

Abstract
Basic and clinical studies suggest that neurokinin 1 (NK1) receptor antagonists have efficacy in the treatment of affective disorders through effects on the dorsal raphe nucleus (DR), a source of forebrain-projecting serotonin (5-HT) neurons that has also been implicated in affective disorders. To investigate the regulation of the DR-5-HT system by NK1 receptors, the effects of substance P (an NK1 agonist) on rat DR neuronal activity were characterized. Most of the DR neurons (83%; n = 47 total) were inhibited by substance P microinfusion into the DR, and in some cases (17%) this was preceded by a brief activation. Pure excitation was observed in a small population of neurons (17%) that were localized in the dorsal DR, where NK1 receptors are most dense. Sendide, a selective NK1 antagonist, attenuated the effects of substance P, indicating that they were mediated by NK1 receptor activation. The selective 5-HT1A antagonist, WAY 100635, administered systemically or into the DR, prevented the inhibitory effects of substance P, implicating DR 5-HT1A receptors in this response. Finally, microinfusion of the excitatory amino acid antagonist, kynurenic acid, into the DR prevented both excitatory and inhibitory effects. The results suggest that NK1 receptor activation in the DR excites a population of 5-HT neurons via glutamatergic transmission. This results in 5-HT release throughout the DR, activation of 5-HT1A receptors, and subsequent inhibition. Interactions between NK1 and 5-HT1A receptors within DR neural networks may contribute to the mechanism of action of novel antidepressants acting at NK1 receptors.
Keywords: serotonin, neurokinin 1, anxiety, glutamate, substance P, dorsal raphe nucleus, sendide
Introduction
Substance P, acting primarily at the neurokinin 1 (NK1) receptor, has been well studied for its role in pain (Severini et al., 2002). Substance P neurotransmission has also been associated with aversion and anxiety in several behavioral models (Gradin et al., 1992; Aguiar and Brandao, 1994), however, and recent interest has focused on the potential utility of NK1 receptor antagonists as antidepressants and anxiolytics (Kramer et al., 1998; Rupniak and Kramer, 1999; Stout et al., 2001).
Evidence suggests that the potential therapeutic effects of NK1 antagonists are mediated by the dorsal raphe nucleus (DR), a major source of forebrain serotonin (5-HT) that has been implicated in affective disorders. Thus, NK1 receptor immunoreactivity is robustly expressed in dendrites in the DR (Maeno et al., 1993; Froger et al., 2001; Commons and Valentino, 2002). NK1 receptor disruption, either by antagonist treatment or by genetic alteration, results in desensitization of the autoinhibitory 5-HT1A receptor and enhanced serotonergic neurotransmission (Froger et al., 2001; Santarelli et al., 2001). Because these effects are similar to those produced by administration of classic antidepressants that directly interact with serotonergic neurotransmission, it has been suggested that they are integral to the therapeutic efficacy of these drugs (Froger et al., 2001; Haddjeri and Blier, 2001; Santarelli et al., 2001).
Given evidence that NK1 antagonists exhibit anti-anxiety/antidepressant potential through activation of the DR-5-HT system, it might be predicted that anxiogenic effects associated with NK1 receptor activation are mediated by inhibition of this system; however, few studies have examined the effects of NK1 receptor activation on the DR-5-HT system. One recent study demonstrated that NK1 receptor activation increased the frequency of EPSCs in DR neurons in vitro through an excitatory amino acid mechanism (Liu et al., 2002). Although this is consistent with the reported localization of NK1 receptor immunoreactivity on glutamatergic neurons in the DR (Commons and Valentino, 2002), it suggests that NK1 agonists activate rather than inhibit the DR-5-HT system. To further elucidate the regulation of the DR-5-HT system by the NK1 receptor that may contribute to the pathophysiology of affective disorders, the present study characterized the effects of the endogenous NK1 agonist, substance P, on activity of rat DR neurons in vivo.
