

Abstract
Parkinson's disease (PD) is a neurodegenerative disorder characterized by the dysfunction of the nigrostriatal dopaminergic pathway. Although its etiology is not yet fully understood, an interaction of genetic predisposition and environmental factors is frequently discussed. The neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) can evoke PD-like symptoms and neuropathological changes in various species, including mice. It was found repeatedly that mouse strains differ in their susceptibility to MPTP, which might serve as a model for genetic predisposition to neurodegeneration of the nigrostriatal system. In the present study, F2 intercross mice, derived from parental strains with high (C57BL/6J) versus low (BALB/cJ) MPTP susceptibility, were treated with MPTP and phenotyped for dopamine (DA) loss in the neostriatum, a highly sensitive marker of nigrostriatal dysfunction. A subsequent quantitative trait loci analysis revealed a gender-dependent locus for DA loss on chromosome 15 and a putative locus on chromosome 13. A number of potential candidate genes, including the membrane dopamine transporter, are located in the respective areas. Several mechanisms that are possibly involved in the control of the action of MPTP on the nigrostriatal system are discussed.
Keywords: MPTP, quantitative trait loci, Parkinson's disease, dopamine, basal ganglia, F2 intercross, inbred strains, mouse
Introduction
Parkinson's disease (PD) is a disorder of motor control with the main symptoms akinesia, tremor, and rigidity. These symptoms are primarily caused by a dopamine (DA) deficit in the putamen and caudate nucleus that in turn is caused by dysfunction and neurodegeneration of the dopaminergic neurons in the substantia nigra (Gerlach and Riederer, 1996). The underlying causes of the neurodegeneration are only partly understood. Etiological models assuming an interaction of environmental and genetic factors have been widely discussed (Checkoway et al., 1998; McNaught et al., 1998; Mizuno et al., 1998; Jenner, 1998; Feldman and Ratner, 1999). Mutations in the α-synuclein gene and the parkin gene can induce familial parkinsonism (Polymeropoulos et al., 1997; Kitada et al., 1998), but they obviously do not explain the majority of PD cases. The identification of other risk genes in humans has remained controversial (Nicholl et al., 1999; Tan et al., 2000).
Valuable insights into the processes that lead to the dysfunction and neurodegeneration of the nigrostriatal system have been derived from studying the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) in laboratory animals (Przedborski et al., 2000). MPTP reproduces virtually all symptoms of PD in various animals, including mice (Gerlach and Riederer, 1996). When applied systemically, it passes the blood-brain barrier and is converted by monoamine oxidase B (MAO-B) to the active toxin 1-methyl-4-phenylpyridine (MPP+), which is transported via the membrane dopamine transporter (DAT) into dopaminergic cells, where it evokes a cascade of events finally leading to apoptosis or necrosis (Gerlach and Riederer, 1996; Przedborski et al., 2000). There is evidence that the action of MPTP can be influenced by genetic background because the degree of nigrostriatal damage produced by a defined MPTP regimen differs between strains of mice (Giovanni et al., 1991; German et al., 1996). This is further supported by examinations of F1 cross-breedings of inbred mice (Hamre et al., 1999; Sedelis et al., 2000a). Numerous experiments have been undertaken to examine separately the potential influence of single factors such as (neuro)melanin, α-synuclein, and MAO-B, yet no candidate mechanism has been identified unequivocally so far (Hofele et al., 2001; Rathke-Hartlieb et al., 2001).
Quantitative trait loci (QTL) analysis is a genome screen method that has been a powerful tool in neuroscience research, allowing identification of chromosomal regions involved, e.g., in alcohol and drug sensitivity (Crabbe et al., 1999) or seizure response to epileptogenic compounds (Ferraro et al., 1997), thereby setting the stage for the search for the underlying genes. In the present study, a QTL design was used that aimed to identify loci related to MPTP neurotoxicity. We produced an F2 intercross from C57BL/6 and BALB/c, i.e., two inbred strains that differ in their susceptibility to MPTP (Sedelis et al., 2000b). F2 animals were treated with MPTP, phenotyped by neurochemical examination of their neostriatal DA levels, which are highly sensitive markers for nigrostriatal damage (Schwarting and Huston, 1996b), and genotyped to allow for QTL analysis. The results of this approach might help to generate new insights into the specific genetic mechanisms involved in MPTP neurotoxicity in the mouse and in neurodegeneration in PD.
Materials and Methods
Animals. Male and female (C57BL/6 × BALB/c)F2 hybrids were bred at the Tierversuchsanlage of the University of Düsseldorf, Germany. The C57BL/6J and BALB/cJ founder animals were originally obtained from RCC (Itingen, Switzerland). The mating crosses used to generate the F1 and F2 animals were reciprocal. Thus, the Y chromosome (Chr.) is equally represented from C57BL/6J and BALB/cJ strains. During the experiment, mice were housed singly in plastic cages under a 12 hr light/dark cycle at an ambient temperature of 25 ± 2°C and had ad libitum access to food and water.
Treatment. MPTP solution was prepared by dissolving MPTP-HCl (RBI, Köln, Germany) in saline. The animals received four injections of MPTP-HCl solution, which was injected intraperitoneally in a volume of 1 ml/100 gm body weight, equivalent to 12.5 mg/kg free base per injection; the interval between injections was 2 hr. (C57BL/6 × BALB/c)F2 controls received four injections with vehicle (saline), respectively. The age at which animals were treated was held constant in a range of 8-10 weeks for all animals used in this experiments.
Neurochemistry. Seven days after drug treatment, the animals were killed by cervical dislocation. The brains were cut coronally using a rodent brain matrix (ASI Instruments, Warren, PA), and slices of 1 mm thickness anterior to the chiasma opticum were obtained. Tissue from the neostriatum was then quickly dissected out manually from these slices. Dissection was performed on an ice-cold plate, using a standard mouse brain atlas as a reference (Franklin and Paxinos, 1997). Anatomical landmarks for the identification of the borders of the neostriatum were the lateral ventricle, the external capsule, and the anterior commissure. The respective samples from the left and right hemispheres were pooled to one sample per animal that was weighed, sonicated, centrifuged, filtered, and examined for its contents of DA using HPLC with electrochemical detection according to the method of Schwarting and Huston (1987).
