

Abstract
Nerve injury-induced afferent discharge is thought to elicit spinal sensitization and consequent abnormal pain. Experimental neuropathic pain, however, also depends on central changes, including descending facilitation arising from the rostral ventromedial medulla (RVM) and upregulation of spinal dynorphin. A possible intersection of these influences at the spinal level was explored by measuring evoked, excitatory transmitter release in tissues taken from nerve-injured animals with or without previous manipulation of descending modulatory systems. Spinal nerve ligation (SNL) produced expected tactile and thermal hyperesthesias. Capsaicin-evoked calcitonin gene-related peptide (CGRP) release was markedly enhanced in lumbar spinal tissue from SNL rats when compared with sham-operated controls. Enhanced, evoked CGRP release from SNL rats was blocked by anti-dynorphin A(1-13) antiserum; this treatment did not alter evoked release in tissues from sham-operated rats. Dorsolateral funiculus lesion (DLF) or destruction of RVM neurons expressing μ-opioid receptors with dermorphin-saporin, blocked tactile and thermal hypersensitivity, as well as SNL-induced upregulation of spinal dynorphin. Spinal tissues from these DLF-lesioned or dermorphin-saporin-treated SNL rats did not exhibit enhanced capsaicin-evoked CGRP-IR release. These data demonstrate exaggerated release of excitatory transmitter from primary afferents after injury to peripheral nerves, supporting the likely importance of increased afferent input as a driving force of neuropathic pain. The data also show that modulatory influences of descending facilitation are required for enhanced evoked transmitter release after nerve injury. Thus, convergence of descending modulation, spinal plasticity, and afferent drive in the nerve-injured state reveals a mechanism by which some aspects of nerve injury-induced hyperesthesias may occur.
Keywords: neuropathic pain, descending facilitation, RVM, primary afferent, dynorphin, CGRP release
Introduction
Peripheral nerve injury is accompanied initially by spontaneous firing of afferent fibers (Kirk, 1974; Wall and Gutnick, 1974a,b), resulting in spinal “sensitization.” Increased excitatory transmitter release from the afferents may occur, although this has not been directly demonstrated. These processes are considered an important mechanism driving neuropathic pain (Devor, 1991a,b; Woolf and Thompson, 1991; Kajander and Bennett, 1992). Whereas the onset of spontaneous discharges coincides with the onset of neuropathic behaviors (Han et al., 2000; C. N. Liu et al., 2000; X. Liu et al., 2000), spontaneous activity decreases dramatically by postinjury day 5 (Chaplan et al., 1994; Han et al., 2000; C. N. Liu et al., 2000; X. Liu et al., 2000). Nevertheless, once established, neuropathic pain persists for many months, suggesting that additional mechanisms are necessary to sustain pain (Malan et al., 2000; Burgess et al., 2002a).
Although injury-induced discharge may drive the initiation of neuropathic pain, its maintenance depends on descending facilitation from the rostral ventromedial medulla (RVM) (Ossipov et al., 2001; Porreca et al., 2001; Burgess et al., 2002a). Thus, RVM lidocaine blocks neuropathic pain without altering acute nocifensive responses (Kovelowski et al., 2000; Burgess et al., 2002a). Similarly, destruction of μ-opioid receptor-expressing RVM neurons, possibly pain facilitation cells (Heinricher et al., 1992), also blocks neuropathic pain (Porreca et al., 2001; Burgess et al., 2002b). Finally, dorsolateral funiculus (DLF) lesion, a major conduit of spinopetal projections from the RVM, ipsilateral to the nerve injury also abolishes neuropathic behavior (Ossipov et al., 2000). These studies support the importance of descending facilitation in the neuropathic state, but the specific mechanisms by which these actions occur remain unknown.
Peripheral nerve injury elicits an upregulation of spinal dynorphin (Cox et al., 1985; Faden et al., 1985; Kajander et al., 1990; Draisci et al., 1991; Dubner and Ruda, 1992; Wagner and Deleo, 1996; Malan et al., 2000; Porreca et al., 2001; Burgess et al., 2002a). The maximal increase in spinal dynorphin content occurs relatively late, i.e., approximately postinjury day 10 (Malan et al., 2000). Prodynorphin “knock-out” mice with spinal nerve ligation (SNL) develop neuropathic pain initially, but the pain resolves within 7-10 d after injury, whereas SNL wild-type mice sustain abnormal pain (Wang et al., 2001). These and other studies led to the suggestion that pathological levels of dynorphin are pronociceptive (Claude et al., 1999; Malan et al., 2000) and that SNL-induced increases in spinal dynorphin are essential for the sustained expression of neuropathic pain (Wang et al., 2001). Critically, nerve injury-induced alteration of spinal dynorphin expression may be secondary to neuroplastic changes occurring within the RVM. Lesions of the DLF or dermorphin-saporin deletion of RVM μ-opioid receptor-expressing cells prevent nerve injury-induced upregulation of spinal dynorphin (Burgess et al., 2002a).
The present study searched for possible intersection of these diverse mechanisms that are apparently critical for the expression of experimental neuropathic pain by evaluating evoked release of an excitatory transmitter from primary afferents in tissues taken from nerve-injured rats with or without previous manipulations of descending modulatory pathways. Our data show convergence of nerve injury-induced afferent input, spinal plasticity, and descending modulation, revealing a potential mechanism by which some aspects of experimental neuropathic pain may occur.
Materials and Methods
Male Sprague Dawley rats (Harlan Sprague Dawley, Indianapolis, IN) were maintained in a climate-controlled room on a 12 hr light/dark cycle (lights on at 6:00 A.M.), with food and water available ad libitum. The body weights for the rats used in this study depended on the type(s) of surgeries received and the times of testing after surgery. At the time of surgery, rats that received either sham operation or SNL surgery alone weighed between 200 and 225 gm. Rats that received previous lesions of the DLF weighed between 275 and 300 gm at the time of SNL or sham surgery. Rats with cannulas in the RVM underwent either sham operation or SNL surgery after 28 d and weighed 325-350 gm. All testing was performed in accordance with the policies and recommendations of the International Association for the Study of Pain and the National Institutes of Health guidelines for the handling and use of laboratory animals and received approval from the Institutional Animal Care and Use Committee of the University of Arizona.
L5/L6 spinal nerve ligation surgery
SNL injury was produced as described by Chung and colleagues (Kim and Chung, 1992). Anesthesia was induced with 2% halothane in O2 at 2 l/min and maintained with 0.5% halothane in O2. The dorsal vertebral column from L4 to S2 was exposed, and the L5 and L6 spinal nerves were identified and carefully isolated. The L5 and L6 spinal nerves were tightly ligated distal to the dorsal root ganglion with 4 - 0 silk suture. The incision was closed, and the animals were observed for uneventful recoveries. Rats that exhibited motor deficiency (<10%) were excluded from additional testing. Sham SNL rats underwent the same surgery and handling as the experimental animals but without SNL.
Microinjection into the RVM
Rats were prepared for bilateral RVM drug administration by placing anesthetized (80 mg/kg ketamine and 12 mg/kg xylazine, i.p.) animals in a stereotaxic head holder. For intracranial bilateral drug administrations, the skull was exposed, and two 26-gauge guide cannulas separated by 1.2 mm (Plastics One, Roanoke, VA) were directed toward the lateral portions of the RVM (anteroposterior, -11.0 mm from bregma; lateral, ±0.6 mm from midline; H, -8.5 mm from the cranium) (Paxinos and Watson, 1986) and cemented in place. Drug administrations into the RVM were performed by slowly expelling 0.5 μl of drug solution or saline through a 33 gauge injection cannula inserted through the guide cannula and protruding an additional 1 mm into fresh brain tissue to prevent backflow of drug into the guide cannula. At the termination of the experimental procedures, pontamine blue was injected into the site of the RVM injections, and cannula placement was verified histologically; no animals were excluded because of cannula placement in the current study. Dermorphin, saporin, dermorphin-saporin (Advanced Targeting Systems, San Diego, CA), or saline vehicle were administered as a single dose of 3 pmol into the RVM (1.5 pmol in 0.5 μl each side) as described previously (Porreca et al., 2001).
Behavioral tests
Behavioral responses to thermal and tactile stimuli were determined before surgery and again 2 and 10 d after SNL to establish the presence of experimental neuropathic pain.
Thermal hyperalgesia. The method of Hargreaves et al. (1988) was used to assess paw-withdrawal latency to a thermal nociceptive stimulus. Rats were allowed to acclimate within a Plexiglas enclosures on a clear glass plate maintained at 30°C. A radiant heat source (i.e., high-intensity projector lamp) was activated with a timer and focused onto the plantar surface of the hindpaw. Paw-withdrawal latency was determined by a motion detector that halted both lamp and timer when the paw was withdrawn. A maximal cutoff of 40 sec was used to prevent tissue damage. A significant reduction in paw-withdrawal latency from the pre-SNL baseline value was interpreted as thermal hyperalgesia. Pairwise comparisons were performed with Student's t test. Significance was set at the level of p ≤ 0.05.
