

Abstract
Apoptotic pathways and DNA synthesis are activated in neurons in the brains of individuals with Alzheimer disease (AD). However, the signaling mechanisms that mediate these events have not been defined. We show that expression of familial AD (FAD) mutants of the amyloid precursor protein (APP) in primary neurons in culture causes apoptosis and DNA synthesis. Both the apoptosis and the DNA synthesis are mediated by the p21 activated kinase PAK3, a serine-threonine kinase that interacts with APP. A dominant-negative kinase mutant of PAK3 inhibits the neuronal apoptosis and DNA synthesis; this effect is abolished by deletion of the PAK3 APP-binding domain or by coexpression of a peptide representing this binding domain. The involvement of PAK3 specifically in FAD APP-mediated apoptosis rather than in general apoptotic pathways is suggested by the facts that a dominant-positive mutant of PAK3 does not alone cause neuronal apoptosis and that the dominant-negative mutant of PAK3 does not inhibit chemically induced apoptosis. Pertussis toxin, which inactivates the heterotrimeric G-proteins Go and Gi, inhibits the apoptosis and DNA synthesis caused by FAD APP mutants; the apoptosis and DNA synthesis are rescued by coexpression of a pertussis toxin-insensitive Go. FAD APP-mediated DNA synthesis precedes FAD APP-mediated apoptosis in neurons, and inhibition of neuronal entry into the cell cycle inhibits the apoptosis. These data suggest that a normal signaling pathway mediated by the interaction of APP, PAK3, and Go is constitutively activated in neurons by FAD mutations in APP and that this activation causes cell cycle entry and consequent apoptosis.
Keywords: Alzheimer disease, apoptosis, cell cycle, amyloid precursor protein, p21 activated kinase, heterotrimeric G-proteins, APP intracellular domain
Introduction
All individuals with Alzheimer disease (AD) experience a progressive loss of cognitive function, resulting from selective neurodegeneration. Alzheimer disease can occur as a “sporadic” event, it can result from the possession of an extra copy of chromosome 21 (Down syndrome), or it can be caused by mutations in the amyloid precursor protein (APP) gene on chromosome 21 or in the presenilin genes on chromosomes 1 and 14.
The mechanism by which neurons die in the disease remains to be defined. Failure of regulation of the cell cycle has been observed in AD brain (for review, see Arendt, 2002). Notably, ectopic expression of cdc2, cdk4, p16, Ki-67, cyclin B1, and cyclin D has been reported in pathologically affected or vulnerable neurons in AD brain (Liu et al., 1995; Smith and Lippa, 1995; Vincent et al., 1996, 1997; Arendt et al., 1996; Vincent et al., 1996, 1997; McShea et al., 1997; Busser et al., 1998). Busser et al. (1998) found abnormal appearance of cell cycle markers in regions of AD brain where cell death is extensive, and Chow et al. (1998) found increases in expression of genes encoding cell cycle proteins in single neurons in late-stage relative to early-stage AD brain. A number of the cell cycle regulators have been detected in vulnerable neurons before lesion formation (Kondratick and Vandre, 1996; Busser et al., 1998; Vincent et al., 1998). Patrick et al. (1999) have shown that p25, a truncated form of p35, the regulatory subunit of Cdk5, is increased in AD brain. One of the consequences of the aberrant expression of cell cycle proteins for the neuropathology of AD appears to be that vulnerable neurons in AD brain re-enter the cell cycle. Yang et al. (2001) have demonstrated that a significant number of neurons in affected regions of AD brain have undergone full or partial DNA replication, showing that they have completed the S phase.
Activation of cell cycle proteins in neurons can, in some cases, lead to a form of cell suicide called apoptosis (for review, see Copani et al., 2001). The notion that apoptosis contributes to the neuropathology of AD was first proposed by Su et al. (1994), when they reported evidence for DNA fragmentation in neurons in AD brain; this observation subsequently was confirmed by others. Notably, Guo et al. (1998) found that levels of a marker of apoptosis, Par-4 (prostate apoptosis response-4) protein, are elevated in vulnerable neurons in AD brain. In recent years, several groups have reported the presence of activated caspase 3 and downstream caspases in AD brain (for review, see Roth, 2001). These findings suggest strongly that apoptotic pathways may be activated in AD.
Overexpression of wild-type APP in neurons causes apoptosis (Bursztajn et al., 1998; Nishimura et al., 1998; McPhie et al., 2001), and familial AD (FAD) mutants of APP cause DNA fragmentation and apoptosis in cell lines and neurons (Yamatsuji et al., 1996; Zhao et al., 1998; McPhie et al., 2001). These data suggest that APP may participate in the apoptotic events that have been observed in AD brain. We have used herpes simplex virus (HSV)-mediated delivery of genes into neurons to demonstrate that FAD mutants of APP cause apoptosis in primary neurons and that FAD APP-mediated neuronal apoptosis is more severe than that caused by overexpression of wild-type APP. Infection of neurons with FAD APP viral vectors causes DNA synthesis also to occur in neurons. We show that both the apoptosis and the DNA synthesis are mediated by the p21 activated kinase (PAK) 3, which interacts with APP to activate these pathways. Treatment with pertussis toxin blocks FAD APP-mediated apoptosis and DNA synthesis, implicating Go in the pathways as well. Sustained exposure of neurons to a ligand mimetic for APP, the antibody 22C11, causes both apoptosis and DNA synthesis. Neuronal DNA synthesis precedes apoptosis in neurons infected with HSVFAD APP vectors; and inhibition of neuronal entry into the cell cycle inhibits the apoptosis. These data suggest the existence of a neuronal signaling pathway involving APP, PAK3, and Go that becomes constitutively activated and causes cell cycle entry and apoptosis after overexpression of APP or FAD mutants of APP.
Materials and Methods
Plasmid construction. All plasmid constructs were made in the pHSVPr-pUC vector using standard techniques and were verified by sequence analysis.
Apoptosis assays. Primary cultures were plated on poly-d-lysine-coated glass or ACLAR (Ted Pella, Inc., Redding, CA) coverslips. Five days after plating, neurons were infected with the appropriate viruses at a multiplicity of infection (MOI) of one per virus; 16 hr later (except in the cases of HSV-C57 and HSV-C31 infections, in which the cells were processed 6 hr after infection), the cells were fixed for 20 min in freshly made 4% paraformaldehyde. Bisbenzamide assays were performed as described previously (Bursztajn et al., 1998). Ten random fields of 200-300 cells each were analyzed for each condition. The number of cells with condensed nuclei relative to the total number of cells per field was calculated and expressed as a percentage. One-way ANOVA and post hoc unpaired t tests with the Bonferroni multiple comparisons test were used for data analysis. For a number of experiments, apoptosis was also measured using the terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) assay, as described (Bursztajn et al., 1998). In every case, the data obtained using the TUNEL assay were identical to those obtained using bisbenzamide staining. Three replicates of each experiment were done; a representative experiment is shown in each figure. Cells with condensed nuclei were deemed to be neuronal, on the basis of their morphology.
DNA synthesis assays. BrdU (Sigma, St. Louis, MO) was added to the neuronal cultures 5 d after plating at a final concentration of 10 μm at the time of infection with HSV vectors. Sixteen hours later (except in the cases of HSV-C57 and HSV-C31 infections, in which the cells were processed 6 hr after infection), cells were fixed in cold 70% ethanol for 30 min and processed according to the protocol provided with the Zymed (San Francisco, CA) BrdU labeling kit. Ten random fields of cells were counted per condition, and the number of BrdU-positive cells was expressed as a percentage of the total number of cells. Data analysis was performed as above. Three replicates of each experiment were done; a representative experiment is shown in each figure. Staining of the infected cultures with an antibody to the neuron-specific antigen NeuN has shown that ∼20% of the cells in the cultures at the time of infection are non-neuronal and that the induction of DNA synthesis occurs selectively in neurons (data not shown).
Antibodies and immunoblots. A goat polyclonal antibody to the N terminus of PAK3 was purchased from Santa Cruz Biotechnology (Santa Cruz, CA), and a rabbit polyclonal to mPAK3 was purchased from Up-state Biotechnology (Lake Placid, NY). The polyclonal antibody 369 (Buxbaum et al., 1990) was raised against a region from amino acids 645-694 of human APP-695 and was a generous gift of Dr. Sam Gandy (Farber Institute of Neuroscience, Philadelphia, PA). Antibody C8 was raised against the C terminal 20 amino acids of APP and was a generous gift of Dr. Dennis Selkoe (Harvard Medical School, Boston, MA). Anti-rab5A(S-19) and anti-rab7(C19) were from Santa Cruz. Antibody 6E10 was from Signet (Dedham, MA). Anti-rab11 was from Transduction Laboratories (Lexington, KY). The anti-myc antibody 9E10 and the anti-APP antibody 22C11 were from Roche Products, Hertforshire, UK. For immunoblots demonstrating expression of transgenes from HSV vectors, infected primary neurons were homogenized in lysis buffer (100 mm Tris-HCl, 20 mm NaCl, 10 mm EDTA, 10 mm EGTA, and 1% SDS with these protease inhibitors: 10 μg/ml leupeptin, 10 μg/ml aprotinin, 1 mm Na3VO4, 1 mm PMSF, 1 mm benzamidine, and 10 mm β-glycerol phosphate), and proteins in the lysates were separated by SDS-PAGE. Immunoblots were performed as described previously (McPhie et al., 1997).
Isolation of PAK3 cDNAs.35S-APP-C100 was prepared in vitro as described (Kozlowski et al., 1992), except that transcription and translation reagents were purchased from Ambion (Austin, TX). After the incubation, N-acetyl-d-glucosamine (Sigma, St. Louis, MO) was added to each reaction at a final concentration of 10 mm. The 35S-APP-C100 [1.2 ml lysate/50 ml binding buffer (see below)] was used as a ligand to screen a rat fetal [embryonic day 18 (E18)] brain expression cDNA library (Neve et al., 1987) in λgt11. After a wash in binding buffer [in mm: 50 Tris, pH 7.7, 50 NaCl, 2 MgCl2, and 1 dithiothreitol (DTT)], filters containing library proteins were incubated with 35S-APP-C100 at 3 nM for 2 hr at 4°C. After three 3 min washes at 4°C, consisting of (1) binding buffer, (2) binding buffer plus 0.1% NP-40, and (3) binding buffer, filters were air-dried and exposed to X-OMAT RP film for 6-14 d. Positive plaques were isolated and taken through six further rounds of binding and purification, yielding finally a single positive plaque-pure cDNA clone, which was used for additional screens of the cDNA library to isolate the full-length coding sequence.
Coimmunoprecipitation. Primary cortical neurons were seeded at 4 × 106 viable cells per dish in 60 mm poly-D-lysine-coated dishes. Neurons were infected with the appropriate viral vectors; 16 hr after infection, the cells were lysed in the following buffer: 50 mm Tris, pH 8.0, 100 mm NaCl, 3 mm MgCl2, 10% glycerol, 1% IGEPAL-CA630 (Sigma), and 0.5% sodium deoxycholate with the following protease and phosphatase inhibitors: 50 mm NaF, 1 mm PMSF, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 1 mm Na3VO4, 1 mm iodoacetamide, 1 mm benzamidine, and 10 mm β-glycerol phosphate. The lysates were homogenized in a Dounce homogenizer, 10 times on ice, and then allowed to sit on ice for 1 hr. Lysates were then centrifuged at 16,000 × g to remove insoluble material. An aliquot from each lysate was set aside for immunoblot analysis of protein expression. Lysates were precleared with protein A beads in a 1:1 slurry in lysis buffer for 1 hr, after which antibody 369 or rabbit preimmune serum was added, and the mixture was tumbled overnight at 4°C. Protein A was then added for 1 hr to precipitate the immune complexes. The immune complexes were washed three times with lysis buffer. The proteins in the samples were separated by SDS-PAGE. Immunoblots were performed as described previously (McPhie et al., 1997).
