

Abstract
The effect of the human immunodeficiency virus-1 protein Tat was investigated on neurotransmitter release from human and rat cortical nerve endings. Tat failed to affect the release of several neurotransmitters, such as glutamate, GABA, norepinephrine, and others, but it evoked the release of [3H]ACh via increase of cytosolic [Ca2+]. In human nerve terminals, the Tat effect partly depends on Ca2+ entry through voltage-sensitive Ca2+ channels, because Cd2+ halved the Tat-evoked release. Activation of group I metabotropic glutamate receptors (mGluR) and mobilization of Ca2+ from IP3-sensitive intraterminal stores are also involved, because the Tat effect was prevented by mGluR antagonists 2-methyl-6-(phenylethynyl)pyridine hydrochloride and 7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxylate ethyl ester and by the IP3 receptor antagonists heparin and xestospongin C. Furthermore, the group I selective mGlu agonist (RS)-3,5-dihydroxyphenylglycine enhanced [3H]ACh release. In rat nerve terminals, the Tat-evoked release neither depends on external Ca2+ ions entry nor on IP3-mediated mechanisms. Tat seems to cause mobilization of Ca2+ from ryanodine-sensitive internal stores because its effect was prevented by both 8-bromo-cyclic adenosine diphosphate-ribose and dantrolene. The Tat-evoked release from human synaptosomes was mimicked by the peptide sequences Tat 32-62, Tat 49-86, and Tat 41-60. In contrast, the Tat 49-86 and Tat 61-80 fragments, but not the Tat 32-62 fragment, were active in rat synaptosomes. In conclusion, Tat elicits Ca2+-dependent [3H]ACh release by species-specific intraterminal mechanisms by binding via discrete amino acid sequences to different receptive sites on human and rat cholinergic terminals.
Keywords: HIV-1, Tat, acetylcholine release, human cerebrocortex, rat cerebrocortex, calcium, inositoltrisphosphate receptors, ryanodine receptors, mGlu receptor
Introduction
During human immunodeficiency virus (HIV-1) infection, ∼50% of patients develop motor, cognitive, and behavioral impairments, the origins of which are poorly understood. Because neurons can rarely, if ever, be infected by the virus, it is hypothesized that indirect mechanisms are primarily involved in the neuronal deficits and in the neurotoxic events that often occur during acquired immunodeficiency syndrome (AIDS) infection (Kaul and Lipton, 1999; Valle et al., 2000; Bezzi et al., 2001; Kaul et al., 2001; Mollace et al., 2001), although direct effects of HIV-1 proteins have been reported as well (see below).
The occurrence of neurotoxic events is sustained by postmortem histological observations made in cerebral tissue from HIV-1-positive patients, where loss of neurons has clearly been described (Masliah et al., 1992, 1997; Adle-Biassette et al., 1995; Glass et al., 1995; Petito and Roberts, 1995; Fox et al., 1997; Everall et al., 1999). These dramatic effects may, however, not be necessary to explain the above-mentioned neuropsychiatric deficits; in fact, relatively subtle changes in central neurotransmission induced by HIV-1 itself or by viral proteins might be at the basis of various functional impairments.
In the last decade, evidence has been provided suggesting that HIV-1 proteins, in particular the coat protein gp120, could directly influence central neuronal functions, causing changes in neurotransmitter release or affecting receptor-mediated mechanisms (Müller et al., 1992; Savio and Levi, 1993; Ushijima et al., 1993; Pittaluga and Raiteri, 1994; Lannuzel et al., 1995; Nath et al., 1995; Pittaluga et al., 1996; Fontana et al., 1997; Pattarini et al., 1998; Kaul and Lipton, 1999; Xin et al., 1999; Gemignani et al., 2000; Bezzi et al., 2001; Kaul et al., 2001).
Increasing attention has been focused on Tat, a nonstructural viral protein known to be produced and actively released by HIV-1-infected cells (Ensoli et al., 1993). Tat has been detected in the brain of animals with viral encephalitis, and its encoding mRNA has been found in the CNS of HIV-1-infected patients exhibiting AIDS-dementia symptoms (Hudson et al., 2000; Valle et al., 2000).
Tat can apparently induce both neurological impairments and neurotoxicity because it was shown both to excite and kill human and rodent cerebrocortical neurons (Nath et al., 1996; Kruman et al., 1998) by mechanisms involving neuronal Ca2+ homeostasis (Haughey et al., 1999, 2001). The protein binds to and depolarizes neuronal membranes (Sabatier et al., 1991), allowing increases in cytosolic Ca2+ through receptor-operated channels and mobilization from IP3-sensitive internal stores (Nath et al., 1996; Haughey et al., 1999, 2001).
We investigated whether Tat can affect brain neurotransmission by affecting transmitter release from fresh human and rat cortical nerve endings. We first explored the release of several transmitters present in brain cortical areas and potentially involved in neuronal functions that can be impaired during HIV-1 infection (McGaugh, 1989; Hasselmo and Bower, 1993; Collingridge and Bliss, 1995; Garg, 1999; Gruslin et al., 1999; Pascual Sedano et al., 1999). Most of the work then focused on ACh, the release of which appears to be selectively targeted by Tat.
Materials and Methods
Human brain tissue samples. Samples of human cerebral cortex were obtained from informed and consentient HIV-1-negative patients undergoing neurosurgery, each on a different day, to reach deeply seated tumors. The samples represented parts of frontal (n = 9) and temporal (n = 7) lobes obtained from 11 women and 5 men (age, 30-70 years). Immediately after removal, the tissue was placed in a physiological salt solution at 2-4°C, and crude synaptosomal fractions were prepared within 90 min. The experimental procedures were approved by the Ethical Committee of the University of Genoa.
Rat brain tissue samples. Adult male rats (Sprague Dawley; 200-250 gm) were housed at constant temperature (22 ± 1°C) and relative humidity (50%) under a regular light/dark schedule (light, 7:00 A.M. to 7:00 P.M.). Food and water were freely available. The animals were killed by decapitation, and the tissue was rapidly removed. The experimental procedures were approved by the Department Ethical Committee, in accordance with European legislation (European Communities Council directive of November 24, 1986; 86/609/EEC).
Preparation of synaptosomes. Crude synaptosomal fractions were prepared essentially according to Raiteri et al. (1984). Briefly, the tissue was homogenized in 40 volumes of 0.32 m sucrose, buffered with phosphate to pH 7.4; the homogenate was first centrifuged (5 min at 1000 × g), and the synaptosomal fraction was isolated from the supernatant by centrifugation (20 min at 12,000 × g). In a set of experiments, the tissue was homogenized in buffered sucrose containing 1 mm BAPTA or 40 μm heparin, to entrap these membrane-impermeant agents into subsequently isolated synaptosomes (Raiteri et al., 2000). Based on estimates made by entrapping [3H]sucrose, the intrasynaptosomal concentration of the compounds are ∼5% of the original concentration in the homogenization medium. The synaptosomal pellets were resuspended in a physiological medium having the following composition (in mm): 125 NaCl, 3 KCl, 1.2 MgSO4, 1.2 CaCl2, 1 NaH2PO4, 22 NaHCO3, and 10 glucose (aerated with 95% O2 and 5% CO2), pH 7.2-7.4. Synaptosomal protein contents were determined according to Bradford (1976).
Release experiments. Human or rat cortical synaptosomes were labeled with [3H]choline (final concentration, 30-60 nM; in experiments of [3H]ACh release), with [3H]norepinephrine ([3H]NE; final concentration, 30-50 nm), or with [3H]D-aspartate (final concentration, 40-60 nm). In other experiments, synaptosomes were incubated without radioactive markers, when the release of endogenous aspartate, glutamate, GABA, or serine had to be monitored. Incubation was performed at 37°C, for 15 min, in a rotary water bath, and in an atmosphere of 95% O2 and 5% CO2. Incubation with [3H]NE was performed in the presence of the transporter blockers citalopram (0.1 μm) and GBR 12909 (0.1 μm) to avoid false labeling with [3H]NE of serotonergic and dopaminergic nerve terminals, respectively. After the labeling period, identical portions of the synaptosomal suspensions were distributed on a set of parallel superfusion chambers maintained at 37°C (Raiteri and Raiteri, 2000). Superfusion was then performed at a flow rate of 0.5 ml/min, using media aerated with 95% O2 and 5% CO2, for a total period of 51 min.
