regulate gene transcription and phosphorylating Bcl-2-associated death (BAD) protein to inhibit apoptosis (Brunet et al., 1999; Rodgers and Theibert, 2002). Akt inactivates glycogen synthase kinase-3β (GSK3β), which promotes cellular metabolism, proliferation, and survival (Cross et al., 1995). Activated PI3-kinase also promotes GABAergic transmission in the prefrontal cortical pyramidal neurons that play a key role in learning and memory (Ma et al., 2003). Akt also blocks the accumulation of β-amyloid plaques and neurofibrillary tangles that are associated with Alzheimer's disease (Kaytor and Orr, 2002).
Alzheimer's disease is characterized by progressive memory loss, impaired language, and behavior that culminate in death. Epidemiological studies suggest that insulin resistance and type 2 diabetes increase the risk for age-related cognitive decline (Finch and Cohen, 1997; Carantoni et al., 2000). Although the long-term sequelae of hyperglycemia, including vascular complications, are frequently implicated as the culprit, defects in the insulin-IGF-signaling system might contribute directly to memory loss and dementia (Stolk et al., 1997; Lovestone, 1999; Ott et al., 1999). Like diabetes, Alzheimer's disease occurs in many forms, in which most instances are late onset and sporadic, whereas well defined autosomal mutations cause early onset familial Alzheimer's disease (Hoyer, 2002). The disorder is ordinarily characterized by the accumulation of extracellular deposits of amyloid β-peptide that promotes inflammation and the intracellular accumulation of neurofibrillary tangles composed of paired helical filaments assembled from hyperphosphorylated forms of the microtubule-associated protein tau (Lucas et al., 2001; Selkoe, 2001; Weggen et al., 2001; Hardy and Selkoe, 2002). However, tau neurofibrillary tangles cause frontotemporal dementia without detectable β-amyloid plaques, suggesting that β-amyloid toxicity might be tau dependent (Selkoe and Podlisny, 2002). Insulin-IGF-signaling pathways might protect neurons from inflammation caused by β-amyloid by decreasing intracellular concentrations of β-amyloid (Solano et al., 2000; Gasparini et al., 2001). Neurons in transgenic mice overexpressing a mutant form of human amyloid precursor protein are resistant to apoptosis and develop β-amyloid deposits slowly, resulting from, at least in part, upregulation of insulin-like growth factor-2 and activation of the insulin-IGF-signaling cascade (Stein and Johnson, 2002). Tau phosphorylation is mediated by several kinases, including GSK3β (Hong and Lee, 1997; Lovestone and Reynolds, 1997). Thus, tau phosphorylation might be a direct consequence of reduced insulin-IGF signaling that occurs during aging.
Here, we reveal two important roles for the Irs2 branch of the insulin-IGF-signaling cascade in the brain. First, Irs2 signaling promotes neuronal proliferation that increases brain size during development. Second, Irs2 signaling promotes the dephosphorylation of tau. Thus, failure of the Irs2 branch of the insulin-IGF-signaling pathway might link neurodegeneration with dysregulated nutrient homeostasis caused by insulin resistance and β-cell failure.
Materials and Methods
Mice. The generation of Irs1 and Irs2 knock-out mice has been described previously (Withers et al., 1998, 1999). Intercrosses with mice lacking one copy of the Igf1 receptor were created, and genotyping of the animals was performed by Southern blotting as described previously (Withers et al., 1999). Mice were maintained on a normal light/dark cycle and handled in accordance with Joslin Diabetes Center Care and Use Committee protocols.
Histology and immunostaining. For immunostaining, animals were perfused for 60 sec with PBS and fixed for 30 min by transcardiac perfusion with 4% paraformaldehyde (PFA). The brains were dissected from perfused animals and postfixed in 4% PFA for 4 hr. Brains were either paraffin embedded or cryoprotected in 30% sucrose, and 5-30 μm sections were prepared. Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) assays were performed using the DNA FragEL Kit (Oncogen Research Products, San Diego, CA). Nissl and hematoxylin-eosin (HE) staining as well as immunostaining using anti-glial fibrillary acidic protein (GFAP) (GP-52; Progen, Darra, Queensland, Australia) or anti-bromodeoxyuridine (BrdU) (Boehringer Mannheim, Indianapolis, IN) as primary antibody were performed as described previously. For quantitation, sections were viewed using a Ziess Axiovert S100 microscope and video camera. Counts and analysis were done using Openlab software (Improvision, Lexington, MA).
BrdU incorporation. BrdU (100 μg/gm body weight; Boehringer Mannheim) was injected intraperitoneally into pregnant mice [embryonic day (E) 14] or postnatal day (P) 6 pups. Tissues were harvested after 48 hr and fixed in 4% PFA.
Isolation and culture of cerebellar granule cells. Cerebellar granule neurons were isolated from 5-d-old mouse litters. All manipulations were performed at 4°C unless indicated otherwise. Individual cerebella were isolated, the meninges were removed using a dissecting microscope, and the cerebella were washed three times in HHGN solution (1× HBSS, 2.5 mm HEPES, pH 7.4, 35 mm glucose, 4 mm sodium bicarbonate). Cerebella were then incubated in trypsin solution (10 mg/ml of trypsin, 100 μg/ml of DNase, in HHGN, pH 7.0, with 0.1N NaOH) for 15 min at room temperature (RT). Cerebella were placed on ice, washed three times in HHGN, and then triturated ∼25 times with 1 ml of DNase solution [10 μg/ml of DNase in basal medium eagle (BME)]. The cells were allowed to settle for 5 min at room temperature, the supernatant was transferred into fresh tubes, and the remaining pellet was triturated with an additional 1 ml of DNase solution for another 25 times. After settling, the supernatants were combined and the cells were centrifuged for 5 min at 1000 × g. Cell pellets were suspended in BME containing 10% fetal bovine serum, 100 U of penicillin-streptomycin, 2mm glutamine, and 25 mm KCl (culture medium), counted, and plated on poly-d-lysine-coated 96-well plates. After 24 hr, 10 μm cytosine arabinoside (araC) was added to the cultures to inhibit proliferation of non-neuronal cells.
Neuronal apoptosis. Apoptosis assays were performed after 96 hr (4 d in vitro). Cerebellar granule cells were washed with serum-free BME containing 0.2% BSA. Cells were then incubated with the indicated factors (IGF1 or BDNF) in BME containing 100 U of penicillin-streptomycin, 2 mm glutamine, 0.2% BSA, and 10 μm araC for 24 hr at 37°C. For experiments using LY294002 (LY; 25 μm) and PD98059 (PD; 40 μm), neurons were washed as described above and preincubated with either LY or PD for 30 min at 37°C. The cells were fixed directly in the wells using 10% (w/v) paraformaldehyde [4% (w/v) final], washed three times with PBS, permeabilized with 0.2% (v/v) Triton X-100, washed twice with PBS, and then incubated with Hoechst dye 33342 (10 μg/ml) in PBS for 10 min at RT.
Immunoblotting. For immunoblotting, brains were homogenized in a polytron containing 50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% (v/v) NP40, 5 mm EDTA, 5% (v/v) glycerol, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 1 mm phenylmethylsulfonyl fluoride, and 1 mm Na3VO4. Protein expression was determined from whole-brain lysates (50 μg) dissolved in Laemmli buffer and resolved on 7.5 or 10% SDS-PAGE gels. Proteins were transferred to nitrocellulose; the membrane was blocked with 3% dry milk solution and incubated with the appropriate antisera. Primary antibodies against GFAP (GP-52; Serotec, Indianapolis, IN), protein phosphatase (PP) 2A (multiple subunits; Santa Cruz Biotechnology, Santa Cruz, CA), PP2B catalytic subunit (Santa Cruz Biotechnology), tau 1/2 (Santa Cruz Biotechnology), AT8, and AT180 (Innogenetics, Gent, Belgium) were used. After incubation with protein A-HRP or HRP-labeled secondary antibodies (Santa Cruz Biotechnology), signals were detected using ECL reagents and exposure to x-ray film.
