

Abstract
Alteration by β-amyloid (Aβ) of signaling via nicotinic acetylcholine receptors (nAChRs) has been implicated in the early stages of Alzheimer's disease. nAChRs function both post- and presynaptically in the nervous system; however, little is known about the functional consequence of the interaction of Aβ with these receptors, particularly those on presynaptic nerve terminals. In view of the strong correlation between loss of synaptic terminals and dementia, together with the reduction in nAChRs in Alzheimer's disease, the possibility exists that presynaptic nAChRs may be targets for Aβ. To explore this possibility, we assessed the effect of Aβ peptides on nicotine-evoked changes in presynaptic Ca2+ level via confocal imaging of isolated presynaptic nerve endings from rat hippocampus and neocortex. Aβ1-42 appeared to inhibit presynaptic nAChR activation by nicotine. Surprisingly, picomolar Aβ1-42 was found to directly evoke sustained increases in presynaptic Ca2+ via nAChRs, revealing that the apparent inhibitory action of Aβ1-42 was the result of an occlusion of nicotine to further stimulate the receptors. The direct effect of Aβ was found to be sensitive to α-bungarotoxin, mecamylamine, and dihydro-β-erythroidine, indicating involvement of α7-containing nAChRs and non-α7-containing nAChRs. Prior depolarization strongly attenuated subsequent Aβ-evoked responses in a manner dependent on the amplitude of the initial presynaptic Ca2+ increase, suggesting that nerve activity or Ca2+ channel density may control the impact of Aβ on presynaptic nerve terminal function. Together, these results suggest that the sustained increases in presynaptic Ca2+ evoked by Aβ may underlie disruptions in neuronal signaling via nAChRs in the early stages of Alzheimer's disease.
Keywords: nicotinic receptor, amyloid, presynaptic, hippocampus, calcium imaging, Alzheimer's disease
Introduction
One prominent feature of Alzheimer's disease is the presence of neuritic plaques containing β-amyloid (Aβ) peptides. Aβ peptides (39-43 amino acids in length) are generated by proteolytic cleavage of the Aβ precursor protein, a transmembrane glycoprotein present in multiple isoforms (Selkoe, 1998; Walter et al., 2001). The dominant peptide fragment present within the neuritic plaques, as insoluble fibrils, is the 42-residue species Aβ1-42 (Iwatsubo et al., 1994; Gravina et al., 1995; Selkoe, 1998). The Aβ fibrils most likely result from self-aggregation of the Aβ1-42 (Teplow, 1998; Huang et al., 2000). There is, however, only a weak correlation between fibrillar Aβ content and cognitive dysfunction (Lue et al., 1999; McLean et al., 1999). In contrast, the severity of dementia does correlate with the degree of loss of presynaptic terminals (Terry et al., 1991; Sze et al., 1997) as well as with the total load of soluble Aβ (Lue et al., 1999; McLean et al., 1999). Moreover, transgenic strains exist (e.g., Tg2576) wherein Aβ levels are elevated without plaque formation or nerve cell loss, yet learning and memory deficits are evident (Irizarry et al., 1997; Kotilinek et al., 2002; Westerman et al., 2002). Consequently, it has been hypothesized that Aβ may be largely acting in a soluble form (dimers and/or small oligomers, also referred to as ADDLs) (Garzon-Rodriguez et al., 1997) to disrupt neuronal signaling (Lambert et al., 1998; Klein et al., 2001), particularly at an early stage in Alzheimer's disease, with nicotinic acetylcholine receptors (nAChRs) as a major target (Auld et al., 1998). Other possible targets have been suggested, such as microglial scavenger receptors (El Khoury et al., 1998) and the receptor for advanced glycation end products (Lue et al., 2001).