Tactile hypersensitivity. The paw-withdrawal thresholds of the hindpaws of the rats were determined in response to probing with eight calibrated von Frey filaments (Stoelting, Wood Dale, IL) in logarithmically spaced increments ranging from 0.41 to 15 gm (4-150 mN). Each filament was applied perpendicularly to the plantar surface of the paw of rats kept in suspended wire-mesh cages. Withdrawal threshold was determined by sequentially increasing and decreasing the stimulus strength (“up and down” method) and calculated with a Dixon nonparametric test (Dixon, 1980; Chaplan et al., 1994). Data are expressed as mean paw-withdrawal threshold in grams. A significant reduction in paw-withdrawal threshold from the pre-SNL baseline value indicated tactile hypersensitivity. Pairwise comparisons were performed with Student's t test. Significance was set at the level of p ≤ 0.05.
Spinal DLF lesions
Unilateral spinal lesions at the T8 level were performed in naive rats under halothane anesthesia as described previously (Burgess et al., 2002a). A laminectomy was made at the T8 level to expose the spinal cord. Lesions of the DLF ipsilateral to the side of nerve injury were performed by crushing the area with fine forceps under a dissecting microscope. Sham DLF surgery was performed by exposing the vertebrae and performing the laminectomy but without cutting neuronal tissue. Hemostasis was confirmed, and the wound over the exposed spinal cord was packed with Gelfoam and closed. All lesions were verified histologically at the termination of the experiment. Only rats with appropriately placed DLF lesions, determined post hoc, were included in the subsequent data analysis. Less than 10% of the rats were excluded from this study. The DLF lesions were performed 3 d before SNL or sham surgery.
Tissue extraction and preparation
Rats were deeply anesthetized with halothane and decapitated. The spinal column was cut at the pelvic girdle. Hydraulic extrusion was performed by inserting a 16 gauge blunt needle into the sacral vertebral canal and expelling with ice-cold saline. The spinal cord was immediately placed on ice in a glass Petri dish, and the dorsal half of the lumbar cord was dissected. The dorsal half was further dissected into sections ipsilateral and contralateral to SNL. Tissue samples that were to be quantified for dynorphin content were then immediately frozen in liquid nitrogen and stored at -70°C until assayed. Tissues for use in the calcitonin gene-related peptide (CGRP) release assay were weighed and minced into sections 0.2 mm in width with a McIlwain tissue chopper (Mickle Laboratory Engineering, Gomshall, UK).
CGRP-IR release assays
Immunoreactive CGRP release was measured using methods described by Vasko and colleagues (Chen et al., 1996). Minced tissue samples were placed in a 1 cc chamber and continuously superfused with oxygenated modified Krebs' buffer (135 mm NaCl, 3.5 mm KCl, 1 mm MgCl2,20mm NaHCO3,1mm NaHPO4, 2.5 mm CaCl2, 3.3 mm dextrose, 0.1 mm ascorbic acid, 10 mm thiorphan, and 0.1% bovine serum albumin) maintained at 37°C, pH 7.4. The buffer was maintained at a flow rate of 0.5 ml/min with a Brandel Superfusion Pump (Brandel, Gaithersburg, MD). The tissue was allowed to equilibrate for 45 min. Superfusate was collected in 3 min intervals into test tubes using a fraction collector (Gilson, Middleton, WI). A total of four to five fractions (12-15 min) were collected before the addition of capsaicin to the perfusate. Capsaicin was added to the perfusate to result in a concentration of 1 μm for 6 min (two fractions). The superfusate was then collected for an additional 27-30 min (9-10 fractions).
Radioimmunoassay for CGRP in superfusate
Each tube of superfusate obtained from the release assay was preincubated with 100 μl of a C-terminally directed anti-CGRP antibody (Peninsula Laboratories/Bachem, San Carlos, CA) for 24 hr at 4°C. The samples were each mixed with 100 μl of [125I-Tyr0]CGRP28-37 (at 20,000 - 25,000 cpm per assay tube) and 50 μl of goat anti-rabbit antiserum coupled to ferric beads and incubated for an additional 24 hr. The [125I]CGRP bound to the CGRP antibody was separated from the free tracer through immunomagnetic separation (PerSeptive Diagnostics, Cambridge, MA). The immunoprecipitates were determined by gamma counting. Standard curves were generated, and CGRP content was determined through logit-log analysis. This assay has a minimal detection limit of 1-3 fmol/tube. The CGRP antiserum used in these experiments binds near the C-terminal end of CGRP and does not cross-react with cholecystokinin, neuropeptide Y, or other peptides with similar C-terminal residues (Gardell et al., 2002a). The CGRP-IR concentrations were plotted against time in 3 min intervals. Evoked release was calculated as the total amount of CGRP released (i.e., CGRP-IR) in response to the capsaicin infusion above the basal release of CGRP-IR.
Total CGRP-IR content
Tissues were placed in 2 ml of 0.01N HCl and homogenized using a Polytron, followed by centrifugation at 2500 × g for 20 min. The supernatant was diluted 1:400 in modified Krebs' buffer and then assayed for total CGRP-IR content using the radioimmunoassay described above. Pairwise comparisons between treatments were detected using Student's t test. Significance was determined at the level of p ≤ 0.05.
Dynorphin immunoassay
Tissues were placed in 1N acetic acid and homogenized with a Polytron. The homogenates were incubated for 30 min at 95°C and centrifuged at 14,000 × g for 20 min (4°C). The supernatants were lyophilized and stored at -70°C. Protein concentrations were determined using the bichinchoninic acid method with bovine serum albumin as a standard. Immunoassay was performed using a commercial enzyme immunoassay system for dynorphin A(1-17) (Peninsula Laboratories/Bachem, San Carlos, CA). The antibody to dynorphin A(1-17) used in these experiments does not cross-react the following peptides: dynorphin A(1-8), dynorphin A(1-13), dynorphin B, and nociceptin (orphanin FQ) (Gardell et al., 2002a,b). Standard curves were constructed using the software Prism (GraphPad, San Diego, CA). Pairwise comparisons between treatments were detected using Student's t test. Significance was determined at the p ≤ 0.05 level.
The anti-dynorphin antiserum that was used to neutralize dynorphin in the release experiments was raised against dynorphin A(1-13) (Peninsula Laboratories/Bachem, San Carlos, CA). This antiserum demonstrates 100% cross-reactivity with dynorphin A(1-17) and dynorphin A(1-12) fragments. Control serum was produced by incubating the dynorphin A(1-13) antiserum with agarose protein A (Vector Laboratories, Burlingame, CA) to adsorb all of the IgG from the antiserum. Packed agarose protein A was mixed with an equal volume of reconstituted dynorphin A(1-13) antiserum (1 mg/100 ml) and incubated on a reciprocal shaker for 24 hr at 4°C. After incubation, the suspension was centrifuged at 5000 × g for 5 min to pellet the agarose protein A. The supernatant “control serum” is thus devoid of IgG. This “subtracted” or “preadsorbed” fraction of the antiserum was used as a control for any possible nonspecific, IgG independent behavioral effects of the intrathecal treatment of dynorphin A(1-13) antiserum as described previously (Gardell et al., 2002a,b).
Immunohistochemistry
For immunohistochemistry, rats were anesthetized with 80 mg/kg ketamine and 12 mg/kg xylazine intraperitoneally and then perfused transcardially with 0.1 m PBS until the exudate ran clear and then with either 4% paraformaldehyde or with 10% Formalin and 0.05% glutaraldehyde in 0.1 m PBS, pH 7.4, for ∼15 min. Lumbar spinal cords were harvested and postfixed in 10% Formalin overnight and cryoprotected with 20% sucrose in 0.1 m PBS. The postfixed tissues were transferred to a 30% sucrose in 0.1 m PBS solution until the tissues sank to the bottom of the containers. Tissues were then embedded in O.C.T (Tissue-Tek Optimal Cutting Temperature Compound; Sakura, Torrance, CA) compound and DRG and spinal cord sections were cut at 10 and 20 μm, respectively, with a cryostat maintained at -20°C. Sections of DRG were cut serially and placed onto slides such that each slide contained an ordered series of sections through the ganglion. Sections of spinal cord were placed in cell culture plate wells containing 0.1 m PBS (five sections per well).