Results
Brain growth in Irs1- and Irs2-deficient mice
The size of nonolfactory brain components varies predictably among vertebrates and is closely related to body size (Finlay and Darlington, 1995). For 230 different species of mice, the brain-body ratio is 2.1%, and our population of wild-type C57BL/6 mice was indistinguishable from this average value (Table 1). However, before diabetes developed between 6 and 8 weeks of age, male Irs2-/- mice displayed a nearly normal body size with unexpectedly small brains (Fig. 1, Table 1). The small brains were detected in Irs2-/- embryos at E16 (Irs2-/-, 51 ± 6 mg; wild types, 74 ± 1 mg; p < 0.05) (Fig. 1b), and 3-4 weeks after birth, the brain/body ratio was only 1.4% (Table 1). Deletion of Irs2 had a global effect on brain size because the forebrain, cerebellum, and brainstem decreased proportionally (Table 1). In contrast, the brains in Irs1-/- mice were only moderately smaller, and the brain/body ratio was 4.2% (Fig. 1, Table 1), similar to the effect of growth hormone receptor knock-out (Lupu et al., 2001). In each case, the smaller skull might limit brain growth. Although male Irs2-/- mice die between 10 and 15 weeks of age because of extreme hyperglycemia, female Irs2-/- mice develop diabetes more slowly and survive for ≥6 months (Burks et al., 2000). However, longitudinal studies with female mice revealed that brain size remained proportionately small until the experiment was terminated after 5 months (data not shown).
Table 1.
Body and brain weights from mice 3-4 weeks of age
|
|
Wild-type |
Irs1−/− |
Irs2−/− |
|---|---|---|---|
| Body weight (gm) | 21.2 ± 1.4 | 8.9 ± 2.1 | 22.4 ± 3.7 |
| Brain weight (gm) | 0.45 ± 0.02 | 0.36 ± 0.02 | 0.28 ± 0.02 |
| Brain/body (%) | 2.1 ± 0.2 | 4.2 ± 0.9 | 1.3 ± 0.2 |
| (p = 0.94) | (p < 0.001) | (p < 0.001) | |
| Forebrain weight (gm) | 0.25 ± 0.02 | 0.23 ± 0.02 | 0.17 ± 0.03 |
| Cerebellum weight (gm) | 0.061 ± 0.004 | 0.05 ± 0.01 | 0.046 ± 0.007 |
| Cell surface (μm2) (motor neurons of the hypoglossal nucleus)
|
349 ± 33.9
|
350 ± 29
|
353 ± 48
|
Forebrains and cerebella were dissected carefully and weighed separately. Cell surface areas of the motor neurons (150 cells per group) of the hypoglossal nucleus were measured using OpenLab software (Improvision). All measurements represent the average ± SD of six animals per group; p values for the brain/body ratio are relative to the mean brain/body ratio (2.1 ± 0.29) reported for 230 different mouse strains (available at http://www.nervenet.org/main/databases.html).
Figure 1.

Brains of Irs2-/- mice are reduced in size. a, Brains of Irs2-/-, Irs1-/-, and wild-type (wt) mice at 4 weeks of age. b, Brains of Irs2-/- and wild-type mice at E16. Embryos were collected from timed pregnant mothers and fixed in 4% paraformaldehyde. All visible brain regions of the Irs2-/- appear small but normal. c, HE-stained sections of Irs2-/- and wild-type brains showing a global reduction in all regions. d, Total cell number of the cerebella of Irs2-/- and wild-type mice at P5. Cerebella were dispersed and the total cell number was determined (n = 6 per group).
We investigated various aspects of CNS development and structure but failed to reveal specific brain defects. Hematoxylin-eosin-stained serial brain sections from Irs2-/- or Irs1-/- mice at 4 weeks of age did not reveal gross structural malformations (Fig. 1c). The cytoarchitecture of the cortical layers was unchanged in Irs2-/- and Irs1-/- mice compared with wild-type controls (data not shown). Moreover, the cell surface area of motor neurons in the hypoglossal nucleus and large cell bodies in layer V of the motor cortex of Irs2-/- and Irs1-/- mice was identical to wild-type controls (Table 1). Immunostaining using antibodies against GFAP at P5, P7, P9, 3 weeks, 6 weeks, and 8 weeks of age revealed no pathological astrocyte activation in Irs2-/- mice (data not shown). These data excluded changes in cell size or brain inflammation as the cause of the small brains in Irs2-/- mice. Because cell density was normal in the different cortical regions of Irs2-/- mice, we hypothesized that the small Irs2-/- brains contained fewer cells and confirmed this hypothesis by counting the cells in dispersed preparations of P5 cerebella (Fig. 1d).
Apoptosis in IRS protein-deficient neurons
We postulated that the decreased number of cells in the Irs2-/-brain might be attributable to increased apoptosis, because previous studies reveal that IGF1 signaling strongly inhibits neuronal apoptosis (Kaytor and Orr, 2002). Surprisingly, the number of apoptotic cells detected by TUNEL assays was not increased in Irs2-/- cerebella during prenatal (E12, E14, and E16) or postnatal (P5, P6, P7, P9, 3 weeks, 6 weeks, and 8 weeks) development (Fig. 2a) (data not shown). Apoptosis in situ can be difficult to quantify, so we investigated cultured granule cells obtained from the cerebella to determine whether the deletion of Irs1 or Irs2 accelerates apoptosis. Previous results suggest that activation of the PI3-kinase→ Akt cascade promotes neuronal survival by promoting phosphorylation of GSK3β, Foxo1, and BAD (Brunet et al., 1999; Kaytor and Orr, 2002). As shown previously, withdrawal of serum and the reduction of KCl to 5 mm stimulated apoptosis of cultured granule cells (Fig. 2b). However, apoptosis increased similarly in wild type, Irs1-/-, or Irs2-/- granule cells, suggesting that Irs2 expression was not especially important (Fig. 2b). IGF1 and BDNF added separately restored normal survival, whereas IGF1 and BDNF added together were even more effective (Fig. 2b). Moreover, IGF1 was equipotent in supporting the survival of granule cells from wild-type, hemizygous (Irs1+/- or Irs2+/-), or homozygous (Irs1-/- or Irs2-/-) mice (Fig. 2c,d), and dose-response studies revealed no differences in IGF1 sensitivity (data not shown). The protective effect of IGF1 was abolished in all genotypes by the PI3-kinase inhibitor LY294002 but not by the MAP kinase kinase inhibitor PD98059 (data not shown). Because apoptosis was not enhanced in Irs2-/- neurons, these data exclude cell death as the molecular basis of the reduced cell number and brain size and suggest that either Irs1 or Irs2 signaling is sufficient or another signaling pathway might contribute.
Figure 2.