Immunocytochemistry and confocal microscopy. HSV-infected cells were fixed for 45 min in freshly made 4% paraformaldehyde, rinsed with PBS, blocked for 30 min at room temperature in PBS with 0.1% Triton X-100 and 5% normal goat serum, and incubated for 2 hr at room temperature with primary antibody in PBS with 5% normal goat serum. Cells were rinsed with PBS and incubated for 90 min at room temperature with secondary antibody (Cappel, Cochranville, PA) in PBS with 5% normal goat serum. After a final rinse in PBS, cells on coverslips were mounted onto glass microscope slides using Gel/Mount (Biomeda, Hayward, CA). Omission of the primary antibodies resulted in only nonpunctate background fluorescence. Confocal images were generated using a Leica (Nussloch, Germany) TCS-NT laser confocal microscope. In double- and triple-labeling immunofluorescence experiments, bleed-through was controlled for by observing the absence of fluorescence of a given fluorochrome-labeled secondary antibody when it was illuminated using the alternative fluorochrome excitation wavelength.
Fractionation of endosomes. The fractionation of endosomes was based on the method of Seemann et al. (1997). Briefly, 10 15 cm plates of primary neurons were rinsed twice with ice-cold PBS. The cells in each dish were scraped up in 1 ml of PBS, after which the cells from the 10 dishes were combined and pelleted at 1300 × g for 10 min. The pellet was resuspended in 1.5 ml of homogenization buffer (HB) plus protease inhibitors (250 mm sucrose, 3 mm imidazole, pH 7.4, 1 mm EDTA plus 1 mm PMSF, 1 mm sodium orthovanadate, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 1 mm benzamidine, and 10 mm β-glycerolphosphate). The pellet was then homogenized 10 strokes in a Dounce homogenizer and passed three times through a 25 gauge needle. After a 1300 × g spin, the postnuclear supernatant (PNS) was adjusted to 40.6% sucrose, 3 mm imidazole, pH 7.4, with 62% sucrose and placed at the bottom of a Beckman Instruments (Fullerton, CA) SW41 tube. This was overlaid successively with 4 ml 35% sucrose, 3 mm imidazole, pH 7.4, 3 ml of 25% sucrose, 3 mm imidazole, pH 7.4, and 2 ml of HB. All sucrose solutions contained the same protease inhibitor mixture as the HB. The step gradient was centrifuged at 160,000 × g for 90 min. Bands were pulled at the following interfaces: 25%-HB (membranes enriched in late endosomes, endosome carrier vesicles), 25-35% (membranes enriched in early endosomes), and 35-40.6% (heavy membranes). Twenty micrograms of each sample were loaded onto 10-20% Tris-glycine SDS-PAGE gels. After transfer to polyvinylidene difluoride membranes, replicate blots of the three fractions were probed with the antibodies C-8 (1:6000), anti-mPAK3 (1:1000), anti-rab5 (1:1000), and anti-rab11 (1:1000), and the blots were visualized with enhanced chemiluminescence as described in McPhie et al. (1997).
RNA blot analysis and in situ hybridization histochemistry. Methods for doing RNA blots have been described previously (Neve et al., 1987). For in situ hybridization histochemistry, tissues were fresh-frozen and cut in 12-μm-thick sections on a cryostat. Sections were fixed in buffered 4% paraformaldehyde for 10 min at room temperature, rapidly air-dried, and stored desiccated at -70°C. At the time of use, sections were blocked with 0.1 m glycine, rinsed, and acetylated with 0.25% acetic anhydride in 0.1 m triethanolamine, pH 8.0. Sections were washed in 2× SSC and delipidated by treatment with ethanol and chloroform. Sections were then partially rehydrated and incubated overnight at 60°C with 10,000 cpm/μl 35S-labeled riboprobes (synthesized using a template representing bp 24-230 of the PAK3 cDNA) in a solution containing 50% form-amide, 10% dextran sulfate, 20× Denhardt's solution, 300 μg/ml sheared salmon sperm DNA, 150 μg/ml tRNA, 2× SSC, and 20 mm β-mercaptoethanol. After hybridization, sections were treated with RNase A and were washed with increasing stringency (final stringency: 0.1× SSC at 60°C). Sections were dried, dipped in a 1:1 dilution of Eastman Kodak (Rochester, NY) NTB2 emulsion with water, and developed after 5 weeks. Some of the sections were exposed to Amersham Biosciences (Arlington Heights, IL) βMax Hyperfilm. At least four independent sets of hybridizations were performed. Controls (not shown) included hybridizations with sense probes.
Chemically induced apoptosis. The staurosporine and etoposide experiments were performed as described by Bursztajn et al. (1998).
Protein kinase assays. PAK kinase assays were performed as described by Joneson et al. (1998) with the following modifications. Briefly, 16 hr after infection, neurons were lysed in lysis buffer (10 mm HEPES, pH 7.5, 150 mm NaCl, 10% glycerol, 0.6% Triton X-100 plus the following protease and phosphatase inhibitors: 1 mm Na3VO4, 50 mm NaF, 1 mm benzamidine, 1 mm PMSF, 10 μg/ml aprotinin, and 10 μg/ml leupeptin). After the lysates were precleared with protein G (Pierce, Rockford, IL), anti-myc antibody 9E10 was added to precipitate the myc-PAKs, and the lysates were incubated overnight at 4°C with tumbling. Protein G was then added to precipitate the immune complexes, and the mixture was tumbled for 1 hr. The beads were washed four times with lysis buffer and once with kinase buffer (20 mm HEPES, pH 7.6, 20 mm MgCl2, 20 mm β-glycerol phosphate, 0.1 mm Na3VO4, and 2 mm DTT). Forty microliters of final kinase mixture (20 μm ATP, 10 μCi 33P-ATP, and 3 μg of histone H4 in kinase buffer) was added to each tube. After a 30 min incubation at 37°C, the reactions were terminated by the addition of Laemmli Sample Buffer and were boiled for 5 min before electrophoresis on 10-20% Tris-HCl gels (Bio-Rad, Hercules, CA). The gels were fixed, dried, and exposed to film to visualize incorporated radioactivity.
Pertussis toxin treatment. Pertussis toxin (RBI, Natick, MA) was added to the cultures at a final concentration of 100 ng/ml 2 hr before infection with HSV recombinants. The construction of a pertussis toxin-insensitive mutant of Gαo (Gαo*) cDNA in which a serine replaces a cysteine four residues from the C terminus has been described previously (Taussig et al., 1992). This Gαo* cDNA was generously provided by Dr. Ronald Taussig (University of Michigan, Ann Arbor, MI).
Mimosine, deferoxamine, and pan-caspase inhibitor treatment. Mimosine and deferoxamine (Sigma) were added at a final concentration of 400 μm and 1 mm, respectively, at the time of HSV infection. BOC-Asp(OMe)-fluoromethyl ketone (Boc-D-FMK; Calbiochem, La Jolla, CA) was added at a final concentration of 50 μm at the time of infection.
Results
HSV-mediated expression of FAD mutants of APP causes neuronal apoptosis and DNA synthesis
We prepared replication-defective HSV vectors expressing APP-695 and -751, the Swedish mutant of APP-695 (HSV-SWE695), and the V642I mutant of APP-751 (HSV-V642I-751) as described (McPhie et al., 1997). Primary E18 rat cortical cultures at 5d in vitro were infected with the viruses at an MOI of 1 per virus. Sixteen hours later, the infected cells were harvested, and their proteins were analyzed by immunoblot analysis to confirm equal expression of the transgenes (Fig. 1A, inset). HSV expressing Escherichia coli β-galactosidase (HSV-Lac) was used as a control.
Figure 1.
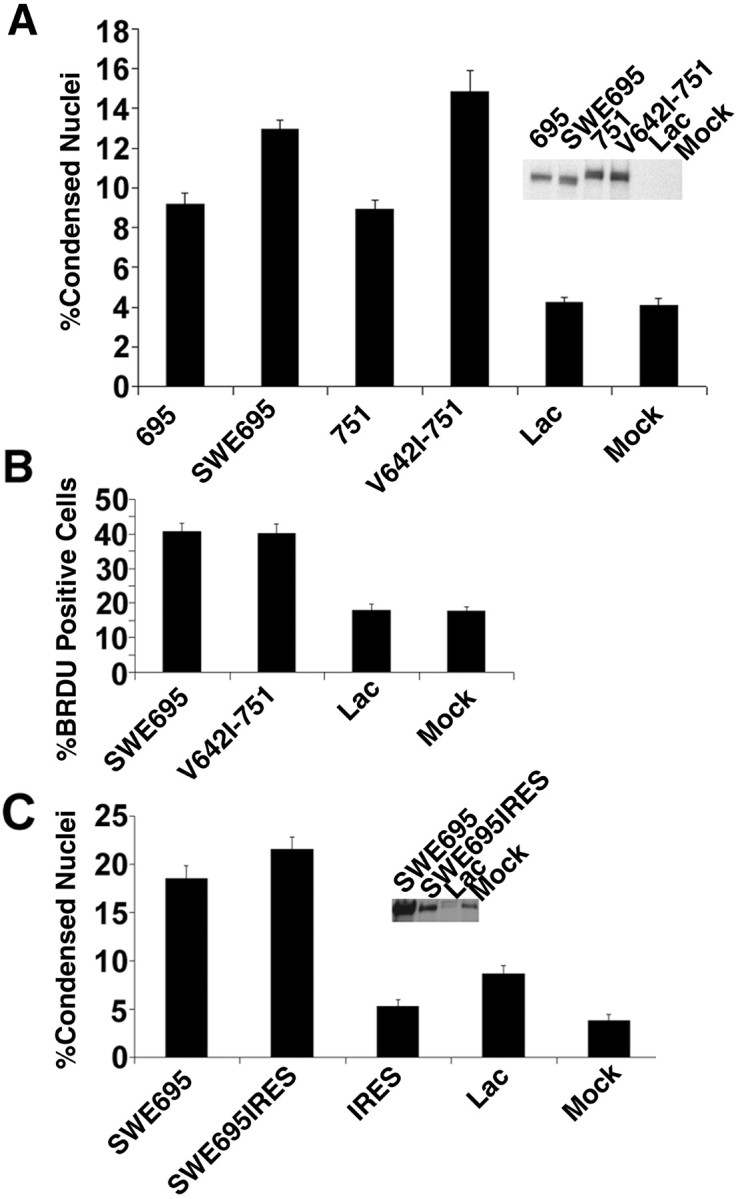
HSV-mediated expression of FAD mutants of APP causes neuronal apoptosis and DNA synthesis; these effects are independent of expression level. A, Quantitative analysis of APP-mediated apoptosis in neurons demonstrates that FAD APP-mediated apoptosis is significantly more severe than that mediated by wild-type APP. Data are expressed as percentages (mean ± SEM). Significant between-group differences were confirmed by a single-factor ANOVA (F(5,53) = 32.9; p < 0.0001). Post hoc t tests were done using Bonferroni multiple comparisons test. Significant differences were seen between the following groups: 695 versus lac or mock, 695 versus SWE 695, 751 versus lac or mock, 751 versus V642I-751 (p < 0.01 for all). All other relevant comparisons were not significant (NS). The inset shows an immunoblot of expression of wild-type and FAD APPs expressed by the different HSV vectors. B, Quantitative analysis of DNA synthesis in cortical neurons expressing FAD APPs shows a significant increase in the number of BrdU-positive nuclei relative to controls. Significant between-group differences were confirmed by a single-factor ANOVA (F(3,36) = 44.9; p < 0.0001). Post hoc t tests were done using the Bonferroni multiple comparisons test. Significant differences were seen between SWE 695 versus lac or mock and V642I-751 versus lac or mock (p < 0.001). All other relevant comparisons were NS. C, Equal titers of either high- or low-expressing HSV-APP vectors cause the same level of neuronal apoptosis 16 hr after infection. There was no significant difference in the number of condensed nuclei between cultures infected with high-expressing HSV-SWE695 or the low-expressing vector HSV-SWE695-IRESGFP (SWE695IRES). Significant between-group differences were confirmed by a single-factor ANOVA (F(4,44) = 70.4; p < 0.0001). Post hoc t tests were done using Bonferroni multiple comparisons test. Significant differences were seen between SWE695 and lac or mock, and between SWE695-IRES and lac or mock (p < 0.001 for all comparisons). There was no significant difference in the number of condensed nuclei among the HSV-IRESGFP (IRES), HSV-Lac, and mock groups.