Mounted DRG sections and free-floating spinal cord sections were used. The sections were extensively rinsed and then blocked for 2 hr in 10% normal goat serum in 0.1 m PBS at room temperature, followed by 24 hr in the primary reagents (i.e., 2% normal goat serum, 0.3% Triton X-100, and a primary antiserum in 0.1 m PBS) at 4°C. For prodynorphin immunostaining, the sections were washed with PBS and incubated for 2 hr in the secondary antiserum solution composed of Alexa Fluor 594-conjugated goat anti-guinea pig IgG (1:1000; Molecular Probes, Eugene, OR) and 2% normal goat serum in 0.1 m PBS. For CGRP immunostaining, the sections were washed in 0.1 m PBS and then incubated with a rabbit anti-CGRP antiserum (1:40,000; Peninsula Laboratories, Belmont, CA) in 0.1 m PBS with 5% normal goat serum overnight at 4°C, followed by washing and secondary incubation with a Cy3-conjugated goat anti-rabbit IgG (1:500; Jackson ImmunoResearch, West Grove, PA) for 2 hr. The sections were rinsed and mounted in Vectashield (Vector Laboratories). Fluorescence digital images were captured with a Nikon (Tokyo, Japan) E800 fluorescence microscope equipped with standard filters for immunofluorescence detection. Images were acquired with a Hamamatsu C5810 color CCD camera and its proprietary Image Processor software (Hamamatsu, Bridgewater, NJ). The acquired images were then processed with Adobe PhotoShop (Abode Systems, San Jose, CA).
Data analysis
Data collected within each release assay group were calculated as femtomoles of CGRP per milligram of tissue. To account for intrinsic variations in baseline values across assays, the data were converted to percentage of an appropriate within-group control (e.g., sham-operated vs SNL) as described previously (Chen et al., 1996; Kilo et al., 1997; Ulrich-Lai et al., 2001; Gardell et al., 2002a,b). Means are compared with the reference control group by ANOVA and, where significance was indicated, followed by the post hoc Fisher's least significant difference test.
Results
Evaluation of tactile and thermal thresholds after sham or L5/L6 SNL
The L5/L6 SNL surgeries reliably produced behavioral signs of neuropathic pain, indicated by hypersensitivity to light tactile and noxious thermal stimuli. The mean paw-withdrawal threshold to probing of the hindpaw with von Frey filaments was 15 ± 0 gm before any treatment. Rats with SNL demonstrated a significant (p ≤ 0.05) reduction in paw-withdrawal threshold to 3.68 ± 0.48 gm within 2 d of SNL, indicating tactile hypersensitivity. The mean paw-withdrawal threshold at day 10 after SNL was 3.21 ± 0.44 gm. In contrast, sham-operated rats did not show any decreases in paw-withdrawal thresholds. The mean paw-withdrawal thresholds on days 2 and 10 after sham surgery were both 15 ± 0 gm.
The mean paw-withdrawal latency to noxious radiant heat was 21.4 ± 0.25 sec before any manipulations. Rats with SNL demonstrated a significant (p ≤ 0.05) decrease in withdrawal latency to 14.9 ± 0.33 sec by day 2 after SNL, indicating thermal hyperalgesia. The mean paw-withdrawal latency was also significantly (p ≤ 0.05) depressed on day 10 after SNL, as indicated by a value of 14.3 ± 0.35 sec. In contrast, sham-operated rats did not demonstrate any significant changes in paw-withdrawal latencies from pretreatment baseline values. The mean paw-withdrawal latencies of the sham-operated group were 21.0 ± 0.33 and 21.4 ± 0.36 sec on days 2 and 10, respectively, after surgery.
Evaluation of CGRP-like immunoreactivity in spinal cord and L5 DRG
Rats with L5/L6 SNL were confirmed to exhibit behavioral neuropathic pain and were killed 10 d after the surgery. Spinal cord and DRG sections ipsilateral to surgery were taken from rats with sham surgery or SNL for immunofluorescence examination of CGRP distribution (Fig. 1A). The number of neurons expressing immunofluorescence for CGRP in the DRG of rats with SNL compared with that DRG obtained from sham-operated rats was decreased (Fig. 1A). An observer blinded to the treatments evaluated a total of 1102 DRG cells from the sham-operated group and 1119 DRG cells from the SNL group by counting randomly selected fields from six sections taken from the L5 DRG. A mean of 50 ± 2.72% of the sham-operated DRG neurons were CGRP-positive, whereas that value was significantly reduced to 30 ± 2.99% in the SNL group (p ≤ 0.05) (Fig. 1B). There was also an apparent reduction in the intensity of immunofluorescence for CGRP in the ipsilateral spinal dorsal horn after SNL (Fig. 1A). These observations are consistent with previous studies that indicate a downregulation of CGRP in DRG and laminas I-II of the dorsal spinal cord after sciatic axotomy (Noguchi et al., 1990; Hokfelt et al., 1999). In contrast to the immunofluorescent evidence, radioimmunoassay did not reveal a significant change in lumbar CGRP content after SNL (Fig. 1C). The CGRP content measured in dorsal lumbar quadrant of spinal tissue ipsilateral to the sham surgery was 110 ± 6% that of the contralateral side (Fig. 1B); these values were not significantly different. Similarly, the CGRP of the spinal dorsal quadrant ipsilateral to the SNL surgery was 96 ± 4% that of the CGRP content of the contralateral side (Fig. 1C); these values were not significantly different.
Figure 1.
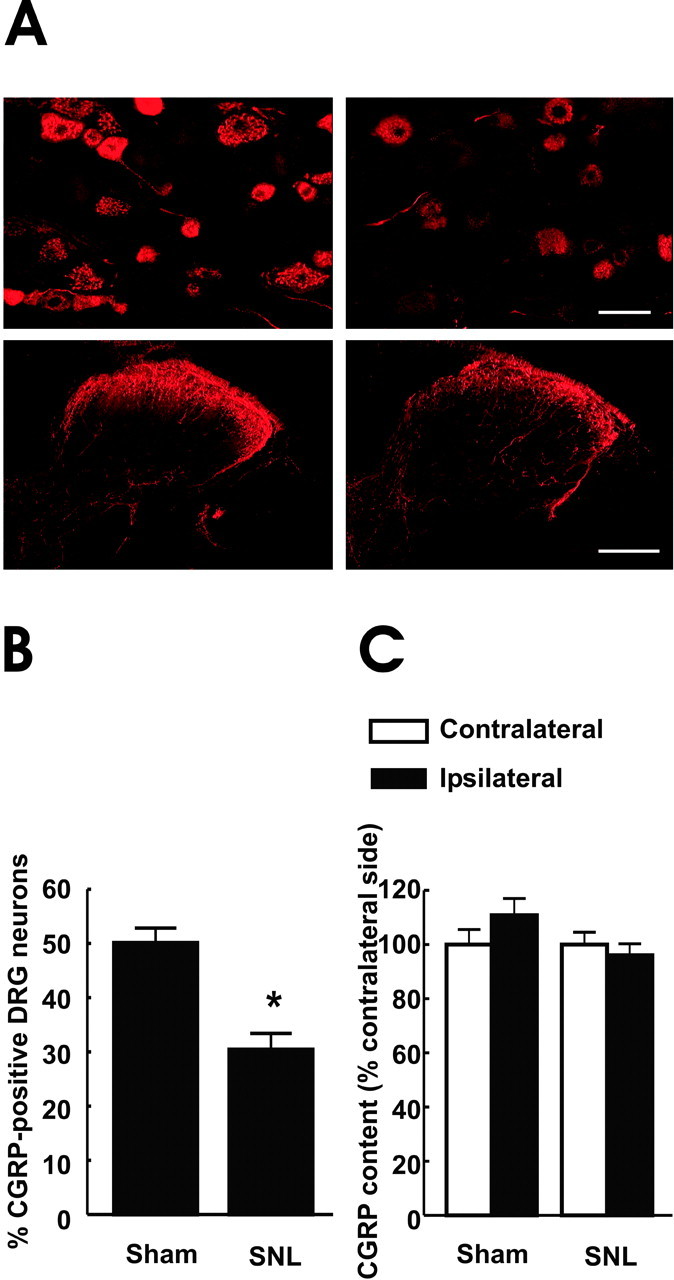
A, Visualization of CGRP-IR in the DRG and spinal dorsal horn of sham-operated and SNL rats 10 d after surgery. There is an apparent decrease in CGRP-like immunoreactivity in the L5 DRG of the group that received SNL. Scale bar, 50 μm. CGRP-IR also appears reduced in the L5 dorsal horn of the SNL group compared with the sham-operated spinal dorsal horn section. Scale bar, 200 μm. B, The percentage of DRG neurons that showed CGRP-IR after SNL is significantly (*p ≤ 0.05) diminished when compared with the sham-operated group. Radioimmunoassay for CGRP was performed with the dorsal spinal quadrants ipsilateral and contralateral to sham or SNL surgeries. Data are presented as percentage of the contralateral side to account for between-assay differences. C, No differences in CGRP content was seen between the sham-operated and SNL groups. n = 6-8 rats per group.