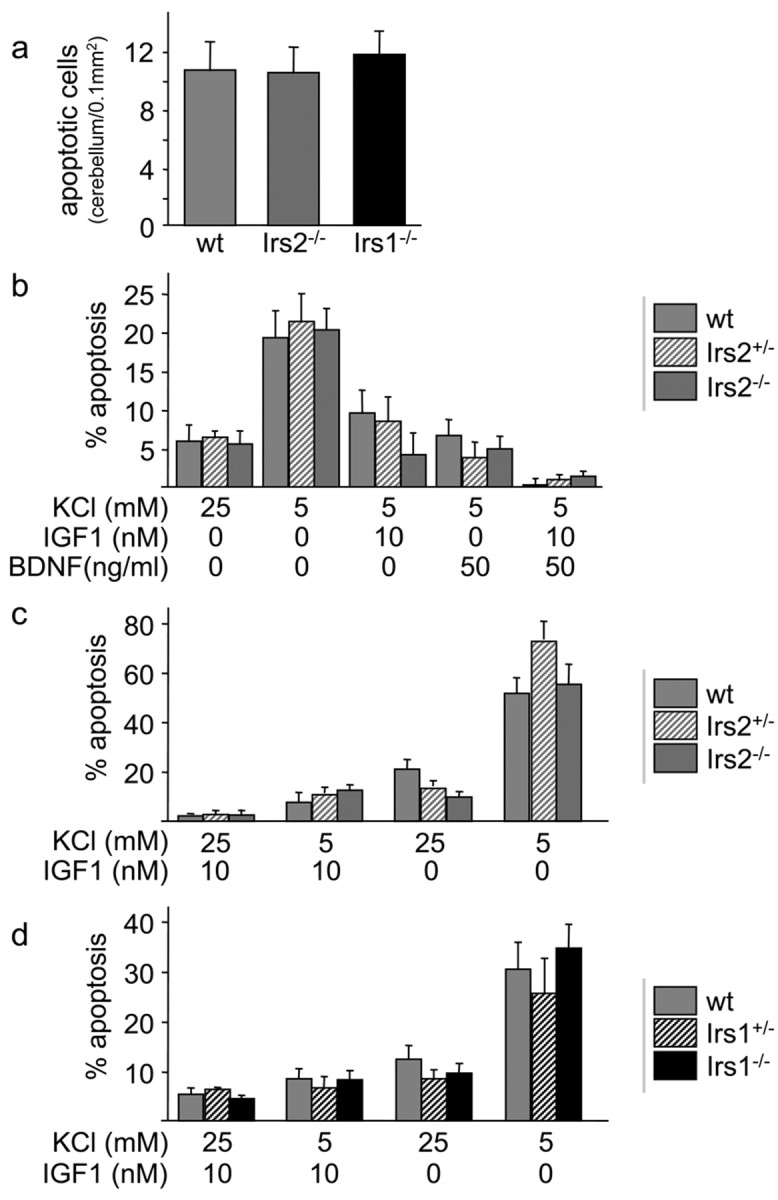
Apoptosis in brains and cultured neurons from mice lacking IRS proteins. a, TUNEL assay of the cerebellum at P6 reveals no differences between Irs2-/-, Irs1-/-, and wild-type (wt) mice. Quantification of apoptotic nuclei showed the same number per area in all genotypes. b, IGF1 and BDNF protect against neuronal apoptosis in the absence of Irs2. Cerebellar granule cells (P5) were cultured for 4 d in medium containing normal serum. Neurons were then incubated for an additional 24 hr in serum-free media containing 5 mm KCl, 5 mm KCl plus 10 nm IGF1, 5 mm KCl plus 50 ng/ml BDNF, or 25 mm KCL. Cells were stained with Hoechst 33342 and counted. Percentage apoptosis was calculated (n = 3 per genotype and experiment; 3 independent experiments showed similar results). c, Cerebellar granule cells from Irs2+/- × Irs2+/- litters (Irs2+/- or Irs2-/- cells) were cultured for 4 d, followed by incubation for 24 hr in serum-free media containing 25 mm KCl plus 10 nm IGF1 (+KCl, +IGF1), 10 nm IGF1 (-KCl, +IGF1), 25 mm KCl (+KCl, -IGF1), or minimal medium (-KCl, -IGF1) (n = 2 per genotype and experiment; 3 independent experiments showed similar results). d, Cerebellar granule cells from Irs1+/- × Irs1+/- litters (Irs1+/- or Irs1-/- cells) were cultured for 4 d, followed by incubation for 24 hr as described in c.
IRS2 promotes neuronal proliferation
Proliferation of neuronal precursors is critical during brain development, and defects in this process will alter brain size and function. To compare neuronal proliferation within the frontal cortex of wild-type and Irs2-/- mice, BrdU was injected into pregnant females at E14, and the embryonic tissue was harvested 48 hr later. Under these experimental conditions, the total number of BrdU-labeled neurons in Irs2-/- mice decreased 33% compared with wild-type controls (Irs2-/-, 281 ± 67 cells per 0.1 mm2; wild type, 419 ± 72 cells per 0.1 mm2; p = 0.002); experiments conducted at E12 showed similar results (data not shown). Although reduced, the distribution of BrdU-labeled neurons within the developing cortex was normal, suggesting that Irs2 was not required for the migration of newly formed neurons (Fig. 3a,d).
Figure 3.
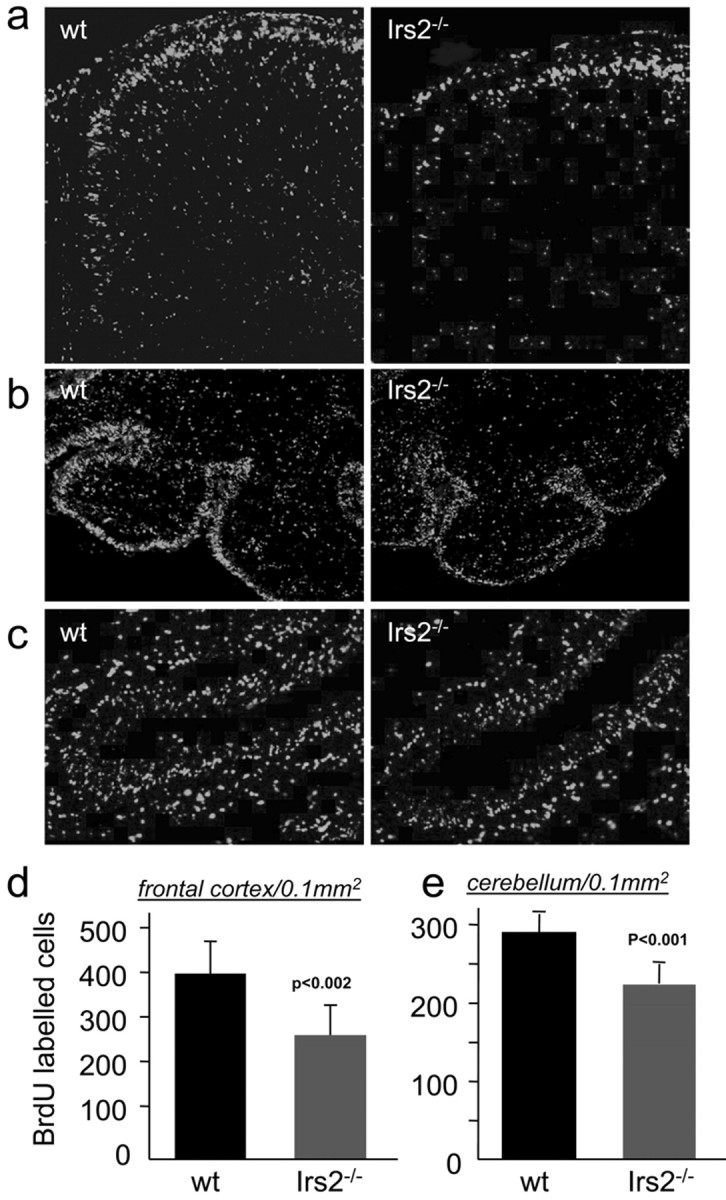
Neuronal proliferation is impaired in Irs2-/- brains. a, Four pregnant mice were injected intraperitoneally with BrdU at E14, and the embryos were harvested after 48 hr. Serial sections of the frontal cortex were obtained, and the total number of BrdU-labeled neurons per area was quantified using the Openlab software (Improvision). b, Mice (P6) were injected with BrdU, brains were fixed after 24 hr, and cryostat sections of the cerebellum were obtained (n = 4 per group). Sections were stained using an anti-BrdU antibody (Boehringer Mannheim). Labeled neurons were counted using the Openlab software (Improvision). c, Enlargement of b, demonstrating labeled neurons in the outer and inner granule layer. d,e, Quantification of BrdU-labeled cells in the frontal cortex (d) and cerebellum (e) from wild-type (wt) or Irs2-/- sections.
To study the proliferation of granule cells in neonates, BrdU was injected at P6, and cerebella were harvested 2 d later. The number of BrdU-labeled granule cells in Irs2-/- brains was reduced 23% compared with wild-type mice (Irs2-/-, 224 ± 25 cells per 0.1 mm2; wild type, 291 ± 24 cells per 0.1 mm2; p < 0.001) (Fig. 3e). Interestingly, the relative decrease in BrdU labeling (∼37% reduction in the frontal cortex) paralleled the reduction in total brain weight, consistent with reduced proliferation as the cause for decreased Irs2-/- brain size. Depth of the folia was also reduced in the Irs2-/- cerebella sections, consistent with reduced neurogenesis (Fig. 3b,c). Similar experiments with Irs1-/- mice revealed a highly variable reduction in proliferation of granule cells (13.7 ± 6.2%) that was disproportionate to the 50% reduced body size.