Cortical cells infected with the HSV vectors or mock-infected cells were fixed 16 hr after infection, and bisbenzamide staining was used to detect apoptotic nuclei. The results of a representative experiment were quantified and are shown in Figure 1A. Cells infected with HSV vectors expressing the Swedish mutant of APP-695 or a London mutant (V642I) of APP-751 showed a significant increase over control (∼13 and ∼15% apoptotic nuclei, respectively, vs ∼4%) in the number of apoptotic cells. Cells infected with HSV vectors expressing wild-type APP-695 or APP-751 gave an intermediate phenotype, with ∼9% apoptotic nuclei. This intermediate apoptotic effect of overexpression of wild-type APP is consistent with our previous data (Bursztajn et al., 1998).
In mammalian neurons, certain types of death, including apoptosis, have been linked to the re-expression of cell cycle markers (for review, see Copani et al., 2001). Moreover, Yang et al. (2001) have demonstrated that a significant number of neurons in vulnerable regions of AD brain have undergone full or partial DNA replication, showing that they have completed the S phase. Therefore, we tested whether FAD APPs could cause entry of neurons into the cell cycle, as evidenced by DNA synthesis. This turned out to be the case, as shown in Figure 1B. Primary neurons were infected with HSV expressing the Swedish or the London mutant of APP in the presence of BrdU, and 16 hr later they were fixed and stained with an antibody to BrdU (Zymed). A significant increase in BrdU-positive cells is seen after infection with HSV-SWE695 or HSV-V642I-75 (∼40%) relative to HSV-Lac- or mock-infected controls (∼18%). To confirm that the enhancement of BrdU incorporation by FAD APP expression is caused by DNA replication rather than DNA repair, we repeated the experiment in the presence or absence of cytosine arabinoside (Ara-C), a DNA replication inhibitor. In the presence of Ara-C (10 μm), which is incorporated into DNA to terminate DNA duplication, FAD APP-induced BrdU incorporation was reduced to control levels (data not shown).
Because the HSV vectors were overexpressing FAD APPs at quite high levels, the possibility existed that we might be causing artifactual neuronal death. To test this possibility, we created vectors expressing FAD APPs at lower levels, to test these vectors for their ability to cause apoptosis. We placed the FAD APP cDNAs upstream of an IRES-GFP cassette taken from the Stratagene (La Jolla, CA) vector pIRES-hrGFP-2a, since we had previously observed (unpublished data) reduced expression of cDNAs cloned into that position. We infected primary cortical neurons with equal MOIs of HSV-SWE695 and HSV-SWE695-IRES-GFP, and 16 hr later we harvested the neurons for immunoblot analysis (Fig. 1C, inset). It can be seen, by comparison with the HSVlac- and mock-infected lanes (which express only endogenous APP), that the new vector conferred lower levels of expression of APP relative to endogenous than did the original HSV-SWE695 vector (left lane). We therefore tested these constructs for their ability to cause neuronal apoptosis (Fig. 1C). When the cultures were all infected at an MOI of 1 per virus and stained with bisbenzamide after 16 hr, the lower-expressing vectors, which expressed a level of transgene approximately equal to endogenous levels of APP, were shown to cause the same degree of neuronal apoptosis as did the higher-expressing vectors. We conclude that the neuronal apoptosis caused by the original vectors is not an artifact of high expression but reflects a specific effect of expression of FAD APP on the neurons. It is interesting that the decreased expression does not cause a lower level of apoptosis. We infer from these data that a threshold level of FAD APP expression beyond endogenous will trigger apoptosis in a given neuron and that levels of expression that exceed this threshold will not cause a greater number of neurons to show DNA fragmentation or nuclear condensation at this particular snapshot in time (16 hr after infection). Remember that these vectors decrease the amount of transgene that is expressed per cell but do not decrease the numbers of neurons that are expressing the transgene, because we infected with an equal MOI.
The C terminus of APP interacts with PAK3
We sought to identify proteins that interacted with the intracellular C-terminal end of APP, which might mediate the apoptosis and DNA synthesis caused by FAD mutants of APP. The portion of the human APP cDNA encoding the C-terminal 100 amino acids of APP (APP-C100) was used for in vitro transcription and translation as described (Kozlowski et al., 1992). The resulting radiolabeled APP-C100 was used as a ligand to screen expression rat brain cDNA libraries. Successive rounds of screening and purification of positive clones yielded a single rat brain cDNA encoding a putative binding protein for APP-C100. The 1965 bp cDNA contained a partial (1352 bp) open reading frame followed by a presumptive 3′ untranslated region (3′ UTR). It was used to reprobe rat brain cDNA libraries, and numerous cDNA clones overlapping with the original clone and extending it at the 5′ end were isolated.
At least three of these cDNAs contained a full-length coding sequence. Sequence analysis of these clones revealed a 1632 bp open reading frame encoding a 544 amino acid protein identical to the previously reported β-PAK (Manser et al., 1997), the rat homolog of the mouse cDNA encoding mPAK-3 (Bagrodia et al., 1995); this protein is now most commonly referred to as PAK3. The putative APP binding protein is therefore a member of the p21(Cdc42/Rac)-activated kinase (PAK) family, which includes the Saccharomyces cerevisiae STE20 gene product (Leberer et al., 1992; Ramer and Davis, 1993), α-PAK (also known as PAK1; Manser et al., 1995), and γ-Pak (Teo et al., 1995), which corresponds to hPAK65 (Martin et al., 1995) and PAK2 (Jakobi et al., 1996). The greatest homology among the members of the family occurs in the C-terminal serine-threonine kinase domain (70% between PAK3 and STE20; >90% between PAK3 and PAK1). In addition, these kinases share, in their N-terminal domains, a peptide motif (amino acids 70-85) representing the p21-binding domain (Burbelo et al., 1995). The PAK proteins are activated by the Rho family p21 proteins Cdc42 and Rac1, which participate in cytoskeletal-mediated events in the cell.
We performed solid phase binding assays to confirm the interaction of PAK3 with APP. In “pull-down” assays (Chow et al., 1996), glutathione S-transferase (GST) fusion proteins of APP-695, APP-751, and APP-C100 were immobilized on glutathione-agarose beads and incubated with in vitro-synthesized radiolabeled PAK3. The beads were pelleted, washed, and subjected to SDS-PAGE. Autoradiograms of the gels (Fig. 2A, lanes 1-3) demonstrate the precipitation of radiolabeled PAK3 by GST-APP-695 and GST-APP-C100, but not by GST alone. GST-APP-751 also precipitated radiolabeled PAK3 (data not shown).
Figure 2.

PAK3 interacts with APP both in vitro and in vivo. A, Lanes 1-3, Precipitation of radiolabeled PAK3 by APP-695 and APP-C100 GST fusion proteins. The 61 kDa PAK3 in vitro translation product is indicated. Lane 4, Precipitation of radiolabeled C-terminal-truncated PAK3-13.5 with the APP-751-GST fusion protein. The band at 31 kDa likely represents a dimer of PAK3-13.5. B, PAK3 specifically coimmunoprecipitates with APP in neurons. Primary cortical neurons were coinfected with HSV-myc-PAK3 alone or together with HSV vectors expressing FAD APPs. Immunoprecipitations were with anti-APP antibody 369. The blot was probed with 9E10, specific for the myc epitope. The specific myc-PAK3 band immunodetected by 9E10 is indicated with an arrow and comigrates with the myc-PAK3 band in the lysate from a culture infected with HSV-SWE695+HSV-myc-PAK3. Note the absence of the band in the preimmune serum control. HSV-myc-rab5 was used as a control to demonstrate that antibody 369 does not interact with the myc tag. The bottom panel shows the expression levels of the different myc-tagged proteins in the lysates before immunoprecipitation.
The original cDNA retrieved by screening the library with APP-C100 encoded amino acid residues 93-544 of PAK3, comprising a short region adjacent to but not including the Rac1/Cdc42 binding domain (residues 70-85), together with the C-terminal kinase domain of PAK3. To determine whether the binding site for APP-C100 lay in the former region, we synthesized in vitro a C-terminal-truncated version of PAK3 (amino acid residues 1-126, termed PAK3-13.5 because of its calculated molecular mass of 13.5 kDa) that includes the Rac1/Cdc42 binding domain and the region immediately C-terminal to it but not the kinase domain. PAK3-13.5 was precipitated by the APP-GST fusion proteins (Fig. 2A, lane 4), suggesting that APP binds to PAK3 in the region immediately adjacent to the Rac1/Cdc42 binding domain, between amino acid residues 93 and 126.
To demonstrate a physiologically relevant interaction of APP with PAK3 in neurons, we expressed PAK3 together with APP-695 or APP-751 in rat primary cortical neurons in culture using HSV vectors. Rat cortical cultures were coinfected with HSV-SWE695 or HSV-V642I-751 and HSV-myc-PAK3 (PAK3 with an N-terminal myc epitope), or with HSV-SWE695 or HSV-V642I-751 and an HSV vector expressing an irrelevant myc-tagged protein (myc-rab5). APP was immunoprecipitated with anti-APP antibody 369, and the immunoprecipitated material was blotted with the anti-myc antibody 9E10 (Fig. 2B). 9E10 immunodetected a specific protein band in the immunoprecipitated fraction (arrow) that comigrates with myc-PAK3 in the lysate from a culture infected with HSV-SWE695+HSV-myc-PAK3 (leftmost lane). This band was not detected in immunoprecipitates from cultures infected with HSV-SWE695 or HSV-V642I-751 together with HSV-myc-rab5. The presence of the band in cultures infected with HSV-myc-PAK3 alone indicates that PAK3 interacts with endogenous wild-type APP. These data suggest that PAK3 interacts with APP in cells in rat primary cortical cultures.
APP and PAK3 colocalize in rab5- and rab11-positive structures in neurons
To determine whether APP and PAK3 were present in the same subcellular compartments, we coinfected primary cultures of rat cortical neurons with HSV vectors expressing each of the two proteins. Immunocytochemical analysis of the cultures with antibodies to APP and PAK3 revealed significant overlap in their subcellular distributions, primarily in punctate endosome-like structures (Fig. 3). To identify the compartments in which they colocalized, we performed triple-labeling immunocytochemistry of HSV-APP-695- and HSV-PAK3-infected neuronal cultures, using APP and PAK3 antibodies together with a battery of antibodies for specific cellular organelles. The greatest degree of colocalization of APP and PAK3 (Fig. 3A) was seen in rab11-positive endosomes, which are a subset of rab5-containing endosomes (Ulrich et al., 1996; Ren et al., 1998). Subcellular fractionation of uninfected primary cortical neurons confirmed the localization of APP and PAK3 in the fractions that were enriched in rab11-containing endosomes (Fig. 3B). A significant but lower degree of localization of APP and PAK3 in fractions enriched in rab5-containing endosomes was also observed. Little colocalization was seen in the other subcellular locations examined (data not shown).
Figure 3.