Materials and Methods
Subjects. Adult male Sprague Dawley rats (Taconic Farms, Germantown, NY) (300 gm) were housed three to a cage (20°C, 12 hr light/dark cycle; lights on at 6:00 A.M.) with food and water available ad libitum. Protocols for care and use of animals were approved by the Children's Hospital of Philadelphia Institutional Animal Care and Use Committee and in accordance with the NIH Guide for the Use and Care of Laboratory Animals.
Surgery. Rats were anesthetized with a 1-1.5% halothane/air mixture and positioned in a stereotaxic instrument with the head at a 20o angle, nose down. Body temperature was maintained at 36.5-37°C. The skull was exposed and a hole was drilled, exposing the dura and associated superficial transverse and sagittal sinuses. The sagittal sinus and dura were ligated, transected, and reflected to allow for a midline approach toward the DR with minimal blood loss.
Recording and microinfusion. Double-barrel micropipettes were used to record single-unit neuronal discharge and simultaneously microinfuse substance P. This technique has been characterized previously (Akaoka et al., 1992). The recording pipette was filled with 2% Pontamine Sky Blue (PSB) dye in 0.5 m sodium acetate. The infusion pipette was angled (30-45o) with its tip adjacent to the tip of the recording pipette but 100 μm dorsal. This was filled with substance P [1 mg/ml in artificial CSF (ACSF)] and connected to a source of solenoid-activated pneumatic pressure (Picospritzer, General Valve, Fairfield, NJ). This infusion pipette was calibrated such that 1 mm displacement = 60 nl volume. To examine interactions between substance P and sendide, kynurenic acid, or WAY 100635 within the DR, triple-barrel micropipettes, constructed in a manner similar to that described above, were used.
Micropipettes were advanced toward the DR. Microelectrode signals were filtered and amplified and monitored with an oscilloscope and audio monitor. DR neurons were identified during recording by their relatively slow and regular spontaneous discharge rate. When a stable, unitary action potential was isolated, an amplitude trigger was used to convert the occurrence of each action potential into a digital pulse that was integrated on-line into a recording of mean frequency using a Cambridge Electronics Design 1401 data analysis system and Spike-2 software.
Experimental protocol. DR discharge rate was recorded for 5-10 min before substance P microinfusion (10-30 nl). Intraraphe microinfusions were made by applying pressure pulses (10-30 psi, 20-40 msec duration) to the infusion pipette. Movement of solution through the calibrated pipette was observed during pressure application to assure that it was being ejected. Typically, effects occurred immediately after application of only one or two pulses of pressure, amounting to one-half of a division or less (≤30 nl). Neuronal activity was recorded until recovery (usually 5-10 min). At this time substance P was again microinfused to assess reproducibility or an antagonist was administered, and the effects of substance P were determined 5-10 min after antagonist administration. Triple-barrel micropipettes were used to examine the interaction between substance P and different antagonists locally within the DR. Kynurenic acid (3 mm, 30 nl), sendide (10 pg/nl, 30 nl), WAY 100635 (0.1 μm), or ACSF was microinfused into the DR 5 min before substance P, as described above. In some cases (n = 4), a solution containing both substance P (1 ng/nl) and WAY 100635 (0.1 m) was microinfused into the DR. The effects of systemic administration of WAY 100635 (1 mg/kg, s.c.), 5 min before microinfusion of substance P, were also examined. The mean discharge rates (expressed as a percentage of the baseline rate) were calculated during five 1 min time epochs after the injection, corresponding to a time when substance P had its peak effect.
Histology. The recording site was marked by iontophoresis of PSB (-15 mA, 10 min). Rats were injected with pentobarbital (100 mg/kg), and the brains were removed, frozen in isopentane, and stored at -70°C. Frozen 40 μm sections were cut, mounted on glass slides, and stained with neutral red for visualization of the PSB spot. The locations of recording sites were reproduced on corresponding atlas plates of brain maps (Swanson, 1992).