DNA isolation. Mouse tail tissue, weighing 4-6 mg, was placed in 300 μl of cell lysis buffer (Puregene, Gentra Systems, Minneapolis, MN), and 1.5 μl of proteinase K solution (20 mg/ml) was added after overnight incubation at 45°C. RNA was removed by adding 1.5 μl of RNase solution (Roche) (0.5 mg/ml) to the cell lysate, after incubation at 37°C for 45 min. Protein precipitation was achieved by addition of 100 μl of protein precipitation solution (Puregene, Gentra Systems) and subsequent centrifugation at 14,000 × g for 5 min. DNA was precipitated by pouring the supernatant into a fresh tube containing 300 μl of 2-propanol. After 15 sec centrifugation was performed to make DNA adhere to the bottom, supernatant was poured off and 300 μl of ethanol (70%) was added to wash the DNA pellet. After 15 sec centrifugation, the tube was drained and the pellet allowed to dry for 2 hr. Thereafter, 300 μlofTE' buffer was added, and DNA was rehydrated overnight at 45°C and then stored at 4°C until genotyping.
Genotyping. The genotyping method of Buck et al. (1997) was used, using microsatellite markers from the MIT set (Research Genetics, Huntsville, AL). In short, the signal was amplified in a standard 40 cycle PCR reaction using AmpliTAQ Gold (Roche) as polymerase. The PCR products were subsequently separated by running them on agarose gels (4%) at 180 V for 45-150 min, depending on the marker. Ethidium bromide staining allowed visualization of the bands under UV light. Bands were imaged in a fluorescence scanner using QuantityOne Software (Bio-Rad), printed, and identified manually by comparing them with C57BL/6, BALB/c, and C57BL/6 + BALB/c heterozygous control bands.
QTL analysis. QTL detection was performed in two steps. First, from a total of n = 311 animals that survived MPTP treatment, the animals with the most extreme phenotypes were selected (i.e., the animals in the first and tenth decile of the DA depletion distribution, separately for each gender). This subsample of mice (n = 62) was genotyped with a total of 68 markers, covering the genome at intervals of ∼20 centimorgans (cM) as part of step 1. The MapManager software (Macintosh version b28) (Manly and Olson, 1999) was used to detect QTLs. LOD scores (df = 2) were estimated as -log10(p). A permutation test (Churchill and Doerge, 1994) implemented within MapManager was used to estimate the empirical significance and suggestive thresholds for this genome-wide search, in which the significance threshold denotes acceptance of a 5% chance of even one false positive, whereas the suggestive threshold denotes an average of one false positive in a full genome search. The threshold for significance was p = 0.0003 (LOD 3.5), and for suggestive it was p = 0.008 (LOD 2.1) on the basis of 10,000 permutations.
In addition, the error check routine in Mapmaker/QTL (Lincoln and Lander, 1992) was used to screen the data set for potential occurrence of erroneous genotyping, and interval mapping was performed by the linear least squares method of Haley and Knott (1992) using MapManager QT.
The first step data were also analyzed for QTLs and for interactions (epistasis) using the Pseudomarker 0.9 PAIRSCAN program written for the MATLAB (Mathworks Inc., Natick, MA) programming environment (Sen and Churchill, 2001) (obtained from http://www.jax.org/research/churchill) using default settings except where noted otherwise. This program uses Monte Carlo computer sampling to estimate the most likely values of unknown parameters (QTL genotypes, locations, missing data) using Bayesian methods. Sixty-four imputations (samplings) of unknown genotypes based on the observed marker data were made at 5 cM intervals, which approximate interval mapping at this resolution. The PAIRSCAN program assessed the phenotypic effects of pairs of marker (or intervals) taken jointly, which allows a genome-wide search for epistasis, or gene-gene interactions, as described below.
For tests of epistasis, we required that the combined (or full model) effects of a marker pair, which reflect the main effects of both markers plus their interaction, exceed the threshold for significance (5% genome-wide error rate) estimated by a permutation test performed using the Pseudomarker PAIRSCAN program. In this study the threshold was set at LOD 7.3 (df = 8; p = 5 × 10-5) on the basis of 500 permutations. When the combined effects of a marker pair were significant, we then tested for their interaction at a significance threshold of p < 0.01 (Sen and Churchill, 2001). This approach represents a conditional search for epistasis (Chase et al., 1997) because only marker pairs attaining significance for their joint effects on a trait (the condition) are tested for possible interactions. The main protection against false-positive interactions rests on the stringent criterion set for the combined or joint effects of marker pairs (which includes their interaction), which markedly reduces the number of locus pairs tested directly for interactions alone (only two pairs in this study). This is in contrast to an unconditional search for epistasis, in which all mathematically possible pairs of loci are tested for interactions (∼3000 marker pairs). Only an unconditional search can detect interactions between markers, neither of which has appreciable main effects on the phenotype; however, an unconditional search was not used here because it greatly increases the rate of false positives to the point at which little power to detect interactions remains with our limited sample size (Hood et al., 2001).
In a second step, all remaining MPTP-treated animals (i.e., those with a nonextreme phenotype) were genotyped with those markers that attained p < 0.05 in the first step on the basis of MapManager QT results. For this second step, the p value was set to 0.0002 on the basis of permutation tests run as described for the first step. This procedure was chosen to address the problem of type I error (identification of a false QTL) versus type II error (failure to detect an actual QTL) (Belknap et al., 1996).
Consensus standards for assessing the significance of gender-specific QTLs do not exist. We therefore used a Bonferroni correction to correct for the nine QTL regions (see Table 2) that we subjected to QTL analysis by gender. This yields a threshold of p = 0.05/9 = 0.005 (two-tailed) for the gender difference. Therefore, gender specificity of QTLs was judged to be significant when the ratio of p values per gender differed by at least 2.3 orders of magnitude. This corresponds to p < 0.005 or LOD 2.3 for the gender difference.