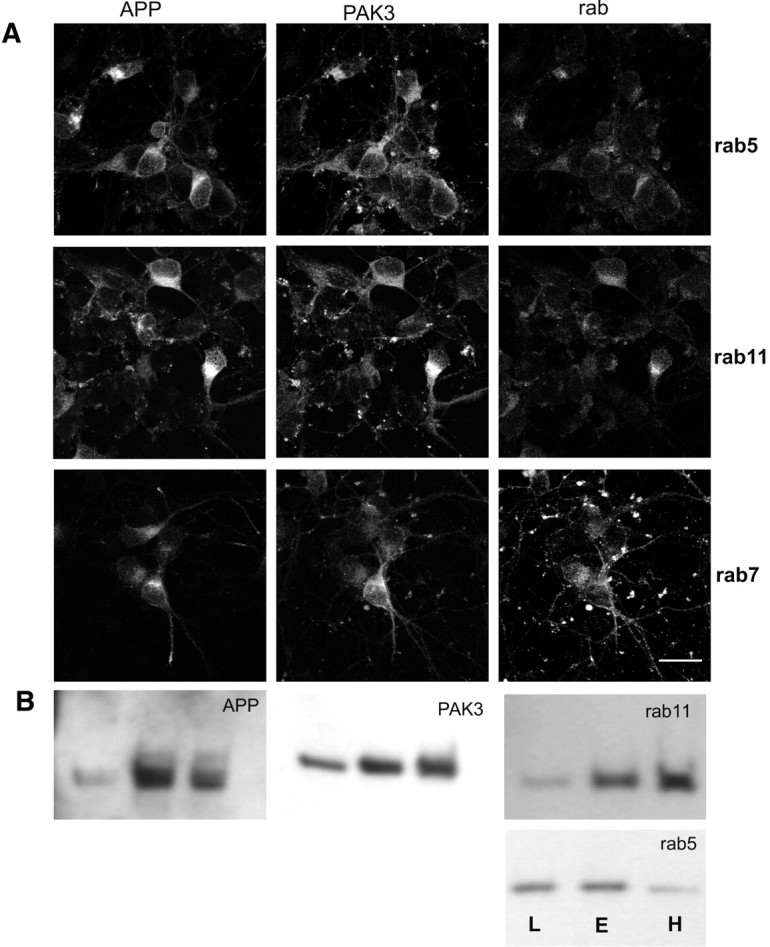
APP and PAK3 colocalize in rab5- and rab11-positive structures in neurons. A, Neuronal cultures were infected with both HSV-PAK3 and HSV-APP-695. The first column of cells represents APP immunofluorescence, the middle column PAK3 immunofluorescence, and the last column rab5, rab7, or rab11 immunofluorescence, as indicated by the row label. Note overall colocalization of APP and PAK3. Rab11 shows slightly more specific colocalization with APP than does rab5. Rab7 localization shows minimal overlap with that of APP or PAK3. Scale bar, 20 μm. B, Immunoblots of endosomal fractionation of rat brain lysates demonstrates that PAK3 and APP cofractionate into rab-11 and rab-5 positive fractions. The three lanes of each blot are designated L, E, and H: L, the membrane fraction that is enriched in late endosomes; E, the membrane fraction that is enriched in early endosomes; H, the fraction that contains heavy membranes.
PAK3 is expressed selectively in the nervous system
PAK3 mRNA is enriched in the brain in rodents (Manser et al., 1995). To determine the tissue specificity of PAK3 mRNA expression in the human, its cDNA was used to probe an RNA blot containing RNA from a range of human fetal (20-22 week) tissues (Fig. 4A). An ∼11 kb mRNA was detected only in the brain, confirming that expression of the PAK3 gene is restricted to the nervous system in the human as well as in the rodent.
Figure 4.
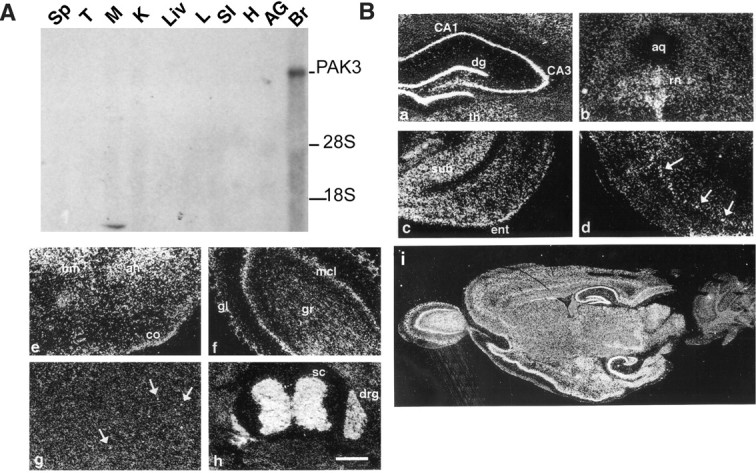
RNA blot and in situ hybridization analyses of PAK3 gene expression in the nervous system. A, The PAK3 cDNA was used to probe an RNA blot containing RNA from a range of human fetal (20-22 week) tissues (Sp, spleen; T, thymus; M, muscle; K, kidney; Liv, liver; L, lung; SI, small intestine; H, heart; AG, adrenal gland; Br, brain). B, In situ hybridization analysis of PAK3 mRNA expression in the rat nervous system. Panels show coronal sections of adult brain (a-d), sagittal sections of adult brain (e-g), a coronal section through E18 body (h), and a low-power sagittal section of rat brain (i). a, Hippocampus with intense signal in CA1 through CA4; b, brainstem; c, temporal cortex; d, cerebral cortex with cell layer containing intense signal (arrows); e, amygdala; f, olfactory bulb; g, striatum containing dispersed, heavily labeled cells (arrows); h, spinal cord; i, sagittal section through postnatal day 21 rat brain. dg, Dentate gyrus; th, lateral dorsal thalamic nucleus; aq, aqueduct; rn, raphe nucleus; ent, entorhinal cortex; sub, subiculum; bm, basomedial amygdaloid nucleus; ah, amygdalohippocampal area; co, posterolateral cortical amygdaloid nucleus; gl, glomerular cell layer; mcl, mitral cell layer; gr, granule cell layer; sc, spinal cord; drg, dorsal root ganglion. Scale bar, 500 μm.
To localize PAK3 mRNA more precisely in the rat brain, in situ hybridization was performed using an antisense riboprobe corresponding to the region of PAK3 cDNA that shares least homology with the α-PAK cDNA. Consistent with the expression of PAK3 mRNA observed on blots, the expression of PAK3 mRNA in rat brain was robust in some areas and restricted in others (Fig. 4i). Very high levels of PAK3 mRNA were observed in brain regions including the hippocampus, amygdala, piriform and entorhinal cortex, and olfactory bulb. A robust signal was also observed in discrete brainstem nuclei, including the raphe nucleus. Moderate levels of signal were found in neocortex and thalamus. In some brain regions with an overall low level of signal, including the caudate nucleus, dispersed intensely labeled cells were nonetheless observed. The cerebellum was devoid of detectable signal. Control hybridizations with sense riboprobe gave no specific signal (data not shown).
To determine whether PAK3 mRNA was localized outside the brain, in situ hybridization of antisense PAK3 riboprobe to coronal sections of E18 rats was performed. PAK3 mRNA was found in spinal cord and dorsal root ganglia, as well as in brain (Fig. 4h), suggesting that it is expressed in both the central and peripheral nervous systems. Non-neural tissues, including gut, bone, and muscle, were not labeled (data not shown).
Coexpression of a dominant-negative mutant of PAK3 blocks FAD APP-mediated neuronal apoptosis and DNA synthesis; this block is independent of p21 binding and is specific to PAK3
To find out whether the interaction between APP and PAK3 plays a role in FAD APP-mediated apoptosis and/or DNA synthesis, we tested the effects of dominant-negative (K297R) and dominant-positive (T421E) mutants (Sells et al., 1997) of PAK on FAD APP-induced apoptosis and DNA synthesis. Primary rat cortical cultures at 5 d in vitro were infected with HSV vectors expressing myc-tagged PAK and the mutant PAKs; 16 hr later the infected cells were harvested, and their proteins were analyzed by immunoblot analysis to confirm equal expression of the transgenes (Fig. 5A, inset). We then coinfected neurons with specific combinations of APP and PAK3 vectors and quantified apoptotic nuclei and BrdU incorporation in the infected neurons (Fig. 5A-C). Coexpression of the dominant-negative mutant of PAK3 (K297R) with wild-type and FAD APPs caused a significant inhibition of APP-induced apoptosis, almost to basal levels (Fig. 5A). Coexpression of the dominant-positive mutant of PAK3 (T421E) slightly enhanced the apoptosis caused by the Swedish mutant of APP, but did not significantly increase the apoptosis caused by the V642I mutant of APP. Expression of wild-type or mutant PAK3 cDNAs alone did not cause a significant change in apoptosis relative to basal levels (Fig. 5B).
Figure 5.

Coexpression of a dominant-negative mutant (K297R) of PAK3 blocks FAD APP-mediated neuronal apoptosis and DNA synthesis. A, Quantitative analysis of condensed nuclei in neurons coinfected with HSV-SWE695 or HSV-V642I-751 and HSV vectors expressing myc-PAK3 or mutants of myc-PAK3 demonstrates that the dominant-negative mutant of PAK3, PAK3-K297R, protects against apoptosis caused by FAD mutants of APP. A dominant-positive mutant (T421E) of PAK3 enhances the apoptosis caused by FAD APPs. Wild-type PAK3 has no effect on the apoptosis caused by FAD APPs. Note that HSV-Lac was used to equalize virus amounts among the conditions. Data are expressed as percentages (mean ± SEM). The inset shows an immunoblot of expression in neurons of myc-tagged PAK3 and PAK3 mutants. Significant between-group differences were confirmed by a single-factor ANOVA (F(9,90) = 26.4; p < 0.0001). Post hoc t tests done using the Bonferroni multiple comparisons test reveal significant differences in the number of cells with condensed nuclei between the following groups: SWE695+lac or SWE695+myc-PAK3 versus SWE695+myc-PAK3-K297R (p < 0.001), SWE695+lac versus SWE695+myc-PAK3-T421E (p < 0.05), V642I-751+ lac or V642I-751+myc-PAK3 versus V642I-751+myc-PAK3-K297R (p < 0.001), SWE695+lac or V642I-751+lac versus lac or mock (p < 0.001). All other relevant comparisons were NS. B, Expression of wild-type or mutant PAK3 alone does not result in apoptosis. Data are expressed as in A. A single-factor ANOVA showed no significant differences among the groups. C, Quantitative analysis of BrdU-positive nuclei in neurons coinfected with HSV-SWE695 or HSV-V642I-751 and HSV-myc-PAK3-K297R demonstrates a significant reduction in the number of BrdU-stained nuclei in neurons coexpressing myc-PAK3-K297R. Significant between-group differences were confirmed by a single-factor ANOVA (F(5,53) = 41.9; p < 0.0001). Post hoc t tests done using the Bonferroni multiple comparisons test reveal significant differences in the number of BrdU-positive nuclei between the following groups: SWE695+lac versus SWE695+myc-PAK3-K297R, V642I-751+lac versus V642I-751+myc-PAK3-K297R, and SWE695+lac or V642I-751+lac versus lac or mock (p < 0.001 for all comparisons). Other relevant comparisons were NS. D, Abrogation of FAD APP-mediated apoptosis by dominant-negative PAK3 is independent of p21 binding. Dominant-negative mutants of PAK3 both with and without functional p21 binding domains both significantly decreased the number of condensed nuclei in cultures expressing SWE-695 (HSV-SWE695 added in lanes with plus signs). Significant differences were confirmed by ANOVA (F(4,44) = 34.6; p < 0.0001). Post hoc t tests done using the Bonferroni multiple comparisons test reveal significant reductions in the number of condensed nuclei in cultures coexpressing SWE695+myc-PAK3-K297R or SWE695+myc-PAK3-K297R (no p21) versus SWE695+lac (p < 0.001). E, There is no change in in vitro PAK kinase activity after coexpression of wild-type or FAD APPs with either myc-PAK1 (top panel, lanes with plus signs) or myc-PAK3 (bottom panel, lanes with plus signs). The final lane in each panel demonstrates that the dominant-negative of each isoform of PAK is in fact kinase-dead. F, Coexpression of dominant-negative (K299R) PAK1 has the opposite effect of dominant-negative PAK3 on FAD APP-mediated apoptosis in cortical neurons, whereas coexpression of dominant-negative (K278R) PAK2 has no effect, indicating individual isoform-specific effects of PAKs on FAD APP-mediated apoptosis. Quantitative analysis of condensed nuclei in neurons coinfected with HSV-V642I-751 (lanes with plus signs) and HSV vectors expressing myc-tagged wild-type or dominant-negative PAK1, PAK2, or PAK3 demonstrates a significant reduction in condensed nuclei in neurons coexpressing wild-type myc-PAK1, but no reduction in condensed nuclei in neurons coexpressing myc-PAK1-K299R. Neither wild-type or dominant-negative PAK2 affects apoptosis mediated by V642I-751. Significant differences were confirmed by ANOVA (F(12,116) = 28.1; p < 0.0001). Post hoc t tests done using the Bonferroni multiple comparisons test reveal significant differences in the number of condensed nuclei between V642I-751+lac versus V642-751+myc-PAK3-K297R or V642I-751+myc-PAK1 (p < 0,001). All other relevant comparisons were NS.