Evidence for a strong interaction between Aβ and specific nAChRs, particularly α7 subunit-containing nAChRs, has accumulated (Dineley et al., 2001; Liu et al., 2001; Pettit et al., 2001; Wang et al., 2000a,b). Studies examining binding of Aβ1-42 to nAChRs expressed on clonal cell lines indicated that Aβ has a picomolar affinity for α7-containing nAChRs (Wang et al., 2000a). In both rat hippocampal slices (Pettit et al., 2001) and cultured neurons (Liu et al., 2001), nanomolar Aβ was shown to inhibit nicotine-evoked currents, including α7-AChRs and non-α7-AChRs, in a reversible, apparently noncompetitive manner. In addition, acute treatment with Aβ was found to activate the MAP kinase cascade in mouse hippocampal slices via α7-nAChRs, whereas chronically elevated Aβ in a mouse model of Alzheimer's disease led to downregulation of MAP kinase with concomitant upregulation of α7-nAChRs in aged animals (Dineley et al., 2001). These latter data suggest that Aβ may first activate and then inhibit nAChRs, although no direct activation by Aβ of nAChRs was noted in any of the aforementioned reports using primary tissue.
To determine the consequence of Aβ action on presynaptic nicotinic receptors on nerve terminal signaling, we investigated the effects of Aβ peptides on nicotine-induced Ca2+ responses in individual isolated nerve terminals from rat hippocampus and neocortex.
Materials and Methods
Materials. Synthetic Aβ peptides were purchased from BACHEM (King of Prussia, PA). For all experiments, Aβ peptides were suspended in physiological saline at 100-1000× stock concentration, thoroughly bath sonicated, and then immediately diluted for use. Under these conditions, the final solutions (picomolar-nanomolar peptide) remain clear, with no visible particles when viewed by phase-contrast microscopy (Lorenzo and Yankner, 1994), for the course of an experiment (15-30 min). Fluo-3 and fluo-4 were obtained from Molecular Probes, Inc. (Eugene, OR) and are kept as 1 mm stock solutions in DMSO at -20°C. Cell-Tak was purchased from Collaborative Biomedical Products (Bedford, MA). Nicotine, m-chlorophenyl biguanide, α-bungarotoxin, mecamylamine, and dihydro-β-erythroidine were purchased from Sigma (St. Louis, MO). Conotoxins and agatoxin-TK were purchased from Alomone Labs (Jerusalem, Israel). All drugs and toxins were suspended in physiological saline just before use.
Synaptosome preparation. Hippocampi, striata, or cortices were dissected out of brains from adult male Sprague Dawley rats (Taconic Farms, Germantown, NY) and immediately placed in ice-cold 0.32 m sucrose, following a protocol approved by the Drexel University College of Medicine (formerly MCP Hahnemann University) Institutional Animal Care and Use Committeee. Tissue was then homogenized in 0.32 m sucrose using a glass Teflon tissue grinder. Synaptosomes were isolated according to the method described by Dunkley et al. (1986). The preparations were washed into oxygenated HEPES-buffered saline [HBS; composition (in mm): 142 NaCl, 2.4 KCl, 1.2 K2PO4, 1 MgCl2, 5 d-glucose, and 10 HEPES, pH 7.4]. This procedure yields synaptosomes, ∼90% of which have shown to be intact and functional, based on the stability and consistency of Ca2+ responses of dye-loaded synaptosomes to multiple rounds of stimulation as gauged using confocal imaging (Nayak et al., 2000, 2001).
Calcium imaging. Synaptosomes were loaded with fluorescent Ca2+ indicator dye (fluo-3 or fluo-4) at 5 μm by incubating with the acetoxymethyl ester derivative in HBS for 30-45 min at 37°C. Dye-loaded preparations were washed in HBS containing 1 mm CaCl2 and then plated onto Cell-Tak-coated coverslips. Relative changes in internal Ca2+ in individual synaptosomes were assessed using confocal imaging (Rondé and Nichols, 1998; Nayak et al., 2001) via a Nikon PCM 2000 laser-scanning confocal imaging system connected to a Nikon Diaphot 300 microscope. In brief, the preparations on coverslips were mounted in a rapid-exchange Warner (36 μl volume) perfusion system attached to the microscope and subjected to perfusion with HBS containing Ca2+ at 3-5 ml/min. Imaging was commenced, and after obtaining a baseline series of five images, stimulatory agents were applied by rapid switching between manifolds on the perfusion system. Complete exchange of the perfusion chamber took place in <1 sec. Images were typically collected at 4-sec intervals, although in several experiments 15-sec intervals were used. A given experiment corresponded to a series of images captured from a single preparation. The fluorescent intensities associated with a given structure, determined from digitized images using OPTIMAS image analysis software (Optimas Co., Seattle, WA), were expressed as normalized values (F/F0; where F0 = fluorescence intensity at t0). All time series were corrected for photobleaching.