Table 2.
Loci identified using a conservative significance level by separate QTL analyses for each gender
|
Marker |
p value, males |
p value, females |
Ratio of p values |
|---|---|---|---|
| D13Mit91 | 0.00542 | 0.17491 | 0.0310 |
| D15Mit100 | 0.00904 | 9.2 × 10-6 | 0.0010 |
| D15Mit68 | 0.00275 | 1.2 × 10-6 | 0.0004 |
| DXMit144 | 0.11325 | 0.18151 | 0.6239 |
| DXMit67
|
0.03653
|
0.09527
|
0.3834
|
This table lists associations between the phenotype and those MIT markers, which were significant at p < 0.0002 in a previous QTL analysis applied to the entire sample of n = 311 mice (see Results). QTL analyses were here performed separately for males (n = 179) and females (n = 132). A difference between genders was considered significant if the ratio of p values per gender differed by at least 2.3 orders of magnitude, equivalent to p < 0.005 for the gender difference.
Results
Lethality
Male (n = 179) and female (n = 193) (C57BL/6 × BALB/c)F2 hybrids were treated with MPTP. A number of animals treated with MPTP did not survive after the application of the neurotoxin. This MPTP-related lethality was strictly gender dependent, because the mortality rate was 31.6% in females (n = 61) but 0% in males.
Sex differences in phenotype
Seven days after treatment, neostriatal DA levels were determined. This phenotypic marker could be obtained only from those animals that survived the MPTP treatment.
In addition, the mean neostriatal dopamine content of 23 male and 24 female (C57BL/6 × BALB/c)F2 hybrid mice, which had been treated with saline and thus served as controls for our F2 cross-breeding, was determined separately for each gender. For each MPTP-treated individual the neostriatal DA depletion was calculated and defined as loss in percentage of mean control levels. (Absolute numbers of DA control levels and their variability are given in the legend of Fig. 1.)
Figure 1.
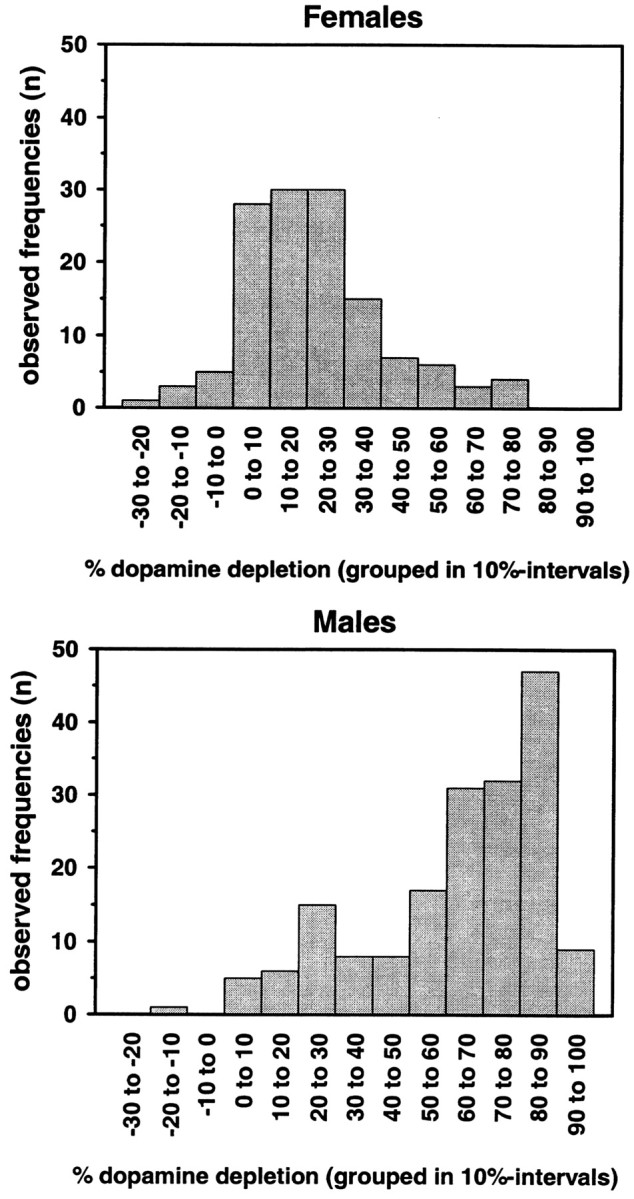
Distribution pattern of the phenotype “neostriatal dopamine depletion after MPTP administration.” (C57BL/6 × BALB/c)F2 hybrids were treated with four doses of 15 mg MPTP-HCl/g body weight in 2 hr intervals, and neostriatal DA levels were determined postmortem by HPLC, with electrochemical detection 7 d later. Absolute numbers of animals are shown in 10% intervals of DA depletion separately for males (n = 179) and females (n = 132). “Depletion” is defined as dopamine loss in percentage compared with controls. DA depletion was significantly higher in males than in females (t test; t = 16.1; df = 309; p < 0.001). Saline-treated controls, which are C57BL/6 × BALB/c)F2 hybrids, had neostriatal dopamine levels of 15.23 ± 0.41 and 15.28 ± 0.40 for males (n = 23) and females (n = 24), respectively (nanograms of DA per milligram of wet tissue weight; means ± SEM).
The phenotype “DA depletion” showed a gender-dependent pattern (Fig. 1). Male mice were significantly more susceptible to MPTP neurotoxicity than female mice (t test; t = 16.1; df = 309; p < 0.001).
Simulation: if males had died
To test for the scenario that the lower mean DA depletion in females could be a result of the possibility that just the most affected females, in terms of DA depletion, might have died after MPTP treatment, we simulated a comparable situation in the male subsample. Of males (n = 57) at the upper end of the DA depletion distribution, corresponding to the actual lethality rate in females, 31.6% were excluded from the sample, and the mean DA depletion was calculated again (Fig. 2). This simulated DA depletion mean is ∼10% points lower than the actual DA depletion mean for the males, but the gender difference persists, because the mean DA loss is still considerably higher than in females.