As described above, neuronal apoptosis caused by FAD mutants of APP can be blocked by a dominant-negative mutant of PAK3. Can this same mutant prevent DNA synthesis caused by FAD mutants of APP? Neurons were coinfected with HSV vectors expressing the FAD mutants of APP together with a vector expressing PAK3 in the presence of BrdU, and 16 hr later they were fixed and stained with an antibody to BrdU. The results (Fig. 5C) show that a dominant-negative mutant of PAK3 prevents the increase in DNA synthesis caused by the FAD APPs, suggesting that entry into the cell cycle caused by FAD APPs is mediated by the same signal transduction pathway that mediates the neuronal apoptosis caused by FAD mutants of APP.
The APP binding site in PAK3 is adjacent to the p21 binding site; this raises the question of whether the binding of the Rho GTPases Rac1 or Cdc42 to PAK3 modulates its functional role in mediating FAD APP-induced apoptosis. One way to test this is to ask whether the protection against FAD APP-induced apoptosis afforded by PAK3 is affected by p21 binding to PAK3. We made mutated PAK3 so that leucine residues were substituted for two highly conserved histidine residues (his78 and his81) within the p21-binding domain. These mutations abolish binding of PAK3 to Cdc42 or Rac1 (Sells et al., 1997). Neurons were coinfected with HSV vectors expressing the Swedish mutant of APP together with a vector expressing dominant-negative PAK3 (K297R) or dominant-negative PAK3 plus the mutated histidine residues [K297R (no p21)]. The results (Fig. 5D) indicate that the inhibition of FAD APP-mediated apoptosis by PAK3 is independent of p21 binding. Dominant-negative PAK3, either with or without a functional p21 binding domain, significantly decreased the number of condensed nuclei in cultures expressing the Swedish mutant of APP.
The APP binding domain of PAK3 overlaps with a PAK3 autoinhibitory domain (amino acids 78-144). Thus, one explanation for our results is that competition for the autoinhibitory domain of PAK3 by APP renders PAK active. We tested this possibility by determining whether PAK activity is influenced by APP coexpression (Fig. 5E). Neurons were infected with HSV vectors expressing myc-PAK1 or myc-PAK3, either alone or in combination with wild-type or FAD APP. myc-PAK1 and myc-PAK3 were immunoprecipitated from the cell lysates and incubated with histone H4, a substrate for PAKs, in the presence of 33P-ATP. The reactions were subjected to SDS-PAGE, after which the gels were fixed, dried, and exposed to film to visualize incorporated radioactivity. As shown in Figure 5E, there is no change in in vitro PAK kinase activity after coexpression of wild-type or FAD APPs with either myc-PAK1 (top panel) or myc-PAK3 (bottom panel). Furthermore, note that neither myc-PAK3(101-126), which represents the APP binding domain, nor myc-PAK3(78-144), which represents the PAK3 autoinhibitory domain, affects the kinase activity of coexpressed PAK3. Note also that these data confirm that the dominant-negative mutants of PAK1 and PAK3 are, as expected, kinase-dead.
PAK3 is a member of a family of proteins that contains at least six members (for review, see Jaffer and Chernoff, 2002). We made dominant-negative mutants of members of the group I PAKs (PAK1, PAK2, PAK3) to test whether they also protected against FAD APP-mediated neuronal apoptosis. Neurons were infected with HSV vectors expressing a London mutant (V642I) of APP-751 together with wild-type or dominant-negative mutants of PAK1, PAK2, and PAK3. The results (Fig. 5F) indicate that coexpression of dominant-negative (K299R) and wild-type PAK1 has the opposite effect of dominant-negative (K297R) and wild-type PAK3 on FAD APP-mediated apoptosis in cortical neurons: wild-type PAK1 protects against the apoptosis, whereas dominant-negative PAK1 has no effect on the apoptosis. Coexpression of dominant-negative (K278R) or wild-type PAK2 has no effect on FAD APP-mediated apoptosis. These data suggest that there are individual isoform-specific effects of PAKs on FAD APP-mediated apoptosis.
Dominant-negative PAK3 does not block chemically induced neuronal apoptosis
To ascertain whether the dominant-negative mutant of PAK3 has a general effect on apoptotic pathways, we tested the effect of expression of K297R-PAK3 on neurons treated with the apoptosis-inducing agents etoposide and staurosporine (Fig. 6A). Both etoposide and staurosporine induced a significant increase in DNA fragmentation in cortical cells. This increase in DNA fragmentation was not prevented by coinfection with HSV expressing the dominant-negative mutant of PAK3. These data suggest either that etoposide and staurosporine act in a distinct apoptotic pathway or that their effects on apoptosis are distal to the effect of PAK3 in the same pathway.
Figure 6.

Expression of the PAK3 dominant-negative mutant K297R does not block chemically induced neuronal apoptosis, deletion of the APP binding site in dominant-negative (K297R) PAK3 abrogates its ability to inhibit FAD APP-mediated apoptosis, and coexpression of a peptide representing the APP binding site in PAK3 blocks FAD APP-mediated apoptosis. A, Quantitative analysis of condensed nuclei in neurons treated with staurosporine (S) or etoposide (E) and coinfected with myc-PAK3-K297R shows no significant reduction in the number of condensed nuclei as compared with neurons treated with staurosporine or etoposide alone. Data are expressed as percentages (mean ± SEM). B, Coexpression of myc-PAK3-K297R with SWE695 significantly blocks the FAD APP-mediated apoptosis. However, deletion of the APP binding site (amino acids 101-126) in PAK3-K297R abrogates its ability to inhibit the FAD APP-mediated apoptosis. Expression of myc-PAK3-K297R (del101-126) alone does not result in apoptosis (data not shown). No significant difference in the number of condensed nuclei is seen between SWE695+lac versus SWE695+ PAK3-K297R (del101-126). Coexpression of GFP- or myc-tagged APP binding site peptide PAK (101-126) with the Swedish mutant of APP-695 significantly blocked FAD APP-mediated apoptosis. Data are expressed as percentages (mean ± SEM). Significant differences were confirmed by ANOVA (F(6.62) = 29.1; p < 0.0001). Post hoc t tests done using the Bonferroni multiple comparisons test reveal significant differences between SWE695+lac versus SWE695+myc-PAK3-K297R. Significant differences also are seen between SWE695+myc-PAK (101-126) or SWE695+GFP-PAK (101-126) versus SWE695+lac (p < 0.001 for both). The comparison of SWE695+lac versus SWE695+myc-PAK3-K297R (del101-126) was NS. All other relevant comparisons were NS. The insets show immunoblots of expression of epitope-tagged PAK (101-126) peptides and of the myc-tagged PAK3 mutations.
Binding of APP to PAK3 regulates FAD APP-induced apoptosis
We then performed a set of experiments using two different strategies to test whether an interaction between FAD APP and PAK3 was necessary for FAD APP-induced apoptosis (Fig. 6B). First, a deletion of the putative APP-binding domain of PAK3 [101-126; these residues were chosen to not interfere with p21 binding to PAK3 (Zhao et al., 1998)] was introduced into the dominant-negative mutant of PAK3. This double mutant, unlike the K297R mutant alone, did not inhibit apoptosis caused by the Swedish mutant of APP-695. Then we coexpressed with the Swedish mutant of APP-695, myc- or GFP-tagged peptides representing the PAK3-binding domain of APP (101-126). Expression of the 101-126 peptide, whether it was fused to the myc tag or to GFP, inhibited FAD APP-induced apoptosis to the same degree that it was inhibited by expression of the dominant-negative mutant of PAK3.
Pertussis toxin blocks FAD APP-mediated neuronal apoptosis and DNA synthesis
It has been shown that APP interacts with the G-protein Go (Nishimoto et al., 1993; Brouillet et al., 1999), specifically via Gβγ (Giambarella et al., 1997), and that members of the STE20/PAK family interact with heterotrimeric G-proteins (Leeuw et al., 1998). Therefore we tested whether pertussis toxin, an inhibitor of Go and Gi, had an effect on APP- or FAD APP-induced apoptosis (Fig. 7A). In the presence of pertussis toxin, no significant increase in apoptosis above basal levels was detected in cultures infected with HSV recombinants expressing FAD (Swedish-695 or V642I-751) mutants of APP. If the pertussis toxin-treated cultures were coinfected with a pertussis toxin-insensitive mutant of Goα (Goαptx), the apoptosis caused by the Swedish mutant of APP was partially “rescued” (Fig. 7C), suggesting that it is the specific inhibition of Go, rather than that of Gi, by pertussis toxin that blocks FAD APP-mediated neuronal apoptosis. Pertussis toxin inhibits not only FAD APP-mediated apoptosis, but also FAD APP-mediated DNA synthesis in neurons (Fig. 7B). These data suggest that Go interaction with APP is essential for both apoptosis and DNA synthesis mediated by FAD APPs.
Figure 7.

Pertussis toxin blocks FAD APP-mediated neuronal apoptosis and DNA synthesis. This block is prevented by coexpression of a pertussis toxin-insensitive mutant (Gαo*), by deletion of the Go binding domain of APP, or by deletion of the putative Go binding domain of PAK3. A, Pertussis toxin (PTX) treatment of cultures infected with HSV vectors expressing FAD mutants of APP significantly reduces apoptosis to basal levels. Data are expressed as percentages (mean ± SEM). Significant differences were confirmed by ANOVA (F(7,72) = 14.9; p < 0.0001). Post hoc t tests done using the Bonferroni multiple comparisons test reveal significant differences between SWE695 versus SWE695+PTX and between V642I-751 versus V642I-751+PTX (p < 0.001). Other relevant comparisons were NS. B, PTX treatment of cultures infected with HSV vectors expressing FAD mutants of APP significantly reduces DNA synthesis to basal levels. Data are expressed as percentages (mean ± SEM). Significant differences were confirmed by ANOVA (F(7,71) = 34.1; p < 0.0001). Post hoc t tests done using the Bonferroni multiple comparisons test reveal significant differences between SWE695 versus SWE695+PTX and between V642I-751 versus V642I-751+PTX (p < 0.001). All other relevant comparisons were NS. C, Coexpression of the PTX-insensitive mutant Gαo* significantly prevents the PTX block of FAD APP-mediated apoptosis. Significant differences were confirmed by ANOVA (F(3,35) = 12.3; p < 0.0001). Post hoc t tests done using the Bonferroni multiple comparisons test reveal significant differences between the following groups: SWE695+lac versus SWE695+lac+PTX and SWE695+lac+PTX versus either SWE695+Gαo* or SWE695+ Gαo*+PTX (p < 0.001). SWE695+lac versus either SWE695+Gαo* or SWE695+Gαo*+PTX were NS. D, Deletion of the Go binding domain (amino acids 657-676) in APP or the putative Go binding domain in PAK3 significantly blocks FAD APP-mediated apoptosis. Significant differences were confirmed by ANOVA (F(9,88) = 18.1; p < 0.0001). Post hoc t tests done using the Bonferroni multiple comparisons test reveal significant differences between the following groups: SWE695 versus SWE695 (del657-676), V642I-751 versus V642I-751 (del657-676), SWE695+lac versus SWE695+mycPAK3 (noGo), or V642I-751+ lac versus V642I-751+mycPAK3 (no Go) (p < 0.001 for all comparisons). All other relevant comparisons were NS.