Synaptosomes were first equilibrated during 36 min of superfusion; five consecutive 3-min fractions (termed “b1 to b5”) were then collected. Tat or its fragments were introduced at the end of the first fraction (b1; t = 39 min), whereas glutamate receptor antagonists xestospongin C, dantrolene, oligomycin, or 8-bromo-cyclic adenosine diphosphateribose (Br-cADP-ribose) were present from 8 min before Tat or its fragments until the end of superfusion.
In a series of experiments, performed to study the Ca2+ dependency of the Tat-induced release, the superfusion medium was replaced, starting from t = 20 min, with a medium containing 0.1 mm Ca2+ and in which the Mg2+ concentration was increased to 10 mM to prevent the entry of Ca2+ through voltage-sensitive Ca2+ channels (VSCCs).
Fractions collected and superfused synaptosomes were finally counted for radioactivity or analyzed for their endogenous amino acid content.
Endogenous amino acid determination. Endogenous amino acids were determined by HPLC analysis (Waters 715 Ultrawisp; Waters Associates, Milford, MA) after automatic derivatization with o-phthalaldehyde and resolution through a C18 reverse-phase chromatographic column (Chrompack, Middleburg, The Netherlands; 10 × 4.6 mm, 3 μm; 30°C), coupled with fluorimetric detection (Shimadzu, Tokyo, Japan; RF551; excitation wavelength, 350 nm; emission wavelength, 450 nm). The compounds were eluted with a Waters 600 MS gradient system after a ternary gradient program, as described previously (Pittaluga et al., 1997). Homo-serine was used as internal standard.
Endogenous cADP-ribose determination. Rat cortical synaptosomes (0.3 mg of protein/tube) were incubated for 8 min at 37°C in a rotary water bath in the absence (control) or in the presence of Tat (1 nm). At the end of the incubation period, HClO4 (final concentration, 0.6 mm) was added to stop the enzymatic reactions, and samples were rapidly frozen until cADP-ribose determination. Synaptosomal c-ADP-ribose content was analyzed according to Graeff and Lee (2002).
Denaturation or immunoprecipitation of Tat. Tat was denaturated by heating at 90°C for 30 min. Immunoprecipitation was performed by incubating the protein (100 nm) with a rabbit anti-Tat polyclonal antibody (1:500) in the presence of goat anti-rabbit IgG agarose for 90 min at room temperature. The suspension was then centrifuged, and the supernatant was immediately used in release experiments.
Calculations. The amounts of endogenous amino acids released into each superfusate fraction were expressed as picomoles per milligram of protein. The effects of Tat on the release of endogenous transmitters were evaluated by calculating the ratio between the amino acid content in the fraction in which the maximal effect was reached (usually b4, the fourth fraction collected) and the amount in the first fraction (b1) and comparing this ratio with the corresponding ratio obtained under control conditions (no drug added).
The amount of radioactivity released into each superfusate sample was expressed as a percentage of the total synaptosomal radioactivity content at the start of the fraction collected (fractional efflux). When the time course of the Tat effect is described, the amount of radioactivity into each superfusate fraction collected was expressed as the percentage of the first fraction collected (b1). When results are expressed as percentage of increase over basal release, drug effects were evaluated from the ratio between the percentage of release in b4 and that in b1. This ratio was compared with the corresponding ratio obtained under control conditions (no drug added). ANOVA was performed, followed by Dunnett's test or Newman-Keuls multiple comparisons test, as appropriate.
The amount of endogenous c-ADP-ribose present in each sample was expressed as picomoles per milligram of protein. ANOVA was performed by direct comparisons with two-tailed Student's t test.
Drugs. [methyl-3H] Choline chloride (specific activity, 83 Ci/mmol), [3H]NE (specific activity, 38 Ci/mmol), and [3H]D-aspartate (specific activity, 16.3 Ci/mmol) were obtained from Amersham Radiochemical Centre (Buckinghamshire, UK). (RS)-3,5-Dihydroxyphenylglycine (3,5-DHPG), AMPA, 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo [f]quinoxaline-7-sulfonamide (NBQX), 2-methyl-6-(phenylethynyl)pyridine hydrochloride (MPEP), and 7-(hydroxyimino)cycloprop-a[b]chromen-1a-carboxylate ethyl ester (CPCCOEt) were obtained from Tocris Cookson (Bristol, UK). Heparin, dantrolene, oligomycin, Br-cADP-ribose, and goat anti-rabbit IgG agarose (IgG fraction of antiserum) were from Sigma (St. Louis, MO); BAPTA was from Fluka Biochemika (Milan, Italy), and xestospongin C was from Calbiochem (Darmstadt, Germany). The following drugs were kindly provided by the companies indicated: cis-4-(phosphonomethyl)-2-piperidinecarboxilic acid (CGS19755; Novartis Pharmaceutical Corporation, Summit, NJ); (R)-5-(4-aminophenyl)-7-methylcarbamoyl-8-methyl-8,9-dihydro-7H-1,3-dioxolo[4,5-h][2,3]-benzodiazepine (LY303070; Eli Lilly, Indianapolis, IN); citalopram (H. Lundbeck A/S, Copenhagen, Denmark); GBR 12909 (Gist-Brocades, Delft, The Netherlands).
Recombinant Tat (HIV-1 strain HxB2) and rabbit anti-Tat (HIV-1 strain HxB2) polyclonal antibody were from Intracell (London, UK). Tat peptide 32-62 (HIV-1 strain LAI), Tat peptide 49-86 (HIV-1 strain LAI), and the 10 amino acids overlapping Tat 20mers (HIV-1 strain LAI) were kindly donated by the “AIDS Reagent Project” (ARP 7004.2 and 7005) and by the European program EVA (779.1-779.1-8) (NIBSC, Hertfordshire, UK). The amino acid sequence of the Tat protein strain LAI perfectly overlaps that of Tat strain HxB2.
Results
Tat selectively enhances [3H]ACh release
We first investigated the effects of Tat on the release of different transmitters from human and rat cortical superfused synaptosomes. As shown in Table 1, Tat, added to the superfusion medium at 1 or 3 nm, did not affect the release of endogenous aspartate, glutamate, GABA, or serine. The protein also had no effect on the release of previously taken up [3H]NE or [3H]D-aspartate from either human or rat synaptosomes. On the contrary, Tat evoked the release of [3H]ACh from human and rat synaptosomes prelabeled with [3H]choline. The releasing effect of 1 or 3 nm Tat was significantly higher in human than in rat nerve endings (Table 1; see also Fig. 1).
Table 1.
Effects of Tat on the spontaneous release of endogenous or preloaded tritiated neurotransmitters from human and rat cortical synaptosomes
|
|
Human synaptosomes |
Rat synaptosomes |
||||
|---|---|---|---|---|---|---|
| Neurotransmitter
|
1 nm Tat
|
3 nm Tat
|
1 nm Tat
|
3 nm Tat
|
||
| Aspartate | −4.8 ± 6.2 (3) | −9.5 ± 3.7 (3) | 6.9 ± 3.6 (4) | 3.9 ± 4.6 (4) | ||
| Glutamate | −0.3 ± 6.9 (3) | 9.8 ± 3.9 (3) | 7.9 ± 2.9 (4) | −0.8 ± 0.9 (4) | ||
| GABA | −3.3 ± 5.9 (3) | 10.5 ± 7.1 (3) | 11.8 ± 4.5 (4) | 3.7 ± 3.4 (4) | ||
| Serine | −14.2 ± 5.4 (3) | −8.7 ± 8.4 (3) | 2.6 ± 7.6 (4) | 10.1 ± 6.6 (4) | ||
| [3H]d-Aspartate | 6.5 ± 8.3 (5) | 10.2 ± 4.1 (3) | 0.4 ± 6.5 (5) | −1.8 ± 4.7 (5) | ||
| [3H]ACh | 88.2 ± 16.0 (10) | 68.9 ± 16.6 (3) | 35.3 ± 4.6 (15) | 36.2 ± 7.9 (4) | ||
| [3H]NE
|
−1.9 ± 7.5 (5)
|
−7.9 ± 7.5 (4)
|
4.7 ± 2.9 (5)
|
0.6 ± 0.3 (6)
|
||
The transmitters released in the first 3 min fraction (b1) collected (see Materials and Methods) during superfusion of human synaptosomes amounted to aspartate = 41.0 ± 4.5 pmol/mg protein; glutamate = 126.1 ± 7.6 pmol/mg protein; GABA = 45.3 ± 8.1 pmol/mg protein; serine = 95.5 ± 6.7 pmol/mg protein; [3H]d-aspartate = 3.40 ± 0.29%; [3H]ACh = 3.52 ± 0.26%; and [3H]NE = 2.22 ± 0.18%. The release in b1 during superfusion of rat synaptosomes amounted to aspartate = 105.3 ± 9.7 pmol/mg protein; glutamate = 230 ± 35.7 pmol/mg protein; GABA = 104.0 ± 9.1 pmol/mg protein; serine = 197.3 ± 9.6 pmol/mg protein; [3H]d-aspartate = 1.07 ± 0.09%; [3H]ACh = 1.80 ± 0.11%; and [3H]NE = 1.67 ± 0.16%. The release in b1 is expressed as picomoles per milligram of protein in the case of endogenous transmitters and as a percentage of the total synaptosomal tritium content at the onset of the collection period in the case of the radiolabeled transmitters. Synaptosomes were exposed to Tat from the end of b1 throughout superfusion. The effect of Tat is expressed as the percentage of increase over basal. Data are means ± SEM of the number of experiments (in parentheses) run in triplicate (three superfusion chambers for each experimental condition).
