

Abstract
It is generally believed that consolidation of long-term memory requires activation of protein kinases, transcription of genes, and new protein synthesis. However, little is known about the signal cascades involved in the extinction of memory, which occurs when the conditioned stimulus is no longer followed by the unconditioned stimulus. Here, we show for the first time that an intra-amygdala injection of transcription inhibitor actinomycin D at the dose that blocked acquisition failed to affect extinction of a learned response. Conversely, protein synthesis inhibitor anisomycin blocked both acquisition and extinction. Extinction training-induced expression of calcineurin was blocked by anisomycin but not by actinomycin D. NMDA receptor antagonist, phosphatidylinositol 3-kinase (PI-3 kinase), and MAP kinase inhibitors that blocked the acquisition also blocked the extinction of conditioned fear. Likewise, PI-3 kinase inhibitor blocked fear training-induced cAMP response element-binding protein (CREB) phosphorylation as well as extinction training-induced decrease in CREB phosphorylation, the latter of which was associated with calcineurin expression and could be reversed by a specific calcineurin inhibitor. Thus, molecular processes that underlie long-term behavioral changes after acquisition and extinction share some common mechanisms and also display different characteristics.
Keywords: learning and memory, amygdala, calcineurin, synaptic plasticity, fear conditioning, fear-potentiated startle
Introduction
It is a normal and adaptive reaction that animals learn to express a fear response when encountering a predictive or conditioned stimulus (CS) that was previously paired with a noxious or unconditioned stimulus (US) (Pavlov, 1927). This Pavlovian fear conditioning has been widely used as an animal model of post-traumatic stress disorder and as a leading behavioral paradigm for studying the neural mechanisms through which emotional memory is formed and stored (Davis, 2000; LeDoux, 2000). In contrast, if animals are exposed only to the CS without pairing with the US, previously acquired fear responses will gradually decrease (Falls et al., 1992; Rescorla, 2001; Santini et al., 2001; Myers and Davis, 2002). This fear inhibition known as fear extinction receives increasing attention because it could become an effective intervention for the treatment of fear-related disorders. Converging evidence indicates that enhancement of synaptic strength in the lateral amygdala (LA) and basolateral amygdala (BLA) underlies the encoding of fear memory (McKernan and Shinnick-Gallagher, 1997; Rogan et al., 1997; Maren, 1999; Schafe et al., 2001). Recent studies have identified the first messengers (glutamate acts on NMDA receptors) (Miserendino et al., 1990; Lee and Kim, 1998), the second messengers [Ca2+, protein kinase A (PKA), MAP kinase (MAPK)] (Atkins et al., 1998; Huang et al., 2000; Schafe and LeDoux, 2000; Schafe et al., 2000), and the third messengers [cAMP response element-binding protein (CREB), zif268] (Impey et al., 1998; Hall et al., 2001a,2001b; Josselyn et al., 2001; Ressler et al., 2002) responsible for consolidation of fear memory. In contrast to fear conditioning, much less information is available regarding the signal pathways underlying fear inhibition.
It has been shown that extinction of memory could be blocked by the NMDA receptor antagonists and MAPK kinase (MEK) inhibitors in the amygdala, as assessed in a fear-potentiated startle paradigm (Falls et al., 1992; Lu et al., 2001), and in the hippocampus, as measured with one-trial inhibitory avoidance task (Szapiro et al., 2003). In contrast, extinction of a conditioned taste aversion appeared not to depend on NMDA receptors and MAPK cascade in the insular cortex (Berman and Dudai, 2001). Extinction of auditory fear memory was impaired in cannabinoid receptor 1-deficient mice (Marsicano et al., 2002) or in protein phosphatase 1-inhibited mice (Genoux et al., 2002). In the present study, we aimed to accomplish two goals: (1) to differentiate the signal cascades that underlie long-term behavioral changes between consolidation of conditioning and consolidation of extinction of fear memory; and (2) to construct the causal relationship among protein synthesis, calcineurin expression, CREB phosphorylation, and extinction of a learned response.
Materials and Methods
Surgery
Male Sprague Dawley rats weighing 150-220 gm were used. They were housed in group cages of four rats each in the air-conditioned vivarium, with ad libitum access to food and water. Throughout the study, a 12/12 hr light/dark cycle was maintained, with light on at 8:00 A.M. Behavioral tests were performed in the light phase. All procedures adhered to the Guidelines for Care and Use of Experimental Animals of the National Cheng-Kung University. Wortmannin, LY294002, U0126, anisomycin, and actinomycin D were obtained from Sigma (St. Louis, MO). FK 506 was obtained from Calbiochem-Novabiochem International (San Diego, CA).
Rats were anesthetized with sodium pentobarbital (50 mg/kg, i.p.). When fully anesthetized, they were mounted on a stereotaxic apparatus (David Kopf Instruments, Tujunga, CA), and two cannulae made of 22 gauge stainless steel tubing (C313G; Plastic Products) were implanted bilaterally into the LA or BLA. A 28 gauge dummy cannula was inserted into each cannula to prevent clogging. The coordinates were AP (-2.3 mm), ML (±4.5 mm), and DV (-7.0 mm) according to Paxinos and Watson (1986). Three jewelry screws were implanted over the skull serving as anchors, and the whole assembly was affixed on the skull with dental cement. The rats were monitored and handled daily and were given 7 d to recover. The drugs were administered bilaterally in a volume of 0.8-1.0 μl at a rate of 0.5 μl/min, using 28 gauge infusion cannulas that extended 1.0 mm from the base of the guide. The infusion cannulas were attached to a 10 μl Hamilton syringe, and, after the infusion, the cannulas were left in place for 1-3 min to allow diffusion of the drugs from the tip.
Fear conditioning
The startle response was measured in a startle apparatus (San Diego Instrument, San Diego, CA) as described in previous studies (Lu et al., 2000; Lin et al., 2003). Briefly, rats were constrained in a Plexiglas cyclidrical tube (length, 20 cm; diameter, 10 cm) with an accelerometer sensor attached on the base. The whole setup was enclosed in a ventilated, sound-attenuating cabinet (length, 38 cm; width, 38 cm; height, 55 cm). The acoustic startle stimulus was 50 msec white noise at the intensity of 95 dB delivered by a speaker 30 cm above the animal. The visual CS was a 3.7 sec light produced by an 8 W fluorescent bulb. The US was a 0.6 mA footshock with a duration of 0.5 sec. All data were reserved for off-line analyses.
Acclimation. On 3 consecutive days, rats were placed in the startle test boxes for 10 min and returned to their home cages.
Matching. On 2 consecutive days, rats were placed in the startle box and 3 min later presented with 10 startle stimuli at 2 min intertrial interval (ITI). On the basis of their mean startle amplitudes in the second of these two sessions, rats were matched into groups with similar response levels.