The Go binding domain within APP [amino acids 657-676 using the numbering of APP-695, as defined by Nishimoto et al. (1993) and Lang et al. (1995)], was deleted from the Swedish and London mutants of APP. These mutants were expressed in neurons using HSV vectors; 16 hr after infection the neurons were fixed and stained with bisbenzamide to detect condensed nuclei. As seen in Figure 7D, deletion of the Go binding domain from either of the FAD APPs reduces their ability to cause neuronal apoptosis almost to control levels. These data suggest that interaction of FAD APPs with Go is required for them to cause neuronal apoptosis and are consistent with our data demonstrating that pertussis toxin inhibits FAD APP-mediated apoptosis.
The data described thus far suggest that APP interacts with PAK3 both directly, at a region between amino acids 93 and 126, and indirectly, via activation of Go and presumably Gβγ. Furthermore, it appears that both direct and indirect interactions are necessary to get activation, because blocking either one (with a peptide representing amino acids 101-126 of PAK3 or with pertussis toxin) prevents apoptosis. This differs from heptahelical G-protein-coupled receptors, for which there is believed to be only the indirect, G-protein-mediated interaction between the receptor and the effector. The interaction between PAK3 and Gβγ most likely occurs in the last 10-12 residues at the C terminus of PAK3 (Leeuw et al., 1998). To test whether a direct interaction between PAK3 and Gβγ is also necessary for FAD APP-mediated apoptosis to occur, we constructed a mutant of PAK3 [PAK3(del Go)] with the last 12 amino acids deleted. This mutant should be unable to interact with Gβγ. Neurons were infected with HSV vectors expressing FAD mutants of APP, both alone and together with an HSV vector expressing PAK3(del Go). Analysis of the results (Fig. 7D) showed that PAK3(del Go) behaved in a dominant-negative manner, inhibiting the apoptosis caused by the FAD mutants of APP. These data indicate that a direct interaction between PAK3 and Gβγ is necessary for FAD APP-mediated apoptosis to occur.
Antibody 22C11, which binds to the extracellular domain of APP, causes neuronal apoptosis that is inhibited by dominant-negative PAK3
Okamoto et al. (1995) showed that an antibody to the extracellular domain of APP (22C11) that may act as a ligand mimetic causes activation of Go, reinforcing the idea that APP may be a G-protein-coupled receptor. Subsequently, Rohn et al. (2000) showed that this same antibody induces neuronal apoptosis. We tested whether 22C11-induced neuronal apoptosis activated the same signaling pathway or pathways that are activated by FAD APPs. The antibody 22C11 (2 μg/ml) was added to primary neuronal cultures for 16 hr, in the presence of HSV vectors expressing dominant-negative PAK3 (K297R) or the PAK3 APP binding domain, after which the cells were fixed and stained with bisbenzamide. Figure 8A shows that 22C11 causes neuronal apoptosis in our paradigm and that this apoptosis is prevented by coexpression of mutants of PAK3 that inhibit FAD APP-mediated apoptosis. 22C11 also causes DNA synthesis to occur in neurons (Fig. 8B). These data indicate that a similar signaling pathway is activated by 22C11 as is activated by FAD mutants of APP. Whereas 22C11 could act by blocking the cell survival promoting effect of secreted forms of APP, this possibility is unlikely because we changed the medium, thereby removing secreted APP, immediately before adding 22C11.
Figure 8.

Treatment of neurons with antibody 22C11 causes apoptosis and DNA synthesis and expression of C57 or C31 causes neuronal apoptosis and DNA synthesis. A, Sustained treatment of neurons with 1 μl/ml of 22C11, a ligand mimetic for APP, causes neuronal apoptosis. Expression of either myc-PAK3-K297R or peptide myc-PAK3 (101-126) significantly abrogates 22C11-mediated neuronal apoptosis. Significant differences were confirmed by ANOVA (F(6,62) = 28.0; p < 0.0001). Post hoc t tests done using the Bonferroni multiple comparisons test reveal significant differences between the following groups: 22C11 or 22C11+lac versus 22C11+mycPAK3K297R, 22C11+mycPAK3 (101-126), 22C11 versus lac (no 22C11), mock (no 22C11) or 9E10 (p < 0.001 for all comparisons). B, Sustained treatment of cortical neurons with 22C11 leads to a significant increase in the number of neurons showing DNA synthesis. Significant differences were confirmed by ANOVA (F(5,53) = 95.4; p < 0.0001). Post hoc t tests done using the Bonferroni multiple comparisons test reveal significant differences between the following groups: 22C11 (1 μl) or 22C11 (2 μl) versus 9E10 (1 μl), 9E10 (2 μl), lac, or mock (p < 0.001 for all comparisons), and 22C11 (1 μl) versus 22C11 (2 μl) (p < 0.01). All other relevant comparisons were NS. C, Infection of neurons with HSV vectors expressing myc-tagged C57 or C31 causes an increase in the number of apoptotic nuclei relative to infection of neurons with control HSV-Lac. Coexpression of dominant-negative PAK3 or addition of PTX causes a significant reduction in the number of apoptotic nuclei in cultures expressing myc-C57 or myc-C31. Significant differences were confirmed by ANOVA (F(8,81) = 34.95; p < 0.0001). Post hoc t tests done using the Bonferroni multiple comparisons test reveal significant differences between the following groups: mycC57 versus mycC57+mycPAK3K297R, mycC57+PTX or lac, and mycC31 versus mycC31+mycPAK3K297R, mycC31+PTX or lac (p < 0.001 for all comparisons). Other relevant comparisons were NS. D, Expression of myc-tagged C57 or C31 leads to a significant increase in the number of neurons showing DNA synthesis. Significant differences were confirmed by ANOVA (F(3,36) = 12.98; p < 0.0001). Post hoc t tests done using the Bonferroni multiple comparisons test demonstrated a significant increase in the number of BrdU-positive nuclei in cultures infected with HSV-myc-C57 or HSV-myc-C31 versus lac or mock (p < 0.001). Other relevant comparisons were NS.
C57 and C31 cause apoptosis that is mediated by PAK3 and Go
We suggested previously (McPhie et al., 2001) that the C-terminal 31 amino acids of APP (C31), which is generated by intracellular caspase cleavage, is responsible for the apoptosis caused by FAD mutants of APP. Moreover, it has been shown that the APP intracellular domain (AICD) generated by γ-secretase cleavage of APP can cause apoptosis in neuroglioma cells (Kinoshita et al., 2002). We asked whether apoptosis caused by C31 or C57 (one of the fragments released intracellularly by γ-secretase cleavage) is mediated by PAK3 and Go. Neurons in culture were infected with HSV vectors expressing myc-tagged C57 or C31, alone or in combination with dominant-negative PAK3 or pertussis toxin. The results (Fig. 8C) show that both C57 and C31 directly cause neuronal apoptosis and that both forms of apoptosis are inhibited by dominant-negative PAK3 or pertussis toxin. Thus, PAK3 and Go mediate apoptosis caused by C57 or C31. We then asked whether C57 and C31 also caused neuronal DNA synthesis. The results (Fig. 8D) indicate that both of these fragments, when expressed ectopically in primary neurons in culture, cause DNA synthesis to occur.
FAD APP-mediated DNA synthesis precedes FAD APP-mediated apoptosis in neurons
Is FAD APP-mediated apoptosis caused by entry into the cell cycle, or is the converse true? To begin to answer this question, we treated neurons expressing FAD APPs with the cell cycle G1/S blockers mimosine and deferoxamine (Farinelli and Greene, 1996). First we tested whether these compounds block FAD APP-mediated DNA synthesis. Neurons were infected with HSV vectors expressing the Swedish or the London mutant of APP; at the same time, mimosine or deferoxamine was added to the cultures, along with 10 μm BrdU. 16 hr later the cultures were fixed and stained with antibody to BrdU. The data in Fig. 9A show that mimosine blocks DNA synthesis completely in primary neuronal cultures. Deferoxamine interestingly appears to block FAD APP-induced DNA synthesis but not basal synthesis as measured in mock- or HSV-lacz-infected cultures.
Figure 9.

FAD APP-mediated DNA synthesis precedes FAD APP-mediated apoptosis in neurons. A, The G1/S blockers mimosine and deferoxamine block the FAD APP-mediated increase in DNA synthesis in primary neuronal cultures. A significant decrease in the number of BrdU-positive nuclei is seen after treatment of HSV-FAD APP-infected cultures with either 400 μm mimosine or 1 mm deferoxamine as confirmed by ANOVA (F(11,107) = 54.8; p < 0.0001). Post hoc t tests done using the Bonferroni multiple comparisons test reveal significant differences between the following groups: SWE695 versus SWE695+mimosine, SWE695+deferoxamine, lac or mock; V642I-751 versus V642I-751+mimosine, V642I-751+deferoxamine, lac or mock (p < 0.001). All other relevant comparisons were NS. B, The G1/S phase blockers mimosine and deferoxamine block FAD APP-mediated apoptosis. Addition of 400 μm mimosine or 1 mm deferoxamine to HSV-FAD APP-infected cortical neurons significantly blocked the FAD APP-mediated increase in condensed nuclei. Significant differences were confirmed by ANOVA (F(11,107) = 7.3; p < 0.0001). Post hoc t tests done using the Bonferroni multiple comparisons test reveal significant differences between the following groups: SWE695 versus SWE695+mimosine, SWE695+deferoxamine, lac, or mock; and V642I-751 versus V642I-751+mimosine, V642I-751+ deferoxamine, lac or mock (p < 0.001). All other relevant comparisons were NS. C, The pan-caspase inhibitor Boc-DFMK significantly blocks FAD APP-mediated apoptosis. Significant differences were confirmed by ANOVA (F(7,72) = 40.3; p < 0.0001). Post hoc t tests done using the Bonferroni multiple comparisons test reveal significant differences between the following groups: SWE695+DMSO versus SWE695+BocDFMK, lac+DMSO, or mock+DMSO; and V642I-751+DMSO versus V642I-751+BocDFMK, lac+DMSO or mock+DMSO (p < 0.001). All other relevant comparisons were NS. D, The pan-caspase inhibit or Boc-DFMK does not block FAD APP-mediated DNA synthesis. No significant differences in the number of BrdU-positive nuclei were seen between SWE695+DMSO versus SWE695+BocDFMK or between V642I-751+DMSO versus V642I-751+BocDFMK.
We then assessed whether these same compounds inhibited FAD APP-mediated apoptosis. As shown in Figure 9B, both mimosine and deferoxamine significantly inhibited apoptosis mediated by the Swedish and London mutants of APP. Both compounds appeared to increase basal levels of apoptosis slightly (see the data for mock- and HSV-LacZ-infected cultures), but significantly decrease FAD APP-induced apoptosis. These data suggest that blocking entry into the cell cycle inhibits neuronal apoptosis caused by FAD APPs. Interestingly, the DNA synthesis inhibitor aphidicolin did not block neuronal apoptosis caused by FAD APPs (data not shown), implying that inhibiting DNA synthesis per se does not provide protection against apoptosis.
We then asked whether inhibition of neuronal apoptosis in HSV-FAD-APP-infected cultures would inhibit DNA synthesis. Neurons were infected with HSV vectors expressing the Swedish or the London mutant of APP; at the same time, the pan-caspase inhibitor Boc-D-FMK was added to the cultures (BrdU was also added at this time to samples in which DNA synthesis was to be assessed). Sixteen hours later, the levels of apoptosis or DNA synthesis in the cultures were measured (Fig. 9C). As expected, the pan-caspase inhibitor blocked neuronal apoptosis caused by the FAD APPs. In contrast, however, Boc-D-FMK had no effect on DNA synthesis caused by the Swedish and London mutants of APP (Fig. 9D). The data in Figure 9 show that DNA synthesis precedes and is a prerequisite for apoptosis to occur in neuronal cultures expressing FAD APPs, but that prevention of apoptosis does not affect DNA synthesis.