Figure 1.

Effects of Tat on [3H]ACh release from human (A) and rat (B) cortical synaptosomes: time course of release and inactivation after Tat immunoprecipitation or heat shock denaturation. Gray circles, Control (no Tat added); solid circles, Tat (1 nm); open triangles, heat shock-denatured Tat (1 nm); open squares, Tat (1 nm) after immunoprecipitation. For the sake of clarity, basal release from rat synaptosomes (B) is not indicated. Results are expressed as percentages of the first superfusion fraction collected (b1). Data are means ± SEM of three to nine experiments run in triplicate (3 superfusion chambers for each experimental condition). Insets (C,D), Concentration-dependent effects of Tat on the release of [3H]ACh from human (C) and rat (D) cortical synaptosomes. Results are expressed as the percentage of increase over basal release. Data are means of three to six experiments run in triplicate. *p < 0.05 versus control; **p < 0.01 versus control; #p < 0.05 versus Tat; ##p < 0.01 versus Tat.
As shown in Figure 1A, the Tat-induced [3H]ACh release from human cortical synaptosomes reached a maximal effect at b3 (i.e., after 6 min of exposure to the protein); heat shock denaturation or immunoprecipitation of Tat prevented its releasing effect. The protein also elicited release of [3H]ACh from rat cortical synaptosomes (Fig. 1B). Similarly, the releasing effect produced by 1 nm Tat was prevented by immunoprecipitation or heat shock denaturation. When tested at varying concentrations (0.1-3 nm), Tat appeared to reach a maximum effect at ∼1 nm (Fig. 1C,D).
Calcium dependence of the Tat-evoked release from human cortical nerve endings
The [3H]ACh release provoked by 1 nM Tat in human cortical synaptosomes was analyzed for its dependence on external Ca2+ ions. Drastic reduction of Ca2+ (to 0.1 mM) in the superfusion medium and the simultaneous increase of Mg2+ ions to 10 mM prevented the Tat-induced release by ∼55% (Fig. 2A). Moreover, Cd2+ ions (10 μm) diminished to the same extent the releasing effect of the protein in human nerve endings. In contrast, the Tat-induced release was almost completely (∼85%) prevented when the membrane-impermeant Ca2+ chelator BAPTA was entrapped in the synaptosomes, suggesting that Ca2+ release from internal stores also contributes to the releasing effect of Tat from human nerve endings (Fig. 2A).
Figure 2.
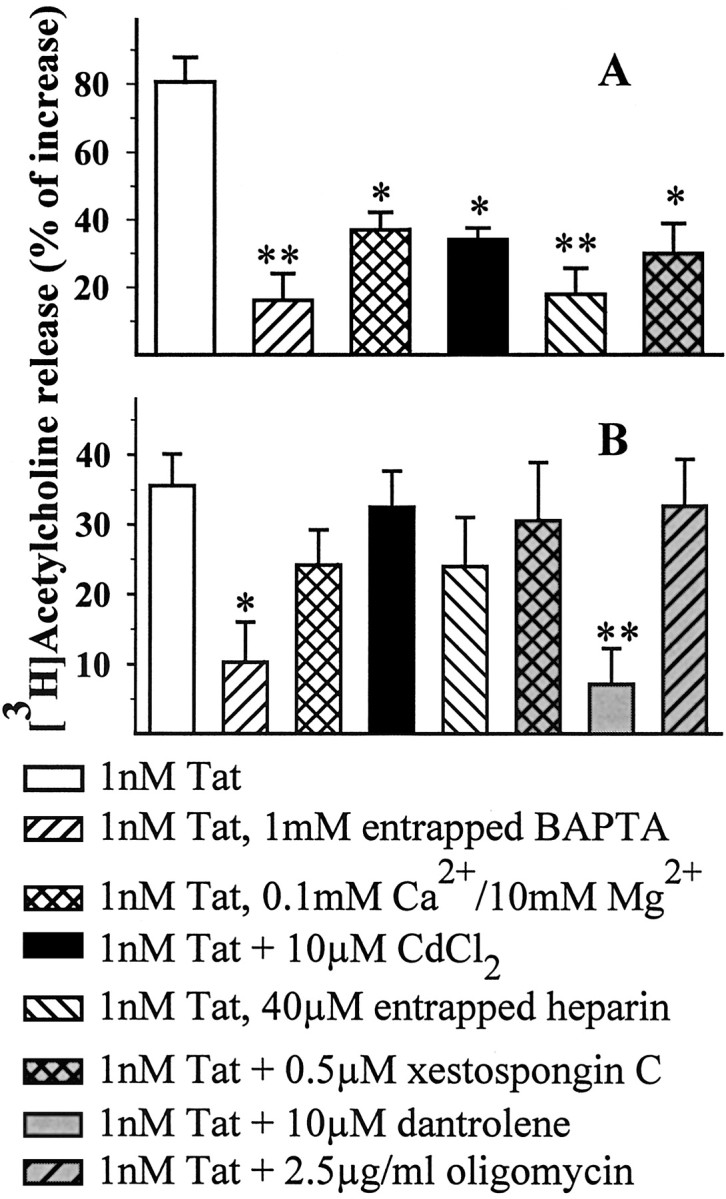
Ca2+ dependency of the Tat-induced release of [3H]ACh from human (A) and rat (B) cortical synaptosomes. Synaptosomes were staffed with BAPTA or heparin during cortical tissue homogenization, whereas antagonists or Cd2+ ions were added during superfusion, 8 min before Tat. At the concentrations used, these drugs did not modify, on their own, the [3H]ACh release (data not shown). Results are expressed as the percentage of increase over basal release; data are means ± SEM of 3-12 experiments run in triplicate. *p < 0.05 versus respective control; **p < 0.01 versus respective control.
Involvement of IP3 in the Tat-evoked release from human nerve endings
Tat has recently been proposed to mobilize Ca2+ from the endoplasmic reticulum through activation of IP3-sensitive receptors (Haughey et al., 1999). To verify the occurrence of such a process in our system, we incorporated the membrane-impermeant IP3 receptor (IP3r) antagonist heparin (40 μm in the homogenization medium; estimated internal concentration, ∼200 nm) into human synaptosomes. As shown in Figure 2A, heparin prevented ∼75% of the Tat-induced [3H]ACh release. At the concentration used, heparin was previously found to prevent the somatostatin-induced NMDA receptor potentiation of NE release from rat hippocampal synaptosomes (Pittaluga et al., 2000). The involvement of IP3-sensitive Ca2+ stores is also indicated by the finding that xestospongin C, a recently available membrane-permeant IP3r antagonist (Gafni et al., 1997), inhibited the Tat-induced release of [3H]ACh from human synaptosomes (Fig. 2A).