Figure 2.

Simulation: neostriatal DA depletion outcome if males severely affected by MPTP had died (in percentage loss of control values; means ± SEM). The black column indicates the actually observed DA depletion after MPTP treatment in the male (C57BL/6 × BALB/c)F2 hybrid mice; the white column indicates the respective mean for the females. A total of 192 females was treated with MPTP, but only 132 of them survived treatment. The DA depletion of the deceased females (n = 61 or 31.6%) could not be determined. None of the males had died. The gray column reflects the simulated outcome if the 31.6% most affected males of our sample would have died as a consequence of MPTP treatment. Note that even after the simulated death of 57 male mice the gender difference is ∼30% points.
Identification of QTL related to MPTP neurotoxicity
In the first step, animals from the bottom and top 10% of the phenotypic distribution were genotyped (males, n = 36; females, n = 26), and a QTL analysis was performed. As documented in Table 1, nine markers showed p < 0.05 associations with striatal DA.
Table 1.
Marker loci attaining p < 0.05 in genotyping step 1
|
Marker |
Chromosomal position |
p value |
|---|---|---|
| D3Mit216 | 58.8 | 0.00442 |
| D13Mit91 | 30.0 | 0.03516 |
| D13Mit233 | 45.0 | 0.01011 |
| D14Mit266 | 60.0 | 0.01904 |
| D15Mit100 | 21.0 | 0.00133 |
| D15Mit68 | 44.1 | 0.01327 |
| D19Mit46 | 24.0 | 0.02109 |
| D19Mit6 | 55.0 | 0.04494 |
| DXMit67
|
60.0
|
0.02480
|
From a total of n = 311 (C57BL/6 × BALB/c)F2 hybrids that had been phenotyped for neostriatal DA depletion after MPTP administration (4 × 15 mg MPTP-HCI/kg body weight), those 10% of animals at each extreme of the phenotypic distribution were chosen. Subsequent genotyping of those animals was performed with PCR using a set of 68 MIT microsatellite markers. The table lists only those associations of the MIT markers with the phenotype attaining p < 0.05. Calculations were done using MapManager QT Software (Manly and Olson, 1999). On the basis of a permutation test, those attaining p < 0.008 were suggestive and p < 0.0003 were significant. Information about the relative positions of the markers on the chromosomes was obtained from The Jackson Laboratory Mouse Genomics Database [http://www.jax.org (May 2002)]. Microsatellite markers used for the genome screen were as follows: Chr. 1: 132, 365, 294, 150, 155; Chr. 2: 58, 498, 230; Chr. 3: 216, 65, 147; Chr. 4: 53, 203, 343; Chr. 5: 13, 58, 136, 168; Chr. 6: 245, 230, 194; Chr. 7: 229, 62, 68; Chr. 8: 145, 320, 156; Chr. 9: 247, 4, 273, 18; Chr. 10: 77, 126, 42, 179; Chr. 11: 78, 174, 41, 338; Chr. 12: 63, 7, 150; Chr. 13: 3, 91, 233, 228; Chr. 14: 121, 69, 266; Chr. 15: 226, 100, 68; Chr. 16: 9, 57, 173, 106; Chr. 17: 198, 193, 221; Chr. 18: 14, 184; Chr. 19: 28, 46, 6; Chr. X: 114, 144, 67.
Subsequently, in the second step, the remaining animals were genotyped with the set of markers listed in Table 1. Then the QTL analysis was repeated using the whole sample size of MPTP-treated animals (n = 311) for these markers, and the significance level was set to p = 0.0002.
This analysis yielded one chromosomal region that was significantly related to the trait. This site was located on chromosome 15 in the proximity of markers D15Mit68 (p = 0.000048) and D15Mit100 (p = 0.00056). A second locus on chromosome 13 (D13Mit91; p = 0.00470) was classified as a suggestive locus according to the Lander and Kruglyak (1995) guidelines.
Because a gender difference in the phenotype was observed, we investigated the possibility that the identified loci might be gender specific. Thus, the QTL analysis was then performed separately for males and females (Table 2). The p values of both genders were compared for each locus. For the locus at D15Mit68, a significant sex difference was revealed, with females having a significantly lower p value (higher LOD scores) than males (Fig. 3).
Figure 3.

The negative logarithm of the p value (LOD score) plotted against map location (cM) along the length of proximal to mid chromosome 15 (Chrom. 15) for both females and males. Numbers along the bottom of the plot are marker numbers, e.g., -226 denotes the map location of D15Mit226. Data are from step 1 involving the extreme scoring tails of the trait distribution because more markers were used than in step 2. A highly significant gender difference is apparent from the 3.7 orders of magnitude difference in p values observed between the two genders.
Remarkably, for both loci at D15Mit68 and D13Mit91, the BALB/c allele was associated with a higher mean DA depletion than the C57BL/6 allele.
Finally, the error check routine of the Mapmaker/QTL software was performed to detect potential genotyping errors. None were found exceeding a LOD score of 2.0. The results of this analysis indicated that no exclusion of data were necessary.
The Pseudomarker PAIRSCAN program identified only one epistatic interaction in our F2 that met the significance criteria set in Materials and Methods (Figs. 4, 5). This was an interaction between a QTL on Chr. 9 (∼29 cM) and another locus on Chr. 3 (∼79 cM) as shown in Figures 4 and 5. This interaction attained p = 0.0008 for the interaction. A second interaction approached significance involving a locus on Chr. 5 (∼65 cM) and another on Chr. 13 (∼65 cM), as shown in Figure 5. This locus pair attained p = 0.0003 for the interaction. Figure 5 also shows all the interactions that attained p < 0.05 in our PAIRSCAN analysis, although there was sufficient power in our limited sample to detect only one of them as significant, as noted above. For plotting purposes, we used a 2.5 cM resolution for Figure 5.