Evaluation of spinal prodynorphin expression and dynorphin A(1-17) content after L5/L6 SNL
Immunofluorescence examination of dorsal horn spinal cord sections taken at the L5 level ipsilateral to SNL at postoperative day 10 showed upregulation in spinal prodynorphin expression when compared with the spinal cord tissue obtained from sham-operated rats (Fig. 2A). Little immunofluorescence for prodynorphin was seen in the spinal dorsal horn of sham-operated rats, and the immunofluorescence that was present was concentrated in the outer laminas of the spinal dorsal horn. After SNL, prodynorphin immunofluorescence was enhanced in the outer laminas of the dorsal horn (Fig. 2A). Moreover, there was clear immunofluorescent evidence of prodynorphin expression into the deeper laminas of the dorsal horn, whereas none was observed in sham-operated rats. These observations are consistent with our previous studies and those of others that suggest an upregulation of spinal dynorphin content in response to peripheral nerve injury (Faden et al., 1985; Kajander et al., 1990; Dubner and Ruda, 1992; Malan et al., 2000; Porreca et al., 2001; Burgess et al., 2002a).
Figure 2.
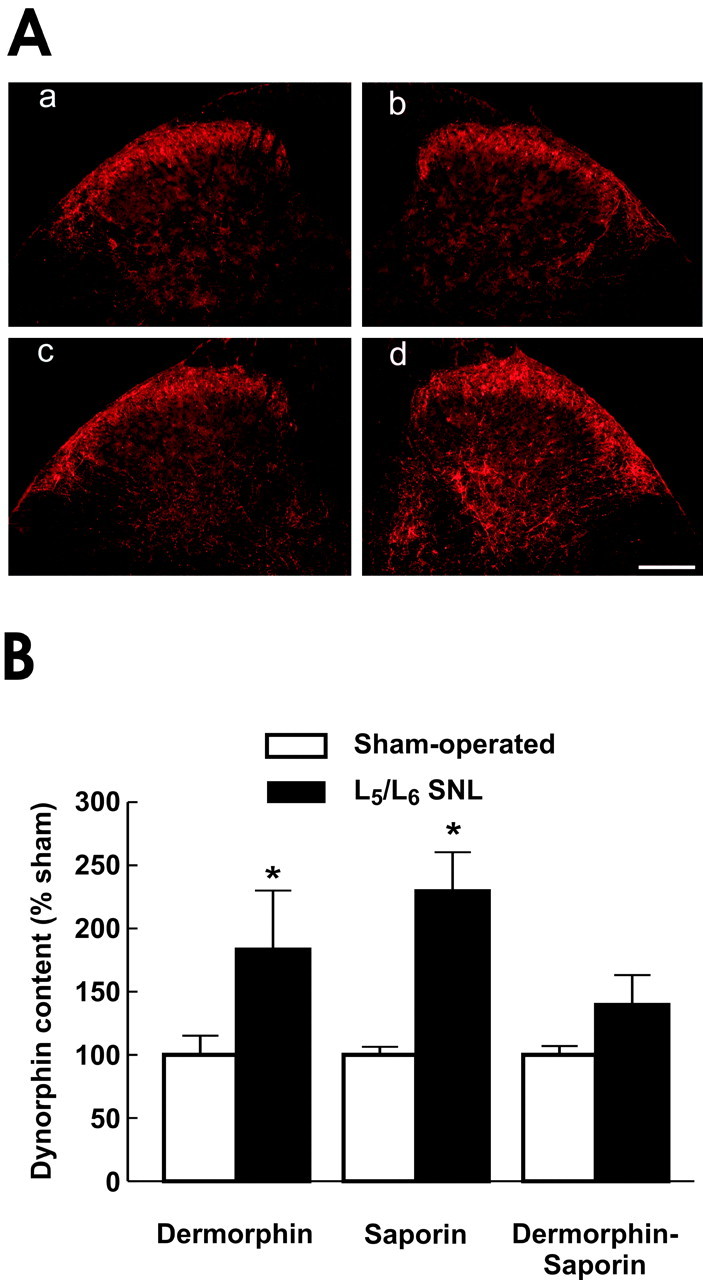
A, Immunofluorescence for prodynorphin in ipsilateral spinal dorsal horn of sham- or SNL-operated rats. Tissues were taken 10 d after surgery after determination of SNL-induced tactile and thermal hypersensitivity. Spinal cord sections from sham-operated rats contralateral (a) and ipsilateral (b) to sham surgery and from SNL rats contralateral (c) and ipsilateral (d) to SNL are shown. Scale bar, 200 μm. There is a clear increase in immunofluorescent intensity for prodynorphin in tissue obtained from the rats with SNL. Moreover, prodynorphin-IR appears in the deeper laminas of the dorsal horn of the SNL tissue but is absent in the same region of sham-operated rats. B, Spinal dynorphin content was determined after RVM pretreatment with saline, saporin, dermorphin, or the dermorphin-saporin conjugate. Rats received sham or SNL surgery 28 d after the RVM pretreatment. On the 10th day after sham or SNL surgery, dynorphin content of the ipsilateral lumbar dorsal quadrant was quantified by radioimmunoassay. Tissue from rats with SNL and pretreated with saline, saporin, or dermorphin in the RVM demonstrated a significantly greater (*p ≤ 0.05) dynorphin content than that from rats with sham surgery. In contrast, dynorphin content of SNL rats pretreated with dermorphin-saporin conjugate was not significantly different from that of the sham-operated group. The data are presented as percentage of the corresponding sham-operated group to account for variations among assay runs. n = 6-8 rats per group.
The immunofluorescence evidence for increased spinal prodynorphin in response to peripheral nerve injury was supported by the quantification of spinal dynorphin content through enzyme immunoassay. Rats were pretreated with a single microinjection of saporin, dermorphin, or the dermorphin-saporin conjugate (3 pmol) into the RVM. After 28 d, rats from each group received either SNL or sham surgery. On the 10th day after surgery (i.e., 38 d after RVM injections), the rats were killed, and the spinal cords were dissected into quadrants for determination of spinal dynorphin content. The spinal dynorphin content of the sham SNL groups receiving RVM treatments did not differ significantly (ANOVA; F(3,36) = 1.72; p = 0.18) from each other or from a naive untreated group, indicating that pretreatment with saporin, dermorphin, or the dermorphin-saporin conjugate in the RVM did not alter spinal dynorphin content of animals without nerve injury. Because these assays were performed separately, but under identical conditions, the data for each experimental group were normalized in terms of percentage of the corresponding sham-operated control group. Thus, the dynorphin content of the sham-operated control groups pretreated with saporin, dermorphin, and dermorphin-saporin conjugate was normalized as 100 ± 6, 100 ± 15, and 100 ± 7%, respectively (Fig. 2B). Rats pretreated with either saporin or dermorphin and subsequently receiving L5/L6 SNL showed significant (p ≤ 0.05) increases in spinal dynorphin content to 230 ± 31 and 183 ± 47% of the corresponding sham-operated control groups. In contrast, the spinal dynorphin content of the group pretreated with the dermorphin-saporin conjugate was 140 ± 22% of the corresponding sham-operated group, a value that was not significantly different (Fig. 2B).
Capsaicin-evoked release of CGRP does not differ among the sham-operated control groups
The level of capsaicin-evoked CGRP release in the sham SNL groups, regardless of pretreatment, did not differ significantly (ANOVA; F(8,58) = 1.01; p = 0.44) from each other, indicating that sham SNL operation did not alter the level of capsaicin-evoked CGRP release of animals without nerve injury. The levels of capsaicin-evoked CGRP release observed in tissues from sham SNL rats ranged from 19.0 ± 8.9 to 36.8 ± 4.6 fmol/mg tissue. Because these assays were performed separately, but under identical conditions, the data for each experimental group were normalized in terms of percentage of the corresponding sham-operated control group (Chen et al., 1996; Kilo et al., 1997; Ulrich-Lai et al., 2001; Gardell et al., 2002). These results demonstrate that neither DLF lesions nor pretreatment with saporin, dermorphin, or the dermorphin-saporin conjugate in the RVM significantly altered capsaicin-evoked release of CGRP in rats without nerve injury.
Capsaicin-evoked release of CGRP is increased after SNL in a time-dependent manner
Spinal sections were obtained from the lumbar dorsal quadrant of rats 2 or 10 d after either sham surgery or L5/L6 SNL and assessed for capsaicin-evoked release of CGRP. There were no differences in basal release of CGRP among groups of tissues within each treatment group at either time point.
The release of CGRP evoked by 1 μm capsaicin from tissue obtained from sham-operated rats 2 d after surgery was normalized as 100 ± 23% (Fig. 3A). The capsaicin-evoked release of CGRP, measured 2 d after SNL, was 80 ± 16% of that of the sham-operated group; these values were not significantly different (Fig. 3). The evoked release obtained from the sham-operated group at day 10 after surgery was normalized as 100 ± 16%; this value was not significantly different from that seen at day 2 after SNL. The capsaicin-evoked release of CGRP from spinal tissue with SNL 10 d after the injury was significantly (p ≤ 0.05) elevated to 172 ± 33% (Fig. 3A).
Figure 3.