Calcium dependence of the Tat-evoked release from rat cortical nerve endings
Surprisingly, the Tat-induced release of [3H]ACh from rat cortical synaptosomes seems not to depend on the presence of extra-terminal Ca2+ ions. As shown in Figure 2B, superfusion with a Ca2+-deprived Mg2+-enriched medium did not significantly prevent the Tat-mediated effect. Moreover, the addition of Cd2+ did not modify the Tat-induced release. In contrast, entrapping BAPTA into rat nerve endings blocked the[3H]ACh release elicited by Tat by ∼70% (Fig. 2B), suggesting that Tat provokes Ca2+ mobilization from internal stores and that the resulting increase in cytosolic Ca2+ facilitates release of [3H]ACh. Finally, neither entrapped heparin nor exposure to xestospongin C prevented the effect of Tat on [3H]ACh release in the rat cortex (Fig. 2B). Because intraterminal Ca2+ may originate from sources other than IP3-sensitive stores (for review, see Berridge, 1998; Mattson et al., 2000), we investigated the effect of dantrolene, an antagonist at ryanodine receptors (RYRs), and of oligomycin, an inhibitor of the mitochondrial ATPase. As shown in Figure 2B, dantrolene dramatically reduced the Tat-evoked release from rat synaptosomes, whereas oligomycin had no effect. At the concentrations used, dantrolene and oligomycin were previously found to inhibit the external Ca2+-independent K+-induced GABA release from rat cortical synaptosomes (Raiteri et al., 2002) as well as NGF-mediated potentiation of K+-induced glutamate release (Raiteri et al., 2003).
The Tat-evoked [3H]ACh release from human and rat cortical nerve endings does not involve activation of glutamate ionotropic receptors
It had been proposed that Tat can directly excite human and rodent neurons via activation of non-NMDA receptors (Magnuson et al., 1995). We found that human cortical nerve endings are endowed with AMPA/kainate receptors, activation of which causes [3H]ACh release; this effect is prevented by NBQX, an AMPA/kainate-preferring antagonist (100 μm AMPA = 28.4 ± 2.1%; 100 μm AMPA + 10 μm NBQX = 0.3 ± 5.8%; p < 0.01; results were expressed as the percentage of increase over basal release). Release-mediating AMPA/kainate receptors also are present on rat cortical nerve endings (Pittaluga et al., 1997).
We investigated whether the effect of Tat on [3H]ACh release from human and rat cortical synaptosomes could depend, at least in part, on the activation of ionotropic glutamatergic receptors; however, the selective AMPA/kainate receptor antagonists NBQX (10 μm) and LY303070 (10 μm), as well as the competitive NMDA receptor antagonist CGS19755 (10 μm), failed to significantly affect the Tat-induced release (Table 2). At the concentrations indicated, the antagonists did not affect, on their own, the spontaneous release of tritium.
Table 2.
Lack of antagonism by NBQX, LY303070, or CGS19755 on the Tat-induced [3H]ACh release from superfused human or rat cortical synaptosomes
|
|
Human synaptosomes |
Rat synaptosomes |
|---|---|---|
| Tat (1 nm) | 78.6 ± 5.3 | 36.2 ± 3.6 |
| Tat (1 nm) + NBQX (10 μm) | 71.8 ± 6.8 | 27.4 ± 6.3 |
| Tat (1 nm) + LY303070 (10 μm) | 73.4 ± 5.3 | 32.4 ± 4.3 |
| Tat (1 nm) + CGS19755 (10 μm)
|
67.5 ± 7.2
|
31.9 ± 5.2
|
Antagonists were added 8 min before Tat and maintained until the end of the superfusion period. Results are expressed as the percentage of increase over basal. Data are means ± SEM of three to five experiments run in triplicate.
The Tat-evoked [3H]ACh release involves activation of group I glutamate metabotropic receptors in human, but not in rat cortical nerve endings
Because of the involvement of IP3-sensitive Ca2+ stores in the Tat-releasing effect from human nerve endings, we evaluated whether IP3-linked metabotropic receptors of group I metabotropic glutamate (mGlu) might mediate [3H]ACh release. Human cortical synaptosomes prelabeled with [3H]choline were exposed to Tat in the presence of MPEP or CPCCOEt, two antagonists at IP3-linked mGlu receptors (mGluRs) of group I. As reported in Table 3, MPEP (1 μm) and CPCCOEt (5 μm) inhibited by ∼68% and 62%, respectively, the Tat-evoked release of [3H]ACh; the antagonist effects were not additive. At the concentrations used, the antagonists did not affect, on their own, the basal release of [3H]ACh.
Table 3.
Effects of the mGluR antagonists MPEP and CPCCOEt on the Tat-induced [3H]ACh release from human cortical synaptosomes
|
|
Basal release |
Tat-induced release |
|---|---|---|
| Control | 66.9 ± 10.3 | |
| MPEP (1 μm) | 8.9 ± 6.6 | 19.7 ± 5.1a |
| CPCCOEt (5 μm) | 9.9 ± 4.4 | 25.2 ± 3.6a |
| MPEP (1 μm) + CPCCOEt (5 μm)
|
7.6 ± 3.5
|
24.4 ± 4.7a
|
Antagonists were added 8 min before 1 nm Tat and maintained until the end of the superfusion period. Results are expressed as the percentage of increase over basal. Data are means ± SEM of four experiments run in triplicate.
p < 0.01 versus Tat.
Consistent with these results, the compound 3,5-DHPG, a selective agonist at group I mGluR, concentration-dependently released [3H]ACh from human cortical nerve endings as shown in Table 4; the releasing effect induced by 3,5-DHPG (100 μm) was almost totally antagonized by MPEP (1 μm) and CPCCOEt (5 μm). Of note, 3,5-DHPG (100 μm) did not cause the release of [3H]ACh from rat cortical nerve endings (30 μm 3,5-DHPG = 1.5 ± 2.4; 100 μm 3,5-DHPG = 7.4 ± 4.5; results were expressed as the percentage of increase over basal release).
Table 4.
Effects of the group I mGluR agonist 3,5-DHPG on the release of [3H]ACh from human cortical synaptosomes: reversal by MPEP and CPCCOEt
|
|
[3H]ACh-induced release |
|---|---|
| 3,5-DHPG (10 μm) | 0.8 ± 8.9 |
| 3,5-DHPG (30 μm) | 29.6 ± 6.2a |
| 3,5-DHPG (100 μm) | 83.2 ± 17.3b |
| 3,5-DHPG (100 μm) + MPEP (1 μm) | 13.3 ± 5.3a |
| 3,5-DHPG (100 μm) + CPCCOEt (5 μm)
|
16.5 ± 7.8a
|
Antagonists were added 8 min before the agonist and maintained until the end of the superfusion period. Results are expressed as the percentage of increase over basal. Data are means ± SEM of four experiments run in triplicate.
p < 0.05 versus respective control
p < 0.01 versus respective control.
The Tat-evoked release from rat nerve endings involves cADP-ribose
RYRs are thought to be endogenously activated by cADP-ribose (Galione et al., 1993; Fossier et al., 1999; Higashida et al., 2001; Lee, 2001). Data are present in the literature demonstrating that in the cholinergic system of frog motor nerve terminals (Brailoiu and Miyamoto, 2000) and Aplysia buccal ganglion neurons (Mothet et al., 1998), the injection of cADP-ribose facilitates ACh release in a Ca2+-dependent manner. To investigate whether the Tat effect in rat cerebrocortex synaptosomes is mediated by cADP-ribose, synaptosomes were exposed to Tat in the presence of Br-cADP-ribose, a membrane-permeant competitive antagonist of the cADP-ribose-binding site (Walseth and Lee, 1993). As reported in Table 5, Br-cADP-ribose prevented the Tat-induced release of [3H]ACh. At the concentrations used, the compound did not affect, on its own, the release of [3H]ACh (data not shown). Exposure of rat synaptosomes to Tat (1 nm) caused a moderate, but significant, increase in the cADP-ribose content [control: 2.49 ± 0.12 pmol/mg protein, n = 8; with added Tat (1 nm): 3.18 ± 0.24, n = 10; p < 0.05]. This effect is probably underestimated, if one assumes that the cADP-ribose increase essentially occurs in cholinergic synaptosomes, representing only a small percentage of the cortical population of nerve endings. The cADP-ribose detected in the synaptosomal fraction is consistent with the cADP-ribose level reported in the CNS by Walseth et al. (1991).