Figure 4.

The degree of dopamine depletion by genotype for the D9Mit4 marker on Chr. 9 as a function of genotype at another locus (D3Mit147) on Chr. 3. An interaction is demonstrated by the fact that the phenotypic effect of the Chr. 9 QTL reverses direction as a function of the Chr. 3 modifier locus genotype at D3Mit147. Neither locus had a significant main effect in the Map-Manager analysis, which does not assess interactions, but emerged as significant only in the PAIRSCAN analysis mainly because of the strength of the interaction shown above (p = 0.0008).
Figure 5.

Two-dimensional plot of PAIRSCAN results for the nine chromosomes of greatest interest using a 2.5 cM grid. These chromosomes were chosen for display either because they contained a significant or suggestive QTL or because they were involved in an interaction attaining p < 0.01. The bottom half shows the LOD scores for the full model (df = 8) for marker pairs, which includes the main effects of each marker plus their interaction, whereas the top half shows LOD scores for the interaction alone (df = 4). Areas in white failed to reach p < 0.05 (LOD 3.4, bottom plot; LOD 2.1, top plot), whereas those exceeding this relaxed criterion are shaded from gray to black as a function of the LOD score scale shown at right. For visual clarity, the LOD scores in the top half were increased by 1.64-fold. Although all interactions attaining p < 0.05 are shown, only one attained significance (Chr. 3 × 9) and another closely approached significance (Chr. 5 × 13). Both are denoted by arrows. The 5% genome-wide significance thresholds estimated by permutation tests were LOD 7.4 (p = 4 × 10-5) in the bottom plot and 4.6 (p = 3 × 10-4) in the top one.
Discussion
Treatment of (C57BL/6 × BALB/c)F2 mice with the neurotoxin MPTP resulted in a depletion of DA in the neostriatum, as described previously for other strains of mice (Sundström et al., 1987; Hoskins and Davis, 1989; Giovanni et al., 1991). In our genetically heterogeneous sample, we observed considerable variance for this phenotype, i.e., the actual amount of DA loss varied within the F2, with some animals showing only minimal loss, whereas others were severely affected. Furthermore, a gender difference was found in that males showed a much higher DA loss than females. Surprisingly, we did not find such gender differences in the founding strains C57BL/6 or BALB/c (Sedelis et al., 2000b), but we did find some differences in their F1 generation (Sedelis et al., 2000a). It is known that the estrogen hormone can provide some protection from DA depletion in the MPTP mouse model (Dluzen et al., 1996; Arvin et al., 2000). If this explanation applied to the gender difference found in the present study, however, it would remain unclear why females in our parental generation could not benefit from this protection. In addition to the neurotoxic effects, our regimen of MPTP caused lethality in a number of females, whereas this phenomenon was virtually absent in males. From pathological examinations in several deceased animals (data not shown) we concluded that a potential cause of death might have been a pulmonary perivascular edema, which can be observed after MPTP treatment (Johannessen et al., 1986). Because estrogen is known to promote formation of edemas (Hadley, 1996), it is tempting to speculate that the lethality in females might be linked to their higher estrogen levels.
It could be argued that the females' lethality in our study might have selectively affected the most DA-depleted individuals, the striatal DA levels of which could not be determined. Simulating a comparable situation in the male sample only slightly lowered the mean DA loss, however, thereby suggesting that the gender difference is most probably caused by factors other than lethality.
For a QTL approach, the choice of an appropriate phenotype is critical. From a number of possible variables, we considered a neurochemical measure, neostriatal DA depletion, to be the most useful parameter of dysfunction of the nigrostriatal system, which is sensitive in a broad range from minimal to massive neuronal damage. The sensitivity is reflected in the fact that some degree of DA loss could be observed in most of our animals. In contrast, a loss of nigral DA neurons occurs only in severely affected animals (German et al., 1996) and thus is less useful to cover the full spectrum of the actions of MPTP on the nigrostriatal system. The same limitation applies to behavioral measures; mice seem to have highly effective compensatory mechanisms, preventing longer-lasting symptoms below DA losses of at least 80% (Sedelis et al., 2001). It could be argued that a higher MPTP regimen should have been applied to obtain a more pronounced neurotoxic effect; however, lethality is a severely limiting factor at only moderately higher MPTP doses in the parental strains (Schwarting et al., 1999), so that the dose used in our study reflects a compromise between acceptable survival rates and considerable neurotoxic effects.
Performing QTL analysis in a two-step approach while pooling data from males and females yielded two loci on separate chromosomes, suggestive of an involvement in susceptibility to MPTP neurotoxicity. The first region of interest was located on chromosome 15 at marker D15Mit68. Interval mapping including D15Mit100 did not narrow the position of the contributing gene. When the data for males and females were calculated separately, the p value for females was significantly lower (LOD score was higher) compared with that of the males. Thus, this locus is gender specific, which means that it contributes to the phenomenon of MPTP susceptibility to a markedly different degree in males and females. Possibly, these results indicate the existence of a protective gene at this locus that is of higher benefit for females than for males. Interestingly, the Mouse Genome Database (MGD) (The Jackson Laboratory, Bar Harbor, ME; http://www.jax.org) lists several QTL in the region of D15Mit68 that are related to dopamine receptor binding, cocaine-related behaviors, and, most interestingly, neostriatal DAT densities (Jones et al., 1999). This transporter plays a crucial role in MPTP neurotoxicity because it provides a carrier mechanism for the active toxic metabolite of MPTP, MPP+, into dopaminergic cells. It was found that DAT mRNA levels were high in those midbrain neurons vulnerable to MPTP (Sanghera et al., 1997) and that the absence of DAT virtually abolished MPTP neurotoxicity in DAT knock-out mice (Gainetdinov et al., 1997). A hypothetical regulative mechanism on chromosome 15 for DAT densities could therefore influence the ability of MPP+ and similar neurotoxins to specifically enter dopaminergic neurons.