Capsaicin-evoked release of CGRP from minced lumbar sections obtained from the ipsilateral dorsal quadrants of rats undergoing sham or SNL surgery (A), which received control serum or anti-dynorphin antiserum (B), previous lesions of the DLF (3 d before sham or SNL surgery) at T8 (C), or were pretreated 38 d previously with RVM saporin (SAP), dermorphin (DERM), or dermorphin-saporin conjugate (D; Derm-Sap). After a 45 min equilibration period in the perfusion chambers, followed by a 12 min collection of perfusate for determination of baseline values, capsaicin (1 μm) was added to the perfusion medium. CGRP content was quantified by radioimmunoassay. Evoked release was defined as the amount of CGRP above the basal values, and data are shown as percentage of the evoked release from sham-operated rats in the same assay. Capsaicin-evoked CGRP release did not differ between the sham-operated rats SNL rats on postsurgery day 2. In contrast, tissue from SNL rats demonstrated a significantly (*p ≤ 0.05) enhanced capsaicin-evoked CGRP release relative to the sham-operated group on postsurgery day 10 (A). Addition of control serum (i.e., dynorphin antiserum with IgG removed) to the perfusate to achieve a concentration of 1 mg/100 ml had no significant effect on capsaicin-evoked release in either the sham-operated or SNL groups. Addition of dynorphin antiserum (1 mg/100 ml) to the perfusate prevented the enhanced evoked CGRP release in SNL tissues (B). The capsaicin-evoked release of CGRP from the sham DLF/SNL group was significantly (*p ≤ 0.05) greater than that of the sham DLF/sham SNL group. Capsaicin-evoked CGRP release in DLF/SNL rats was not significantly different from that of the sham SNL/DLF lesions. Capsaicin evoked CGRP release in DLF/sham SNL lesion did not differ from that in sham DLF/sham SNL rats (C). Tissues from rats with SNL that were pretreated with either saporin or dermorphin in the RVM demonstrated a significantly greater (*p ≤ 0.05) capsaicin-evoked CGRP release than those tissues taken from rats with the same RVM pretreatment and sham surgery. In contrast, RVM pretreatment with dermorphin-saporin prevented the SNL-induced enhancement of capsaicin-evoked CGRP release in tissues from SNL rats. The capsaicin-evoked release of CGRP from the SNL group pretreated with the dermorphin-saporin conjugate was not different from that of the sham-operated group and was not different from that seen in tissues from RVM saline-pretreated sham-operated rats. The data were converted to percentage of the evoked release from sham-operated rats to account for interassay variability. n = 4-20 rats per group.
Enhanced capsaicin-evoked CGRP release in SNL tissues is blocked by anti-dynorphin antiserum
Spinal sections were obtained from the lumbar dorsal quadrant of rats 10 d after L5/L6 SNL or sham surgery. Either preabsorbed control serum or antiserum to dynorphin (Gardell et al., 2002a,b) were added to the perfusion medium at a concentration of 1 mg/100 ml. In these experiments, capsaicin-evoked release obtained from spinal tissue from the SNL group was significantly (p ≤ 0.05) increased to 172 ± 33% of the sham-operated group (100 ± 16%) (Fig. 3B). The addition of 1 mg/100 ml antiserum to dynorphin to the perfusate produced a complete block of the enhancement of the capsaicin-evoked release of CGRP from nerve-injured spinal tissue. The evoked release in SNL tissues in the presence of anti-dynorphin antiserum was 91 ± 14% of the corresponding sham-operated group, which was normalized as 100 ± 11% (Fig. 3B). These values were not significantly different. In contrast to the anti-dynorphin antiserum, the addition of the control (i.e., IgG-free) serum to the perfusion medium did not block enhanced capsaicin-evoked CGRP release in SNL rats. The capsaicin-evoked release of CGRP of spinal tissue taken from SNL rats in the presence of control serum was significantly (p ≤ 0.05) increased to 192 ± 28% of the sham-operated group, which was evaluated as 100 ± 21%. Neither control serum nor anti-dynorphin antiserum produced any change in basal release of CGRP in tissues taken 10 d after SNL or sham SNL surgery (data not shown).
Enhanced evoked CGRP release in tissues from SNL rats is blocked by lesions of the DLF
Tissues were taken from rats undergoing DLF or sham DLF lesion (3 d before peripheral nerve injury) at day 10 after SNL or sham SNL surgery. No differences in baseline release were observed when DLF and sham DLF tissues were compared in sham SNL tissues. Spinal cord tissue obtained from rats with SNL and sham DLF lesions showed capsaicin-evoked release of 173 ± 16% of the corresponding control group (i.e., sham DLF and sham SNL), which was 100 ± 27%, demonstrating a significant (p ≤ 0.05) enhancement of capsaicin-evoked CGRP release (Fig. 3C). Capsaicin-evoked release of CGRP after DLF lesion and SNL was 115 ± 7% of the sham SNL and DLF group, which was 100 ± 23% (Fig. 3C); these values were not significantly different.
Enhanced capsaicin-evoked release of CGRP in tissues from SNL rats is blocked by pretreatment with RVM dermorphin-saporin
Twenty-eight days after receiving a single RVM saporin, dermorphin, or dermorphin-saporin conjugate pretreatment, rats received either sham or L5/L6 SNL surgery. Tissues were taken for evaluation of evoked CGRP release after an additional 10 d, i.e., 38 d after the RVM pretreatment. Spinal cord sections at the L5 level were removed, and capsaicin-evoked release of CGRP was performed as described above. Each set of data within each of the pretreatment groups were compared with the sham-operated control of the corresponding group. Baseline release of CGRP was unaltered by any of the RVM pretreatments. The RVM pretreatment with either unconjugated saporin or dermorphin did not alter the expected enhanced capsaicin-evoked release of CGRP resulting from peripheral nerve injury. The capsaicin-evoked CGRP release of saporin- or dermorphin-pretreated, sham-operated groups were 100 ± 33 and 100 ± 21%, respectively (Fig. 3D). Both the dermorphin- or saporin-pretreated groups with SNL demonstrated significant (p ≤ 0.05) increases in capsaicin-evoked CGRP release of 223 ± 21 and 167 ± 18%, respectively (Fig. 3D). In contrast, the selective lesioning of RVM neurons that express the μ-opioid receptor by microinjection of the dermorphin-saporin conjugate prevented the injury-induced enhancement of capsaicin-evoked CGRP release. The evoked release obtained from the SNL group was 78 ± 6%, which was not significantly different from the value of 100 ± 16% obtained from the corresponding sham-operated control group (Fig. 3D).
Discussion
Our data demonstrate that spinal tissues taken at day 10, but not day 2, after SNL show increased capsaicin-evoked CGRP release, suggesting a mechanism that may be partly responsible for the exaggerated response to noxious sensory stimuli seen in experimental neuropathic pain. Additionally, however, enhanced, evoked release at this time after nerve injury depends on pathological expression of spinal dynorphin. Sustained experimental neuropathic pain has been shown to depend on upregulation of spinal dynorphin (Malan et al., 2000; Porreca et al., 2001; Wang et al., 2001), which in turn is secondary to descending facilitatory mechanisms from the RVM (Burgess et al., 2002a; this study). We extend these observations by demonstrating that manipulations that block SNL-induced pain by impeding the pathophysiologic activity of spinal dynorphin through a specific antiserum, or blockade of its injury-induced upregulation via DLF or RVM dermorphin-saporin lesion, also blocked the enhanced capsaicin-evoked CGRP release seen in SNL tissues. These findings suggest that the function of primary afferents at this time point after injury, like behavioral manifestations of neuropathy, are under similar neuroplastic controls. Collectively, these data reveal a convergence of different features of the neuropathic state represented by enhanced afferent input to the spinal cord, spinal plasticity as shown by upregulation of dynorphin expression and descending modulatory systems from the RVM.
Increased afferent excitability as a mechanism of neuropathic pain is supported by the clinical benefit produced by substances that attenuate neuronal excitability (Rowbotham et al., 1996; Fields et al., 1997; Abdi et al., 1998; Sindrup and Jensen, 1999). Although aberrant injured and/or uninjured afferent discharge is thought to be essential for expression of experimental neuropathic pain (Devor and Seltzer, 1999; Wu et al., 2001, 2002), how this occurs is not known. Presumably, postinjury central sensitization results from increased excitatory transmission from injury-induced afferent discharges. However, increased excitatory transmitter release in the absence of an evoked stimulus has not been demonstrated after nerve injury. Although our data show no differences in baseline CGRP release in tissues from SNL or sham-operated rats, release of peptidic and nonpeptidic transmitters may differ (Lundberg et al., 1980; Lundberg et al., 1981). Additionally, our assay conditions were not optimal for detection of excitatory transmitter release that might occur in vivo.