Table 5.
Effect of Br-cADP-ribose on the Tat-induced [3H]ACh release from rat cortical synaptosomes
|
|
[3H]ACh release |
|---|---|
| Tat (1 nm) | 29.2 ± 3.8 |
| Tat (1 nm) + Br-cADP-ribose (1 μm) | 26.9 ± 15.2 |
| Tat (1 nm) + Br-cADP-ribose (5 μm) | 13.6 ± 3.5 |
| Tat (1 nm) + Br-cADP-ribose (10 μm)
|
2.4 ± 1.6a
|
The cADP-ribose antagonist was added 8 min before Tat and maintained until the end of the superfusion period. Results are expressed as the percentage of increase over basal. Data are means ± SEM of three experiments run in triplicate.
p < 0.05 versus Tat.
In contrast, the Tat-evoked release of [3H]ACh from human cortical nerve endings was insensitive to Br-cADP-ribose (1 nm Tat = 77.3 ± 4.5; 1 nm Tat in the presence of 10 μm Br-cADP-ribose = 86.6 ± 8.7; results are expressed as the percentage of increase over basal release).
Different peptide fragments mimic Tat in human versus rat nerve endings
To identify the peptide sequences responsible for the effects of Tat on the release of [3H]ACh, different fragments of the protein were tested on human and rat cortical synaptosomes prelabeled with [3H]choline.
Figure 3A shows that Tat 32-62 (1 nm) and Tat 49-86 (1 nm) elicited [3H]ACh release from human synaptosomes. In contrast, rat synaptosomes only released [3H]ACh when exposed to 1 nM Tat 49-86, whereas the response to Tat 32-62 was very weak and barely significant (Fig. 3B). Indeed, Tat 49-86 (1 nm) increased [3H]ACh release from rat beginning at 0.1 nM, whereas Tat 32-62 was unable to release [3H]ACh, even when up to 10 nM were added (Fig. 3B, inset).
Figure 3.
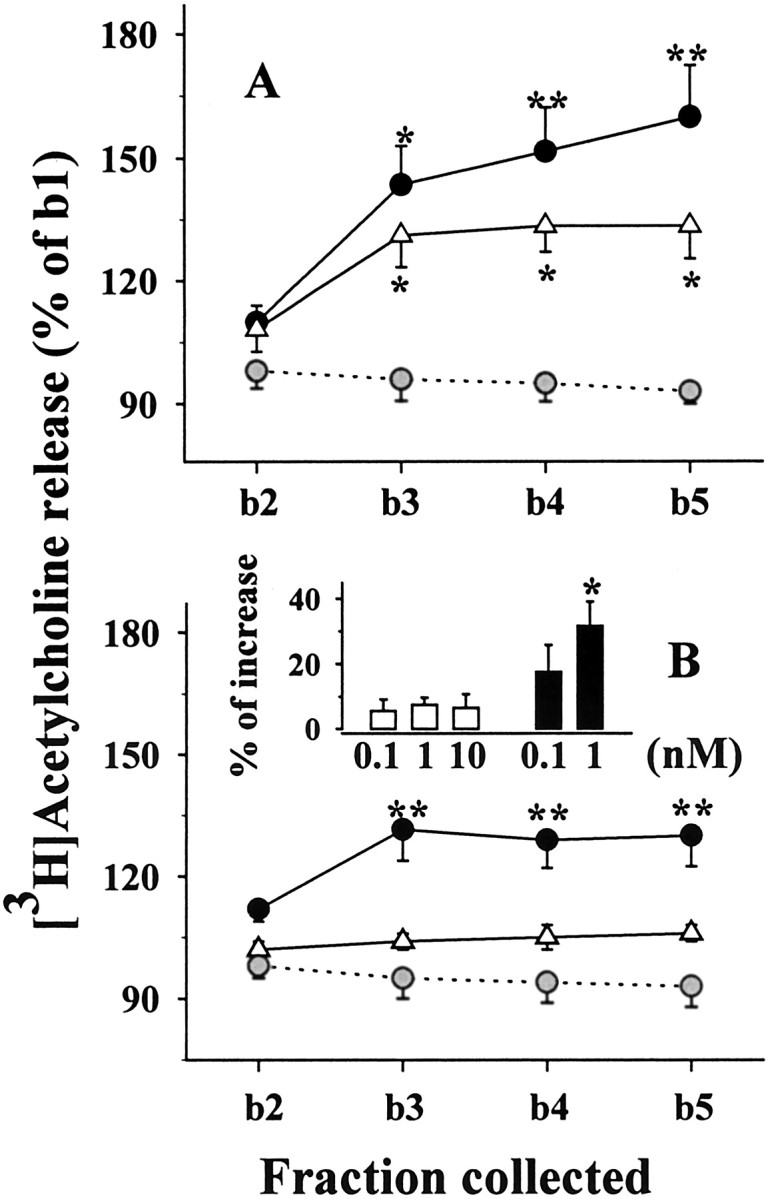
Effects of Tat 32-62 and Tat 49-86 on [3H]ACh release from human (A) and rat (B) cortical synaptosomes. Gray circles, Control (no Tat added); open triangles, Tat 32-62 (1 nm); solid circles, Tat 49-86 (1 nm). Results are expressed as the percentage of the first fraction collected (b1). Data are means ± SEM of three to seven experiments run in triplicate. B, Inset, Concentration dependency of the effect of Tat 32-62 (white bars) or Tat 49-86 (black bars) on [3H]ACh release from rat cortical synaptosomes. Results are expressed as the percentage of increase over basal release; data are means ± SEM of five experiments run in triplicate. *p < 0.05 versus respective control; **p < 0.01 versus respective control.
Finally, rat cortical synaptosomes were exposed to different partially overlapping amino acid fragments collectively spanning the entire Tat sequence. All peptides were applied at the final concentration of 1 nm. As shown in Figure 4, the [3H]ACh release was significantly increased only by Tat 61-80, whereas the other peptides were ineffective. The effects of four consecutive 20mers spanning the entire Tat sequence were also evaluated on the release of [3H]ACh from human cortical synaptosomes. Figure 4 shows that only the sequence Tat 41-60 was able to evoke [3H]ACh release, whereas the other sequences, including Tat 61-80, were much less effective or ineffective.
Figure 4.

Effects of eight peptidic fragments (spanning the entire Tat sequence) on the release of [3H]ACh from human and rat cortical synaptosomes. Human (empty bars) and rat (black bars) synaptosomes were exposed to peptides (1 nm). Results are expressed as the percentage of increase over basal. Data are means ± SEM of three to nine experiments run in triplicate. *p < 0.05 versus respective control.
Discussion
Tat interacts with human and rodent brain neuronal membranes, leading to depolarization and increase in cytosolic [Ca2+] (Sabatier et al., 1991; Cheng et al., 1998; Haughey et al., 1999, 2001). In nerve endings, such events could activate transmitter release (Berridge, 1998). To investigate this possibility, human and rat synaptosomes were exposed to Tat, and the release of various neurotransmitters was monitored.
The first finding of this work is that Tat can increase the basal release of ACh from both human and rat nerve endings. The characteristics of the technique used to monitor release (up-down superfusion of thin layers of synaptosomes) (Raiteri and Raiteri, 2000) permit the exclusion of indirect effects and lead to the conclusion that Tat acts by binding to sites located on cholinergic axon terminals.
Tat exhibits neuronal selectivity as determined by the protein-evoked release of ACh but was unable to elicit release of glutamate, aspartate, GABA, serine, and [3H]NE. These results indicate that, among the neuronal families examined, only cholinergic neurons possess Tat recognition sites, the activation of which leads to transmitter release. Note that the concentrations of Tat (<1 nm) able to significantly enhance the release of ACh approach those proposed to be present extracellularly during HIV-1 infection (Gurwell et al., 2001).