Drugs. Substance P (Bachem, Torrance, CA) was dissolved in water (1 mg/ml), and 10 μl aliquots were concentrated using a Savant Speed Vac concentrator. The aliquots were stored at -70°C and dissolved in ACSF on the day of the experiment. Sendide (Bachem) was dissolved in water, concentrated, and aliquoted as described above. Kynurenic acid (Sigma, St. Louis, MO) was prepared on the day of the experiment. This was dissolved in 10 μl of 10 m NaOH and diluted with ACSF to a concentration of 100 mm, and the pH was adjusted to 7-8. This stock solution was diluted with ACSF to 3.0 mm. WAY 100635 (RBI/Sigma, Natick, MA) was dissolved in saline (1 mg/ml) and administered subcutaneously or was dissolved in ACSF (0.1 μm) and administered into the DR.
Results
Substance P has heterogeneous effects on DR neuronal activity in vivo
Activity was recorded from 69 neurons (n = 64 rats), and 47 of these were localized to the DR (Fig. 1A). The discharge rates of these neurons ranged between 0.3 and 3.1 Hz with a mean frequency of 1.16 ± 0.11 Hz, comparable with previous reports (Aghajanian et al., 1968, 1970). DR neurons exhibited three types of responses to substance P microinfusion. The most prevalent response was a short-onset, pure inhibition that occurred in 31 neurons (66%) (Fig. 1B). Neuronal activity often ceased for a short time (n = 12 cells) and then began to recover within 5 min of the microinfusion (Figs. 2B,C, 3B). Inhibition produced by substance P was reproducible when tested on the same neuron (Fig. 2A,C) or on different neurons in the same electrode track. The mean maximum inhibition produced by substance P in neurons exhibiting pure inhibition was a 77.8 ± 4.3% decrease in discharge rate, and this typically occurred within 2 min after the injection. In some DR neurons (8 of 47) substance P microinfusion elicited a brief activation followed by a longer period of inhibition (Figs. 1C, 2A, 3A). Like the pure inhibitory response, this was reproducible on the same neuron (Fig. 2A). Finally, pure excitatory responses were observed in 8 of 47 neurons (Fig. 1D).
Figure 1.

Topography of effects of substance P on DR neuronal discharge rate. A, Recording sites were plotted in the caudal (top), middle third (middle), and rostral third (bottom) of the DR. Atlas representations were modified from Swanson (1992). The predominant effect was inhibition (black circles) (B). A brief excitation followed by inhibition (gray triangles) (C) or brief excitation (open circles) (D) also occurred. Excitatory effects were observed most often in the dorsal half of the DR. y-axis is firing rate in Hertz; x-axis is time in seconds. Numbers in the right-hand corner of B-D indicate the percentages of neurons that exhibited the shown response.
Figure 2.

The effects of substance P were blocked by the selective NK1 receptor antagonist sendide. A, Substance P was applied two times (SP-1 and SP-2), producing a consistent excitation followed by a brief inhibition. After the antagonist (A-1) sendide was applied, the effect of subsequent substance P applications was markedly attenuated (SP-3 to SP-5) or blocked (SP-6) after an additional application of antagonist (A-2). Asterisk indicates recording artifact. B, Substance P (SP-1) produces a pronounced inhibition, which is completely blocked by antagonist (A). The asterisks indicate injection artifacts. C, Vehicle (V) application has no effect on repeated substance P administration (SP-1 and SP-2).
Figure 3.