Igf1 receptor→Irs2 signaling promotes brain growth
Many studies show that Igf1 promotes brain growth and survival (Recio-Pinto et al., 1984; Dudek et al., 1997). To determine the relationship between Igf1 receptor (Igf1r) and Irs2 signaling during neurogenesis, we crossed Igf1r+/-::Irs2+/- compound heterozygous mice to generate the following five viable genotypes: Igf1r+/-, Irs2+/-, Irs2+/-::Igf1r+/-, Irs2-/-, and Irs2-/-::Igf1r+/-; Igf1r-/- mice die at birth and were not studied (Liu et al., 1993). Heterozygosity for either Irs2 or Igf1r did not influence brain size, whereas brain size was reduced slightly in Irs2+/-::Igf1r+/- mice (Fig. 4a). The Irs2-/-::Igf1r+/- brains were 15% smaller than the Irs2-/- brains (Fig. 4a); however, the corresponding decrease in brain/body ratio was not significant (Fig. 4b). Consistent with these results, ∼46% less Irs2-/- cerebella granule cells were labeled by BrdU in P6 brains and further reduced in Irs2-/-::Igf1r+/- mice (Fig. 4c).
Figure 4.

Igf1r heterozygosity causes a further reduction in brain size in Irs2-/- mice. a, Brain size of 6-week-old wild-type (wt), Igf1r+/-, Irs2+/-, Irs2+/-Igf1r+/-, Irs2-/-, and Igf1r+/-::Irs2-/- mice. b, Brain-body ratio (%) of 6-week-old mice of the different genotypes. c, BrdUincorporation in cerebella granule cells at P6 (48 hr survival) from wild type, Irs2-/-, or Irs2-/-Igf1r+/-. Cell counts and average cell size were calculated with NIH ImageJ. d, Apoptosis in the cerebella granule cells from Igf1r+/-, Irs2+/-Igf1r+/-, and Irs2-/-Igf1r+/- mice (P5). Granule cells were cultured for 4 d and then incubated for an additional 24 hr in serum-free medium containing 5 mm KCl,5mm KCl plus 10 nm IGF1,5 mm KCl plus 50 ng/ml BDNF. pvalues were calculated using Sigmaplot (Sigma, St. Louis, MO) version 8.1.
Apoptosis, assessed by TUNEL assays at various stages of development (E14, E16, P5, P8, 3 weeks, and 6 weeks of age), was not altered in any of the viable genotypes. Moreover, the ability of Igf1 and BDNF to prevent apoptosis was unchanged in cultured cerebella granule cells from these mice (Fig. 4d). Viable embryos (E16) deficient for both Igf1r and Irs2 did not show a further reduction in brain size compared with Igf1r-/- embryos (Igf1r-/-, 42 ± 4 mg; Irs2-/-Igf1r-/-, 42 ± 5 mg), suggesting that Igf1r signaling stimulates neuronal proliferation through the Irs2 branch of the insulin-IGF-signaling cascade. Moreover, the average size of the labeled cells was indistinguishable among wild-type, Irs2-/-, and Igf1r+/-Irs2-/- mice, suggesting that the Igf1r→Irs2 signal did not control cell size (Fig. 4c).
Tau is phosphorylated in the Irs2-/- brain
Alzheimer's disease is characterized by dysregulated metabolism that includes the accumulation of amyloid β-peptide and neurofibrillary tangles containing phosphorylated tau protein (Gasparini et al., 2002; Hardy and Selkoe, 2002; Hoyer, 2002). Phosphorylated tau accumulates in the hippocampus of patients with Alzheimer's disease and is thought to contribute to the neuronal degeneration (Hardy and Selkoe, 2002). Recent evidence suggests that dysfunction of the insulin-Igf-signaling cascade might contribute to this disorder (Gasparini et al., 2002; Liolitsa et al., 2002). Because activation of the insulin-IGF-signaling cascade promotes dephosphorylation of tau, we determined whether tau phosphorylation was increased in Irs2-/- brains. Tau was detected by immunoblotting in both wild-type and Irs2-/- brain extracts at 6 weeks of age. Tau isolated from the Irs2-/- brain migrated more slowly during SDS-PAGE, suggesting that it might be highly phosphorylated (Fig. 5a). Tau is phosphorylated at multiple sites including Ser202 and Thr231, but phosphorylation of Ser202, detectable with phosphospecific antibody AT8, correlates closely with neurodegeneration (Kaytor and Orr, 2002). The phosphorylation of Ser202 was significantly increased in Irs2-/- extracts, whereas phosphorylation of Thr231 was not changed (Fig. 5a). Ser202 phosphorylation was not increased at 4 weeks of age but was readily detected at 6 weeks of age (Fig. 5b).
Figure 5.

Immunoblotting with various antibodies conducted with whole-brain lysates. a, Lysates from 6-week-old mice were immunoblotted with antibodies against tau1/2 protein, the AT8 antibody against tau phosphorylated at Ser202, and the AT180 antibody against tau phosphorylated at Thr231. b, Lysates from 4.5- and 6-week-old mice were immunoblotted with AT8 antibody against tau phosphorylated at Ser202. c, Lysates from 6-week-old mice were immunoblotted with antibodies against phospho-GSK3β (Ser9) and GSK3β protein. d, Lysates from 6-week-old mice were immunoblotted with antibodies against PP2A scaffold subunit (PR65) and PP2A catalytic subunits. wt, Wild type.
Tau is phosphorylated on Ser202 by GSK3β, and insulin-Igf inhibits tau phosphorylation by inhibiting GSK3β activity through Akt-mediated phosphorylation on Ser9 (Kaytor and Orr, 2002). However, GSK3β is probably not directly phosphorylating tau in Irs2-/- mice, because its expression was normal at 6 weeks of age, and the inhibitory Ser9 phosphorylation was increased (Fig. 5c). Other kinases that might be involved, including CDK5 (cyclin-dependent kinase 5), MAPK (mitogen-activated protein kinase), or casein kinase-1, were not investigated. Protein phosphatase 2A dephosphorylates Ser202 on tau, and its activity might be essential to block the accumulation of phospho-tau (Sontag et al., 1996, 1999). PP2A is composed of a regulatory and catalytic subunit associated with a scaffold subunit (PR65) that stabilizes the catalytic complex. Consistent with increased phosphorylation of Ser202, the level of PP2A catalytic subunit was significantly reduced in Irs2-/- brain extracts, and PR65 was barely detected (Fig. 5d). Thus, disruption of the PP2A complex might contribute to tau phosphorylation in Irs2-/- brains.
Phosphorylated tau accumulates in old Irs2-/- brains
It is difficult to validate whether hyperphosphorylated tau forms neurofibrillary tangles in old Irs2-/- brains because Irs2-/- mice die from diabetes between 10 and 15 weeks of age (Withers et al., 1998). To study the effect of Irs2 deficiency in the brains of older mice, we restored β-cell function by crossing Irs2-/- mice with transgenic mice expressing Pdx1 (Dutta et al., 2001). Pdx1 is a transcription factor that is critical for β-cell function because it regulates pancreas development and promotes β-cell function in adults (Kushner et al., 2002). Pdx1 expression is significantly reduced in Irs2-/- β-cells, but transgenic expression of Pdx1 in Irs2-/- pancreas restores β-cell function and glucose-stimulated insulin secretion (Kushner et al., 2002). Thus, glucose tolerance of Irs2-/-::Pdx1tg improves significantly, and the mice survive for nearly 2 years (Fig. 6a).
Figure 6.