Statistics. Data sets were compared using matched Student's t tests. Significance was indicated when p was minimally <0.05.
Results
Nicotine induces robust increases in Ca2+ level in a subpopulation (10-25%) of isolated nerve terminals (synaptosomes), which slowly decay (several minutes) depending on concentration and receptor subtype (Nayak et al., 2001). The nicotine-induced Ca2+ responses in striatal synaptosomes were found to be effectively independent of voltage-gated Ca2+ channels, indicating that the sustained changes in internal Ca2+ likely parallel, although certainly with some delay, the influx of Ca2+ via the nicotinic receptor channel. Nicotine-induced Ca2+ responses in hippocampal or cortical synaptosomes, in contrast, display significant dependence on voltage-gated Ca2+ channels, as gauged by sensitivity to Ca2+ channel toxins wherein responses were inhibited to 30-40% of controls, indicating a strong depolarizing component to the responses in the nerve terminals in these brain regions. Sustained elevation in internal Ca2+ seems to be a common feature underlying responses in nerve terminals to activation of presynaptic nicotinic receptors (McGehee et al., 1995; Gray et al., 1996; Coggan et al., 1997; Léna and Changeux, 1997; Mansvelder and McGehee, 2000; Kiyosawa et al., 2001; Diíaz-Hernández et al., 2002), as well as the closely related 5-HT3 serotonin receptors when expressed on presynaptic nerve endings (Rondé and Nichols, 1998, 2001).
Treatment with Aβ1-42 appeared to strongly inhibit nicotine-induced Ca2+ responses in individual hippocampal synaptosomes in a readily reversible manner (Fig. 1A, left), with the nicotine-induced Ca2+ responses recovering to a significant degree as soon as 30 sec after washing out the Aβ1-42 (Fig. 1B). The action of Aβ1-42 on nicotinic receptors present on the presynaptic terminals appears to be specific, because it had no significant effect on presynaptic 5-HT3 serotonin receptor-induced Ca2+ responses in the same synaptosomes (Fig. 1A, right), as assessed using the highly selective 5-HT3 receptor agonist m-chlorophenyl biguanide. The latter conclusion was made based on the finding that 5-HT3 serotonin receptors colocalize with nicotinic receptors at presynaptic sites (Nayak et al., 2000). The lack of significant effect of Aβ1-42 on 5-HT3 receptors is also consistent with previous reports (Wang et al., 2000a; Liu et al., 2001). In addition, Aβ1-42 has been shown to have no effect on glutamate receptors in a hippocampal preparation (Pettit et al., 2001). The inhibitory effect of Aβ1-42 was concentration dependent, with low nanomolar levels producing near complete blockade of the nicotine-induced Ca2+ responses, whereas significant inhibition was evident down into the picomolar range (IC50, ∼10 pm; Fig. 1C). Finally, a control peptide having a reversed sequence (40-1; ′Aβ′40-1 used at 100 nm) from that of Aβ1-42 had no significant effect on nicotine-induced Ca2+ responses in the hippocampal synaptosomes (Fig. 1D). In contrast, 100 nm Aβ12-28 was nearly as effective as 100 nm Aβ1-42 in inhibiting the nicotine-induced responses (Fig. 1D). Aβ12-28 was previously shown to interact strongly with α7-nAChRs (Wang et al., 2000b), suggesting that it may contain the nicotinic receptor-binding motif of the Aβ peptides. Aβ12-28 was also shown to block nicotine-induced currents in hippocampal neurons, as, if not more, effectively as Aβ1-42 (Pettit et al., 2001). These results indicate that presynaptic nicotinic receptors are selectively inhibited by soluble Aβ. However, because each Ca2+ response is normalized to baseline, the question arises as to whether Aβ has a direct effect on presynaptic Ca2+ levels, resulting in an occlusion of the nicotine-induced Ca2+ responses, in contrast to direct inhibition of the presynaptic nicotinic receptor. As described later, the inhibitory effect of Aβ was indeed because of an occlusion, i.e., full activation of the nAChRs, preventing further activation by nicotine.