Conditioning training. Rats were placed in the startle boxes and, after a 5 min-acclimation period, received 10 light-footshock pairings with an ITI of 2 min. Unpaired controls received the same number of light and footshock presentations, but in a pseudorandom manner in which the US could occur at anytime, except at the 3.2 sec after the CS.
Preextinction test. Twenty-four hours after training, rats were tested for fear-potentiated startle. This involved 30 startle-eliciting noise bursts presented alone (noise-alone trial) and 30 noise bursts presented 3.2 sec after onset of the 3.7 sec light (light-noise trials). The two trial types were presented in a balanced mixed order (ITI, 30 sec). The percentage of fear-potentiated startle was computed as: [(startle amplitude on CS-noise - noise-alone trials)/(noise-alone trials)] × 100.
Extinction training. Ten minutes after the preextinction test, rats returned to the startle box and received three sessions of 10 presentations of the 3.7 sec light in the absence of either shock or the startle-elicited noise burst (light-alone trials). Each session was separated by 10 min with an ITI of 1 min. The context control group remained in the box for an equivalent period of time without receiving any stimulation.
Postextinction test. Twenty minutes after extinction training, rats were tested for fear-potentiated startle in a procedure identical to preextinction test.
Western blots analysis
Rats were given 10 pairings of light and footshock and were tested 24 hr later (pretest). Subsequently, rats received light-alone trials and were sacrificed by decapitation immediately after the trials. The lateral and basolateral subregions of the amygdala were sonicated briefly in ice-cold buffer (50 mm Tris-HCl, pH 7.5, 0.3 m sucrose, 5 mm EDTA, 2 mm sodium pyrophosphate, 1 mm sodium orthovanadate, 1 mm PMSF, 20 μg/ml leupeptin, and 4 μg/ml aprotinin). After sonication, the samples were centrifuged at 7,500 rpm for 15 min, and the supernatant was obtained after pelleting the crude membrane fraction by centrifugation at 50,000 rpm for 1 hr at 4°C. Protein concentration in the soluble fraction was then measured using a Bradford assay, with BSA as the standard. Equivalent amounts of protein for each sample were resolved in 8.5% SDS-polyacrylamide gels, blotted electrophoretically to Immobilon and blocked overnight in TBS buffer (50 mm Tris-HCl, pH 7.5, and 150 mm NaCl) containing 3% BSA. For detection of the protein level of calcineurin, blots were incubated with anti-calcineurin (1:2000; BD Transduction Laboratories, Los Angeles, CA) antibody. An ECL kit (NEN Life Science Products, Boston, MA) was used for detection. Western blots were developed in the linear range used for densitometry. The density of the immunoblots was determined by an image analysis system installed with software BIO-ID (Vilber Lourmat, France).
Nuclear extract preparation
Rats were decapitated, the brains were rapidly removed, and the LA and BLA were dissected out using a Dounce grinder with loose pestle in iced-chilled buffer (15 mm HEPES, 60 mm KCl, 1 mm NaCl, 0.25 m sucrose, 5 mm EDTA, 1 mm EGTA, 1 mm PMSF, 10 μg/ml aprotinin, 15 μg/ml leupeptin, 2 mm NaF, and 1 mm sodium orthovanadate). The homogenate was centrifuged for 10 min at 2,000 rpm, and pellet was resuspended in buffer (10 mm HEPES, pH 7.2, 15 mm MgCl2,10mm KCl, 1 mm PMSF, 2 mm NaF, 15 μg/ml leupeptin, and 1 mm sodium orthovanadate). After a brief vortexing, they were incubated on ice for 10 min and lysated with tight pestle. The homogenate was centrifuged at 4,000 rpm for 10 min. The pelleted nuclei were resuspended in 40-60 μl of extraction buffer consisting of 100 mm HEPES, pH 7.2, 1.5 mm MgCl2, 1mm EDTA, 0.8 M NaCl, 15% glycerol, 2 mm NaF, 1 mm PMSF, 15 μg/ml leupeptin, and 1 mm sodium orthovanadate and were incubated on ice for 2-4 hr. The nuclear suspension was centrifuged at 14,000 rpm for 30 min at 4°C, and the supernatant was saved. Equal amounts of nuclear extracts were subjected to SDS-PAGE (10% gels), and immunoblots were performed with anti-CREB and anti-pCREB antibodies (New England Biolabs, Beverly, MA), respectively.
Results
Fear conditioning has been shown to induce phosphorylation of protein kinases in the rat amygdala (Schafe et al., 2000; Lin et al., 2001). The increase in the protein phosphorylation was abrogated after extinction training, which was accompanied by an increase in the protein level and enzymatic activity of calcineurin (Lin et al., 2003). We noted previously that the expression of cytosolic calcineurin peaked immediately (≤10 min) after extinction training that was rapid and not usual, implicating that mRNA that is used to encode the proteins for extinction of memory is likely already present in the amygdala neurons. We, therefore, investigated whether extinction of fear memory required new mRNA or protein synthesis by pharmacologically blocking transcription with actinomycin D and translation with anisomycin. Rats were given 10 pairings of light and footshock and were tested 24 hr later (pretest). After initial training, animals exhibited fear of the light, as demonstrated by an increase in startle responses. Subsequently, rats were given an intra-amygdala injection of anisomycin (125 μg dissolved in 1.6 μl of vehicle; 0.8 μl per side), actinomycin D (10 μg dissolved in 1.6 μl of vehicle; 0.8 μl per side), or vehicle before receiving light-alone trials (Fig. 1A). Figure 1B shows that application of anisomycin blocked extinction. Fear-potentiated startles in animals treated with anisomycin were not different from their preextinction tests (t(5) = 0.41; p = 0.70; paired t test). Conversely, rats given actinomycin D or vehicle displayed a normal decrease in startle after light-alone trials (p < 0.001). Furthermore, as an independent test to determine the specific effect of anisomycin on extinction of fear in the conditioned rats, anisomycin was administered into the unpaired rats and was found to have no effect on the startle response. Cannula tip placements from rats infused with vehicle, actinomycin D, or anisomycin are shown in Figure 1C.
Figure 1.