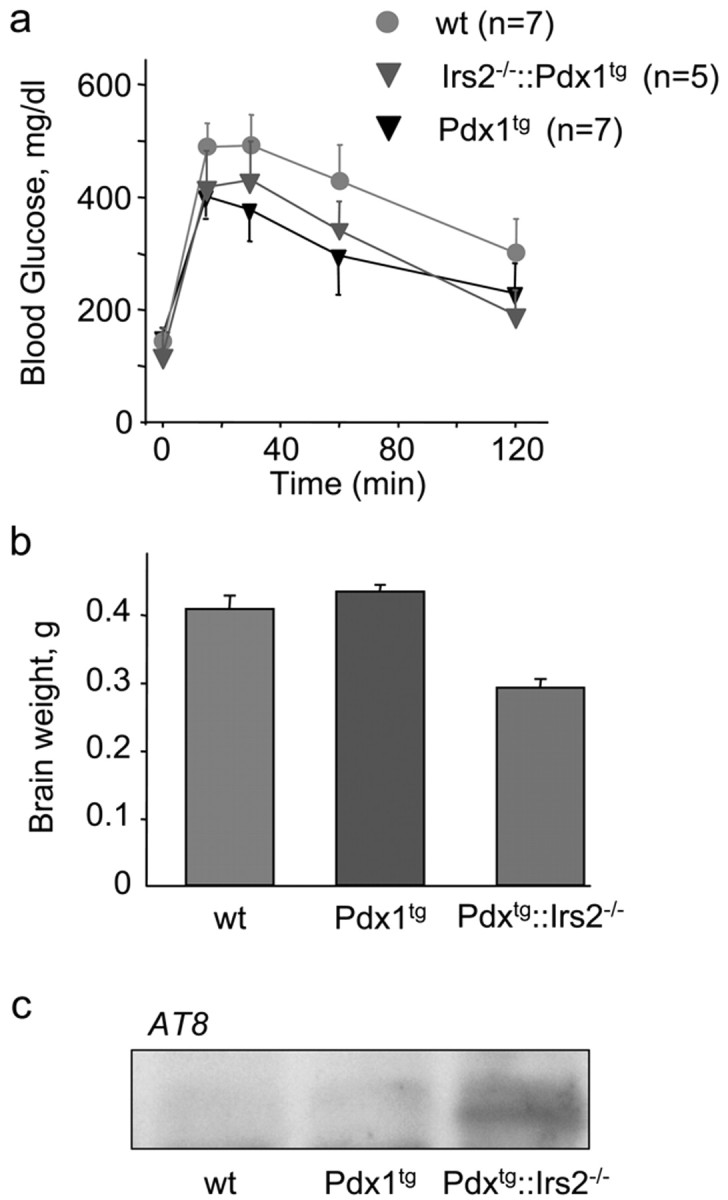
Glucose tolerance, brain size, and tau phosphorylation in wild-type (wt), Pdx1tg, and Irs2-/-::Pdx1tg mice. a, Glucose tolerance was measured at 14 months of age. b, Brain weight at ∼14 months of age. c, Western blot analysis using phospho-tau Ser202 antibody (AT8) of whole-brain lysates at 16 months of age.
Similar to young Irs2-/- mice, Irs2-/-::Pdx1tg mice have small brains (Fig. 6b). Moreover, whole-brain lysates from 16-month-old Irs2-/-::Pdx1tg mice contain elevated levels of phosphorylated tau, compared with age-matched wild-type and Pdx1 transgenic mice (Fig. 6c). Immunostaining, using the AT8 antibody, revealed increased axonal staining and cytoplasmic deposits of phosphorylated tau in Irs2-/-::Pdx1tg hippocampus sections compared with wild type and Pdx1tg between 12 and 16 months of age (Fig. 7). Although tau was hyperphosphorylated in young Irs2-/-::Pdx1tg mice, cytoplasmic deposits were only detected after 12 months and almost entirely in the hippocampus (Fig. 7). Thus, Irs2 deficiency was associated with increased tau phosphorylation that progressed to cytoplasmic deposits during aging, even when diabetes was prevented. However, up to 16 months of age, TUNEL assays and glial fibrillary acidic protein staining did not reveal increased apoptosis, suggesting that the accumulation of hyperphosphorylated tau in the Irs2-/- brain did not reach cytotoxic levels in our experiments.
Figure 7.

AT8 immunostaining from 12- or 16-month-old brains of wild-type (wt), Pdx1tg, and Irs2-/-::Pdx1tg mice. Pyramidal cells of the CA1 region or coronal sections through the corpus striatum (Striatum) were stained with the AT8 antibody (CA1; original magnification, 200×).
Discussion
The insulin-IGF1-signaling system has been recognized for a long time to play a critical role in the determination of body size (Birnbaum, 2002). Insulin and IGF1 signals are coordinated by tyrosine phosphorylation of homologous insulin receptor substrates Irs1 and Irs2 (White, 2002). Although both substrates have a similar composition, the Igf1r→ Irs2 branch of the pathway is entirely responsible for the effects of IGF1r on brain size. Moreover, Irs2 protects the aging brain from accumulation of phosphorylated tau that might form neurofibrillary tangles. These results are consistent with the role of Igf1 for neuronal proliferation in vivo and in vitro (DiCicco-Bloom and Black, 1988; Zackenfels et al., 1995; Ye et al., 1996; Anlar et al., 1999; Dentremont et al., 1999; Aberg et al., 2000; Pixley et al., 2000) and the ability of insulin or Igf1 to reduce tau phosphorylation in cultured neurons through a PI3-kinase-dependent mechanism (Hong and Lee, 1997). Because Irs2 signaling also mediates peripheral insulin action and promotes pancreatic β-cell function, dysregulation of Irs2 signaling provides a molecular basis for understanding the relationship between neurodegeneration and peripheral insulin resistance and diabetes (Gasparini et al., 2002).
We were surprised to find that brain and body growth diverge at the IRS proteins. Thus, upregulation of Irs2 might increase brain size with a minimal effect on the body, providing a mechanism for adaptive increase in brain size without a proportional increase in body size. This mechanism contrasts the growth hormone (GH)/GH receptor → Igf1/Igf1r→ Irs1 pathway that promotes body growth. Because Irs2 is also critical for pancreatic β-cell growth, survival, and function, the increased demand for nutrients caused by a large brain might be compensated by increased β-cell capacity. However, this relationship might be problematic because the failure of Irs2 signaling, resulting from a predetermined genetic program, chronic inflammation, or aging, might link insulin resistance syndromes and diabetes to neurodegeneration.
In cell-based experiments, insulin and IGF1 promote proliferation, increase neurite growth, and stimulate protein synthesis (Recio-Pinto et al., 1984; Mill et al., 1985; Fernyhough et al., 1989; Heidenreich and Toledo, 1989; Dudek et al., 1997). Our results show that Irs2 plays a unique role in the proliferative branch of the Igf1 signal in brain. Embryonic brain size is 55% of normal (N) in mice lacking Igf1r, and Irs2 mediates most or all of this effect. Hemizygous disruption of the Igf1r has no effect on brain size, but without Irs2, the Igf1r+/- brain was also 55%N. Although complete disruption of Irs2 in Igf1r-/- mice is usually embryonic lethal, it does not further reduce brain size at E16, suggesting that the effect of Igf1 on embryonic brain growth is mediated entirely by the Irs2 branch of the pathway.
Insulin and IGF1 strongly promote neuronal survival in vitro (Russell et al., 1998; Anlar et al., 1999). IGF1 activates Akt to promote BAD phosphorylation and its association with 14-3-3 to release and activate Bcl-2 (Datta et al., 1997; Brunet et al., 1999). However, in our experience, the lack of either Irs2 or Irs1 does not reduce survival of cultured cerebella granule cells, and Igf1 or Bdnf prevents apoptosis normally in cerebella granule cells lacking Irs1 or Irs2. Thus, Irs1 and Irs2 might be exactly redundant regarding Igf1-mediated anti-apoptosis, or other Igf-mediated signals might be involved.
Similar to mice, IGF1 deficiency in people causes mental retardation and microcephaly, suggesting that IGF1 has a critical role in human brain development (Woods et al., 1996). Direct evidence that Irs2 plays a role in human brain growth is not available. However, breaks at the distal end of human chromosome 13 (13q) near the IRS2 gene between microsatellite D13S285 and D13S1295 are frequently associated with microcephaly (Towfighi et al., 1987; Bektas et al., 1999). Interestingly, very distal deletions between D13S274 and D13S1311 are associated with microcephaly and neural tube defects, suggesting that partial Irs2 deficiency might contribute at least in part to the observed microcephaly (Luo et al., 2000). Because the elimination of the Igf1r→ Irs2 signaling pathway in people or mice does not cause anencephaly, neurogenesis needed to establish the basic brain pattern, including fibroblast growth factors, sonic hedgehog, and wnt→β-catenin signaling is independent of Igf→ Irs2 signaling cascade (Vaccarino et al., 1999).
The molecular basis for the distinct functions of Irs1 and Irs2 during neuronal proliferation and survival in the brain is not clear. Both proteins have similar composition, including a pleckstrin homology domain and a phosphotyrosine-binding domain that couple to activated receptors, and a tail of tyrosine phosphorylation sites that engage SH2 domains in various signaling proteins (Yenush and White, 1997). Cell-based experimental systems such as 32D myeloid progenitor cells fail to show clear differences in the regulation of downstream signaling pathways, including the PI3-kinase or the ERK cascade (Uchida et al., 2000). In vitro cultured neurons used in this study also failed to reveal the specificity for Irs1 or Irs2 during IGF1-stimulated anti-apoptosis. However, regulatory differences are likely to exist at the level of gene regulation, message stability and translation, protein stability, and interactions with upstream and downstream elements. Controlling these processes might provide ways to treat certain diseases.