Furthermore, the gene for heat shock factor 1 (hsf1), a transcription factor for heat shock proteins, has been mapped to 43.0 cM on chromosome 15. There is experimental evidence that at least one of these proteins, HSP72, can exert a neuroprotective effect in the MPTP mouse model (Freyaldenhoven et al., 1995; Freyaldenhoven and Ali, 1997). It can be speculated that the transcriptional regulation of heat shock proteins might contribute to protection from MPTP-induced neurodegeneration.
On chromosome 13, a locus not reaching significance and therefore classified as “suggestive” was located in the region between marker D13Mit91 and D13Mit233. Three genes in this part of the genome are of particular interest. First, the dopamine receptor D1a gene was found in the putative region; the role of dopamine receptors in compensation of neostriatal DA loss has been well documented (Schwarting and Huston, 1996a,b); in contrast, information about a possible involvement in the control of the neurodegenerative process itself in the MPTP model is scarce. Results from a human association study do not support the idea of such a function at least for the DA receptors DRD2 and DRD4 (Nicholl et al., 1999).
Second, the Ntrk2 gene, encoding a receptor for the brain-derived neurotrophic factor (BDNF), is located at position 36.0 cM (MGD). The internal levels of BDNF are supposed to mediate, at least partly, the differential vulnerabilities to MPTP of the mesolimbic and nigrostriatal systems, and external application of BDNF acts as a neuroprotective agent against MPP+in vivo and in vitro (Hung and Lee, 1996; Son et al., 1999). An approach somewhat complementary to our study was used in a recent study. There, differential gene expression analysis in brain tissue of MPTP-treated mice revealed, among other changes, the up-regulation of two other growth factors, glia cell line-derived neurotrophic factor and nerve growth factor α-subunit (Grünblatt et al., 2001), further supporting the possibility of an involvement of such factors in MPTP susceptibility.
Third, the gene encoding the membrane DAT maps to 41.0 cM. Considering these results, and the potential association of the DAT density QTL on chromosome 15 with the MPTP phenotype in our study, it would be highly interesting to compare DAT functions and polymorphisms in strains of mice known to differ in MPTP susceptibility. In this context, it is exciting that certain amino acid exchanges in the rat DAT gene have been shown in vitro to alter the affinity of the DAT to MPP+ or its uptake velocity, respectively (Mitsuhata et al., 1998). Furthermore, it has been speculated recently to what extent dopamine transporters play a role in human Parkinson's disease (Uhl, 1998; Miller et al., 1999); however, the human association study mentioned above could not establish a significant link between DAT polymorphisms and PD (Nicholl et al., 1999).
Surprisingly, for both the chromosome 15 QTL and the putative chromosome 13 locus, the BALB/c alleles were associated with higher striatal DA depletion, although BALB/c is the more MPTP-resistant parental strain. Assuming a polygenetic influence on MPTP susceptibility, however, it is likely that each of the parental strains carries alleles that enhance and reduce MPTP susceptibility (or, depending on the mechanism, MPTP resistance). Such an assumption would be in line with both our previous findings of a decreased MPTP susceptibility in our F1 generation as compared with the parental generation (Sedelis et al., 2000a) and apparently contradictory findings of a C57BL/6 MPTP susceptibility locus acting in a dominant manner (Hamre et al., 1999). Furthermore, in the present study, analysis of gene interaction demonstrated that the influence of one QTL (marker D9Mit4) on nigrostriatal DA depletion can depend critically on the genotypic configuration at a second locus (D3Mit147); interestingly, the former locus is in close vicinity to the dopamine receptor 2 gene Drd2.
Summing up, our findings support the assumption of a polygenic influence on the effects of MPTP on nigrostriatal neurochemistry. More detailed examination of the identified loci on chromosomes 13 and 15 will be necessary, e.g., by further testing promising candidate mechanisms, such as dopamine transporter functions, in separate experiments. It remains to be determined whether and to what extent genetic mechanisms of susceptibility of the nigrostriatal system identified in the MPTP mouse model might also apply to the neurodegenerative processes found in idiopathic Parkinson's disease.
Footnotes
This work was supported by Grant Hu 306/21-1 from the Deutsche Forschungsgemeinschaft and National Institutes of Health Grants DA10913 and DA05228. We thank Jennifer Dicks, Melinda Helms, Patrizia Kutz, and Ingo Willuhn for excellent technical assistance and Dr. Anke Brumloop for skillful pathological examinations.
Correspondence should be addressed to Dr. John K. Belknap, Research Service (R&D-5), Veterans Affairs Medical Center, Portland, OR 97239. E-Mail: belknajo@ohsu.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/238247-07$15.00/0
References
- Arvin M, Fedorkova L, Disshon KA, Dluzen DE, Leipheimer RE ( 2000) Estrogen modulates responses of striatal dopamine neurons to MPP+: evaluations using in vitro and in vivo techniques. Brain Res 872 : 160-171. [DOI] [PubMed] [Google Scholar]
- Belknap JK, Mitchell SR, O'Toole LA, Helms ML, Crabbe JC ( 1996) Type I and type II error rates for quantitative trait loci (QTL) mapping studies using recombinant inbred mouse strains. Behav Genet 26 : 149-160. [DOI] [PubMed] [Google Scholar]
- Buck KJ, Metten P, Belknap JK, Crabbe JC ( 1997) Quantitative trait loci involved in genetic predisposition to acute alcohol withdrawal in mice. J Neurosci 17 : 3946-3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase K, Adler FR, Lark KG ( 1997) Epistat: a computer program for identifying and testing interactions between pairs of quantitative trait loci. Theor Appl Genet 94 : 724-730. [Google Scholar]
- Checkoway H, Farin FM, Costa MP, Kirchner SC, Costa LG ( 1998) Genetic polymorphisms in Parkinson's disease. Neurotoxicology 19 : 635-643. [PubMed] [Google Scholar]
- Churchill GA, Doerge RW ( 1994) Empirical threshold values for quantitative trait loci mapping. Genetics 138 : 963-971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabbe JC, Phillips TJ, Buck KJ, Cunningham CL, Belknap JK ( 1999) Identifying genes for alcohol and drug sensitivity: recent progress and future directions. Trends Neurosci 22 : 173-179. [DOI] [PubMed] [Google Scholar]
- Dluzen DE, McDermott JL, Liu B ( 1996) Estrogen as a neuroprotectant against MPTP-induced neurotoxicity in C57/Bl mice. Neurotoxicol Teratol 18 : 603-606. [DOI] [PubMed] [Google Scholar]
- Feldman RG, Ratner MH ( 1999) The pathogenesis of neurodegenerative disease: neurotoxic mechanisms of action and genetics. Curr Opin Neurol 12 : 725-731. [DOI] [PubMed] [Google Scholar]
- Ferraro TN, Golden GT, Smith GG, Schork NJ, St.-Jean P, Ballas C, Choi H, Berrettini WH ( 1997) Mapping murine loci for seizure response to kainic acid. Mamm Genome 8 : 200-208. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G ( 1997) The mouse brain in stereotaxic coordinates. San Diego: Academic.