Buffering cytosolic Ca2+ ions with the Ca2+ chelator BAPTA almost completely prevented the releasing effect of Tat from both human and rat synaptosomes. Therefore, it is reasonable to assume that the release of ACh provoked by Tat occurs by vesicular exocytosis dependent on Ca2+ reaching the cytosol when Tat binds to cholinergic terminals. One way to increase cytosolic [Ca2+] is by entry of external Ca2+ ions through activated VSCCs. In human nerve endings, the releasing effect of Tat was about halved in low Ca2+ medium or after the addition of Cd2+ ions, which are known to block VSCCs. These findings indicate that external Ca2+ ions and VSCC activation contribute to the Tat-induced ACh release from human cerebrocortical nerve endings in a consistent manner. In contrast, neither the removal of external Ca2+ nor the addition of Cd2+ ions significantly decreased the release of ACh elicited by Tat from rat synaptosomes, indicating that this BAPTA-sensitive release depends exclusively on Ca2+ mobilization from intraterminal stores.
If intraterminal Ca2+ is the only source used by Tat to evoke ACh release from rat synaptosomes, ∼50% of the Tat-evoked release from human axon terminals depends on this source. Surprisingly, however, the mechanisms involved in the two internal Ca2+-dependent releases clearly differ between human and rat nerve endings.
It is well known that intraneuronal Ca2+ can be mobilized from endoplasmic reticulum, through the activation of IP3-sensitive and RY-sensitive receptors, as well as from mitochondria (for review, see Berridge, 1998; Mattson et al., 2000).
In human cholinergic nerve endings, Tat causes Ca2+ mobilization from IP3-sensitive stores because heparin and xestospongin C, two IP3r antagonists, strongly prevented the Tat-induced release of ACh. Intracellular IP3 production usually requires activation of membrane metabotropic receptors. We found that cholinergic human, but not rat, terminals are endowed with group I mGluRs, mediating the Tat-releasing effect, because antagonists at group I mGluRs potently prevented the Tat-evoked release of [3H]ACh. The finding is compatible with the idea that Tat binds to mGluRs located on human cortical cholinergic terminals and causes IP3-mediated Ca2+ release from intraterminal stores with consequent exocytosis of ACh.
Unlike for human nerve endings, Ca2+ mobilization from IP3-sensitive stores can be excluded when Tat releases ACh from rat terminals because IP3r antagonists were unable to significantly prevent the Tat effect. The release of ACh elicited by Tat from rat nerve terminals was instead almost abolished by dantrolene, a RYR antagonist, indicating that intraterminal Ca2+ in rat cholinergic nerve terminals is essentially mobilized after activation of RYRs. As mentioned previously, the endogenous agonist at RYRs is likely to be cADP-ribose (Galione et al., 1993; Lee, 2001). The involvement of this intracellular second messenger in the Tat effect on rat cholinergic terminals is supported by the findings that (1) Br-cADP-ribose almost totally prevented the Tat-mediated releasing effect, and (2) Tat increased cADP-ribose synthesis in rat cortical synaptosomes. Oligomycin, a mitochondrial ATPase blocker, had no effect on Tat-evoked ACh release, indicating that mitochondrial Ca2+ contributes little, if any, to the effect of Tat.
It had been proposed that Tat 1-72 can induce intracellular Ca2+ mobilization from IP3-sensitive stores in human fetal neurons and astrocytes (Haughey et al., 1999), whereas contradictory results are present in the literature concerning the rodent brain (Kruman et al., 1998). Recently, Haughey et al. (2001) reported that Tat did not affect the basal levels of intracellular calcium on its own but that it facilitated the glutamate-triggered calcium flux through an IP3-dependent mechanism.
Involvement of RYRs in Ca2+ mobilization by Tat has so far not been reported. Haughey et al. (2001) demonstrated that the Tat facilitatory effect on the glutamate-mediated Ca2+ mobilization in rat hippocampal neurons was RYR independent. The inhibition of the Tat-induced ACh release by dantrolene as well as by Br-cADP-ribose observed here indicates that activation of RYRs plays a significant role. However, it remains to be understood how Tat can stimulate cADP-ribose production and consequent Ca2+ mobilization from RY-sensitive stores. Tat can act both intracellularly or extracellularly (Ensoli et al., 1993; Milani et al., 1998; Lindgren et al., 2000), but an intracellular action of the protein seems unlikely in our system because entrapped heparin, a Tat scavenger (Rusnati et al., 1997), failed to prevent the releasing effect. If Tat binds extracellularly, it might be recognized by as yet unidentified sites on rat cholinergic neurons, the activation of which leads to cADP-ribose production.
The Tat-releasing effect on human synaptosomes is retained by the peptide fragments 32-62, 49-86, and 41-60, suggesting that Tat binds at human cortical cholinergic terminals through a sequence encompassing positions between 49 and 60. The three peptides contain the arginine-rich basic region (Tat 49-57) that not only is critical for efficient trans-activation (Ensoli et al., 1993), but is also required to induce depolarization in human fetal neurons (Nath et al., 1996), making it a potential candidate for the effect on ACh release. At variance with this possibility, the effect of Tat on rat cortical nerve endings maps to another region, because Tat 49-86, but not Tat 32-62, potentiated the release of ACh; this ability was retained by the peptide 61-80, but not by the flanking regions 51-70 and 71-80.
Conclusions
In conclusion, we found that the HIV-1 protein Tat can evoke release of ACh from human and rat cerebrocortex cholinergic axon terminals with remarkable neuronal selectivity. The mechanisms involved in the release from human terminals differ strikingly from those in rodent terminals. In human terminals, Tat behaves as an extremely potent agonist at IP3-linked mGluRs of group I, able to mobilize intraterminal Ca2+ and consequently to elicit exocytotic-like ACh release. The binding of Tat to rat terminals leads to ACh release that is also dependent on intraterminal Ca2+ but which is mobilized after RYR activation, possibly via their putative endogenous agonist cADP-ribose. If this is the case, Tat could be considered a potent activator of an as yet unidentified membrane receptor linked to the second messenger cADP-ribose. That Tat binds to different recognition sites on human versus rat cholinergic terminals is also indicated by the finding that the releasing effect of Tat in human terminals is mimicked by peptide fragments different from those mimicking it in rat terminals. Previously, we found that the HIV-1 coat protein gp120 potentiates the release of norepinephrine from human (Pittaluga et al., 1996) and rat (Pittaluga and Raiteri, 1994) cortical axon terminals. In contrast with Tat, gp120 acts, in both human and rat terminals, on glutamate NMDA receptors as a very potent agonist of the glycine site. The present data on Tat indicate that, although the protein elicits ACh release from both human and rat cholinergic terminals, the mechanisms are different and, therefore, the rodent model should be exploited with caution. Of note, the disease state of patients seems not to be involved in the species difference observed here, because data obtained from different patients did not differ significantly from each other. In contrast, species differences among presynaptic receptors mediating neurotransmitter release have been reported to occur in mammalian brains (Hoyer and Middlemiss, 1989).
The pathophysiological significance of the Tat effect on ACh release observed here is presently unknown. The Tat concentrations used are likely to be lower than those causing overt neurotoxicity. However, it must be noted that, once present in the brain extracellular fluid, Tat may not be easily inactivated and may, therefore, provoke release of ACh outside of the physiological range and with abnormal kinetic characteristics, potentially contributing to the cognitive disturbances characteristic of AIDS dementia.
Footnotes
This work was supported by grants from Istituto Superiore di Sanitá (Programma nazionale di ricerca sull'AIDS—Progetto “Patologia, clinica e terapia dell'AIDS”) and from Center of Excellence, University of Genoa (to M.R.). We thank AIDS Reagent Project and National Institute for Biological Standards and Control for the kind gift of Tat peptides and Tat 20mers, respectively. We thank Maura Agate for excellent assistance in preparing this manuscript and Fabio Longordo for skillful technical assistance.
Correspondence should be addressed to Dr. A. Pittaluga, Dipartimento di Medicina Sperimentale, Sezione di Farmacologia e Tossicologia, Università di Genova, Viale Cembrano 4, 16148 Genova, Italy. E-mail: pittalug@pharmatox.unige.it.