Cognitive impairment is common in diabetes, and cerebral atrophy is common among young, otherwise healthy patients with type 1 diabetes (Sharma et al., 2003). Moreover, diabetes is associated with an increased incidence of Alzheimer's disease, suggesting that abnormal insulin signaling might contribute to dementia (Frolich et al., 1999; Blass et al., 2002; Gasparini et al., 2002; Hoyer, 2002). Activation of the insulin signaling machinery might be important for many aspects of neuronal function. In prefrontal cortical pyramidal neurons, insulin signaling promotes GABAergic transmission that facilitates learning and memory (Ma et al., 2003). An important marker of Alzheimer's disease and other brain dysfunctions is the accumulation of neurofibrillary lesions composed of hyperphosphorylated tau (Spillantini and Goedert, 1998). Tau is a neuronal microtubule-associated protein found predominantly in axons, where it promotes tubulin polymerization and stabilizes microtubules (Recio-Pinto et al., 1984; Binder et al., 1985; Drechsel et al., 1992). Because the disruption of Irs2 signaling promotes tau phosphorylation in the mouse hippocampus, dysregulated Irs2 signaling might be a common link between this marker of neurodegeneration and peripheral insulin resistance or diabetes.
A direct role for dysregulated signal transduction in the background of diabetes is difficult to establish because of multiple metabolic changes during peripheral insulin resistance and hyperglycemia. Even male Irs2-/- mice cannot be studied in old age because they die between 12 and 15 weeks of age. Normalization of β-cell function by transgenic expression of Pdx1 in the pancreas of Irs2-/- mice prevents the onset of diabetes (Kushner et al., 2002). However, as the euglycemic Irs2-/-::Pdx1tg mice age, neurofibrillary tangles containing phosphorylated tau accumulate in the hippocampus (Kushner et al., 2002). Thus, failure of Irs2 signaling in peripheral tissues and the brain reveals a potential mechanism to link insulin resistance and neurodegeneration without diabetes (Hoyer, 2002).
Several kinases are reported to phosphorylate tau, including Gsk3β, Cdk5, and probably others (Lee et al., 2000; Kaytor and Orr, 2002). Gsk3β is thought to play an important role in tau phosphorylation, but its activity is not elevated in Irs2-/- brains. Gsk3β transcript level [estimated from Affymetrix (Santa Clara, CA) MGU74v2 arrays; data not shown] and protein level were normal, and its specific activity is probably reduced because of elevated phosphorylation of Ser9 (we did not assess a possible role for Cdk5). However, tau phosphorylation might be increased by relative inactivation of PP2A in Irs2-/- brains. PP2A associates with tau and dephosphorylates Ser202 (Garcia et al., 2000), and in Irs2-/- brains, its catalytic and scaffold subunits are significantly reduced. However, direct experiments are required to establish the biochemical mechanism of tau hyperphosphorylation in Irs2-/- brains.
The accumulation of β-amyloid deposits is also an important characteristic of Alzheimer's disease, and several reports suggest that amyloid β-peptide plaques are associated with the accumulation of phosphorylated tau (Hardy and Selkoe, 2002). Amyloid β-peptide plaques activate microglial and astrocytes that cause inflammation in the brain (Selkoe and Podlisny, 2002). Many proinflammatory signaling pathways inhibit the activity of Irs proteins by promoting Jnk-mediated serine phosphorylation or Socs-mediated ubiquitination (Aguirre et al., 2002; Rui et al., 2002). The inhibition of Irs2 signaling byβ-amyloid-induced inflammation might provide a molecular link between these hallmarks of Alzheimer's disease. Because the murine ortholog of human β-amyloid fails to form deposits, we are unable to comment on the possibility that Irs2-/- brains are prone to β-amyloid deposition.
In summary, our results show that Igf1r→ Irs2 signaling is important for brain size by promoting proliferation during development. Moreover, during aging, defects in Irs2 signaling might contribute to the pathobiology of neurodegenerative disorders such as Alzheimer's disease. Because Irs2 signaling is also required for pancreatic β-cell growth and function, failure of Irs2 might be a common link between dysregulated peripheral nutrient homeostasis and neurodegeneration. Lifestyle changes or drugs that promote Irs2 signaling might provide a rational approach to treat the complications of insulin resistance, including diabetes and neurodegeneration.
Footnotes
This work was supported by National Institutes of Health Grants DK43808 and DK38712. We are grateful to C. Cahill for preparation of ultrathin sections and procedural help and K. Copps for discussion and editorial help. We thank M. Montminy for the Pdx1 transgenic mouse.
Correspondence should be addressed to Dr. Morris F. White, Howard Hughes Medical Institute, Joslin Diabetes Center, 1 Joslin Place, Boston, MA 02215. E-mail: morris.white@joslin.harvard.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/237084-09$15.00/0
M.S., D.P.B., and D.J.B. contributed equally to this work.
References
- Aberg M, Aberg N, Hedbacker H, Oscarsson J, Eriksson P ( 2000) Peripheral infusion of IGF-1 selectively induces neurogenesis in the adult rat hippocampus. J Neurosci 20: 2896-2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre V, Werner ED, Giraud J, Lee YH, Shoelson SE, White MF ( 2002) Phosphorylation of ser307 in insulin receptor substrate-1 blocks interactions with the insulin receptor and inhibits insulin action. J Biol Chem 277: 1531-1537. [DOI] [PubMed] [Google Scholar]
- Airey DC, Lu L, Williams RW ( 2001) Genetic control of the mouse cerebellum: identification of quantitative trait loci modulating size and architecture. J Neurosci 21: 5099-5109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anlar B, Sullivan KA, Feldman EL ( 1999) Insulin-like growth factor-I and central nervous system development. Horm Metab Res 31: 120-125. [DOI] [PubMed] [Google Scholar]
- Bektas A, Warram JH, White MF, Krolewski AS, Doria A ( 1999) Exclusion of insulin receptor substrate 2 (IRS-2) as a major locus for early-onset autosomal dominant type 2 diabetes. Diabetes 48: 640-642. [DOI] [PubMed] [Google Scholar]
- Bernal D, Almind K, Yenush L, Ayoub M, Zhang Y, Rosshani L, Larsson C, Pedersen O, White MF ( 1998) IRS-2 amino acid polymorphisms are not associated with random type 2 diabetes among Caucasians. Diabetes 47: 976-979. [DOI] [PubMed] [Google Scholar]
- Binder LI, Frankfurter A, Rebhun LI ( 1985) The distribution of tau in the mammalian central nervous system. J Cell Biol 101: 1371-1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnbaum MJ ( 2002) RhoGAP: the next big thing for small mice? Dev Cell 2: 521-523. [DOI] [PubMed] [Google Scholar]
- Bjornholm M, He AR, Attersand A, Lake S, Liu SC, Lienhard GE, Taylor S, Arner P, Zierath JR ( 2002) Absence of functional insulin receptor substrate-3 (IRS-3) gene in humans. Diabetologia 45: 1697-1702. [DOI] [PubMed] [Google Scholar]
- Blass JP, Gibson GE, Hoyer S ( 2002) The role of the metabolic lesion in Alzheimer's disease. J Alzheimers Dis 4: 225-232. [DOI] [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME ( 1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96: 857-868. [DOI] [PubMed] [Google Scholar]
- Bruning JC, Michael MD, Winnay JN, Hayashi T, Horsch D, Accili D, Goodyear LJ, Kahn CR ( 1998) A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol Cell 2: 559-569. [DOI] [PubMed] [Google Scholar]
- Burks DJ, de Mora JF, Schubert M, Withers DJ, Myers MG, Towery HH, Altamuro SL, Flint CL, White MF ( 2000) IRS-2 pathways integrate female reproduction and energy homeostasis. Nature 407: 377-382. [DOI] [PubMed] [Google Scholar]