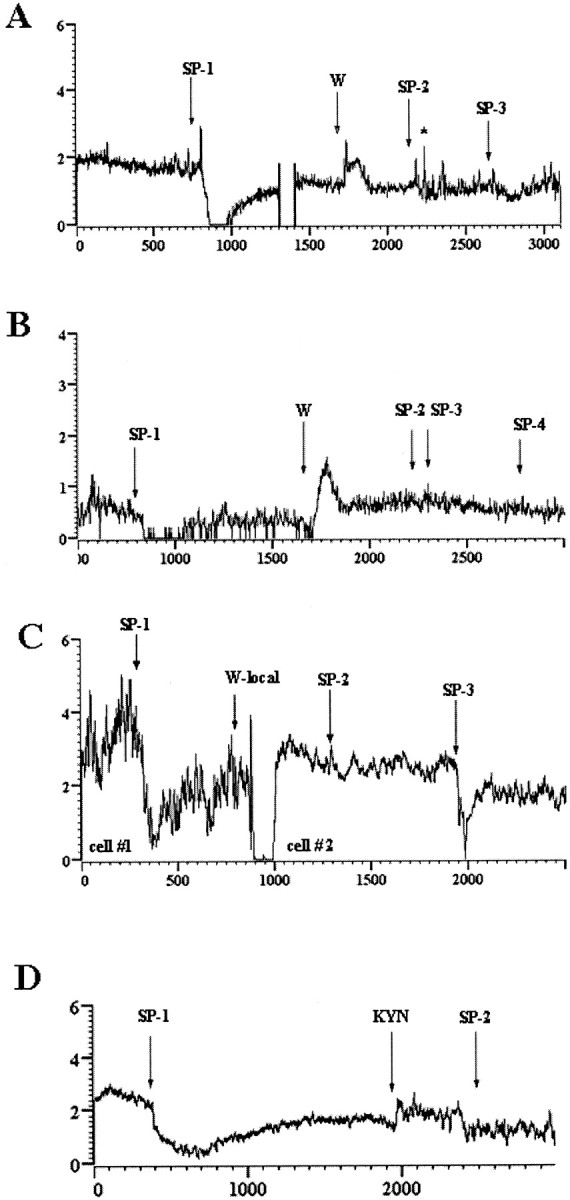
5-HT1A and glutamate antagonists block the effects of substance P. A, Excitation followed by inhibition produced by substance P (SP-1) is converted to a brief excitation (SP-2, SP-3) after systemic administration of WAY 100635 (W). Time break and asterisk indicate recording artifacts. B, Inhibition produced by substance P (SP-1) is reversed after systemic administration of WAY 100635 (W), and subsequent administration of substance P (SP-2 to SP-4) is ineffective. C, Inhibition produced by substance P (cell #1) was followed by WAY 100635 microinfusion. Activity from cell 1 was lost (900 sec), and another neuron in the vicinity was recorded (cell #2). Application of substance P (SP-2) to cell 2 500 sec after WAY 100635 had no effect but inhibited cell 2 when applied at a much later time, which is indicative of recovery. D, Inhibition produced by substance P (SP-1) is blocked at SP-2 after local application of kynurenic acid (KYN).
Figure 1A shows the location of the recorded neurons within the DR with respect to responses to substance P. Neurons that exhibited pure inhibition were generally localized more ventrally and rostrally. In contrast, DR neurons that exhibited pure excitation or a mixed response were typically more dorsal.
The effects of substance P were prevented by previous microinfusion of the NK1 antagonist sendide into the DR. Figure 2A shows a recording from a neuron that exhibited a reproducible mixed response to substance P (SP-1 and SP-2). A single microinfusion of sendide did not alter DR discharge but attenuated the effect of substance P applied 200 sec later and produced a greater attenuation of the effect of substance P applied 15 min after antagonist administration (Fig. 2A). In this case, a second microinfusion of the antagonist was sufficient to completely prevent the effect. Microinfusion of sendide also prevented effects of substance P that were purely inhibitory (Fig. 2B) (n = 4) or excitatory (data not shown; n = 1). In contrast, microinfusion of ACSF (30 nl) before substance P did not alter DR responses to substance P (Fig. 2C) (n = 4). In all, previous microinfusion of sendide prevented the effects of substance P on each of six neurons tested in six different rats. The mean maximum inhibition produced by substance P in subjects pretreated with sendide versus ACSF was 5 ± 3 and 86 ± 12%, respectively.