Copyright © 2003 Society for Neuroscience 0270-6474/03/236810-09$15.00/0
References
- Adle-Biassette H, Levy Y, Colombel M, Poron F, Natchev S, Keohane C, Gray F ( 1995) Neuronal apoptosis in HIV infection in adults. Neuropathol Appl Neurobiol 21: 218-227. [DOI] [PubMed] [Google Scholar]
- Berridge MJ ( 1998) Neuronal calcium signaling. Neuron 21: 13-26. [DOI] [PubMed] [Google Scholar]
- Bezzi P, Domercq M, Brambilla L, Galli R, Schols D, De Clercq E, Vescovi A, Bagetta G, Kollias G, Meldolesi J, Volterra A ( 2001) CXCR4-activated astrocyte glutamate release via TNFa: amplification by microglia triggers neurotoxicity. Nat Neurosci 4: 702-710. [DOI] [PubMed] [Google Scholar]
- Bradford MM ( 1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein dye binding. Anal Biochem 72: 248-254. [DOI] [PubMed] [Google Scholar]
- Brailoiu E, Miyamoto MD ( 2000) Inositol trisphosphate and cyclic adenosine diphosphate-ribose increase quantal transmitter release at frog motor nerve terminals: possible involvement of smooth endoplasmic reticulum. Neuroscience 95: 927-931. [DOI] [PubMed] [Google Scholar]
- Cheng J, Nath A, Knudsen B, Hochman S, Geiger JD, Ma M, Magnuson DSK ( 1998) Neuronal excitatory properties of human immunodeficiency virus type 1 Tat protein. Neuroscience 82: 97-106. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Bliss TV ( 1995) Memories of NMDA receptors and LTP. Trends Neurosci 18: 54-56. [PubMed] [Google Scholar]
- Ensoli B, Buonaguro L, Barillari G, Fiorelli V, Gendelman R, Morgan RA, Wingfield P, Gallo RC ( 1993) Release, uptake, and effects of extracellular human immunodeficiency virus type 1 Tat protein on cell growth and viral transactivation. J Virol 67: 277-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everall IP, Heaton RK, Marcotte TD, Ellis RJ, McCutchan JA, Atkinson JH, Grant I, Mallory M, Masliah E ( 1999) Cortical synaptic density is reduced in mild to moderate human immunodeficiency virus neurocognitive disorder. Brain Pathol 9: 209-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana G, Valenti R, Raiteri M ( 1997) gp120 can revert antagonism at the glycine site of NMDA receptors mediating GABA release from cultured hippocampal neurons. J Neurosci Res 49: 732-738. [DOI] [PubMed] [Google Scholar]
- Fossier P, Tauc L, Baux G ( 1999) Calcium transients and neurotransmitter release at an identified synapse. Trends Neurosci 22: 161-166. [DOI] [PubMed] [Google Scholar]
- Fox L, Alford M, Achim C, Mallory M, Masliah E ( 1997) Neurodegeneration of somatostatin-immunoreactive neurons in HIV encephalitis. J Neuropathol Exp Neurol 56: 360-368. [DOI] [PubMed] [Google Scholar]
- Gafni J, Munsch JA, Lam TH, Catlin MC, Costa LG, Molinski TF, Pessah IN ( 1997) Xestospongins: potent membrane permeable blockers of the inositol 1,4,5-trisphosphate receptor. Neuron 19: 723-733. [DOI] [PubMed] [Google Scholar]
- Galione A, White A, Willmott N, Turner M, Potter BVL, Watson SP ( 1993) cGMP mobilizes intracellular Ca2+ in sea urchin eggs by stimulating cyclic ADP-ribose synthesis. Nature 365: 456-459. [DOI] [PubMed] [Google Scholar]
- Garg RK ( 1999) HIV infection and seizures. Postgrad Med J 75: 387-390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemignani A, Paudice P, Pittaluga A, Raiteri M ( 2000) The HIV-1 coat protein gp120 and some of its fragments potently activate native cerebral NMDA receptors mediating neuropeptide release. Eur J Neurosci 12: 2839-2846. [DOI] [PubMed] [Google Scholar]
- Glass JD, Fedor H, Wesselingh SL, McArthur JC ( 1995) Immunocytochemical quantitation of human immunodeficiency virus in the brain: correlations with dementia. Ann Neurol 38: 755-762. [DOI] [PubMed] [Google Scholar]
- Graeff R, Lee HC ( 2002) A novel cycling assay for cellular cADP-ribose with nanomolar sensitivity. Biochem J 361: 379-384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruslin E, Descombes S, Psarropoulou C ( 1999) Epileptiform activity generated by endogenous acetylcholine during blockade of GABAergic inhibition in immature and adult rat hippocampus. Brain Res 835: 290-297. [DOI] [PubMed] [Google Scholar]
- Gurwell JA, Nath A, Sun Q, Zhang J, Martin KM, Chen Y, Hauser KF ( 2001) Synergistic neurotoxicity of opioids and human immunodeficiency virus-1 Tat protein in striatal neurons in vitro. Neuroscience 102: 555-563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselmo ME, Bower JM ( 1993) Acetylcholine and memory. Trends Neurosci 16: 218-222. [DOI] [PubMed] [Google Scholar]
- Haughey NJ, Holden CP, Nath A, Geiger JD ( 1999) Involvement of inositol 1,4,5-trisphosphate-regulated stores of intracellular calcium in calcium dysregulation and neuron cell death caused by HIV-1 protein Tat. J Neurochem 73: 1363-1374. [DOI] [PubMed] [Google Scholar]
- Haughey NJ, Nath A, Mattson MP, Slevin JT, Geiger JD ( 2001) HIV-1 Tat through phosphorylation of NMDA receptors potentiates glutamate excitotoxicity. J Neurochem 78: 457-467. [DOI] [PubMed] [Google Scholar]
- Higashida H, Hashii M, Yokoyama S, Hoshi N, Asai K, Kato T ( 2001) Cyclic ADP-ribose as a potential second messenger for neuronal Ca2+ signaling. J Neurochem 76: 321-331. [DOI] [PubMed] [Google Scholar]
- Hoyer D, Middlemiss DN ( 1989) Species differences in the pharmacology of terminal 5-HT autoreceptors in mammalian brain. Trends Pharmacol Sci 10: 130-132. [DOI] [PubMed] [Google Scholar]
- Hudson L, Liu J, Nath A, Jones M, Raghavan R, Narayan O, Male D, Everall I ( 2000) Detection of the human immunodeficiency virus regulatory protein tat in CNS tissues. J Neurovirol 6: 145-155. [DOI] [PubMed] [Google Scholar]
- Kaul M, Lipton SA ( 1999) Chemokines and activated macrophages in HIV gp120-induced neuronal apoptosis. Proc Natl Acad Sci USA 96: 8212-8216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul M, Garden GA, Lipton SA ( 2001) Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature 410: 988-994. [DOI] [PubMed] [Google Scholar]
- Kruman II, Nath A, Mattson MP ( 1998) HIV-1 protein Tat induces apoptosis of hippocampal neurons by a mechanism involving caspase activation, calcium overload, and oxidative stress. Exp Neurol 154: 276-288. [DOI] [PubMed] [Google Scholar]
- Lannuzel A, Lledo PM, Lamghitnia HO, Vincent JD, Tardieu M ( 1995) HIV-1 envelope proteins gp120 and gp160 potentiate NMDA-induced [Ca2+]i increase, alter [Ca2+]i homeostasis and induce neurotoxicity in human embryonic neurons. Eur J Neurosci 7: 2285-2293. [DOI] [PubMed] [Google Scholar]
- Lee HC ( 2001) Physiological functions of cyclic ADP-ribose and NAADP as calcium messengers. Annu Rev Pharmacol Toxicol 41: 317-345. [DOI] [PubMed] [Google Scholar]
- Lindgren M, Hällbrink M, Prochiantz A, Langel Ü ( 2000) Cell-penetrating peptides. Trends Pharmacol Sci 21: 99-103. [DOI] [PubMed] [Google Scholar]
- Magnuson DSK, Knudsen BE, Geiger JD, Brownstone RM, Nath A ( 1995) Human immunodeficiency virus type 1 Tat activates non-N-methyl-d-aspartate excitatory amino acid receptors and causes neurotoxicity. Ann Neurol 37: 373-380. [DOI] [PubMed] [Google Scholar]