Figure 1.

Inhibitory effect of Aβ on nicotine-evoked increases in Ca2+ level in individual isolated hippocampal nerve endings. A, Successive stimulation with 500 nm nicotine (left) or 100 nm of the 5-HT3 agonist m-chlorophenyl biguanide (mCPBG; right) in the absence or presence of 100 nm Aβ1-42 during an intervening 10-min wash period and the second stimulation. Top, Representative successive Ca2+ responses in individual synaptosomes. Bottom, Averaged responses expressed as means ± SEM before and after incubation of Aβ1-42. Left graph, n = 45; Right graph, n = 15. Reversibility (B), concentration dependence (C), and peptide specificity (D) of the inhibitory effect of 100 nm Aβ1-42 were also examined. In D, peptides were present at 100 nm. Note that for A, B, Ca2+ responses to the second stimulation were renormalized (*), although a maintained Ca2+ increase occurred with Aβ (Fig. 3). In C,D, data are expressed as percentage of control plateau values (% inhibition). B, n = 13; C: 0 pm, n = 26; 10 pm, n = 8; 100 pm, n = 5; 1 nm, n = 8; D: Aβ1-42, n = 73; Aβ12-28, n = 15; ′Aβ′40-1, n = 6.
Nicotine-induced Ca2+ responses in a subset of hippocampal synaptosomes (12 ± 6% SD) seem to involve both α7-containing nAChRs and non-α7-containing nAChRs (Fig. 2A), as gauged by sensitivity of the responses to various nicotinic receptor antagonists (α7, α-bungarotoxin; non-α7, dihydro-β-erythroidine or mecamylamine), consistent with studies of nicotine-induced neurotransmitter release (Clarke and Reuben, 1996; Gray et al., 1996; Radcliffe et al., 1999; Fabian-Fine et al., 2001; Kulak et al., 2001). The profiles of sensitivity of presynaptic nicotine-induced Ca2+ responses were similar between hippocampal and cortical synaptosomes (data not shown). Interestingly, the apparent inhibitory effect of Aβ1-42 was more pronounced with hippocampal and cortical synaptosomes as compared with striatal synaptosomes (Fig. 2B). Although Aβ1-42 has been shown to inhibit non-α7-containing nAChRs (Pettit et al., 2001), it displays the highest affinity for α7-containing nAChRs (Wang et al., 2000b), and α7-containing nAChRs appear to be a minor presence, at best, on striatal nerve terminals (Nayak et al., 2000; cf. Marchi et al., 2002), perhaps explaining the much smaller effect of Aβ1-42 on striatal synaptosomes, particularly when used at 100 pm. Evidence for involvement of α7-containing nAChRs on hippocampal synaptosomes is indicated by α-bungarotoxin sensitivity of direct effects of Aβ1-42 to increase synaptosomal Ca2+ (Fig. 5).
Figure 2.