Experimental neuropathy is characterized by behavioral hypersensitivity to noxious thermal or innocuous mechanical stimuli to the paw ipsilateral to injury. Excitatory afferent transmitter release may also be exacerbated by such noxious or innocuous stimuli. For this reason, the demonstration of enhanced evoked transmitter release would provide a mechanistic basis for the exaggerated behavioral sensitivity to sensory stimuli. Here, enhanced capsaicin-evoked CGRP release was detected in SNL tissues, suggesting a potential mechanism for heat hyperalgesia after nerve injury. It is not known whether these observations also pertain to the enhanced sensitivity to normally innocuous stimuli. In this regard, our previous studies have mechanistically distinguished nerve injury-induced tactile and thermal hypersensitivity. Tactile, but not thermal, hypersensitivity is blocked selectively by microinjection into the nucleus gracilis of lidocaine (Sun et al., 2001; Ossipov et al., 2002) or NPY antagonists (Ossipov et al., 2002) or by dorsal column lesions (Sun et al., 2001), pointing to the role of large myelinated afferents in this response. In contrast, nerve injury-induced thermal hypersensitivity is selectively blocked by desensitization of vanilloid receptor 1 (VR1)-positive afferent fibers by resiniferatoxin (Ossipov et al., 1999), suggesting the predominant importance of C-fibers in this response. Because CGRP is known to be expressed in small- and medium-sized VR1-positive fibers (Carr et al., 1990; Chen et al., 1996), our experimental paradigm is likely to pertain specifically to nerve injury-induced thermal, but not necessarily tactile, hypersensitivity. Our data showed a time dependence in the increased CGRP release in SNL tissues that was evident at postinjury day 10 but not day 2. Because nerve injury-induced thermal hyperalgesia is evident at postinjury day 2 (Burgess et al., 2002a), it seems likely that enhanced evoked CGRP release is not essential in the behavioral hypersensitivity seen at this early time point. However, this possibility should be viewed cautiously given the extremely limited time course represented by only two time points.
Increased evoked CGRP release also occurs after inflammation (Southall et al., 1998) and sustained morphine exposure (Ma et al., 2001; Gardell et al., 2002). Although these conditions upregulate CGRP expression (Smith et al., 1992; Seybold et al., 1995; Weihe et al., 1995; Gardell et al., 2002), the opposite is seen with nerve injury (Kajander and Xu, 1995; Na et al., 2001). Our immunohistochemical findings confirm CGRP downregulation, although no change in overall CGRP content could be detected by radioimmunoassay at the coarse resolution of the spinal dorsal quadrant of the SNL rat. Thus, increased evoked CGRP release after SNL did not likely result from increased CGRP content. Electrical stimulation of afferents at A-fiber strength was demonstrated to release substance P only after nerve transection, raising the possibility of a “phenotypic switch” (i.e., de novo expression of this transmitter in large fibers) (Malcangio et al., 2000). Our results are unlikely to result from a similar mechanism as the number of CGRP-expressing cells decreased after SNL, and these cells did not demonstrate properties of injured large-diameter neurons such as NPY expression (Ossipov et al., 2002) (E. Zhang and F. Porreca, unpublished observations). Furthermore, capsaicin was used to evoke CGRP release specifically through the VR1 receptor (Helliwell et al., 1998; Szallasi et al., 1999), suggesting activation of small, rather than large, fibers.
Because ectopic discharge and neuropathic pain behaviors do not correlate temporally (C. N. Liu et al., 2000; Wu et al., 2001), we suggested differences in processes that initiate and those that maintain neuropathic pain. Thus, at early time points, pain may result from the direct consequences of injury, whereas prolonged expression of pain depends on modulation of primary afferent activity subsequent to central neuroplastic changes, including activation of descending facilitation from the RVM and upregulation of spinal dynorphin (Burgess et al., 2002a). This possibility seems supported here because enhanced evoked CGRP release was observed only at times at which spinal dynorphin is upregulated (i.e., day 10) (Malan et al., 2000). Thus, nerve injury-induced spinal neuroplasticity, represented by the upregulation of spinal dynorphin, has an important influence on primary afferent function only at time points after the initiation phase of the neuropathic state.
Pain modulation is known to occur within the RVM, and neurons expressing the μ-opioid receptor may correlate with those mediating spinopetal facilitation (Heinricher et al., 1994; Roychowdhury and Heinricher, 1997). Because RVM lidocaine blocks behavioral and electrophysiologic indices elicited by nerve injury (Pertovaara et al., 1996; Kovelowski et al., 2000; Pertovaara et al., 2001; Porreca et al., 2001), it is thought that facilitation may result from this region (for review, see Porreca et al., 2002). A critical feature of such RVM facilitation its time dependency, which is revealed by the actions of lidocaine (Burgess et al., 2002a), RVM dermorphin-saporin lesion of μ-opioid-expressing cells (Porreca et al., 2001), or DLF lesions (Burgess et al., 2002a). In all of these cases, RVM facilitation was detected only after postinjury day 3, suggesting the need for SNL-induced plasticity at this site. An important observation was that DLF lesions also blocked injury-induced spinal dynorphin upregulation (Burgess et al., 2002a). These findings have been extended here by demonstrating that dermorphin-saporin lesion of RVM μ receptor-expressing cells, a treatment that blocks neuropathic pain (Porreca et al., 2001), also blocks spinal dynorphin upregulation when measured 10 d after the SNL injury. At the same time, this treatment abolished enhanced capsaicin-evoked CGRP release in SNL tissues. In contrast, dermorphin or saporin treatments, which do not alter neuropathic pain, failed to alter SNL-induced spinal dynorphin upregulation (Porreca et al., 2001; Burgess et al., 2002a) or enhanced, evoked CGRP release; these treatments were inactive in sham-operated groups. Moreover, DLF lesions, known to block abnormal pain and elevated dynorphin content (Burgess et al., 2002a), also blocked the enhanced capsaicin-evoked CGRP release from SNL tissue at postinjury day 10. Thus, enhanced capsaicin-evoked CGRP release occurred only at times that correlated with upregulated spinal dynorphin and the presence of RVM descending facilitation.
Spinal anti-dynorphin antiserum inhibited neuropathic pain (Nichols et al., 1997; Bian et al., 1999; Wang et al., 2001). Pharmacological addition of non-opioid dynorphin fragments enhance capsaicin-evoked, but not basal, CGRP release (Claude et al., 1999; Gardell et al., 2002), as well as increased release of excitatory amino acids or substance P (Faden, 1992; Skilling et al., 1992; Arcaya et al., 1999). Our results link nerve injury-induced increased endogenous dynorphin levels and evoked neurotransmitter outflow. The increased capsaicin-evoked CGRP release after SNL was blocked by dynorphin antiserum but not by control serum. Moreover, the antiserum blocked only the enhanced CGRP release, without affecting capsaicin-evoked CGRP release in SNL tissues, and did not alter evoked CGRP release in sham-operated spinal tissues. Elevated endogenous dynorphin may thus enhance responses to noxious stimuli without directly eliciting nociception.
These results provide evidence supporting the hypothesis that neuropathic pain may be maintained by enhanced responsiveness of primary afferent neurons. These results link with behavioral and electrophysiologic observations, confirming that enhanced neurotransmitter release is a consequence of evoked stimuli after nerve injury. We show that enhanced afferent transmitter release is maintained by increased spinal dynorphin, itself a consequence of descending facilitation from the RVM. Our results synthesize several apparently unrelated consequences of the postinjury state and reveal a possible mechanism by which some aspects of neuropathic pain may occur.
Footnotes
This work was supported by National Institute on Drug Abuse Grant R01 DA11823. We thank Kerry Cook, Kerry Vault, and Chngmin Zhong for their technical assistance.