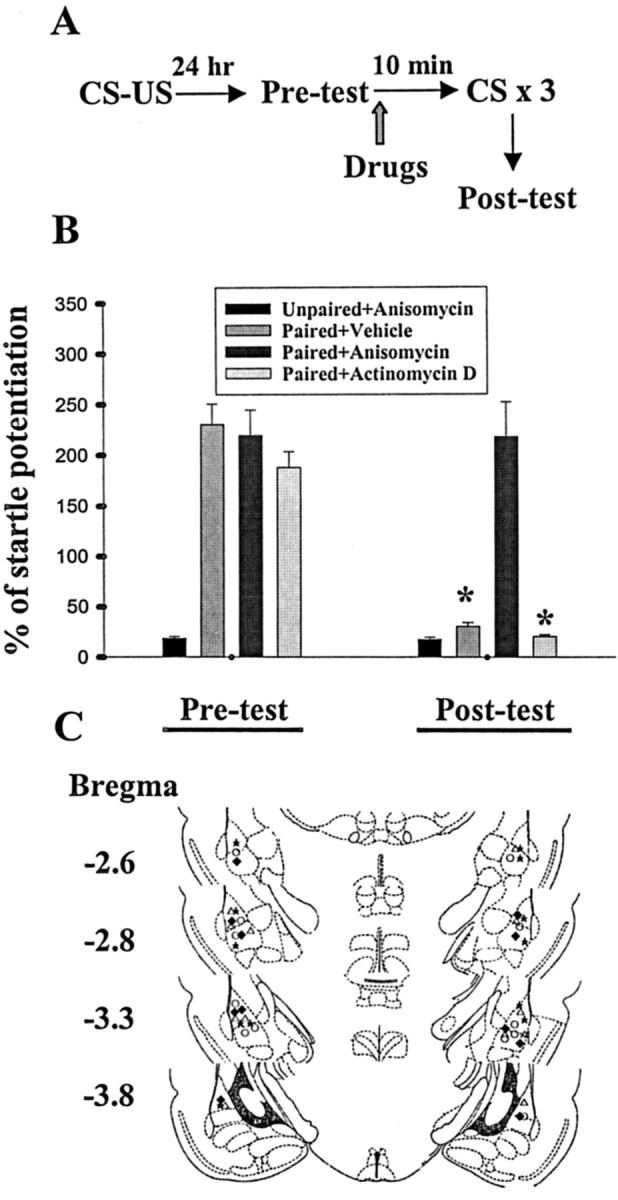
Intra-amygdala administration of anisomycin, but not actinomycin D, blocks extinction of fear memory. A, Behavioral procedure used in experiment B. B, Bilateral amygdala infusion of anisomycin (125 μg dissolved in 1.6 μl of vehicle; 0.8 μl per side) before light-alone trials blocked extinction of fear memory, whereas actinomycin D (10 μg dissolved in 1.6 μl of vehicle; 0.8 μl per side) was without effect. *p < 0.001 versus preextinction tests. C, Cannula tip placements from paired rats infused with vehicle (open circle), anisomycin (open triangle), or actinomycin D (filled diamond). Filled stars represent the tip locations in the unpaired rats given anisomycin.
To determine the causal relationship between protein synthesis inhibition and extinction training-induced calcineurin expression, we bilaterally infused anisomycin or vehicle into the trained rats before light-alone trials (Fig. 2A). Representative immunoblots and densitometric analysis of calcineurin expression are shown in Figure 2B. There was a significant decrease in calcineurin expression; extinction training induced an increase in calcineurin by 124.5 ± 25.8%, which was reduced to only 9.1 ± 2.3% after anisomycin treatment (n = 6 in each group; p < 0.01; unpaired t test). In contrast, the calcineurin level was unaltered in rats treated with actinomycin D (p > 0.1).
Figure 2.
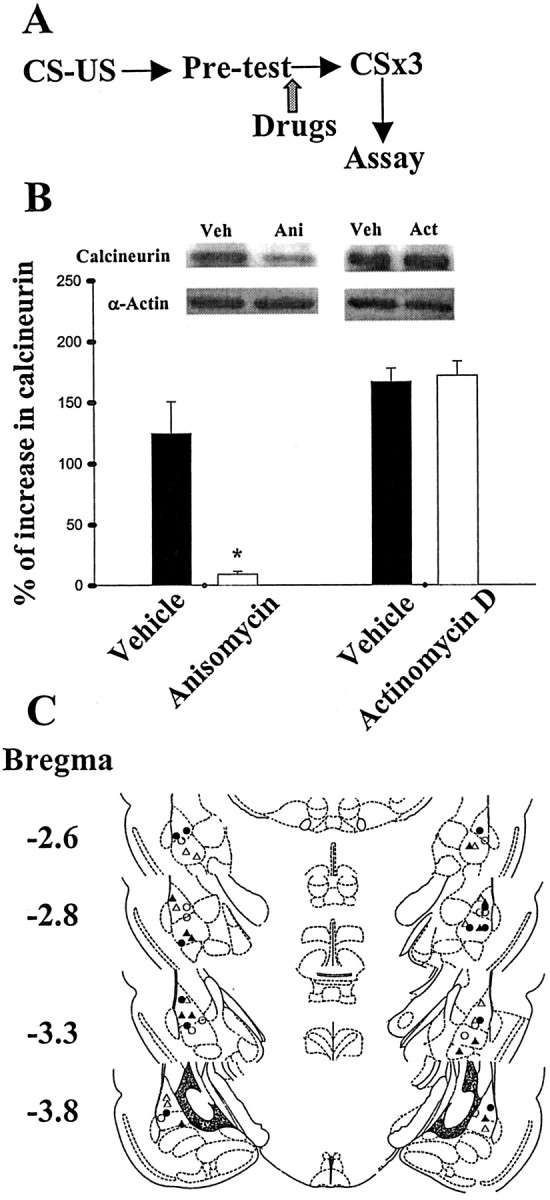
Extinction training-induced calcineurin expression is blocked by anisomycin but not by actinomycin D. A, Behavioral procedure used in experiment B. Extinction-trained rats were killed by decapitation immediately after light-alone trials. The LA and BLA were microdissected, and cytosolic calcineurin levels were measured as described in Materials and Methods. B, Calcineurin levels in the extinction-trained rats given drugs or vehicle were normalized to those of unpaired controls. Light-alone trials induced an increased expression of calcineurin in cytosolic (α-actin as internal control) fraction that could be blocked by anisomycin (125 μg dissolved in 1.6 μl of vehicle; 0.8 μl per side). In contrast, actinomycin D (10 μg dissolved in 1.6 μl of vehicle; 0.8 μl per side) was without effect. *p < 0.01 versus vehicle. C, Cannula tip placements from paired rats infused with vehicle (open circle), anisomycin (filled circle), or actinomycin D (filled triangle).
The lack of the effect of actinomycin D could be accounted for by an insufficient concentration reaching the amygdala to inhibit transcription. We tested this possibility by examining the effect of this drug on acquisition of fear memory. Thus, the same dose of actinomycin D (same concentration and volume) was applied to the amygdala 30 min before training, and the retention of memory was assessed 24 hr later. We found that pretraining infusion of actinomycin D attenuated fear startle (24.7 ± 3.3% of potentiation, n = 6 rats, vs 172.2 ± 10.2% of potentiation in vehicle controls, n = 6 rats; p < 0.01; unpaired t test). A previous study showed that intra-amygdala administration of 2.5 ng/side of actinomycin D (half-dose of the present study) was effective in producing a ∼40% depression of mRNA synthesis (Bailey et al., 1999).