Sensitivity of nicotine-evoked presynaptic Ca2+ increases to various nicotinic antagonists and to Aβ for preparations from various regions of the brain. A, Average maximal Ca2+ responses to 500 nM nicotine in hippocampal synaptosomes in the absence (n = 34) or presence of 5 μm dihydro-β-erythroidine (DHβE;n=26), 500nmα-bungarotoxin (BgTx; n= 9), 10 μm mecamylamine (Mec; n = 18), or BgTx plus Mec (n = 11) during the second stimulation, using the successive stimulation protocol described in the legend to Figure 1. B, Inhibitory effects of 100 nm Aβ1-42 on nicotine-evoked Ca2+ responses in synaptosomes from hippocampus (Hippo; n = 17), cortex (CTX; n = 13), or striatum (Str; n = 10). Qualitatively similar results were obtained using 100 pm Aβ1-42. *p < 0.05; t test with paired control.
Figure 5.
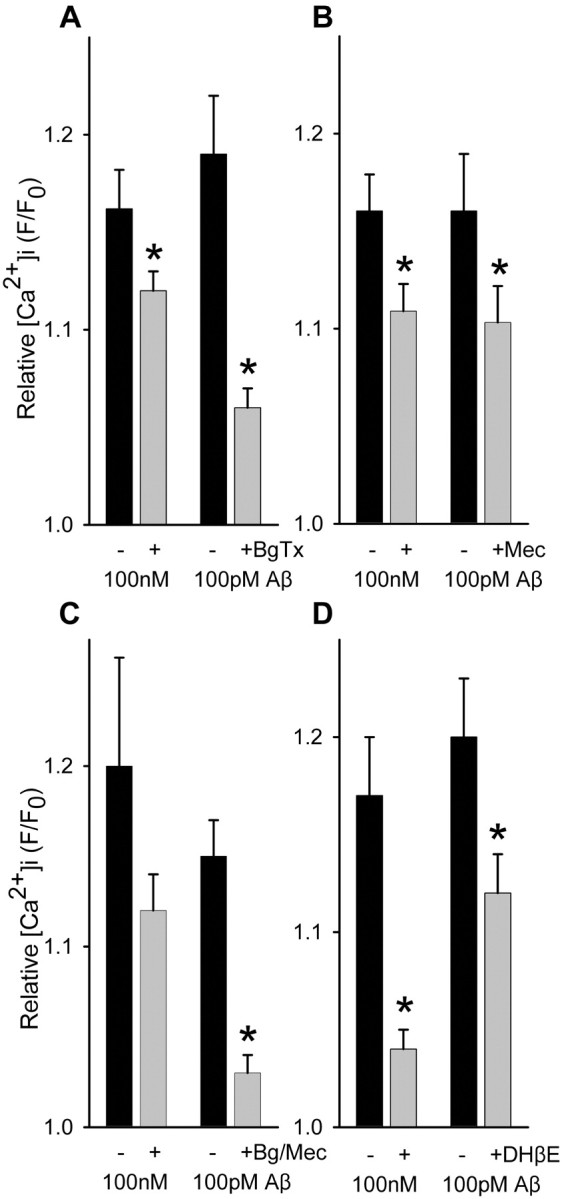
Sensitivity of Aβ-evoked Ca2+ increases in isolated hippocampal nerve endings to nicotinic receptor antagonists. Average maximal responses Ca2+ responses to Aβ1-42 in the absence or presence of 500 nm α-bungarotoxin (BgTx; A) (100 pm, n = 14; 100 nm, n = 22), 10 μm mecamylamine (Mec; B) (100 pm, n = 14; 100 nm, n = 14), 500 nm α-bungarotoxin plus 10 μm mecamylamine (Bg/Mec; C) (100 pm, n = 12; 100 nm, n = 6), or 5 μm dihydro-β-erythroidine (DHβE; D) (100 pm, n = 22; 100 nm, n = 16). *p < 0.05; t test with paired control.