- Freyaldenhoven TE, Ali SF ( 1997) Role of heat shock proteins in MPTP-induced neurotoxicity. Ann NY Acad Sci 825 : 167-178. [DOI] [PubMed] [Google Scholar]
- Freyaldenhoven TE, Ali SF, Hart RW ( 1995) MPTP- and MPP(+)-induced effects on body temperature exhibit age- and strain-dependence in mice. Brain Res 688 : 161-170. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR, Fumagalli F, Jones SR, Caron MG ( 1997) Dopamine transporter is required for in vivo MPTP neurotoxicity: evidence from mice lacking the transporter. J Neurochem 69 : 1322-1325. [DOI] [PubMed] [Google Scholar]
- Gerlach M, Riederer P ( 1996) Animal models of Parkinson's disease: an empirical comparison with the phenomenology of the disease in man. J Neural Transm 103 : 987-1041. [DOI] [PubMed] [Google Scholar]
- German DC, Nelson EL, Liang CL, Speciale SG, Sinton CM, Sonsalla PK ( 1996) The neurotoxin MPTP causes degeneration of specific nucleus A8, A9 and A10 dopaminergic neurons in the mouse. Neurodegeneration 5 : 299-312. [DOI] [PubMed] [Google Scholar]
- Giovanni A, Sieber BA, Heikkila RE, Sonsalla PK ( 1991) Correlation between the neostriatal content of the 1-methyl-4-phenylpyridinium species and dopaminergic neurotoxicity following 1-methyl-4-phenyl-1, 2,3,6-tetrahydropyridine administration to several strains of mice. J Pharmacol Exp Ther 257 : 691-697. [PubMed] [Google Scholar]
- Grünblatt E, Mandel S, Maor G, Youdim MBH ( 2001) Gene expression analysis in N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mice model of Parkinson's disease using cDNA microarray: effect of R-apomorphine. J Neurochem 78 : 1-12. [DOI] [PubMed] [Google Scholar]
- Hadley ME ( 1996) Endocrinology. Englewood Cliffs, NJ: Prentice-Hall.
- Haley CS, Knott SA ( 1992) A simple regression method for mapping quantitative trait loci in line crosses using flanking markers. Heredity 69 : 315-324. [DOI] [PubMed] [Google Scholar]
- Hamre K, Tharp R, Poon K, Xiong XP, Smeyne RJ ( 1999) Differential strain susceptibility following 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) administration acts in an autosomal dominant fashion: quantitative analysis in seven strains of Mus musculus Brain Res 828 : 91-103. [DOI] [PubMed] [Google Scholar]
- Hofele K, Sedelis M, Auburger GW, Morgan S, Huston JP, Schwarting RKW ( 2001) Evidence for a dissociation between MPTP-toxicity and tyrosinase activity based on congenic mouse strain susceptibility. Exp Neurol 168 : 116-122. [DOI] [PubMed] [Google Scholar]
- Hood H, Belknap JK, Crabbe JC, Buck KJ ( 2001) Genome-wide search for epistasis in a complex trait: pentobarbital withdrawal convulsions in mice. Behav Genet 31 : 93-100. [DOI] [PubMed] [Google Scholar]
- Hoskins JA, Davis LJ ( 1989) The acute effect on levels of catecholamines and metabolites in brain, of a single dose of MPTP in 8 strains of mice. Neuropharmacology 28 : 1389-1397. [DOI] [PubMed] [Google Scholar]
- Hung HC, Lee EH ( 1996) The mesolimbic dopaminergic pathway is more resistant than the nigrostriatal dopaminergic pathway to MPTP and MPP+ toxicity: role of BDNF gene expression. Brain Res Mol Brain Res 41 : 14-26. [DOI] [PubMed] [Google Scholar]
- Jenner P ( 1998) Oxidative mechanisms in nigral cell death in Parkinson's disease. Mov Disord 13 [Suppl 1]: 24-34. [PubMed] [Google Scholar]
- Johannessen JN, Adams JD, Schuller HM, Bacon JP, Markey SP ( 1986) 1-Methyl-4-phenylpyridine (MPP+) induces oxidative stress in the rodent. Life Sci 38 : 743-749. [DOI] [PubMed] [Google Scholar]
- Jones BC, Tarantino LM, Rodriguez LA, Reed CL, McClearn GE, Plomin R, Erwin VG ( 1999) Quantitative-trait loci analysis of cocaine-related behaviours and neurochemistry. Pharmacogenetics 9 : 607-617. [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N ( 1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392 : 605-608. [DOI] [PubMed] [Google Scholar]
- Lander ES, Kruglyak L ( 1995) Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet 11 : 241-247. [DOI] [PubMed] [Google Scholar]
- Lincoln SE, Lander ES ( 1992) Systematic detection of errors in genetic linkage data. Genomics 14 : 604-610. [DOI] [PubMed] [Google Scholar]
- Manly KF, Olson JM ( 1999) Overview of QTL mapping software and introduction to Map Manager QT. Mamm Genome 10 : 327-334. [DOI] [PubMed] [Google Scholar]
- McNaught KS, Carrupt PA, Altomare C, Cellamare S, Carotti A, Testa B, Jenner P, Marsden CD ( 1998) Isoquinoline derivatives as endogenous neurotoxins in the aetiology of Parkinson's disease. Biochem Pharmacol 56 : 921-933. [DOI] [PubMed] [Google Scholar]