The inhibitory effects of substance P are mediated by 5-HT 1A receptors
The inhibition of DR activity produced by microinfusion of substance P was antagonized by systemic administration of the 5-HT1A antagonist WAY 100635 in each of six cases (Fig. 3A,B). WAY 100635 both prevented (Fig. 3A) and reversed (Fig. 3B) substance P-elicited inhibition of DR activity, whereas it had less of an effect on the excitatory component of the response (Fig. 3A). Systemic administration of WAY 100635 produced a transient increase in DR discharge rate that returned to baseline within minutes of administration. The mean discharge rate after substance P administration in subjects systemically administered WAY 100635 was 105 ± 11% of the rate determined before substance P administration.
Administration of WAY 100635 directly into the DR also attenuated the effects of substance P whether it was administered from a separate barrel of the micropipette 5 min before substance P (n = 6 cells in three rats) or simultaneously with substance P from the same pipette (n = 4 cells in four rats). Figure 3C shows a representative example of antagonism of the inhibitory effect of substance P by locally administered WAY 100635. The mean maximum inhibition of DR discharge rate after substance P administration in rats treated with WAY 100635 into the DR was 18 ± 6%.
The effects of substance P are mediated by excitatory amino acid receptor activation
Previous microinfusion of kynurenic acid prevented the response to substance P in seven cases and was ineffective in one case. Figure 3D shows that substance P elicited inhibition of a DR neuron before administration of kynurenic acid. Microinfusion of kynurenic acid was associated with a transient increase in discharge rate, and microinfusion of substance P after this was ineffective. The mean maximum decrease in DR discharge rate associated with substance P administration in subjects pretreated with kynurenic acid was 28 ± 13%.
Discussion
In the present study substance P administration into the DR had complex effects on DR neuronal activity that depended on the location of the recorded cell within the nucleus and were sensitive to the selective NK1 antagonist sendide, implicating this receptor subtype in the responses. The predominant response was inhibition that required 5-HT1A receptor activation within the DR because it was attenuated by both systemic and intra-DR pretreatment with the selective 5-HT1A antagonist WAY 100635. Substance P also activated a population of DR neurons, and this was often followed by a period of inhibition. The sensitivity of these effects to an excitatory amino acid antagonist is consistent with the finding that substance P increases EPSCs in DR neurons in vitro via glutamatergic mechanisms (Liu et al., 2002). Taken with evidence supporting the localization of NK1 receptors on glutamatergic dendrites in the dorsomedial DR (Commons and Valentino, 2002), it is proposed that substance P interacts with NK1 receptors on glutamate neurons that are poised to excite 5-HT neurons in the dorsal aspect of the DR (Fig. 4). Release of 5-HT from these neurons can subsequently inhibit neuronal activity through activation of 5-HT1A receptors. Because disruption of NK1 receptor function results in 5-HT1A receptor desensitization and anxiolytic behavior (Santarelli et al., 2001), the effects of substance P reported here may play a role in anxiogenic behaviors.
Figure 4.

Model of substance P regulation of DR activity. Substance P acting NK1 receptor excites a population of glutamate neurons in the DR that subsequently drive a population of 5-HT neurons, many located dorsally in the nucleus. Activation of these neurons then triggers inhibition of neurons throughout the nucleus via 5-HT1A receptor activation.
Technical considerations
DR activity was recorded in the halothane-anesthetized state, which may impact on neuronal activation by substance P. For example, anesthesia abolishes morphine-induced excitation of the DR-5-HT system (Tao and Auerbach, 1994). Although these findings suggest that the excitatory component of the substance P response may be more pronounced in the unanesthetized state, it is noteworthy that robust excitatory responses were still observed in the present study in halothane-anesthetized rats. Future studies involving simultaneous DR unit recording and local DR injection in unanesthetized rats would be required to resolve this issue. An additional caveat of the present study is that the absolute concentration of microinjected substance that reaches the recorded neuron can never be known with certainty. The results of the present study remain notable considering these technical limitations.