Aβ1-42 was found to induce directly substantial increases in Ca2+ level (Fig. 3) in a subset of hippocampal synaptosomes (17 ± 10% SD), with sustained elevation in Ca2+ level evident for over 10 min of incubation (Fig. 3A). Aβ1-42-induced synaptosomal Ca2+ responses were comparable in number and average magnitude to those evoked by nicotine stimulation. The direct effect of Aβ1-42 was concentration dependent, with measurable responses in the low picomolar range (Fig. 3C). Similar findings were obtained using cortical synaptosomes; however, the increases in Ca2+ level in response to 100 pM Aβ1-42 (F/F0, 1.13 ± 0.03 SEM; n = 23) were substantially lower than those seen in response to 100 nm Aβ1-42 (F/F0, 1.45 ± 0.03 SEM; n = 30), in contrast to what was observed for Aβ1-42-evoked responses in hippocampal synaptosomes at these same concentrations, indicating a significantly lower potency of Aβ for cortical presynaptic terminals. These direct effects of Aβ were unaltered by filtration of the Aβ1-42-containing HBS perfusion solution (Fig. 3E), indicating that the Aβ was not acting via an aggregated form. Aβ12-28 was also able to induce directly increases in synaptosomal Ca2+ (Fig. 3B). Interestingly, synaptosomal Ca2+ responses to picomolar Aβ1-42 were dependent on external Ca2+, whereas they were only partially dependent on external Ca2+ for concentrations of Aβ1-42 in the high nanomolar to micromolar range (Fig. 4A). However, Zn2+ (50 μm), known to block alleged Aβ channels that form at relatively high (micromolar) concentrations of Aβ1-42 (Lin et al., 2001), had no effect on synaptosomal Ca2+ responses to Aβ1-42 at any concentration (Fig. 3D). Moreover, the stimulatory effect of Aβ1-42 was partially dependent on voltage-gated Ca2+ channels at 100 pM but not at 100 nM (Fig. 4B), as gauged by the insensitivity of the Aβ1-42-induced Ca2+ responses to inhibition by a mixture of Ca2+ channel blockers (agatoxin-TK, o-conotoxin MVIIC, o-conotoxin GVIA) shown previously to block K+-induced synaptosomal Ca2+ responses (Rondé and Nichols 1998). Similar results were obtained when Ca2+ channels were blocked with micromolar Cd2+ plus Co2+ (data not shown). The relative sensitivity of nicotine-induced Ca2+ responses to Ca2+ blockers was similar to that seen for 100 pm Aβ1-42-induced responses (data not shown). Finally, the direct effect of Aβ1-42 was sensitive to α-bungarotoxin (Fig. 5A), the open-channel blocker mecamylamine (Fig. 5B), and dihydro-β-erythroidine (Fig. 5D), to extents comparable with the inhibitory effects of these nicotinic antagonists on nicotine-induced Ca2+ responses (compare Fig. 2), indicating a significant involvement of α7-containing nAChRs and non-α7-containing nAChRs. Interestingly, the effect of α-bungarotoxin was more pronounced when Aβ1-42 was used at 100 pm, whereas the effect of dihydro-β-erythroidine was more pronounced when Aβ1-42 was used at 100 nm. Combining α-bungarotoxin with mecamylamine led to an inhibition profile similar to that of α-bungarotoxin alone (Fig. 5C). These results indicate that soluble Aβ at picomolar concentration directly activates presynaptic nAChRs predominantly of the α7-containing subtype to increase nerve terminal Ca2+ in a manner dependent on entry of Ca2+ through both the nAChR receptor channel and voltage-gated Ca2+ channels, whereas soluble Aβ at nanomolar concentration directly activates presynaptic nAChRs predominantly of the non-α7-containing subtype to increase nerve terminal Ca2+ largely via the nAChR receptor channel.
Figure 3.

Aβ-evoked Ca2+ increases in individual hippocampal nerve endings. Composites of Ca2+ responses in individual synaptosomes to Aβ1-42 (A) and Aβ12-28 (B) over an extended time course. Top, Representative initial phases of successive Ca2+ responses in an individual synaptosome to 100 nM Aβ1-42. Data sampling were 4-sec intervals. C, Concentration dependence: 1 pm, n = 26; 10 pm, n = 20; 100 pm, n = 116; 100 nm, n = 175. D, Insensitivity to prior treatment with 50 μm ZnCl2 (n = 15). E, Average maximal Ca2+ responses to 100 nM Aβ1-42 (n = 11) or 100 pM Aβ1-42 (n = 14) before or after filtration of the Aβ solutions through 0.2 μm filters.