The results obtained so far indicate that both consolidation of conditioning and consolidation of extinction require new protein synthesis, but they differ in their susceptibility to block by actinomycin D. We next tested whether extinction of memory like that of acquisition involved activation of NMDA receptors (Falls et al., 1992; Lee and Kim, 1998). After being conditioned, rats were given an intra-amygdala injection of either d-APV (25 nmol dissolved in 1.6 μl of artificial CSF; 0.8 μl per side) or vehicle before receiving three sessions of 10 presentations of light in the absence of shock. Figure 3A shows that startle amplitude was not reduced in the d-APV group (t(5) = 0.66; p = 0.54; paired t test). In contrast, rats in the vehicle group exhibited a decrease after extinction training (t(5) = 7.7, p < 0.001). We also examined the effect of selective non-NMDA receptor antagonist CNQX on the extinction of fear. Rats were given an intra-amygdala infusion of CNQX (25 nmol dissolved in 1.6 μl of DMSO; 0.8 μl per side), an equivalent nanomolar dose of d-APV, before light-alone trials. As shown in Figure 3A, CNQX did not affect the extinction of fear. Rats given CNQX showed a normal reduction in fear-potentiated startle from the preextinction to the postextinction tests (p < 0.001).
Figure 3.
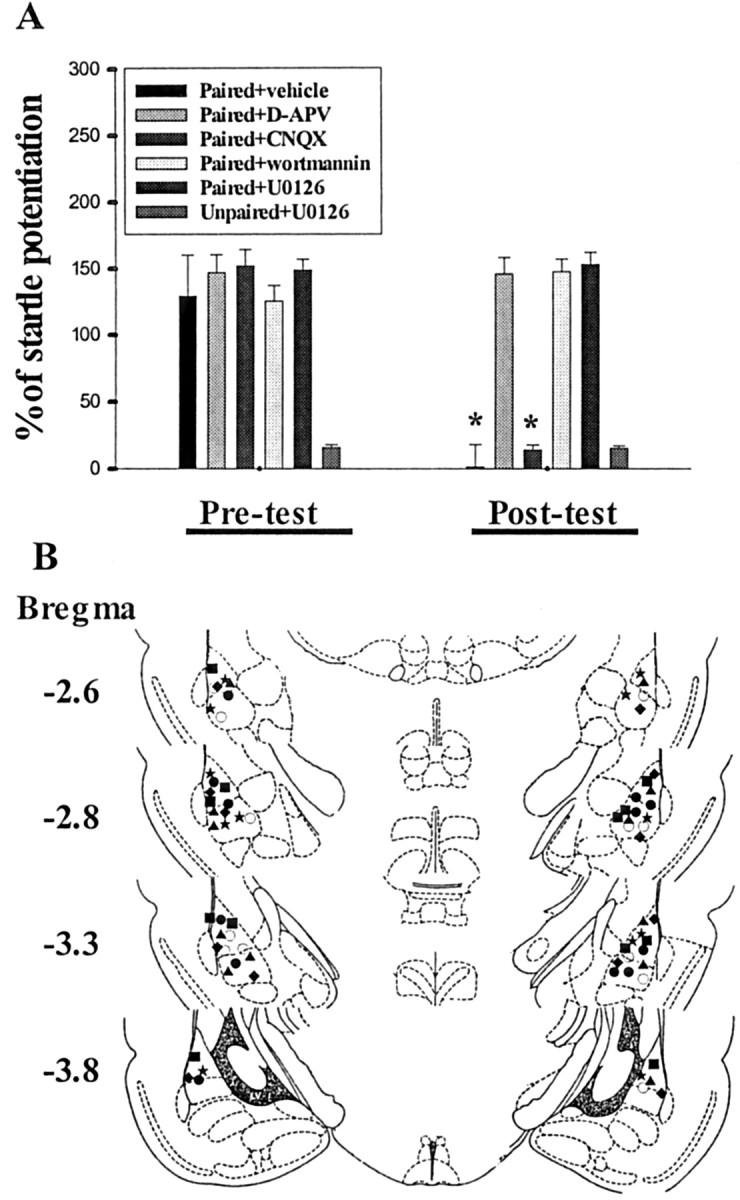
Effects of NMDA and non-NMDA receptors, PI-3 kinase, and MEK inhibitors on the extinction of fear startle. A, Percentage of startle potentiation before (pretest) and after (post-test) three sessions of extinction training in rats given various pharmacological agents before light-alone trials. Extinction of fear memory was blocked by intra-amygdala infusion of d-APV (25 nmol dissolved in 1.6 μl of artificial CSF; 0.8 μl per side), wortmannin (5 μg dissolved in 1.6 μl of DMSO; 0.8 μl per side), or U0126 (2 μg dissolved in 1.6 μl of DMSO; 0.8 μl per side), whereas CNQX (25 nmol dissolved in 1.6 μl of DMSO; 0.8 μl per side) was without effect (n = 6 rats in each group). In addition, a control experiment was conducted in which U0126 was injected into the amygdala of the unpaired rats, and the result showed that this drug did not affect the startle response. *p < 0.001 versus pretest. B, Cannula tip placements from paired rats infused with vehicle (open circle), d-APV (filled square), CNQX (filled star), wortmannin (filled circle), or U0126 (filled triangle). Filled diamonds represent the tip locations in the unpaired rats given U0126.
We have recently shown that fear conditioning resulted in a selective activation of phosphatidylinositol 3-kinase (PI-3 kinase) and MAPK in the amygdala (Lin et al., 2001). It is of interest to see whether extinction of fear responses also requires activation of PI-3 kinase and MAPK (Lu et al., 2000). Animals were trained with fear-potentiated startle paradigm and subsequently were given an intra-amygdala injection of the PI-3 kinase inhibitor wortmannin (5 μg dissolved in 1.6 μl of DMSO; 0.8 μl per side) or the MEK inhibitor U0126 (2 μg dissolved in 1.6 μl of DMSO; 0.8 μl per side) before receiving extinction trials. We found that the startle responses were unaltered after light-alone trials in rats treated with wortmannin (t(5) = 1.75; p = 0.14) or U0126 (t(5) = 0.43; p = 0.69). Thus, an intra-amygdala infusion of wortmannin or U0126 blocked extinction. Cannula tip placements in these experiments are shown in Figure 3B. In summary, these results, together with others, suggest that both acquisition and extinction of fear memory require activation of NMDA receptors, PI-3, and MAPK.