Role of NK1 receptors in anxiety
The function of NK1 receptors has become of great interest because of convergent clinical (Kramer et al., 1998) and basic findings suggesting that NK1 antagonists have anxiolytic and antidepressant potential. Thus, mice with a genetic deletion of NK1 receptors exhibit anxiolytic effects in several models, including the elevated plus maze, novelty suppressed feeding, and maternal separation-induced ultrasonic vocalizations (Santarelli et al., 2001). Additionally, plasma corticosterone levels after exposure to the elevated plus maze were reduced in mutant compared with wild-type mice. Similar to genetic disruption of NK1 receptor function, acute NK1 receptor antagonism is associated with anxiolytic activity in some of the same models and in the social interaction test (Teixeira et al., 1996; File, 1997; Kramer et al., 1998; Santarelli et al., 2001). Conversely, the NK1 receptor agonists have anxiogenic activity in the same models and induce place aversion (Aguiar and Brandao, 1994, 1996; Teixeira et al., 1996).
Role of the DR in NK1 receptor-mediated effects
Several findings suggest that the anxiolytic effects associated with the loss of NK1 receptor function are related to activation of the DR-5-HT system. NK1 receptor immunoreactivity is abundant in dendrites in the DR (Santarelli et al., 2001; Commons and Valentino, 2002; Liu et al., 2002). DR discharge rates are elevated in transgenic mice that do not express NK1 receptors (Santarelli et al., 2001). Acute systemic administration of NK1 antagonists increases DR neuronal activity in mice and guinea pigs (Froger et al., 2001; Haddjeri and Blier, 2001; Santarelli et al., 2001; Conley et al., 2002), and repeated administration elevates DR discharge in rats (Haddjeri and Blier, 2001). Substantial evidence suggests that the enhancement of DR activity in mutant mice and that observed with repeated administration of an NK1 antagonist to rats are the result of selective desensitization of 5-HT1A autoreceptors (Froger et al., 2001; Haddjeri and Blier, 2001; Santarelli et al., 2001). In contrast, DR activation associated with acute administration of NK1 antagonists is not related to 5-HT1A desensitization and has been attributed to effects of the agents outside of the DR (Conley et al., 2002).
Together, the studies discussed above suggest that disruption of NK1 receptor function results in activation of the DR-5-HT system. On the other hand, several studies suggest that activation of NK1 receptors in the DR also produces effects that are consistent with activation of the DR-5-HT system. For example, administration of NK1 agonists into the DR increases 5-HT release and 5-HT-mediated behaviors (Gradin et al., 1992). NK1 agonists increase EPSCs in 5-HT neurons in vitro, and this is mediated by activation of local glutamate neurons (Liu et al., 2002), consistent with the localization of NK1 receptor immunoreactivity within glutamate-immunoreactive dendrites in the DR (Commons and Valentino, 2002).
Model of the NK1 actions in the DR
The present study is the first to characterize the effects of direct administration of an NK1 agonist on rat DR-5-HT neuronal activity in vivo and reconciles the incongruous findings discussed above. Substance P produced both excitatory and inhibitory effects that were prevented by kynurenic acid, consistent with NK1 receptor localization on glutamatergic processes (Commons and Valentino, 2002). The finding that the population of DR neurons that was excited by substance P were preferentially localized in the dorsal part of the nucleus, where NK1 receptor immunoreactivity is most dense, is consistent with in vitro studies demonstrating that substance P increases EPSCs (Liu et al., 2002). Nonetheless, the predominant response to substance P administration was a 5-HT1A-mediated inhibition, which was most apparent in the ventral part of the nucleus. Together, the results suggest that NK1 receptor activation of glutamatergic neurons excites synaptically linked DR neurons (i.e., in the dorsal DR). Subsequent release of 5-HT from these neurons can cause inhibition after excitation of the same neurons and pure inhibition of 5-HT neurons that are synaptically removed from NK1-expressing glutamatergic neurons (i.e., in the ventral DR) (Fig. 4). This model might predict that 5-HT1A antagonists would potentiate excitatory responses to substance P in neurons having a profile of mixed excitation-inhibition. This was not apparent in the present study, although the probability of encountering neurons with this profile was relatively low, and an insufficient number of neurons could be tested to statistically quantify this. It is also possible that the interaction between excitatory and inhibitory components of the response to substance P may be more complex than simple additivity. For example, the responses may be mediated by cellular mechanisms that are mutually exclusive and have time courses with limited overlap. In this case, 5-HT1A antagonism would not alter the excitatory component of the response. This issue may be better addressed in vitro.