Discussion
We have shown that neuronal expression of FAD mutants of APP causes apoptosis and DNA synthesis. Both the apoptosis and the DNA synthesis are mediated by the interaction of the C terminus of APP with PAK3, but not with two other members of the PAK family, PAK1 and PAK2. Inhibition of the heterotrimeric G-protein Go prevents the DNA synthesis and apoptosis caused by FAD mutants of APP. Sustained exposure of neurons to a ligand mimetic for APP, the antibody 22C11, also causes apoptosis and DNA synthesis, via a similar signaling pathway. C57, the APP intracellular domain, or C31, a C-terminal caspase cleavage product of APP, can alone cause neuronal apoptosis and DNA synthesis. FAD APP-mediated DNA synthesis precedes FAD APP-mediated apoptosis in neurons, and inhibition of neuronal entry into the cell cycle inhibits the apoptosis. Our finding that FAD mutants of APP activate apoptotic pathways and DNA synthesis in neurons provides support for the hypotheses that some neurons degenerate via apoptotic pathways in AD (Cotman and Anderson, 1995; Guo et al., 1998) and that entry into the cell cycle may initiate these apoptotic pathways (Yang et al., 2001).
Because the apoptotic process proceeds to completion within 16-24 hr, one might predict a virtually complete loss of HSVFAD APP-infected neurons after 16 hr of infection. Clearly, this does not happen in our paradigm; the percentage of cells entering apoptosis does not exceed 30% at most. Cotman (1998) and recently Raina et al. (2003) have suggested that the induction of compensatory responses to apoptosis in neurons protects them from terminal apoptosis and that a dynamic competition between cell death processes and compensatory responses exists in these terminally differentiated cells, which cannot easily be replenished if lost through apoptosis.
We have identified an APP-interacting protein, PAK3, which together with Go mediates the neuronal apoptosis and DNA synthesis caused by FAD mutant APP. We have confirmed in an in vitro assay that PAK3 binds to APP in its C-terminal domain and that APP binds adjacent to the Cdc42/Rac1 binding site of PAK3. We have shown that PAK3 coprecipitates with APP from neuronal cultures coinfected with recombinant HSV vectors encoding myc-PAK3 and FAD mutants of APP. PAK3 coprecipitates with wild-type APP as well (data not shown). The putative binding of APP to PAK3 in neurons is consistent with the growing body of evidence that APP is a signaling receptor that specifically activates the G-protein Go in a ligand-dependent manner (Nishimoto et al., 1993; Okamoto et al., 1995). The yeast homolog of PAK3, Ste20, is part of a pathway activated by heterotrimeric G-protein βγ subunits. Ste20 is a serine-threonine kinase that acts at an early step of a signal transduction cascade and has been demonstrated to be the direct target of the yeast homolog of the βγ subunit (Leeuw et al., 1998). If PAK3 is similarly situated in mammalian neurons, to interact directly with heterotrimeric G-proteins, this targets it to a molecular location at which APP also may be found, and suggests that mutual interaction with G-protein βγ subunits may mediate or modulate the coupling of PAK3 and APP.
One obvious explanation for the ability of a kinase-dead mutant of PAK3 to inhibit FAD APP-mediated apoptosis and DNA synthesis is that FAD APPs increase the kinase activity of PAK3. Our observation that APP binds to an autoinhibitory domain of PAK3 makes such an explanation even more attractive. However, we showed both that expression of constitutively active PAK3 does not itself cause apoptosis or DNA synthesis in neurons and also that coexpression of either wild-type or FAD APPs with PAK3 does not detectably increase its kinase activity. In fact, basal kinase activity of PAK3 in neurons appears to be relatively robust to begin with. What explanation, then, could account both for the observation that PAK3 kinase activity is necessary for FAD APP-mediated apoptosis and DNA synthesis to occur (as demonstrated by their inhibition by a dominant-negative mutant of PAK3) and also for the observation that FAD APPs do not increase PAK3 activity? One possibility is that binding of APP to the autoinhibitory domain of PAK3 changes its conformation in such a way that enables PAK3 to interact with, and phosphorylate, a substrate with which it normally does not come into contact. Alternatively, binding of APP to PAK3 may draw it into a complex of proteins, or move it into a compartment of the cell, from which it is normally sequestered, thereby exposing it to new substrates. To resolve these possibilities, it will be necessary to identify PAK3 targets both in the basal state and also when it is coexpressed with FAD APPs.
In addition to PAK3 and Go, the presumptive adaptor proteins Fe65 and X11 (for review, see Russo et al., 1998) and APP-BP1 (Chow et al., 1996) have been reported to interact with the C terminus of APP, presumably to initiate intracellular signaling. Although the functions of Fe65 and X11 are not known, Fe65 has the characteristics of adaptor proteins, which are thought to link signal transduction events emanating from plasma membrane receptors to intracellular molecules, by forming complexes of these proteins. APP may be part of a Go protein-centered complex that transduces extracellular signals to the cytoplasm and the nucleus, with Fe65 bringing together the molecules in this complex. We have shown recently (Chen et al., 2000) that APP-BP1 is a cell cycle protein that causes apoptosis when overexpressed in neurons. APP-BP1, and two other proteins reported to interact with the C terminus of APP, histone acetyltransferase Tip60 (Cao and Südhof, 2001) and Jun N-terminal kinase (JNK) interaction protein 1 (JIP1) (Scheinfeld et al., 2002), also may be linked to this pathway.
Either C57, one of the fragments released intracellularly by γ-secretase cleavage of APP, or C31, which can be released intracellularly by caspase cleavage of APP, can alone cause neuronal apoptosis and DNA synthesis. These data suggest that the PAK3 binding site on APP is within the C-terminal 31 amino acids, which also contains the Go binding site (Okamoto et al., 1995). They also show that Aβ is not involved in FAD APP-mediated neuronal apoptosis and DNA synthesis, except insofar as γ-secretase cleavage of APP can generate a fragment C-terminal to Aβ that can mediate these events. These data are consistent with our previous finding that inhibition of γ-secretase cleavage of an FAD mutant of APP does not interfere with the ability of the mutant to cause neuronal apoptosis (McPhie et al., 2001).
Our data reveal that blocking entry into the cell cycle inhibits neuronal apoptosis caused by FAD APPs. The DNA synthesis inhibitor aphidicolin did not block neuronal apoptosis caused by FAD APPs, implying that inhibiting DNA synthesis per se does not provide protection against apoptosis. Thus, although we have used DNA synthesis as a marker for entry of neurons into the cell cycle, we do not consider that DNA synthesis is necessarily the pathological event that mediates apoptosis caused by FAD APPs. Inhibition of apoptosis with a pan-caspase inhibitor did not inhibit DNA synthesis caused by FAD mutants of APP. Thus, DNA synthesis appears to precede and cause apoptosis in neurons, rather than the converse, in our model. These data are consistent with those of Yang et al. (2001), who demonstrated that a significant number of hippocampal pyramidal (4 vs 0% in control hippocampus) and basal forebrain neurons in AD brain have undergone full or partial DNA replication, showing that they have completed the S phase. This is not seen in unaffected regions of AD brain or in control brains. Yang et al. (2001) speculate this entry into the cell cycle eventually is lethal to neurons.
We demonstrated that antibody 22C11, which is thought to act as a ligand mimetic for APP, causes neuronal apoptosis and DNA synthesis. These data are consistent with our hypothesis that APP has an intrinsic signaling function in the neuron that is normally tightly regulated but that causes apoptosis after sustained activation of APP. Presumably FAD mutations of APP have a similar effect by causing APP to signal in a ligand-independent manner.
Deletion of the APP-binding site from a dominant-negative mutant of PAK3 abrogates its ability to inhibit wild-type and FAD APP-mediated apoptosis, suggesting that interaction between APP and PAK3 is necessary for the apoptosis to occur. This suggestion is further supported by the observation that expression of the PAK3 APP-binding domain in neurons strongly inhibits FAD APP-mediated apoptosis, presumably by a competitive mechanism. We hypothesize that a normal, regulated interaction of APP with PAK3 is altered by overexpression of APP or FAD mutant APPs. These data highlight the importance of understanding the normal function of APP in the brain and how this function may be disturbed in AD.
Footnotes
This work was supported by National Institutes of Health Grant AG12954 to R.L.N. K.A.N. was supported by the Veterans Affairs Merit Review and Career Scientist Programs. We thank Dr. Li-Huei Tsai for the generous gift of the wild-type and dominant-negative PAK1 and PAK2 cDNAs.
Correspondence should be addressed to either of the following: Donna L. McPhie, MRC 223, McLean Hospital, 115 Mill Street, Belmont, MA 02478, E-mail mcphie@helix.mgh.harvard.edu; or Rachael L. Neve, MRC 223, McLean Hospital, 115 Mill Street, Belmont, MA 02478, E-mail: neve@helix.mgh.harvard.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/236914-14$15.00/0
References
- Arendt T ( 2002) Dysregulation of neuronal differentiation and cell cycle control in Alzheimer's disease. J Neural Transm Suppl 62: 77-85. [DOI] [PubMed] [Google Scholar]
- Arendt T, Rodel L, Gartner U, Holzer M ( 1996) Expression of the cyclin-dependent kinase inhibitor p16 in Alzheimer's disease. NeuroReport 7: 3047-3049. [DOI] [PubMed] [Google Scholar]
- Bagrodia S, Taylor SJ, Creasy CL, Chernoff J, Cerione RA ( 1995) Identification of a mouse p21cdc42/rac activated kinase. J Biol Chem 270: 22731-22737. [DOI] [PubMed] [Google Scholar]
- Brouillet E, Trembleau A, Galanaud D, Volovitch M, Bouillot C, Valenza C, Prochiantz A, Allinquant B ( 1999) The amyloid precursor protein interacts with Go heterotrimeric protein within a cell compartment specialized in signal transduction. J Neurosci 19: 1717-1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbelo PD, Brechsel D, Hall A ( 1995) A conserved binding motif defines numerous candidate target proteins for both Cdc42 and Rac GTPases. J Biol Chem 270: 29071-29074. [DOI] [PubMed] [Google Scholar]
- Bursztajn S, DeSouza R, McPhie DL, Berman SA, Shioi J, Robakis NK, Neve RL ( 1998) Overexpression in neurons of human presenilin-1 or a presenilin-1 familial Alzheimer disease mutant does not enhance apoptosis. J Neurosci 18: 9790-9799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busser J, Geldmacher DS, Herrup K ( 1998) Ectopic cell cycle proteins predict the sites of neuronal cell death in Alzheimer's disease brain. J Neurosci 18: 2801-2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxbaum J, Thinakaran G, Koliatsos V, O'Callahan J, Slunt HH, Price DL, Sisodia SS ( 1990) Processing of Alzheimer β/A4 amyloid precursor protein: Modulation by agents that regulate protein phosphorylation. Proc Natl Acad Sci USA 87: 6003-6006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X, Südhof TC ( 2001) A transcriptionally active complex of APP with Fe65 and histone acetyltransferase Tip60. Science 293: 115-120. [DOI] [PubMed] [Google Scholar]