The consolidation of fear memory is thought to involve gene expression associated with transactivation of CREB (Bourtchuladze et al., 1994; Bernabeu et al., 1997; Impey et al., 1998; Kida et al., 2002). Using an antibody that detects CREB when it is phosphorylated on the regulated site Ser133 (anti-pCREB), we examined CREB phosphorylation before and after extinction training. Rats were trained with startle paradigm, and memory retention was tested 24 hr later. One hour after behavioral tests, animals were sacrificed, and the tissue of amygdala was removed for analysis. Figure 4A shows that there was a significant increase in pCREB immunoreactivity in nuclear fraction of the LA and BLA from conditioned rats. Subsequent extinction training significantly reduced CREB phosphorylation (p < 0.01 vs preextinction test; n = 6 rats in each group; unpaired t test). In addition, extinction-induced decrease in pCREB was reversed by FK-506 (10 μg dissolved in 1.6 μl of DMSO; 0.8 μl per side) given before extinction, suggesting the involvement of calcineurin in the extinction of fear memory. Therefore, we determined the nuclear calcineurin level, and Figure 4B shows that extinction training induced a significant increase in calcineurin that peaked at 20 min after training and subsided within 60 min (F(5,30) = 29.58; p < 0.001). Newman-Keuls t tests revealed the differences between control and 20, 30, and 40 min time points (p < 0.001). As control, the immunoreactivity for histone was checked and found unaltered.
Figure 4.
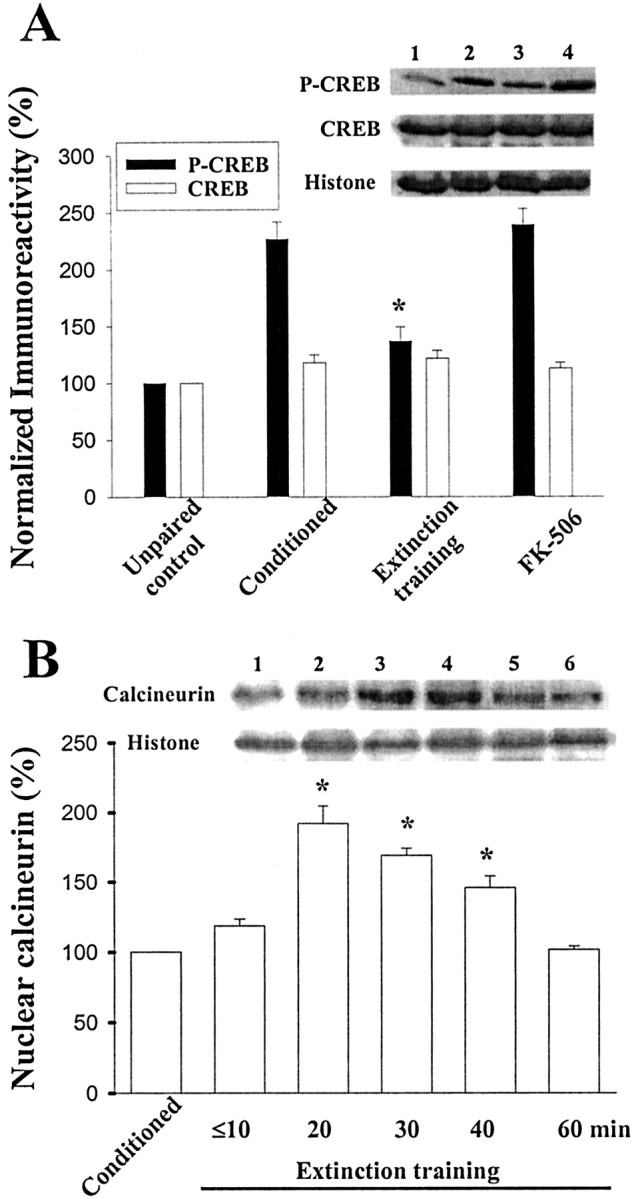
Effect of extinction training on CREB phosphorylation. A, Representative blots and mean ± SE percentage of pCREB immunoreactivity from unpaired control rats (lane 1), conditioned rats (lane 2), and conditioned rats given light-alone training (lane 3). Fear conditioning resulted in an increase in pCREB, whereas extinction training caused a decrease. Bilateral infusion of FK-506 (10 μg dissolved in 1.6 μl of DMSO; 0.8 μl per side) before extinction trials blocked extinction-induced decrease in pCREB (lane 4). *p < 0.01 versus conditioned. B, Time course of calcineurin expression induced by light-alone trials in nuclear fraction. Figure shows the representative blots and mean ± SE of calcineurin immunoreactivities from rats decapitated at various time points (n = 6 rats in each time point) after extinction training (histone used as internal control). *p < 0.001 versus conditioned.
Do conditioning-induced phosphorylation of CREB and extinction-induced decrease in pCREB require PI-3 kinase activation? Rats were infused bilaterally with vehicle, wortmannin (5 μg dissolved in 1.6 μl of DMSO; 0.8 μl per side), or LY 294002 (30 μg dissolved in 1.6 μl of DMSO; 0.8 μl per side) 30 min before training (Fig. 5A). As shown in Figure 5B, the increase in pCREB immunoreactivity after testing was abolished in PI-3 kinase inhibitor-pretreated animals. These results suggest that conditioning-induced CREB phosphorylation is dependent on a PI-3 kinase-mediated cascade. In another group of rats, animals were trained with fear-potentiated startle paradigm and subsequently were given an intra-amygdala injection of wortmannin (5 μg dissolved in 1.6 μl of DMSO; 0.8 μl per side) or vehicle before receiving extinction trials. Immunoblotting for pCREB from nuclear extract was performed 20 min after extinction training (Fig. 5C). Figure 5D shows that light-alone trials reduced CREB phosphorylation in control (conditioning plus extinction training; lane 1) and vehicle (lane 2) groups. In contrast, extinction-induced decrease in pCREB was inhibited in the wortmannin group (lane 3; n = 4 rats in each group; p < 0.05 vs vehicle; unpaired t test). Thus, extinction-induced decrease in pCREB, like that of conditioning-induced phosphorylation of CREB, depends on PI-3 kinase-mediated cascade.
Figure 5.

Block of conditioning-induced CREB phosphorylation and extinction-induced dephosphorylation of pCREB by PI-3 kinase inhibitors. A, Behavioral procedure used in experiment. B, Rats were killed 1 hr after preextinction tests, and the nuclear extracts of the amygdala were analyzed. Conditioned rats treated with vehicle exhibited a greater degree of CREB phosphorylation (lane 2) compared with unpaired controls (lane 1). Bilateral infusion of wortmannin (lane 3) or LY 294002 (lane 4) 30 min before training blocked CREB phosphorylation. **p < 0.01 versus control. C, Behavioral procedure used in experiment D. D, Extinction-induced dephosphorylation of pCREB is blocked by PI-3 kinase inhibitor. Rats were given an intra-amygdala injection of wortmannin (5 μg dissolved in 1.6 μl of DMSO; 0.8 μl per side) or vehicle before receiving extinction trials. Immunoblotting for pCREB from nuclear extract was performed 20 min after extinction training. In contrast to those of the control and vehicle groups, extinction trials did not cause CREB dephosphorylation in wortmannin-treated animals (n = 4 rats in each group). *p < 0.05 versus vehicle.