- Carantoni M, Zuliani G, Munari MR, D'Elia K, Palmieri E, Fellin R ( 2000) Alzheimer disease and vascular dementia: relationships with fasting glucose and insulin levels. Dement Geriatr Cogn Disord 11: 176-180. [DOI] [PubMed] [Google Scholar]
- Chenn A, Walsh CA ( 2002) Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science 297: 365-369. [DOI] [PubMed] [Google Scholar]
- Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA ( 1995) Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378: 785-789. [DOI] [PubMed] [Google Scholar]
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME ( 1997) Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91: 231-241. [DOI] [PubMed] [Google Scholar]
- Dentremont KD, Ye P, D'Ercole AJ, O'Kusky JR ( 1999) Increased insulin-like growth factor-I (IGF-I) expression during early postnatal development differentially increases neuron number and growth in medullary nuclei of the mouse. Brain Res Dev Brain Res 114: 135-141. [DOI] [PubMed] [Google Scholar]
- DiCicco-Bloom E, Black IB ( 1998) Insulin growth factors regulate the mitotic cycle in cultured rat sympathetic neuroblasts. Proc Natl Acad Sci USA 85: 4066-4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drechsel DN, Hyman AA, Cobb MH, Kirschner MW ( 1992) Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol Biol Cell 3: 1141-1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME ( 1997) Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science 275: 661-665. [DOI] [PubMed] [Google Scholar]
- Dutta S, Gannon M, Peers B, Wright C, Bonner-Weir S, Montminy M ( 2001) PDX:PBX complexes are required for normal proliferation of pancreatic cells during development. Proc Natl Acad Sci USA 98: 1065-1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantin VR, Lavan BE, Wang Q, Jenkins NA, Gilbert DJ, Copeland NG, Keller SR, Lienhard GE ( 1999) Cloning, tissue expression, and chromosomal location of the mouse insulin receptor substrate 4 gene. Endocrinology 140: 1329-1337. [DOI] [PubMed] [Google Scholar]
- Fernyhough P, Mill JF, Roberts JL, Ishii DN ( 1989) Stabilization of tubulin mRNAs by insulin and insulin-like growth factor I during neurite formation. Brain Res Mol Brain Res 6: 109-120. [DOI] [PubMed] [Google Scholar]
- Finch CE, Cohen DM ( 1997) Aging, metabolism, and Alzheimer disease: review and hypotheses. Exp Neurol 143: 82-102. [DOI] [PubMed] [Google Scholar]
- Finlay BL, Darlington RB ( 1995) Linked regularities in the development and evolution of mammalian brains. Science 268: 1578-1584. [DOI] [PubMed] [Google Scholar]
- Frolich L, Blum-Degen D, Riederer P, Hoyer S ( 1999) A disturbance in the neuronal insulin receptor signal transduction in sporadic Alzheimer's disease. Ann NY Acad Sci 893: 290-293. [DOI] [PubMed] [Google Scholar]
- Garcia A, Cereghini S, Sontag E ( 2000) Protein phosphatase 2A and phosphatidylinositol 3-kinase regulate the activity of Sp1-responsive promoters. J Biol Chem 275: 9385-9389. [DOI] [PubMed] [Google Scholar]
- Gasparini L, Gouras GK, Wang R, Gross RS, Beal MF, Greengard P, Xu H ( 2001) Stimulation of beta-amyloid precursor protein trafficking by insulin reduces intraneuronal beta-amyloid and requires mitogen-activated protein kinase signaling. J Neurosci 21: 2561-2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparini L, Netzer WJ, Greengard P, Xu H ( 2002) Does insulin dysfunction play a role in Alzheimer's disease? Trends Pharmacol Sci 23: 288-293. [DOI] [PubMed] [Google Scholar]
- Goldowitz D, Hamre K ( 1998) The cells and molecules that make a cerebellum. Trends Neurosci 21: 375-382. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ ( 2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297: 353-356. [DOI] [PubMed] [Google Scholar]
- Heidenreich KA, Toledo SP ( 1989) Insulin receptors mediate growth effects in cultured fetal neurons. I. Rapid stimulation of protein synthesis. Endocrinology 125: 1451-1457. [DOI] [PubMed] [Google Scholar]
- Hong M, Lee VM ( 1997) Insulin and insulin-like growth factor-1 regulate tau phosphorylation in cultured human neurons. J Biol Chem 272: 19547-19553. [DOI] [PubMed] [Google Scholar]
- Horner PJ, Gage FH ( 2000) Regenerating the damaged central nervous system. Nature 407: 963-969. [DOI] [PubMed] [Google Scholar]
- Hoyer S ( 2002) The aging brain. Changes in the neuronal insulin/insulin receptor signal transduction cascade trigger late-onset sporadic Alzheimer disease (SAD). A mini-review. J Neural Transm 109: 991-1002. [DOI] [PubMed] [Google Scholar]
- Kaytor MD, Orr HT ( 2002) The GSK3 beta signaling cascade and neurodegenerative disease. Curr Opin Neurobiol 12: 275-278. [DOI] [PubMed] [Google Scholar]
- Kulkarni RN, Bruning JC, Winnay JN, Postic C, Magnuson MA, Kahn CR ( 1999) Tissue-specific knockout of the insulin receptor in pancreatic β cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell 96: 329-339. [DOI] [PubMed] [Google Scholar]
- Kushner JA, Ye J, Schubert M, Burks DJ, Dow MA, Flint CL, Dutta S, Wright CV, Montminy MR, White MF ( 2002) Pdx1 restores beta cell function in Irs2 knockout mice. J Clin Invest 109: 1193-1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavan BE, Lane WS, Lienhard GE ( 1997) The 60-kDa phosphotyrosine protein in insulin-treated adipocytes is a new member of the insulin receptor substrate family. J Biol Chem 272: 11439-11443. [DOI] [PubMed] [Google Scholar]
- Lee MS, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH ( 2000) Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature 405: 360-364. [DOI] [PubMed] [Google Scholar]
- Liolitsa D, Powell J, Lovestone S ( 2002) Genetic variability in the insulin signalling pathway may contribute to the risk of late onset Alzheimer's disease. J Neurol Neurosurg Psychiatry 73: 261-266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JP, Baker J, Perkins JA, Robertson EJ, Efstratiadis A ( 1993) Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r). Cell 75: 59-72. [PubMed] [Google Scholar]
- Lovestone S ( 1999) Diabetes and dementia: is the brain another site of end-organ damage? Neurology 53: 1907-1909. [DOI] [PubMed] [Google Scholar]
- Lovestone S, Reynolds CH ( 1997) The phosphorylation of tau: a critical stage in neurodevelopment and neurodegenerative processes. Neuroscience 78: 309-324. [DOI] [PubMed] [Google Scholar]
- Lu L, Airey DC, Williams RW ( 2001) Complex trait analysis of the hippocampus: mapping and biometric analysis of two novel gene loci with specific effects on hippocampal structure in mice. J Neurosci 21: 3503-3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas JJ, Hernandez F, Gomez-Ramos P, Moran MA, Hen R, Avila J ( 2001) Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J 20: 27-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig T, Eggenschwiler J, Fisher P, D'Ercole AJ, Davenport ML, Efstratiadis A ( 1996) Mouse mutants lacking the type 2 IGF receptor (IGF2R) are rescued from perinatal lethality in Igf2 and Igf1r null backgrounds. Dev Biol 177: 517-535. [DOI] [PubMed] [Google Scholar]
- Luo J, Balkin N, Stewart J, Sarwark J, Charrow J, Nye J ( 2000) Neural tube defects and the 13q deletion syndrome: evidence for a critical region in 13q33-34. Am J Med Genet 91: 227-230. [DOI] [PubMed] [Google Scholar]
- Lupu F, Terwilliger JD, Lee K, Segre GV, Efstratiadis A ( 2001) Roles of growth hormone and insulin-like growth factor 1 in mouse postnatal growth. Dev Biol 229: 141-162. [DOI] [PubMed] [Google Scholar]