- Chen Y, McPhie DL, Hirschberg J, Neve RL ( 2000) The amyloid precursor protein-binding protein APP-BP1 drives the cell cycle through the S-M checkpoint and causes apoptosis in neurons. J Biol Chem 275: 8929-8935. [DOI] [PubMed] [Google Scholar]
- Chow N, Korenberg JR, Chen XN, Neve RL ( 1996) APP-BP1, a novel protein that binds to the carboxyl-terminal region of the amyloid precursor protein. J Biol Chem 271: 11339-11346. [DOI] [PubMed] [Google Scholar]
- Chow N, Cox C, Callahan LM, Weimer JM, Guo L, Coleman PD ( 1998) Expression profiles of multiple genes in single neurons of Alzheimer's disease. Proc Natl Acad Sci USA 95: 9620-9625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copani A, Uberti D, Sortino MA, Bruno V, Nicoletti F, Merno M ( 2001) Activation of cell-cycle-associated proteins in neuronal death: a mandatory or dispensable path? Trends Neurosci 24: 25-31. [DOI] [PubMed] [Google Scholar]
- Cotman CW ( 1998) Apoptosis decision cascades and neuronal degeneration in Alzheimer's disease. Neurobiol Aging 19: S29-S32. [DOI] [PubMed] [Google Scholar]
- Cotman CW, Anderson AJ ( 1995) A potential role for apoptosis in neurodegeneration and Alzheimer's disease. Mol Neurobiol 10: 19-45. [DOI] [PubMed] [Google Scholar]
- Farinelli SE, Greene LA ( 1996) Cell cycle blockers mimosine, ciclopirox, and deferoxamine prevent the death of PC12 cells and postmitotic sympathetic neurons after the removal of trophic support. J Neurosci 16: 1150-1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giambarella U, Yamatsuji T, Okamoto T, Matsui T, Ikezu T, Murayama Y, Levine MA, Katz A, Gautam N, Nishimoto I ( 1997) G-protein βγ complex-mediated apoptosis by familial Alzheimer's disease mutant of APP. EMBO J 16: 4897-4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Q, Fu W, Xie J, Luo H, Sells SF, Geddes JW, Bondada V, Rangnekar VM, Mattson MP ( 1998) Par-4 is a mediator of neuronal degeneration associated with the pathogenesis of Alzheimer disease. Nature 4: 957-962. [DOI] [PubMed] [Google Scholar]
- Jaffer ZM, Chernoff J ( 2002) p21-activated kinases: three more join the Pak. Int J Biochem Biol 34: 713-717. [DOI] [PubMed] [Google Scholar]
- Jakobi R, Chen CJ, Tuazon PT, Traugh JA ( 1996) Molecular cloning and sequencing of the cytostatic G-protein-activated protein kinase PAK I. J Biol Chem 271: 6206-6211. [DOI] [PubMed] [Google Scholar]
- Joneson T, McDonough M, Bar-Sagi D, Van Aelst L ( 1998) Kinase suppressor of Ras inhibits the activation of extracellular ligand-regulated (ERK) mitogen-activated protein (MAP) kinase by growth factors, activated Ras, and Ras effectors. J Biol Chem 273: 7743-7749. [DOI] [PubMed] [Google Scholar]
- Kinoshita A, Whelan CM, Berezovska O, Hyman BT ( 2002) The γ-secretase-generated carboxyl-terminal domain of the amyloid precursor protein induces apoptosis via Tip60 in H4 cells. J Biol Chem 277: 28530-28536. [DOI] [PubMed] [Google Scholar]
- Kondratick CM, Vandre DD ( 1996) Alzheimer's disease neurofibrillary tangles contain mitosis-specific phosphoepitopes. J Neurochem 67: 2405-2416. [DOI] [PubMed] [Google Scholar]
- Kozlowski MR, Spanoyannis AF, Manly SP, Fidel SA, Neve RL ( 1992) The neurotoxic carboxy terminal fragment of the Alzheimer amyloid precursor binds specifically to a neuronal cell surface molecule: pH dependence of the neurotoxicity and the binding. J Neurosci 12: 1679-1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang J, Nishimoto I, Okamoto T, Regazzi R, Kiraly C, Weller U, Wollheim CB ( 1995) Direct control of exocytosis by receptor-mediated activation of the heterotrimeric GTPases Gi and G(o) or by the expression of their active G alpha subunits. EMBO J 14: 3635-3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leberer E, Dignard D, Harcus D, Thomas DY, Whiteway M ( 1992) The protein kinase homologue Ste20p is required to link the yeast pheromone response G-protein βγ subunits to downstream signalling components. EMBO J 11: 4815-4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeuw T, Wu C, Schrag JD, Whiteway M, Thomas DY, Leberer E ( 1998) Interaction of a G-protein β-subunit with a conserved sequence in Ste20/PAK family protein kinases. Nature 391: 191-196. [DOI] [PubMed] [Google Scholar]
- Liu WK, Williams R, Hall F, Dickson D, Yen SH ( 1995) Detection of a cdc2-related kinase associated with Alzheimer paired helical filaments. Am J Pathol 146: 228-238. [PMC free article] [PubMed] [Google Scholar]
- Manser E, Chong C, Zhao ZS, Leung T, Michael G, Hall C, Lim L ( 1995) Molecular cloning of a new member of the p21-Cdc42/Rac-activated kinase (PAK) family. J Biol Chem 270: 25070-25078. [DOI] [PubMed] [Google Scholar]
- Manser E, Huang HY, Loo TH, Chen XQ, Dong JM, Leung T, Lim L ( 1997) Expression of constitutively active α-PAK reveals effects of the kinase on actin and focal complexes. Mol Cell Biol 17: 1129-1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin GA, Bollag G, McCormick FA, Abo A ( 1995) A novel serine kinase activated by rac/Cdc42Hs-dependent autophosphorylation is related to PAK65 and Ste20. EMBO J 14: 1970-1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPhie DL, Lee RKK, Eckman CB, Olstein DH, Durham SP, Yager D, Younkin SG, Wurtman RJ, Neve RL ( 1997) Neuronal expression of β-amyloid precursor protein Alzheimer mutations causes intracellular accumulation of a C-terminal fragment containing both the amyloid β and cytoplasmic domains. J Biol Chem 272: 24743-24746. [DOI] [PubMed] [Google Scholar]
- McPhie DL, Golde T, Eckman CB, Yager D, Younkin SG, Neve RL ( 2001) The β-secretase cleavage product of the amyloid precursor protein mediates neuronal apoptosis caused by familial Alzheimer's disease mutations. Mol Brain Res 97: 103-113. [DOI] [PubMed] [Google Scholar]
- McShea A, Harris PL, Webster KR, Wahl AF, Smith MA ( 1997) Abnormal expression of the cell cycle regulators P16 and CDK4 in Alzheimer's disease. Am J Pathol 150: 1933-1939. [PMC free article] [PubMed] [Google Scholar]
- Neve RL, Perrone-Bizzozero NI, Finklestein S, Zwiers H, Bird E, Kurnit DM, Benowitz LI ( 1987) The neuronal growth-associated protein GAP-43 (B-50, F1): Neuronal specificity, developmental regulation and regional expression of the human and rat cDNAs. Mol Brain Res 2: 177-183. [DOI] [PubMed] [Google Scholar]
- Nishimoto I, Okamoto T, Matsuura Y, Okamoto T, Murayama Y, Ogata E ( 1993) Alzheimer amyloid protein precursor complexes with brain GTP-binding protein G(o). Nature 362: 75-79. [DOI] [PubMed] [Google Scholar]
- Nishimura I, Uetsuki T, Dani SU, Ohsawa Y, Saito I, Okamura H, Uchiyama Y, Yoshikawa K ( 1998) Degeneration in vivo of rat hippocampal neurons by wild-type Alzheimer amyloid precursor protein overexpressed by adenovirus-mediated gene transfer. J Neurosci 18: 2387-2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto T, Takeda S, Murayama Y, Ogata E, Nishimoto I ( 1995) Ligand-dependent G-protein coupling function of amyloid transmembrane precursor. J Biol Chem 270: 4205-4208. [DOI] [PubMed] [Google Scholar]
- Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai L-H ( 1999) Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 402: 615-622. [DOI] [PubMed] [Google Scholar]
- Raina AK, Hochman A, Ickes H, Zhu X, Ogawa O, Cash AD, Shimohama S, Perry G, Smith MA ( 2003) Apoptotic promoters and inhibitors in Alzheimer's disease: who wins out? Prog Neuropsychopharmacol Biol Psychiatry 27: 251-254. [DOI] [PubMed] [Google Scholar]
- Ramer SW, Davis RW ( 1993) A dominant truncation allele identifies a gene, STE20, that encodes a putative protein kinase necessary for mating in Saccharomyces cerevisiae Proc Natl Acad Sci USA 90: 452-456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren M, Xu G, Zeng, De Lemos-Chiarandini C, Adesnik M, Sabatini DD ( 1998) Hydrolysis of GTP on rab11 is required for the direct delivery of transferrin from the pericentriolar recycling compartment to the cell surface but not from sorting endosomes. Proc Natl Acad Sci USA 95: 6187-6192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohn TT, Ivins KJ, Bahr BA, Cotman CW, Cribbs DH ( 2000) A monoclonal antibody to amyloid precursor protein induces neuronal apoptosis. J Neurochem 75: 2331-2342. [DOI] [PubMed] [Google Scholar]
- Roth KA ( 2001) Caspases, apoptosis, and Alzheimer disease: causation, correlation, and confusion. J Neuropathol Exp Neurol 60: 829-838. [DOI] [PubMed] [Google Scholar]
- Russo T, Faraonio R, Minopoli G, De Candia P, De Renzis S, Zambrano N ( 1998) Fe65 and the protein network centered around the cytosolic domain of the Alzheimer's β-amyloid precursor protein. FEBS Lett 434: 1-7. [DOI] [PubMed] [Google Scholar]
- Scheinfeld MH, Roncarati R, Vito P, Lopez PA, Abdallah M, D'Adamio L ( 2002) Jun NH2-terminal kinase (JNK) interacting protein 1 (JIP1) binds the cytoplasmic domain of the Alzheimer's β-amyloid precursor protein (APP). J Biol Chem 277: 3767-3775. [DOI] [PubMed] [Google Scholar]
- Seemann J, Weber K, Gerke V ( 1997) Annexin I targets S100C to early endosomes. FEBS Lett 413: 185-190. [DOI] [PubMed] [Google Scholar]
- Sells MA, Knaus UG, Bagrodia S, Ambrose DM, Bokoch GM, Chernoff J ( 1997) Human p21-activated kinase (Pak1) regulates actin organization in mammalian cells. Curr Biol 7: 202-210. [DOI] [PubMed] [Google Scholar]
- Smith TW, Lippa CF ( 1995) Ki-67 immunoreactivity in Alzheimer's disease and other neurodegenerative disorders. J Neuropathol Exp Neurol 54: 297-303. [DOI] [PubMed] [Google Scholar]
- Su JH, Anderson AJ, Cummings BJ, Cotman CW ( 1994) Immunohistochemical evidence for DNA fragmentation in neurons in the AD brain. NeuroReport 5: 2529-2533. [DOI] [PubMed] [Google Scholar]
- Taussig R, Sanchez S, Rifo M, Gilman AG ( 1992) Inhibition of the ω-conotoxin-sensitive calcium current by distinct G-proteins. Neuron 8: 799-809. [DOI] [PubMed] [Google Scholar]
- Teo M, Manser E, Lim L ( 1995) Identification and molecular cloning of a p21cdc42/rac1-activated serine/threonine kinase that is rapidly activated by thrombin in platelets. J Biol Chem 270: 26690-26697. [DOI] [PubMed] [Google Scholar]
- Ulrich O, Reinsch S, Urbe S, Zerial M, Parton RG ( 1996) Rab 11 regulates recycling through the pericentriolar recycling endosomes. J Cell Biol 135: 913-924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent I, Rosado M, Davies P ( 1996) Mitotic mechanisms in Alzheimer's disease? J Cell Biol 132: 413-425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent I, Jicha G, Rosado M, Dickson D ( 1997) Aberrant expression of mitotic cdc2/cyclin B1 kinase in degenerating neurons of Alzheimer's disease brain. J Neurosci 17: 3588-3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent I, Zheng JH, Dickson DW, Kress Y, Davies P ( 1998) Mitotic phosphoepitopes precede paired helical filaments in Alzheimer's disease. Neurobiol Aging 19: 287-296. [DOI] [PubMed] [Google Scholar]
- Yamatsuji T, Okamoto T, Takeda S, Fukumoto H, Iwatsubo T, Suzuki N, Asami-Odaka A, Ireland S, Kinane TB, Nishimoto I ( 1996) G-protein-mediated neuronal DNA fragmentation induced by familial Alzheimer's disease-associated mutants of APP. Science 272: 1349-1352. [DOI] [PubMed] [Google Scholar]
- Yang Y, Geldmacher DS, Herrup K ( 2001) DNA replication precedes neuronal cell death in Alzheimer's disease. J Neurosci 21: 2661-2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z-S, Manser E, Chen X-Q, Chong C, Leung T, Lim L ( 1998) A conserved negative regulatory region in αPAK: inhibition of PAK kinases reveals their morphological roles downstream of cdc42 and rac1. Mol Cell Biol 18: 2153-2163. [DOI] [PMC free article] [PubMed] [Google Scholar]