Discussion
Activation of CREB and CRE-mediated gene expression is thought to mediate conversion of short- to long-term memory (Frey et al., 1993; Bourtchuladze et al., 1994; Bernabeu et al., 1997; Impey et al., 1998; Kida et al., 2002). Overexpression of CREB in the amygdala using viral expression vectors enhanced the formation of fear memory (Josselyn et al., 2001), whereas expression of a constitutively active CREB in the hippocampus facilitated the induction of long-lasting long-term potentiation (LTP) in the Schaffer collateral pathway (Barco et al., 2002). Conversely, intrahippocampal infusion of CREB antisense oligonucleotides impaired long-term spatial memory leaving short-term memory intact (Guzowski and McGaugh, 1997). We have previously shown that PI-3 kinase inhibitors blocked tetanic stimulation-, forskolin-, and conditioning-mediated CREB phosphorylation, suggesting an obligatory role for PI-3 kinase in CREB phosphorylation and memory consolidation (Lin et al., 2001). Importantly, in the present study, we found that wortmannin also blocked extinction training-induced decrease in CREB phosphorylation that was associated with calcineurin expression and could be reversed by a specific calcineurin inhibitor. These results, in combination with behavioral tasks showing block of extinction by NMDA receptor antagonists, PI-3 kinase, MEK, and protein synthesis inhibitors clearly indicate that consolidation of conditioning and consolidation of extinction of memory share some common mechanisms. In summary, there are overlapping signal pathways between consolidation of conditioning and consolidation of extinction of fear memory: both processes require activation of NMDA receptors, PI-3 kinase, MAPK, and synthesis of new protein within the amygdala.
Consistent with a previous report (Bailey et al., 1999), we observed that administration of actinomycin D before conditioning resulted in a significant attenuation of fear memory, as assessed in a retention test conducted 24 hr later. However, the intriguing finding here is that intra-amygdala injection of actinomycin D at the same dose that severely disrupted the acquisition did not affect the extinction. Thus, unlike that of acquisition, consolidation of extinction does not involve transcription of new mRNA. It is conceivable that mRNA that is used to encode the protein is already present within the neurons. Two examples of using the existing mRNA to encode proteins for synaptic plasticity have recently been demonstrated in the hippocampal CA1 area (Huber et al., 2000; Manahan-Vaughan et al., 2000). Moreover, extinction-induced calcineurin expression was blocked by anisomycin but not by actinomycin D. It is concluded that extinction is causally related to the expression of calcineurin in which transcription of new mRNA is not necessary. However, because behavioral tests were conducted 20 min but not 24 hr after extinction training, we could not dismiss the possibility that long-term stabilization of the extinction training might require transcription. In summary, in our extinction protocol, extinction differs from acquisition in that consolidation of extinction is associated with the expression of calcineurin and is not sensitive to transcriptional blockade.
Because of spontaneous recovery of memory with time (Baum, 1988), reinstatement after unpaired US presentations (Rescorla and Heth, 1975), and renewal with context change (Bouton and King, 1983), experimental extinction is generally ascribed to a relearning of a new association between the CS and the absence of reinforcement (Myers and Davis, 2002). These results suggest that the original association was intact after extinction trials. Furthermore, preextinction anisomycin infusion inhibited extinction such that animals still responded to the CS, even though it was no longer associated with reinforcement (Berman and Dudai, 2000; Taubenfeld et al., 2001; Vianna et al., 2001). However, there were conflicting results showing that anisomycin did not alleviate the extinction of spatial preference tasks (Lattal and Abel, 2001). In fact, the observation that infusion of anisomycin shortly after memory reactivation produced amnesia has led to the conclusion that memory, when reactivated, became labile and susceptible to disruption by protein synthesis inhibitors (Nader et al., 2000; Sara, 2000; Kida et al., 2002). The reasons for these conflicting data are not known but could be attributable to different tasks or brain areas studied.
Here, we report that extinction training may weaken or erase the original memory. These seemingly conflicting results could be attributable to the extinction protocol we used, which resembled within-session extinction and presumably corresponded to short-term extinction memory. Alternatively, conditioned response may be erased within the amygdala while maintained in other structures. For example, prefrontal cortex and its various subregions have been suggested to play an important role in the development and maintenance of extinction of learned fear (Quirk et al., 2000; Herry and Garcia, 2002).
Our results obtained from the fear-potentiated startle favor an idea of competition between reactivation and destabilization of the original signals (Myers and Davis, 2002; Nader, 2003). Figure 6 is a schematic model showing that both CS-US pairing (conditioning) and CS-alone (extinction) training trigger calcium influx through either NMDA receptors or L-type Ca2+ channels (Weisskopf et al., 1999; Bauer et al., 2002; Cain et al., 2002) in the amygdala. It is hypothesized that the increase in intracellular calcium associated with long-term synaptic depression are both smaller and longer lasting than those associated with LTP (Cho et al., 2001; Cormier et al., 2001). It is likely that, during extinction training, lower levels of calcium influx cause synaptic depression and subsequently extinction of memory, because at these concentrations calcium predominantly activates protein phosphatases rather than protein kinases (Elgersma and Silva, 1999; Winder and Sweatt, 2001). The moderate increase in calcium also results in the activation of protein kinases (e.g., PKA, PI-3 kinase, and MAPK) (Lu et al., 2000; Szapiro et al., 2003), which then translocate into the nucleus in which they phosphorylate CREB to initiate gene transcription and translation. CS-alone training not only reactivates original memory this way but also activates calcineurin and promotes its synthesis. Calcineurin then exerts a negative feedback effect to downregulate kinases and weakens the original memory. These two phenomena compete with each other, and the outcome of the effect on reconsolidation-extinction may depend on which effect predominates. The factors that control the dominance of blocking either reconsolidation or extinction may include the strength of the CS, the types of reinforcement, and the response to reactivation test. For example, anisomycin tended to block extinction in experiments in which reactivation itself caused significant extinction. Conversely, no extinction was induced in studies that reported a block of reconsolidation by anisomycin (Nader, 2003).
Figure 6.

A model of consolidation of conditioning and consolidation of extinction of memory in the amygdala. Both acquisition and extinction trigger calcium influx through either NMDA receptors or L-type calcium channels in the amygdala. The increase in intracellular Ca2+ leads to the activation of protein kinases (e.g., PKA, PI-3 kinase, and MAPK). The activated kinases then translocate into the nucleus in which they phosphorylate CREB to initiate gene transcription and translation. Extinction training not only reactivates original memory this way but also promotes calcineurin synthesis via the existed mRNA. Calcineurin then exerts a negative feedback effect to dephosphorylate kinases and weakens the original memory. Thus, the outcome of the effect of protein synthesis inhibition on the reconsolidation-extinction may depend on a competition between these two processes.