- Ma XH, Zhong P, Gu Z, Feng J, Yan Z ( 2003) Muscarinic potentiation of GABA(A) receptor currents is gated by insulin signaling in the prefrontal cortex. J Neurosci 23: 1159-1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael MD, Kulkarni RN, Postic C, Previs S, Shulman GI, Magnuson MA, Kahn CR ( 2000) Loss of insulin signaling in hepatocyte leads to severe insulin resistance and progressive hepatic dysfunction. Mol Cell 6: 87-97. [PubMed] [Google Scholar]
- Mill JF, Chao MV, Ishii DN ( 1985) Insulin, insulin-like growth factor II, and nerve growth factor effects on tubulin mRNA levels and neurite formation. Proc Natl Acad Sci USA 82: 7126-7130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM ( 1999) Diabetes mellitus and the risk of dementia: the Rotterdam Study. Neurology 53: 1937-1942. [DOI] [PubMed] [Google Scholar]
- Pixley SK, Danogoria NS, Odoms KK, Hastings L ( 2000) Effects of insulin-like growth factor 1 on olfactory neurogensis in vivo and in vitro Ann NY Acad Sci 855: 244-247. [DOI] [PubMed] [Google Scholar]
- Previs SF, Withers DJ, Ren JM, White MF, Shulman GI ( 2000) Contrasting effects of IRS-1 vs IRS-2 gene disruption on carbohydrate and lipid metabolism in vivo J Biol Chem 275: 38990-38994. [DOI] [PubMed] [Google Scholar]
- Recio-Pinto E, Lang FF, Ishii DN ( 1984) Insulin and insulin-like growth factor II permit nerve growth factor binding and the neurite formation response in cultured human neuroblastoma cells. Proc Natl Acad Sci USA 81: 2562-2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers EE, Theibert AB ( 2002) Functions of PI 3-kinase in development of the nervous system. Int J Dev Neurosci 20: 187-197. [DOI] [PubMed] [Google Scholar]
- Rui L, Yuan M, Frantz D, Shoelson S, White MF ( 2002) SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J Biol Chem 277: 42394-42398. [DOI] [PubMed] [Google Scholar]
- Russell JW, Windebank AJ, Schenone A, Feldman EL ( 1998) Insulin-like growth factor-I prevents apopstis in neurons after nerve growth factor withdrawal. J Neurobiol 36: 455-467. [DOI] [PubMed] [Google Scholar]
- Saltiel AR ( 2001) New perspectives into the molecular pathogenesis and treatment of type 2 diabetes. Cell 104: 517-529. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ ( 2001) Alzheimer's disease: genes, proteins, and therapy. Physiol Rev 81: 741-766. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, Podlisny MB ( 2002) Deciphering the genetic basis of Alzheimer's disease. Annu Rev Genomics Hum Genet 3: 67-99. [DOI] [PubMed] [Google Scholar]
- Sharma J, Bakshi R, Lee D, Hachinski V, Chan RKT ( 2003) Cerebral atrophy in young, otherwise healthy patients with type 1 diabetes mellitus. Neurology [Suppl] 35: 3-29. [Google Scholar]
- Solano DC, Sironi M, Bonfini C, Solerte SB, Govoni S, Racchi M ( 2000) Insulin regulates soluble amyloid precursor protein release via phosphatidyl inositol 3 kinase-dependent pathway. FASEB J 14: 1015-1022. [DOI] [PubMed] [Google Scholar]
- Sontag E, Nunbhakdi-Craig V, Lee G, Bloom GS, Mumby MC ( 1996) Regulation of the phosphorylation state and microtubule-binding activity of tau by protein phosphatase 2A. Neuron 17: 1201-1207. [DOI] [PubMed] [Google Scholar]
- Sontag E, Nunbhakdi-Craig V, Lee G, Brandt R, Kamibayashi C, Kuret J, White III CL, Mumby MC, Bloom GS ( 1999) Molecular interactions among protein phosphatase 2A, tau, and microtubules. Implications for the regulation of tau phosphorylation and the development of tauopathies. J Biol Chem 274: 25490-25498. [DOI] [PubMed] [Google Scholar]
- Sordella R, Classon M, Hu KQ, Matheson SF, Brouns MR, Fine B, Zhang L, Takami H, Yamada Y, Settleman J ( 2002) Modulation of CREB activity by the Rho GTPase regulates cell and organism size during mouse embryonic development. Dev Cell 2: 553-565. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Goedert M ( 1998) Tau protein pathology in neurodegenerative diseases. Trends Neurosci 21: 428-433. [DOI] [PubMed] [Google Scholar]
- Stein TD, Johnson JA ( 2002) Lack of neurodegeneration in transgenic mice overexpressing mutant amyloid precursor protein is associated with increased levels of transthyretin and the activation of cell survival pathways. J Neurosci 22: 7380-7388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolk RP, Breteler MM, Ott A, Pols HA, Lamberts SW, Grobbee DE, Hofman A ( 1997) Insulin and cognitive function in an elderly population. The Rotterdam Study. Diabetes Care 20: 792-795. [DOI] [PubMed] [Google Scholar]
- Strom RC, Williams RW ( 1998) Cell production and cell death in the generation of variation in neuron number. J Neurosci 18: 9948-9953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun XJ, Miralpeix M, Myers Jr MG, Glasheen EM, Backer JM, Kahn CR, White MF ( 1992) The expression and function of IRS-1 in insulin signal transmission. J Biol Chem 267: 22662-22672. [PubMed] [Google Scholar]
- Sun XJ, Wang LM, Zhang Y, Yenush L, Myers Jr MG, Glasheen EM, Lane WS, Pierce JH, White MF ( 1995) Role of IRS-2 in insulin and cytokine signalling. Nature 377: 173-177. [DOI] [PubMed] [Google Scholar]
- Towfighi J, Ladda RL, Sharkey FE ( 1987) Purkinje cell inclusions and “atelencephaly” in 13q-chromosomal syndrome. Arch Pathol Lab Med 111: 146-150. [PubMed] [Google Scholar]
- Uchida T, Myers Jr MG, White MF ( 2000) IRS-4 mediates activation of PKB/Akt during insulin stimulation without inhibition of apoptosis. Mol Cell Biol 20: 126-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaccarino F, Schwartz ML, Raballo R, Rhee L, Rhee J, Zhou M, Doetschman TC, Coffin J, Wyland J, Hung Y ( 1999) Changes in cerebral cortex size are governed by fibroblast growth factor during embryogenesis. Nat Neurosci 2: 246-253. [DOI] [PubMed] [Google Scholar]
- Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, Findlay KA, Smith TE, Murphy MP, Bulter T, Kang DE, Marquez-Sterling N, Golde TE, Koo EH ( 2001) A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature 414: 212-216. [DOI] [PubMed] [Google Scholar]
- White MF ( 2002) IRS proteins and the common path to diabetes. Am J Physiol Endocrinol Metab 283: E413-E422. [DOI] [PubMed] [Google Scholar]
- Withers DJ, Gutierrez JS, Towery H, Burks DJ, Ren JM, Previs S, Zhang Y, Bernal D, Pons S, Shulman GI, Bonner-Weir S, White MF ( 1998) Disruption of IRS-2 causes type 2 diabetes in mice. Nature 391: 900-904. [DOI] [PubMed] [Google Scholar]
- Withers DJ, Burks DJ, Towery HH, Altamuro SL, Flint CL, White MF ( 1999) Irs-2 coordinates Igf-1 receptor-mediated beta-cell development and peripheral insulin signalling. Nat Genet 23: 32-40. [DOI] [PubMed] [Google Scholar]
- Woods KA, Camacho-Hubner C, Savage MO, Clark AJL ( 1996) Intrauterine growth retardation and postnatal growth failure associated with deletion of the insulin-like growth factor I gene. N Engl J Med 335: 1363-1367. [DOI] [PubMed] [Google Scholar]
- Yamada M, Ohnishi O, Sano S, Nakatani A., Ikeuchi T, Hatanaka H ( 1997) Insulin receptor substrate (IRS)-1 and IRS-2 are tyrosine-phosphorylated and associated with phosphatidylinositol 3-kinase in response to brain-derived neurotrophic factor cultured cerebral cortical neurons. J Biol Chem 272: 30334-30339. [DOI] [PubMed] [Google Scholar]
- Ye P, Xing Y, Dai Z, D'Ercole AJ ( 1996) In vivo actions of insulin-growth factor-I (IGF-I) on cerebellum development in transgenic mice: evidence that IGF-1 increases proliferation of granule cell progenitors. Brain Res Dev Brain Res 95: 44-54. [DOI] [PubMed] [Google Scholar]
- Yenush L, White MF ( 1997) The IRS-signaling system during insulin and cytokine action. BioEssays 19: 491-500. [DOI] [PubMed] [Google Scholar]
- Zackenfels K, Oppenheim RW, Rohrer H ( 1995) Evidence for an important role of IGF-I and IGF-II for the early development of chick sympathetic neurons. Neuron 14: 731-741. [DOI] [PubMed] [Google Scholar]