- Masliah E, Achim CL, Ge N, DeTeresa R, Terry RD, Wiley CA ( 1992) Spectrum of human immunodeficiency virus-associated neocortical damage. Ann Neurol 32: 321-329. [DOI] [PubMed] [Google Scholar]
- Masliah E, Heaton RK, Marcotte TD, Ellis RJ, Wiley CA, Mallory M, Achim CL, McCutchan JA, Nelson JA, Atkinson JH, Grant I, Chandler JL, Wallace MR, Spector SA, Jernigan T, Hesselink J, Hansen L, Abramson I, Masys D ( 1997) Dendritic injury is a pathological substrate for human immunodeficiency virus-related cognitive disorders. Ann Neurol 42: 963-972. [DOI] [PubMed] [Google Scholar]
- Mattson MP, LaFerla FM, Chan SL, Leissring MA, Shepel PN, Geiger JD ( 2000) Calcium signaling in the ER: its role in neuronal plasticity and neurodegenerative disorders. Trends Neurosci 23: 222-229. [DOI] [PubMed] [Google Scholar]
- McGaugh JL ( 1989) Involvement of hormonal and neuromodulatory systems in the regulation of memory storage. Annu Rev Neurosci 12: 255-287. [DOI] [PubMed] [Google Scholar]
- Milani D, Mazzoni M, Zauli G, Mischiati C, Gibellini D, Giacca M, Capitani S ( 1998) HIV-1 Tat induces tyrosine phosphorylation of p125FAK and its association with phosphoinositide 3-kinase in PC12 cells. AIDS 12: 1275-1284. [DOI] [PubMed] [Google Scholar]
- Mollace V, Nottet HSLM, Clayetee P, Turco MC, Muscoli C, Salvemini D, Perno CF ( 2001) Oxidative stress and neuroAIDS: triggers, modulators and novel antioxidants. Trends Neurosci 24: 411-416. [DOI] [PubMed] [Google Scholar]
- Mothet JP, Fossier P, Meunier FM, Stinnakre J, Tauc L, Baux G ( 1998) Cyclic ADP-ribose and calcium-induced calcium release regulate neurotransmitter release at a cholinergic synapse of Aplysia. J Physiol (Lond) 507: 405-414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller WEG, Schröder HC, Ushijima H, Dapper J, Bormann J ( 1992) Gp120 of HIV-1 induces apoptosis in rat cortical cell cultures: prevention by memantine. Eur J Pharmacol Mol Pharmacol Sect 226: 209-214. [DOI] [PubMed] [Google Scholar]
- Nath A, Padua RA, Geiger JD ( 1995) HIV-1 coat protein gp120-induced increases in levels of intrasynaptosomal calcium. Brain Res 678: 200-206. [DOI] [PubMed] [Google Scholar]
- Nath A, Psooy K, Martin C, Knudsen B, Magnuson DSK, Haughey N, Geiger JD ( 1996) Identification of a human immunodeficiency virus type 1 Tat epitope that is neuroexcitatory and neurotoxic. J Virol 70: 1475-1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual Sedano B, Iranzo A, Marti Fabregas J, Domingo P, Escartin A, Fuster M, Barrio JL, Sambeat MA ( 1999) Prospective study of new-onset seizures in patients with human immunodeficiency virus infection: etiologic and clinical aspects. Arch Neurol 56: 609-612. [DOI] [PubMed] [Google Scholar]
- Pattarini R, Pittaluga A, Raiteri M ( 1998) The human immunodeficiency virus-1 envelope protein gp120 binds through its V3 sequence to the glycine site of N-methyl-d-aspartate receptors mediating noradrenaline release in the hippocampus. Neuroscience 87: 147-157. [DOI] [PubMed] [Google Scholar]
- Petito CK, Roberts B ( 1995) Evidence of apoptotic cell death in HIV encephalitis. Am J Pathol 146: 1121-1130. [PMC free article] [PubMed] [Google Scholar]
- Pittaluga A, Raiteri M ( 1994) HIV-1 envelope protein gp120 potentiates NMDA-evoked noradrenaline release by a direct action at rat hippocampal and cortical noradrenergic nerve endings. Eur J Neurosci 6: 1743-1749. [DOI] [PubMed] [Google Scholar]
- Pittaluga A, Pattarini R, Severi P, Raiteri M ( 1996) Human brain N-methyl-d-aspartate receptors regulating noradrenaline release are positively modulated by HIV-1 coat protein gp120. AIDS 10: 463-468. [DOI] [PubMed] [Google Scholar]
- Pittaluga A, Bonfanti A, Raiteri M ( 1997) Differential desensitization of ionotropic non-NMDA receptors having distinct neuronal location and function. Naunyn Schmiedeb Arch Pharmacol 356: 29-38. [DOI] [PubMed] [Google Scholar]
- Pittaluga A, Bonfanti A, Raiteri M ( 2000) Somatostatin potentiates NMDA receptor function via activation of InsP3 receptors and PKC leading to removal of the Mg2+ block without depolarization. Br J Pharmacol 130: 557-566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raiteri L, Raiteri M ( 2000) Synaptosomes still viable after 25 years of super-fusion. Neurochem Res 25: 1265-1274. [DOI] [PubMed] [Google Scholar]
- Raiteri L, Stigliani S, Zedda L, Raiteri M, Bonanno G ( 2002) Multiple mechanisms of transmitter release evoked by 'pathologically' elevated extracellular [K+]: involvement of transporter reversal and mitochondrial calcium. J Neurochem 80: 706-714. [DOI] [PubMed] [Google Scholar]
- Raiteri L, Bonanno G, Giovedì S, Benfenati F, Raiteri M ( 2003) Cellular mechanisms of the acute increase of glutamate release induced by nerve growth factor in rat cerebral cortex. Neuropharmacology 44: 390-402. [DOI] [PubMed] [Google Scholar]
- Raiteri M, Bonanno G, Marchi M, Maura G ( 1984) Is there a functional linkage between neurotransmitter uptake mechanisms and presynaptic receptors? J Pharmacol Exp Ther 231: 671-677. [PubMed] [Google Scholar]
- Raiteri M, Sala R, Fassio A, Rossetto O, Bonanno G ( 2000) Entrapping of impermeant probes of different size into nonpermeabilized synaptosomes as a method to study presynaptic mechanisms. J Neurochem 74: 423-431. [DOI] [PubMed] [Google Scholar]
- Rusnati M, Coltrini D, Oreste P, Zoppetti G, Albini A, Noonan D, d'Adda di Fagagna F, Presta M ( 1997) Interaction of HIV-1 Tat protein with heparin. J Biol Chem 272: 11313-11320. [DOI] [PubMed] [Google Scholar]
- Sabatier J-M, Vives E, Mabrouk K, Benjouad A, Rochat H, Duval A, Hue B, Bhraoui E ( 1991) Evidence for neurotoxic activity of tat from human immunodeficiency virus type 1. J Virol 65: 961-967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savio T, Levi G ( 1993) Neurotoxicity of HIV coat protein gp120, NMDA receptors, and protein kinase C: a study with rat cerebellar granule cell cultures. J Neurosci Res 34: 265-272. [DOI] [PubMed] [Google Scholar]
- Ushijima H, Kunisada T, Schröder HC, Klöcking P, Müller WEG ( 1993) HIV 1-gp120 and NMDA differentially induce protein kinase C translocation in rat primary neuronal cultures. J Acq Immun Defic Syndr 6: 339-343. [PubMed] [Google Scholar]
- Valle LD, Croul S, Morgello S, Amini S, Rappaport J, Khalili K ( 2000) Detection of HIV-1 Tat and JCV capsid protein, VP1, in AIDS brain with progressive multifocal leukoencephalopathy. J Neurovirol 6: 221-228. [DOI] [PubMed] [Google Scholar]
- Walseth TF, Lee HC ( 1993) Synthesis and characterization of antagonists of cyclic ADP ribose-induced Ca2+ release. Biochim Biophys Acta 1178: 235-242. [DOI] [PubMed] [Google Scholar]
- Walseth TF, Aarhus R, Zeleznikar RJ Jr, Lee HC ( 1991) Determination of endogenous levels of cyclic ADP-ribose in rat tissues. Biochim Biophys Acta 1094: 113-120. [DOI] [PubMed] [Google Scholar]
- Xin KQ, Hamajima K, Hattori S, Cao XR, Kawamoto S, Okuda K ( 1999) Evidence of HIV type 1 glycoprotein 120 binding to recombinant N-methyl-d-aspartate receptor subunits expressed in a baculovirus system. AIDS Res Hum Retroviruses 15: 1461-1467. [DOI] [PubMed] [Google Scholar]