In conclusion, we have investigated the similarities and differences between consolidation of conditioning and consolidation of extinction. Both processes depend on the activation of NMDA receptors, PI-3 kinase, and MAPK and require synthesis of new protein within the amygdala. However, they also display different characteristics, namely the differential sensitivity to block by actinomycin D and the state of CREB phosphorylation affected by calcineurin. These data, together with others, suggest a competition theory in which the outcome of blocking reconsolidation-extinction with anisomycin depends on which of these two processes, the strengthening as opposed to weakening of the original memory, is affected to a greater extent.
Footnotes
This study was supported by the National Health Research Institutes (Grant NHRI-EX92-9202NI) and the Academic Excellence Program of the Ministry of Education (Grant 89-B-FA08-1-4) of Taiwan.
Correspondence should be addressed to Dr. Po-Wu Gean, Department of Pharmacology, College of Medicine, National Cheng-Kung University, Tainan, Taiwan 701. E-mail: powu@mail.ncku.edu.tw.
Copyright © 2003 Society for Neuroscience 0270-6474/03/238310-08$15.00/0
References
- Atkins CM, Selcher JC, Petraitis JJ, Trzaskos JM, Sweatt JD ( 1998) The MAPK cascade is required for mammalian associative learning. Nat Neurosci 7 : 602-609. [DOI] [PubMed] [Google Scholar]
- Bailey DJ, Kim JJ, Sun W, Thompson RF, Helmstetter FJ ( 1999) Acquisition of fear conditioning in rats requires the synthesis of mRNA in the amygdala. Behav Neurosci 113 : 276-282. [DOI] [PubMed] [Google Scholar]
- Barco A, Alarcon JM, Kandel ER ( 2002) Expression of constitutively active CREB protein facilitates the late phase of long-term potentiation by enhancing synaptic capture. Cell 108 : 689-703. [DOI] [PubMed] [Google Scholar]
- Bauer EP, Schafe GE, LeDoux JE ( 2002) NMDA receptors and L-type voltage-gated calcium channels contribute to long-term potentiation and different components of fear memory formation in the lateral amygdala. J Neurosci 22 : 5239-5249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum M ( 1988) Spontaneous recovery from the effects of flooding (exposure) in animals. Behav Res Ther 26 : 185-186. [DOI] [PubMed] [Google Scholar]
- Berman DE, Dudai Y ( 2000) Memory extinction, learning anew, and learning the new: dissociation in the molecular machinery of learning in cortex. Science 291 : 2417-2419. [DOI] [PubMed] [Google Scholar]
- Bernabeu R, Bevilaqua L, Ardenghi P, Bromberg E, Schmitz P, Bianchin M, Izquierdo I, Medina JH ( 1997) Involvement of hippocampal cAMP/cAMP-dependent protein kinase signaling pathways in a late memory consolidation phase of aversively motivated learning in rats. Proc Natl Aca Sci USA 94 : 7041-7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourtchuladze R, Frenguelli B, Blendy J, Cioffi D, Schutz G, Silva AJ ( 1994) Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell 79 : 59-68. [DOI] [PubMed] [Google Scholar]
- Bouton ME, King DA ( 1983) Contextual control of conditioned fear: tests for the associative value of the context. J Exp Psychol [Anim Behav] 9 : 248-256. [PubMed] [Google Scholar]
- Cain CK, Blouin AM, Barad M ( 2002) L-type voltage-gated calcium channels are required for extinction, but not for acquisition or expression, of conditional fear in mice. J Neurosci 22 : 9113-9121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho K, Aggleton JP, Brown MW, Bashir ZI ( 2001) An experimental test of the role of postsynaptic calcium levels in determining synaptic strength using perirhinal cortex of rat. J Physiol (Lond) 532 : 459-466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cormier RJ, Greenwood AC, Connor JA ( 2001) Bidirectional synaptic plasticity correlated with the magnitude of dendritic calcium transients above a threshold. J Neurophysiol 85 : 399-406. [DOI] [PubMed] [Google Scholar]
- Davis M ( 2000) The role of the amygdala in conditioned and unconditioned fear and anxiety. In: The amygdala: a functional analysis (Aggleton JP, ed), pp 213-287. New York: Oxford UP.
- Elgersma Y, Silva AJ ( 1999) Molecular mechanisms of synaptic plasticity and memory. Curr Opin Neurobiol 9 : 209-213. [DOI] [PubMed] [Google Scholar]
- Falls WA, Miserendino JD, Davis M ( 1992) Extinction of fear-potentiated startle: blockade by infusion of an NMDA antagonist into the amygdala. J Neurosci 12 : 854-863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey U, Huang YY, Kandel ER ( 1993) Effect of cAMP simulates a late stage of LTP in hippocampal CA1 neurons. Science 260 : 1661-1664. [DOI] [PubMed] [Google Scholar]
- Genoux D, Haditsch U, Knobloch M, Michalon A, Storm D, Mansuy IM ( 2002) Protein phosphatase 1 is a molecular constraint on learning and memory. Nature 418 : 970-975. [DOI] [PubMed] [Google Scholar]
- Guzowski JF, McGaugh JL ( 1997) Antisense oligodeoxynucleotide-mediated disruption of hippocampal cAMP response element binding protein levels impairs consolidation of memory for water maze training. Proc Natl Aca Sci USA 91 : 5647-5651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall J, Thomas KL, Everitt BJ ( 2001a) Fear memory retrieval induces CREB phosphorylation and Fos expression within the amygdala. Eur J Neurosci 13 : 1453-1458. [DOI] [PubMed] [Google Scholar]
- Hall J, Thomas KL, Everitt BJ ( 2001b) Cellular imaging of zif268 expression in the hippocampus and amygdala during contextual and cued fear memory retrieval: selective activation of hippocampal CA1 neurons during the recall of contextual memories. J Neurosci 21 : 2186-2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herry C, Garcia R ( 2002) Prefrontal cortex long-term potentiation, but not long-term depression, is associated with the maintenance of extinction of learned fear in mice. J Neurosci 22 : 577-583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YY, Martin KC, Kandel ER ( 2000) Both protein kinase A and mitogen-activated protein kinase are required in the amygdala for the macromolecular synthesis-dependent late phase of long-term potentiation. J Neurosci 20 : 6317-6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber KM, Kayser M, Bear MF ( 2000) Role for rapid dendrite protein synthesis in hippocampal mGluR-dependent long-term depression. Science 288 : 1254-1256. [DOI] [PubMed] [Google Scholar]
- Impey S, Smith DM, Obrietan K, Donahue R, Wade C, Storm DR ( 1998) Stimulation of cAMP response element (CRE)-mediated transcription during contextual learning. Nat Neurosci 1 : 595-601. [DOI] [PubMed] [Google Scholar]