Given that neurons in the dorsal and ventral DR have distinct efferent projections, the proposed model suggests the compelling hypothesis that engaging substance P afferents to the DR can regulate 5-HT release in a regionally selective manner, increasing release in targets of the dorsal neurons and decreasing release in targets of ventral neurons. In this regard it is of interest that the central nucleus of the amygdala, which plays a prominent role in the expression of anxiogenic behaviors, is targeted by the dorsomedial DR (Commons et al., 2003).
The present results support an interaction between NK1 and 5-HT1A receptors at the network level within the DR. This is of particular interest, given that 5-HT1A receptor desensitization has been implicated in the therapeutic actions of NK1 antagonists. Moreover, the findings demonstrating topographically organized effects of substance P on DR neuronal activity suggest means for regionally selective regulation of 5-HT release in forebrain that would provide for a unique spectrum of action of NK1 antagonists in the treatment of affective disorders.
Footnotes
This work was supported by Public Health Service Grants MH58250, DA00463, DA14329, and MH02006 and a National Alliance for Research on Schizophrenia and Depression Distinguished Investigator Award.
Correspondence should be addressed to Rita J. Valentino, The Children's Hospital of Philadelphia, 402 Abramson Pediatric Research Center, Thirty-fourth Street and Civic Center Boulevard, Philadelphia, PA 19104. E-mail: valentino@email.chop.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/237155-05$15.00/0
References
- Aghajanian GK, Foote WE, Sheard MH ( 1968) Lysergic acid diethyl amide: sensitive neuronal units in the midbrain raphe. Science 161: 706-708. [DOI] [PubMed] [Google Scholar]
- Aghajanian GK, Foote WE, Sheard MH ( 1970) Action of psychotogenic drugs on single midbrain raphe neurons. J Pharmacol Exp Ther 171: 178-187. [PubMed] [Google Scholar]
- Aguiar MS, Brandao ML ( 1994) Conditioned place aversion produced by microinjections of substance P into the periaqueductal gray of rats. Behav Pharmacol 5: 369-373. [DOI] [PubMed] [Google Scholar]
- Aguiar MS, Brandao ML ( 1996) Effects of microinjections of the neuropeptide substance P in the dorsal periaqueductal gray on the behaviour of rats in the plus-maze test. Physiol Behav 60: 1183-1186. [DOI] [PubMed] [Google Scholar]
- Akaoka H, Saunier C-F, Chergui K, Charlety P, Buda M, Chouvet G ( 1992) Combining in vivo volume-controlled pressure microejection with extracellular unit recording. J Neurosci Methods 42: 119-128. [DOI] [PubMed] [Google Scholar]
- Commons K, Valentino RJ ( 2002) Cellular basis for the effects of substance P in the periaqueductal gray and dorsal raphe nucleus. J Comp Neurol 447: 82-97. [DOI] [PubMed] [Google Scholar]
- Commons KG, Connolley KR, Valentino RJ ( 2003) Anatomical attributes of the dorsomedial raphe nucleus that may underlie a role in affective disorders. Neuropsychopharmacology 28: 206-215. [DOI] [PubMed] [Google Scholar]