Correspondence should be addressed to Dr. Frank Porreca, Department of Pharmacology, College of Medicine, University of Arizona Health Sciences Center, Tucson, AZ 85724. E-mail: frankp@u.arizona.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/238370-10$15.00/0
References
- Abdi S, Lee DH, Chung JM ( 1998) The anti-allodynic effects of amitriptyline, gabapentin, and lidocaine in a rat model of neuropathic pain. Anesth Analg 87 : 1360-1366. [PubMed] [Google Scholar]
- Arcaya JL, Cano G, Gomez G, Maixner W, Suarez-Roca H ( 1999) Dynorphin A increases substance P release from trigeminal primary afferent C-fibers. Eur J Pharmacol 366 : 27-34. [DOI] [PubMed] [Google Scholar]
- Bian D, Ossipov MH, Ibrahim M, Raffa RB, Tallarida RJ, Malan Jr TP, Lai J, Porreca F ( 1999) Loss of antiallodynic and antinociceptive spinal/supraspinal morphine synergy in nerve-injured rats: restoration by MK-801 or dynorphin antiserum. Brain Res 831 : 55-63. [DOI] [PubMed] [Google Scholar]
- Burgess SE, Gardell LR, Ossipov MH, Malan Jr TP, Vanderah TW, Lai J, Porreca F ( 2002a) Time-dependent descending facilitation from the rostral ventromedial medulla maintains, but does not initiate, neuropathic pain. J Neurosci 22 : 5129-5136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess SE, Gardell LR, Xie Y, Ossipov MH, Vanderah TW, Malan Jr TP, Porreca F, Lai J ( 2002b) Dermorphin-saporin targets descending facilitation in the rostral ventromedial medulla (RVM) to block CCK-induced abnormal pain. Soc Neurosci Abstr 28 : 351.311. [Google Scholar]
- Carr PA, Yamamoto T, Nagy JI ( 1990) Calcitonin gene-related peptide in primary afferent neurons of rat: co-existence with fluoride-resistant acid phosphatase and depletion by neonatal capsaicin. Neuroscience 36 : 751-760. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL ( 1994) Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 53 : 55-63. [DOI] [PubMed] [Google Scholar]
- Chen JJ, Barber LA, Dymshitz J, Vasko MR ( 1996) Peptidase inhibitors improve recovery of substance P and calcitonin gene-related peptide release from rat spinal cord slices. Peptides 17 : 31-37. [DOI] [PubMed] [Google Scholar]
- Claude P, Gracia N, Wagner L, Hargreaves KM ( 1999) Effect of dynorphin on ICGRP release from capsaicin-sensitive fibers. Abstr 9th World Congress Pain 9 : 262. [Google Scholar]
- Cox BM, Molineaux CJ, Jacobs TP, Rosenberger JG, Faden AI ( 1985) Effects of traumatic injury on dynorphin immunoreactivity in spinal cord. Neuropeptides 5 : 571-574. [DOI] [PubMed] [Google Scholar]
- Devor M ( 1991a) Neuropathic pain and injured nerve: peripheral mechanisms. Br Med Bull 47 : 619-630. [DOI] [PubMed] [Google Scholar]
- Devor M ( 1991b) Sensory basis of autotomy in rats [editorial]. Pain 45 : 109-110. [DOI] [PubMed] [Google Scholar]
- Devor M, Seltzer Z ( 1999) Pathophysiology of damaged nerves in relation to chronic pain. In: Textbook of pain (Wall PD, Melzack R, eds), pp 129-164. London: Churchill Livingstone.
- Dixon WJ ( 1980) Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol 20 : 441-462. [DOI] [PubMed] [Google Scholar]
- Draisci G, Kajander KC, Dubner R, Bennett GJ, Iadarola MJ ( 1991) Upregulation of opioid gene expression in spinal cord evoked by experimental nerve injuries and inflammation. Brain Res 560 : 186-192. [DOI] [PubMed] [Google Scholar]
- Dubner R, Ruda MA ( 1992) Activity-dependent neuronal plasticity following tissue injury and inflammation. Trends Neurosci 15 : 96-103. [DOI] [PubMed] [Google Scholar]
- Faden AI ( 1992) Dynorphin increases extracellular levels of excitatory amino acids in the brain through a non-opioid mechanism. J Neurosci 12 : 425-429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faden AI, Molineaux CJ, Rosenberger JG, Jacobs TP, Cox BM ( 1985) Increased dynorphin immunoreactivity in spinal cord after traumatic injury. Regul Pept 11 : 35-41. [DOI] [PubMed] [Google Scholar]
- Fields HL, Rowbotham MC, Devor M ( 1997) Excitability blockers: anticonvulsants and low concentration local anesthetics in the treatment of chronic pain. In: Handbook of experimental pharmacology (Dickenson A, Besson JM, eds), pp 93-116. Berlin: Springer.
- Gardell LR, Wang R, Burgess SE, Ossipov MH, Vanderah TW, Malan Jr TP, Lai J, Porreca F ( 2002a) Sustained morphine exposure induces a spinal dynorphin-dependent enhancement of excitatory transmitter release from primary afferent fibers. J Neurosci 22 : 6747-6755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardell LR, Burgess SE, Dogrul A, Ossipov MH, Malan TP, Lai J, Porreca F ( 2002b) Pronociceptive effects of spinal dynorphin promote cannabinoid-induced pain and antinociceptive tolerance. Pain 98 : 79-88. [DOI] [PubMed] [Google Scholar]
- Han HC, Lee DH, Chung JM ( 2000) Characteristics of ectopic discharges in a rat neuropathic pain model. Pain 84 : 253-261. [DOI] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J ( 1988) A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 32 : 77-88. [DOI] [PubMed] [Google Scholar]
- Heinricher MM, Morgan MM, Fields HL ( 1992) Direct and indirect actions of morphine on medullary neurons that modulate nociception. Neuroscience 48 : 533-543. [DOI] [PubMed] [Google Scholar]
- Heinricher MM, Morgan MM, Tortorici V, Fields HL ( 1994) Disinhibition of off-cells and antinociception produced by an opioid action within the rostral ventromedial medulla. Neuroscience 63 : 279-288. [DOI] [PubMed] [Google Scholar]
- Helliwell RJ, McLatchie LM, Clarke M, Winter J, Bevan S, McIntyre P ( 1998) Capsaicin sensitivity is associated with the expression of the vanilloid (capsaicin) receptor (VR1) mRNA in adult rat sensory ganglia. Neurosci Lett 250 : 177-180. [DOI] [PubMed] [Google Scholar]
- Hokfelt T, Broberger C, Diez M, Xu ZQ, Shi T, Kopp J, Zhang X, Holmberg K, Landry M, Koistinaho J ( 1999) Galanin and NPY, two peptides with multiple putative roles in the nervous system. Horm Metab Res 31 : 330-334. [DOI] [PubMed] [Google Scholar]
- Kajander KC, Bennett GJ ( 1992) Onset of a painful peripheral neuropathy in rat: a partial and differential deafferentation and spontaneous discharge in A beta and A delta primary afferent neurons. J Neurophysiol 68 : 734-744. [DOI] [PubMed] [Google Scholar]
- Kajander KC, Xu J ( 1995) Quantitative evaluation of calcitonin gene-related peptide and substance P levels in rat spinal cord following peripheral nerve injury. Neurosci Lett 186 : 184-188. [DOI] [PubMed] [Google Scholar]
- Kajander KC, Sahara Y, Iadarola MJ, Bennett GJ ( 1990) Dynorphin increases in the dorsal spinal cord in rats with a painful peripheral neuropathy. Peptides 11 : 719-728. [DOI] [PubMed] [Google Scholar]
- Kilo S, Harding-Rose C, Hargreaves KM, Flores CM ( 1997) Peripheral CGRP release as a marker for neurogenic inflammation: a model system for the study of neuropeptide secretion in rat paw skin. Pain 73 : 201-207. [DOI] [PubMed] [Google Scholar]
- Kim SH, Chung JM ( 1992) An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain 50 : 355-363. [DOI] [PubMed] [Google Scholar]
- Kirk EJ ( 1974) Impulses in dorsal spinal nerve rootlets in cats and rabbits arising from dorsal root ganglia isolated from the periphery. J Comp Neurol 155 : 165-175. [DOI] [PubMed] [Google Scholar]
- Kovelowski CJ, Ossipov MH, Sun H, Lai J, Malan TP, Porreca F ( 2000) Supraspinal cholecystokinin may drive tonic descending facilitation mechanisms to maintain neuropathic pain in the rat. Pain 87 : 265-273. [DOI] [PubMed] [Google Scholar]
- Liu CN, Wall PD, Ben-Dor E, Michaelis M, Amir R, Devor M ( 2000) Tactile allodynia in the absence of C-fiber activation: altered firing properties of DRG neurons following spinal nerve injury. Pain 85 : 503-521. [DOI] [PubMed] [Google Scholar]
- Liu X, Eschenfelder S, Blenk KH, Janig W, Habler H ( 2000) Spontaneous activity of axotomized afferent neurons after L5 spinal nerve injury in rats. Pain 84 : 309-318. [DOI] [PubMed] [Google Scholar]
- Lundberg JM, Anggard A, Fahrenkrug J, Hokfelt T, Mutt V ( 1980) Vasoactive intestinal polypeptide in cholinergic neurons of exocrine glands: functional significance of coexisting transmitters for vasodilation and secretion. Proc Natl Acad Sci USA 77 : 1651-1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg JM, Anggard A, Emson P, Fahrenkrug J, Hokfelt T ( 1981) Vaso-active intestinal polypeptide and cholinergic mechanisms in cat nasal mucosa: studies on choline acetyltransferase and release of vasoactive intestinal polypeptide. Proc Natl Acad Sci USA 78 : 5255-5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Zheng WH, Powell K, Jhamandas K, Quirion R ( 2001) Chronic morphine exposure increases the phosphorylation of MAP kinases and the transcription factor CREB in dorsal root ganglion neurons: an in vitro and in vivo study. Eur J Neurosci 14 : 1091-1104. [DOI] [PubMed] [Google Scholar]
- Malan TP, Ossipov MH, Gardell LR, Ibrahim M, Bian D, Lai J, Porreca F ( 2000) Extraterritorial neuropathic pain correlates with multisegmental elevation of spinal dynorphin in nerve-injured rats. Pain 86 : 185-194. [DOI] [PubMed] [Google Scholar]
- Malcangio M, Ramer MS, Jones MG, McMahon SB ( 2000) Abnormal substance P release from the spinal cord following injury to primary sensory neurons. Eur J Neurosci 12 : 397-399. [DOI] [PubMed] [Google Scholar]
- Na HS, Kim HJ, Sung B, Back SK, Kim DY, Kim JS, Hong SK ( 2001) Decrease in spinal CGRP and substance P is not related to neuropathic pain in a rat model. NeuroReport 12 : 175-178. [DOI] [PubMed] [Google Scholar]
- Nichols ML, Lopez Y, Ossipov MH, Bian D, Porreca F ( 1997) Enhancement of the antiallodynic and antinociceptive efficacy of spinal morphine by antisera to dynorphin A (1-13) or MK-801 in a nerveligation model of peripheral neuropathy. Pain 69 : 317-322. [DOI] [PubMed] [Google Scholar]
- Noguchi K, Senba E, Morita Y, Sato M, Tohyama M ( 1990) Alpha-CGRP and beta-CGRP mRNAs are differentially regulated in the rat spinal cord and dorsal root ganglion. Brain Res Mol Brain Res 7 : 299-304. [DOI] [PubMed] [Google Scholar]
- Ossipov MH, Bian D, Malan Jr TP, Lai J, Porreca F ( 1999) Lack of involvement of capsaicin-sensitive primary afferents in nerve-ligation injury induced tactile allodynia in rats. Pain 79 : 127-133. [DOI] [PubMed] [Google Scholar]
- Ossipov MH, Hong Sun T, Malan Jr P, Lai J, Porreca F ( 2000) Mediation of spinal nerve injury induced tactile allodynia by descending facilitatory pathways in the dorsolateral funiculus in rats. Neurosci Lett 290 : 129-132. [DOI] [PubMed] [Google Scholar]
- Ossipov MH, Lai J, Malan Jr TP, Vanderah TW, Porreca F ( 2001) Tonic descending facilitation as a mechanism of neuropathic pain. In: Neuropatic pain: pathophysiology and treatment (Hansson PT, Fields HL, Hill RG, Marchettini P, eds), pp 107-124. Seattle: IASP.