- Miller GW, Gainetdinov RR, Levey AI, Caron MG ( 1999) Dopamine transporters and neuronal injury. Trends Pharmacol Sci 20 : 424-429. [DOI] [PubMed] [Google Scholar]
- Mitsuhata C, Kitayama S, Morita K, Vandenbergh D, Uhl GR, Dohi T ( 1998) Tyrosine-533 of rat dopamine transporter: involvement in interactions with 1-methyl-4-phenylpyridinium and cocaine. Mol Brain Res 56 : 84-88. [DOI] [PubMed] [Google Scholar]
- Mizuno Y, Hattori N, Matsumine H ( 1998) Neurochemical and neurogenetic correlates of Parkinson's disease. J Neurochem 71 : 893-902. [DOI] [PubMed] [Google Scholar]
- Nicholl DJ, Bennett P, Hiller L, Bonifati V, Vanacore N, Fabbrini G, Marconi R, Colosimo C, Lamberti P, Stocchi F, Bonuccelli U, Vieregge P, Ramsden DB, Meco G, Williams AC ( 1999) A study of five candidate genes in Parkinson's disease and related neurodegenerative disorders. Neurology 53 : 1415-1421. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL ( 1997) Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 276 : 2045-2047. [DOI] [PubMed] [Google Scholar]
- Przedborski S, Jackson-Lewis V, Djaldetti R, Liberatore G, Vila M, Vukosavic S, Almer G ( 2000) The parkinsonian toxin MPTP: action and mechanism. Restor Neurol Neurosci 16 : 135-142. [PubMed] [Google Scholar]
- Rathke-Hartlieb S, Kahle PJ, Neumann M, Ozmen L, Haid S, Okochi M, Haas C, Schulz JB ( 2001) Sensitivity to MPTP is not increased in Parkinson's disease-associated mutant alpha-synuclein transgenic mice. J Neurochem 77 : 1181-1184. [DOI] [PubMed] [Google Scholar]
- Sanghera MK, Manaye K, McMahon A, Sonsalla PK, German DC ( 1997) Dopamine transporter mRNA levels are high in midbrain neurons vulnerable to MPTP. NeuroReport 8 : 3327-3331. [DOI] [PubMed] [Google Scholar]
- Schwarting R, Huston JP ( 1987) Short-term effects of ether, equithesin and droperidol/fentanyl on catecholamine and indolamine metabolism in the brain of the rat. Neuropharmacology 26 : 457-461. [DOI] [PubMed] [Google Scholar]
- Schwarting RKW, Huston JP ( 1996a) Unilateral 6-hydroxydopamine lesions of meso-striatal dopamine neurons and their physiological sequelae. Prog Neurobiol 49 : 215-266. [DOI] [PubMed] [Google Scholar]
- Schwarting RKW, Huston JP ( 1996b) The unilateral 6-hydroxydopamine lesion model in behavioral brain research. Analysis of functional deficits, recovery and treatments. Prog Neurobiol 50 : 275-331. [DOI] [PubMed] [Google Scholar]
- Schwarting RKW, Sedelis M, Hofele K, Auburger GW, Huston JP ( 1999) Strain-dependent recovery of open-field behavior and striatal dopamine deficiency in the mouse MPTP model of Parkinson's disease. Neurotox Res 1 : 41-56. [DOI] [PubMed] [Google Scholar]
- Sedelis M, Hofele K, Auburger GW, Morgan S, Huston JP, Schwarting RKW ( 2000a) Evidence for resistance to MPTP in C57BL/6 x BALB/c F1 hybrids as compared with their progenitor strains. NeuroReport 11 : 1093-1096. [DOI] [PubMed] [Google Scholar]
- Sedelis M, Hofele K, Auburger GW, Morgan S, Huston JP, Schwarting RKW ( 2000b) MPTP susceptibility in the mouse: behavioral, neurochemical and histological analysis of gender and strain differences. Behav Genet 30 : 171-182. [DOI] [PubMed] [Google Scholar]
- Sedelis M, Schwarting RKW, Huston JP ( 2001) Behavioral phenotyping of the MPTP mouse model of Parkinson's disease. Behav Brain Res 125 : 109-125. [DOI] [PubMed] [Google Scholar]
- Sen S, Churchill GA ( 2001) A statistical framework for quantitative trait mapping. Genetics 159 : 371-387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son JH, Chun HS, Joh TH, Cho S, Conti B, Lee JW ( 1999) Neuroprotection and neuronal differentiation studies using substantia nigra dopaminergic cells derived from transgenic mouse embryos. J Neurosci 19 : 10-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundström E, Strömberg I, Tsutsumi T, Olson L, Jonsson G ( 1987) Studies on the effect of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) on central catecholamine neurons in C57BL/6 mice. Comparison with three other strains of mice. Brain Res 405 : 26-38. [DOI] [PubMed] [Google Scholar]
- Tan EK, Khajavi M, Thornby JI, Nagamitsu S, Jankovic J, Ashizawa T ( 2000) Variability and validity of polymorphism association studies in Parkinson's disease. Neurology 55 : 533-538. [DOI] [PubMed] [Google Scholar]
- Uhl GR ( 1998) Hypothesis: the role of dopaminergic transporters in selective vulnerability of cells in Parkinson's disease. Ann Neurol 43 : 555-560. [DOI] [PubMed] [Google Scholar]