- Conley RK, Cumberbatch MJ, Mason GS, Williamson DJ, Harrison T, Locker K, Swain CJ, Maubach K, O'Donnell R, Rigby M, Hewson L, Smith D, Rupniak NM ( 2002) Substance P (neurokinin 1) receptor antagonists enhance dorsal raphe neuronal activity. J Neurosci 22: 7730-7736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- File SE ( 1997) Anxiolytic action of a neurokinin 1 receptor antagonist in the social interaction test. Pharmacol Biochem Behav 58: 747-752. [DOI] [PubMed] [Google Scholar]
- Froger N, Gardier AM, Moratalla R, Alberti I, Lena I, Boni C, De Felipe C, Rupniak NM, Hunt SP, Jacquot C, Hamon M, Lanfumey L ( 2001) 5-Hydroxytryptamine (5-HT)1A autoreceptor adaptive changes in substance P (neurokinin 1) receptor knock-out mice mimic antidepressant-induced desensitization. J Neurosci 21: 8188-8197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradin K, Qadri F, Nomikos GG, Hillegaart V, Svensson T ( 1992) Substance P injection into the dorsal raphe increases blood pressure and serotonin release in hippocampus of conscious rats. Eur J Pharmacol 218: 363-367. [DOI] [PubMed] [Google Scholar]
- Haddjeri N, Blier P ( 2001) Sustained blockade of neurokinin-1 receptors enhances serotonin neurotransmission. Biol Psychiatry 50: 191-199. [DOI] [PubMed] [Google Scholar]
- Kramer MS, Cutler N, Feighner J, Shrivastava R, Carman J, Sramek JJ, Reines SA, Liu G, Snavely D, Wyatt-Knowles E, Hale JJ, Mills SG, MacCoss M, Swain CJ, Harrison T, Hill RG, Hefti F, Scolnick EM, Cascieri MA, Chicchi GG, et al. ( 1998) Distinct mechanism for antidepressant activity by blockade of central substance P receptors. Science 281: 1640-1645. [DOI] [PubMed] [Google Scholar]
- Liu R, Ding Y, Aghajanian GK ( 2002) Neurokinins activate local glutamatergic inputs to serotonergic neurons of the dorsal raphe nucleus. Neuropsychopharmacology 27: 329-340. [DOI] [PubMed] [Google Scholar]
- Maeno H, Kiyama H, Tohyama M ( 1993) Distribution of substance P receptor (NK-1 receptor) in the central nervous system. Mol Brain Res 18: 43-58. [DOI] [PubMed] [Google Scholar]
- Rupniak NM, Kramer MS ( 1999) Discovery of the antidepressant and anti-emetic efficacy of substance P receptor (NK1) antagonists. Trends Pharmacol Sci 20: 485-490. [DOI] [PubMed] [Google Scholar]
- Santarelli L, Gobbi G, Debs PC, Sibille EL, Blier P, Hen R, Heath MJS ( 2001) Genetic and pharmacological disruption of neurokinin 1 receptor function decreases anxiety-related behaviors and increases serotonergic function. Proc Natl Acad Sci USA 98: 1912-1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severini C, Improta G, Falconieri-Erspamer G, Salvadori S, Erspamer V ( 2002) The tachykinin peptide family. Pharmacol Rev 54: 285-322. [DOI] [PubMed] [Google Scholar]
- Stout SC, Owens MJ, Nemeroff CB ( 2001) Neurokinin 1 receptor antagonists as potential antidepressants. Annu Rev Pharmacol Toxicol 41: 877-906. [DOI] [PubMed] [Google Scholar]
- Swanson LW ( 1992) Brain maps structure of the rat brain. Amsterdam: Elsevier.
- Tao R, Auerbach SB ( 1994) Anesthetics block morphine-induced increases in serotonin release in rat CNS. Synapse 18: 307-314. [DOI] [PubMed] [Google Scholar]
- Teixeira RM, Santos AR, Ribeiro SJ, Calixto JB, Rae GA, De Lima TC ( 1996) Effects of central administration of tachykinin receptor agonists and antagonists on plus-maze behavior in mice. Eur J Pharmacol 311: 7-14. [DOI] [PubMed] [Google Scholar]