- Ossipov MH, Zhang ET, Carvajal C, Gardell L, Quirion R, Dumont Y, Lai J, Porreca F ( 2002) Selective mediation of nerve injury-induced tactile hypersensitivity by neuropeptide Y. J Neurosci 22 : 9858-9867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C ( 1986) The rat brain in stereotaxic coordinates, Ed 2. San Diego: Academic.
- Pertovaara A, Wei H, Hamalainen MM ( 1996) Lidocaine in the rostroventromedial medulla and the periaqueductal gray attenuates allodynia in neuropathic rats. Neurosci Lett 218 : 127-130. [DOI] [PubMed] [Google Scholar]
- Pertovaara A, Keski-Vakkuri U, Kalmari J, Wei H, Panula P ( 2001) Response properties of neurons in the rostroventromedial medulla of neuropathic rats: attempted modulation of responses by [1DMe]NPYF, a neuropeptide FF analogue. Neuroscience 105 : 457-468. [DOI] [PubMed] [Google Scholar]
- Porreca F, Burgess SE, Gardell LR, Vanderah TW, Malan Jr TP, Ossipov MH, Lappi DA, Lai J ( 2001) Inhibition of neuropathic pain by selective ablation of brainstem medullary cells expressing the μ-opioid receptor. J Neurosci 21 : 5281-5288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porreca F, Ossipov MH, Gebhart GF ( 2002) Chronic pain and medullary descending facilitation. Trends Neurosci 25 : 319-325. [DOI] [PubMed] [Google Scholar]
- Rowbotham MC, Davies PS, Verkempinck C, Galer BS ( 1996) Lidocaine patch: double-blind controlled study of a new treatment method for post-herpetic neuralgia. Pain 65 : 39-44. [DOI] [PubMed] [Google Scholar]
- Roychowdhury SM, Heinricher MM ( 1997) Effects of iontophoretically applied serotonin on three classes of physiologically characterized putative pain modulating neurons in the rostral ventromedial medulla of lightly anesthetized rats. Neurosci Lett 226 : 136-138. [DOI] [PubMed] [Google Scholar]
- Seybold VS, Galeazza MT, Garry MG, Hargreaves KM ( 1995) Plasticity of calcitonin gene related peptide neurotransmission in the spinal cord during peripheral inflammation. Can J Physiol Pharmacol 73 : 1007-1014. [DOI] [PubMed] [Google Scholar]
- Sindrup SH, Jensen TS ( 1999) Efficacy of pharmacological treatments of neuropathic pain: an update and effect related to mechanism of drug action. Pain 83 : 389-400. [DOI] [PubMed] [Google Scholar]
- Skilling SR, Sun X, Kurtz HJ, Larson AA ( 1992) Selective potentiation of NMDA-induced activity and release of excitatory amino acids by dynorphin: possible roles in paralysis and neurotoxicity. Brain Res 575 : 272-278. [DOI] [PubMed] [Google Scholar]
- Smith GD, Harmar AJ, McQueen DS, Seckl JR ( 1992) Increase in substance P and CGRP, but not somatostatin content of innervating dorsal root ganglia in adjuvant monoarthritis in the rat. Neurosci Lett 137 : 257-260. [DOI] [PubMed] [Google Scholar]
- Southall MD, Michael RL, Vasko MR ( 1998) Intrathecal NSAIDS attenuate inflammation-induced neuropeptide release from rat spinal cord slices. Pain 78 : 39-48. [DOI] [PubMed] [Google Scholar]
- Sun H, Ren K, Zhong CM, Ossipov MH, Malan TP, Lai J, Porreca F ( 2001) Nerve injury-induced tactile allodynia is mediated via ascending spinal dorsal column projections. Pain 90 : 105-111. [DOI] [PubMed] [Google Scholar]
- Szallasi A, Szabo T, Biro T, Modarres S, Blumberg PM, Krause JE, Cortright DN, Appendino G ( 1999) Resiniferatoxin-type phorboid vanilloids display capsaicin-like selectivity at native vanilloid receptors on rat DRG neurons and at the cloned vanilloid receptor VR1. Br J Pharmacol 128 : 428-434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich-Lai YM, Flores CM, Harding-Rose CA, Goodis HE, Hargreaves KM ( 2001) Capsaicin-evoked release of immunoreactive calcitonin gene-related peptide from rat trigeminal ganglion: evidence for intraganglionic neurotransmission. Pain 91 : 219-226. [DOI] [PubMed] [Google Scholar]
- Wagner R, Deleo JA ( 1996) Pre-emptive dynorphin and N-methyl-d-aspartate glutamate receptor antagonism alters spinal immunocytochemistry but not allodynia following complete peripheral nerve injury. Neuroscience 72 : 527-534. [DOI] [PubMed] [Google Scholar]
- Wall PD, Gutnick M ( 1974a) Properties of afferent nerve impulses originating from a neuroma. Nature 248 : 740-743. [DOI] [PubMed] [Google Scholar]
- Wall PD, Gutnick M ( 1974b) Ongoing activity in peripheral nerves: the physiology and pharmacology of impulses originating from a neuroma. Exp Neurol 43 : 580-593. [DOI] [PubMed] [Google Scholar]
- Wang Z, Gardell LR, Ossipov MH, Vanderah TW, Brennan MB, Hochgeschwender U, Hruby VJ, Malan Jr TP, Lai J, Porreca F ( 2001) Pronociceptive actions of dynorphin maintain chronic neuropathic pain. J Neurosci 21 : 1779-1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weihe E, Nohr D, Schafer MK, Persson S, Ekstrom G, Kallstrom J, Nyberg F, Post C ( 1995) Calcitonin gene related peptide gene expression in collagen-induced arthritis. Can J Physiol Pharmacol 73 : 1015-1019. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Thompson SW ( 1991) The induction and maintenance of central sensitization is dependent on N-methyl-d-aspartic acid receptor activation; implications for the treatment of post-injury pain hypersensitivity states. Pain 44 : 293-299. [DOI] [PubMed] [Google Scholar]
- Wu G, Ringkamp M, Hartke TV, Murinson BB, Campbell JN, Griffin JW, Meyer RA ( 2001) Early onset of spontaneous activity in uninjured C-fiber nociceptors after injury to neighboring nerve fibers. J Neurosci 21 : RC140(1-5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Ringkamp M, Murinson BB, Pogatzki EM, Hartke TV, Weerahandi HM, Campbell JN, Griffin JW, Meyer RA ( 2002) Degeneration of myelinated efferent fibers induces spontaneous activity in uninjured C-fiber afferents. J Neurosci 22 : 7746-7753. [DOI] [PMC free article] [PubMed] [Google Scholar]