- Josselyn SA, Shi C, Carlezon Jr WA, Neve RL, Nestler EJ, Davis M ( 2001) Long-term memory is facilitated by cAMP response element-binding protein overexpression in the amygdala. J Neurosci 21 : 2404-2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kida S, Josselyn SA, de Ortiz SP, Kogan JH, Chevere I, Masushige S, Silva AJ ( 2002) CREB required for the stability of new and reactivated fear memories. Nat Neurosci 5 : 348-355. [DOI] [PubMed] [Google Scholar]
- Lattal KM, Abel T ( 2001) Different requirements for protein synthesis in acquisition and extinction of spatial preferences and context-evoked fear. J Neurosci 21 : 5773-5780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux JE ( 2000) Emotion circuits in the brain. Annu Rev Neurosci 23 : 155-184. [DOI] [PubMed] [Google Scholar]
- Lee H, Kim JJ ( 1998) Amygdalar NMDA receptors are critical for new fear learning in previously fear-conditioned rats. J Neurosci 18 : 8444-8454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CH, Yeh HW, Lin CH, Lu KT, Leu TH, Chang WC, Gean PW ( 2001) A role for the PI-3 kinase signaling pathway in fear conditioning and synaptic plasticity in the amygdala. Neuron 31 : 841-851. [DOI] [PubMed] [Google Scholar]
- Lin CH, Yeh HW, Leu TH, Chang WC, Wang ST, Gean PW ( 2003) Identification of calcineurin as a key signal in the extinction of fear memory. J Neurosci 23 : 1574-1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu KT, Walker DL, Davis M ( 2001) Mitogen-activated protein kinase cascade in the basolateral nucleus of amygdala is involved in extinction of fear-potentiated startle. J Neurosci 21, RC 162: 1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manahan-Vaughan D, Kulla A, Frey U ( 2000) Requirement of translation but not transcription for the maintenance of long-term depression in the CA1 region of freely moving rats. J Neurosci 20 : 8572-8576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maren S ( 1999) Long-term potentiation in the amygdala: a mechanism for emotional learning and memory. Trends Neurosci 22 : 561-567. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, Hermann H, Tang J, Hofmann C, Zieglgansberger W, Di Marzo V, Lutz B ( 2002) The endogenous cannabinoid systen controls extinction of aversive memories. Nature 418 : 530-534. [DOI] [PubMed] [Google Scholar]
- McKernan MG, Shinnick-Gallagher P ( 1997) Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature 390 : 607-610. [DOI] [PubMed] [Google Scholar]
- Miserendino MJ, Sananes CB, Melia KR, Davis M ( 1990) Blocking of acquisition but not expression on conditioned fear-potentiated startle by NMDA antagonists in the amygdala. Nature 345 : 716-718. [DOI] [PubMed] [Google Scholar]
- Myers KM, Davis M ( 2002) Behavioral and neural analysis of extinction. Neuron 36 : 567-584. [DOI] [PubMed] [Google Scholar]
- Nader K ( 2003) Memory traces unbound. Trends Neurosci 26 : 65-72. [DOI] [PubMed] [Google Scholar]
- Nader K, Schafe GE, LeDoux JE ( 2000) Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature 406 : 722-726. [DOI] [PubMed] [Google Scholar]
- Pavlov IP ( 1927) Conditioned reflex: an investigation of the physiological activity of the cerebral cortex. London: Oxford UP. [DOI] [PMC free article] [PubMed]
- Paxinos G, Watson C ( 1986) The rat brain in stereotaxic coordinates. New York: Academic.
- Quirk GJ, Russo GK, Barron JL, Lebron K ( 2000) The role of ventromedial prefrontal cortex in the recovery of extinguished fear. J Neurosci 20 : 6225-6231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rescorla RA ( 2001) Experimental extinction. In: Handbook of contemporary learning theories (Mowrer RR, Klein S, eds), pp 119-154. Mahwah, NJ: Erlbaum.
- Rescorla RA, Heth CD ( 1975) Reinstatement of fear to an extinguished conditioned stimulus. J Exp Psychol Anim Behv Process 1 : 88-96. [PubMed] [Google Scholar]
- Ressler KJ, Paschall G, Zhou XL, Davis M ( 2002) Regulation of synaptic plasticity genes during consolidation of fear conditioning. J Neurosci 22 : 7892-7902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogan MT, Staubli UV, LeDoux JE ( 1997) Fear conditioning induces associative long-term potentiation in the amygdala. Nature 390 : 604-607. [DOI] [PubMed] [Google Scholar]
- Santini E, Muller RU, Quirk GJ ( 2001) Consolidation of extinction learning involves transfer from NMDA-independent to NMDA-dependent memory. J Neurosci 21 : 9009-9017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sara SJ ( 2000) Retrieval and reconsolidation: toward a neurobiology of remembering. Learn Mem 7 : 73-84. [DOI] [PubMed] [Google Scholar]
- Schafe GE, LeDoux JE ( 2000) Memory consolidation of auditory Pavlovian fear conditioning requires protein synthesis and protein kinase A in the amygdala. J Neurosci 20, RC 96: 1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafe GE, Atkins CM, Swank MW, Bauer EP, Sweatt JD, LeDoux JE ( 2000) Activation of ERK/MAPK kinase in the amygdala is required for memory consolidation of Pavlovian fear conditioning. J Neurosci 20 : 8177-8187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafe GE, Nader K, Blair HT, LeDoux JE ( 2001) Memory consolidation of Pavlovian fear conditioning: a cellular and molecular perspective. Trends Neurosci 24 : 540-546. [DOI] [PubMed] [Google Scholar]
- Szapiro G, Vianna MRM, McGaugh JL, Medina JH, Izquierdo I ( 2003) The role of NMDA glutamate receptors, PKA, MAPK, and CAMKII in the hippocampus in extinction of conditioned fear. Hippocampus 13 : 53-58. [DOI] [PubMed] [Google Scholar]
- Taubenfeld SM, Milekic MH, Monti B, Alberini CM ( 2001) The consolidation of new but not reactivated memory requires hippocampal C/EBPβ. Nat Neurosci 4 : 813-818. [DOI] [PubMed] [Google Scholar]
- Vianna MRM, Szapiro G, McGaugh JL, Medina JH, Izquierdo I ( 2001) Retrieval of memory for fear-motivated training initiates extinction requiring protein synthesis in the rat hippocampus. Proc Natl Aca Sci USA 98 : 12251-12254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisskopf MG, Bauer EP, LeDoux JE ( 1999) L-type voltage-gated calcium channels mediate NMDA-independent associative long-term potentiation at thalamic input synapses to the amygdala. J Neurosci 19 : 10512-10519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winder DG, Sweatt JD ( 2001) Roles of serine/threonine phosphatases in hippocampal synaptic plasticity. Nat Rev Neurosci 2 : 461-474. [DOI] [PubMed] [Google Scholar]