Figure 4.

Dependence of Aβ-evoked Ca2+ increases in isolated hippocampal nerve endings on Ca2+ entry and voltage-gated Ca2+ channels. A, Average maximal responses Ca2+ responses to 100 nm (n = 16) or 100 pm (n = 7) Aβ1-42 in the absence or presence of 1 mm external Ca2+. B, Average maximal Ca2+ responses to 100 nM Aβ1-42 (n = 22) or 100 pM Aβ1-42 (n = 17) in the absence or presence of a mixture of voltage-gated Ca2+ channel blockers (toxins: 200 nm agatoxin TK; 500 nm conotoxin GVIA; 500 nm conotoxin MVIIC), which have previously been shown to block completely K+ depolarization-induced synaptosomal Ca2+ responses (Rondé and Nichols, 1998). *p < 0.05; t test with paired control.
Because the apparent inhibitory effect of Aβ1-42 on nicotine-induced responses (Fig. 1) was observed under conditions in which each response was necessarily normalized to the initial baseline, owing to uncertainty over baseline values after extended treatment periods without imaging, sequential addition of Aβ and nicotine was performed while imaging. The addition of 500 nm nicotine just after the initial application of Aβ1-42 to hippocampal synaptosomes resulted in no further increase in Ca2+ level over that obtained in response to Aβ1-42 alone (Fig. 6A), indicating that Aβ1-42 strongly attenuates Ca2+ responses to nanomolar concentrations of nicotine in these nerve endings. In contrast, increasing nicotine to the micromolar range revealed nicotine-induced Ca2+ responses on top of the initial Aβ1-42-induced responses. The control reverse peptide, ′Aβ′40-1, caused no significant change in the synaptosomal Ca2+ level and did not significantly inhibit responses to nicotine added on top of the ′Aβ′40-1 (Fig. 6A). Likewise, the addition of Aβ1-42 just after the initial application of 500 nm nicotine had no significant effect on nicotine-induced Ca2+ responses when applied at 10 pm to 1 nm on top of nicotine, but it did increase synaptosomal Ca2+ when applied at 10-100 nM (Fig. 6C,D). In either case, in which relatively high concentrations of the second stimulatory agent were used (Fig. 3C), all synaptosomes responding to Aβ also responded to nicotine. Taken together, these results indicate that activation of presynaptic nicotinic receptors by picomolar Aβ1-42 occludes the action of nicotine on the same receptors when applied at typical maximal concentration (500 nm) and vice versa. That the occlusion was not the result of a saturating Ca2+ response or dye saturation is evident in the ability of elevated concentrations to elicit responses (Fig. 6B,D). Thus, the apparent inhibitory effect of Aβ on nicotine-induced Ca2+ responses (Fig. 1) was a consequence of prior activation of the nAChRs by Aβ.
Figure 6.

Ca2+ responses in isolated nerve endings to Aβ, followed by various concentrations of nicotine (A, B) compared with Ca2+ responses to nicotine, followed by various concentrations of Aβ (C,D). Averaged responses in A (n = 6) and C (n = 13) are means ± SEM. Averaged maximal responses in B (500 nm, n = 2; 5 μm, n = 4; 50 μm, n = 3; 200 μm, n = 5) and D (10 pm, n = 14;1nm, n = 9; 10 nm, n = 9; 100 nm, n = 8) denote the second agent as a percentage of the maximal response to the first agent. Insets, Representative responses.
Prior depolarization with elevated external K+ resulted in significant attenuation of subsequent Aβ1-42-evoked Ca2+ responses (Fig. 7A) in a manner that was dependent on the amplitude of the initial K+-evoked response (Fig. 7B). Under any condition, application of 30 mm (or higher) KCl will substantially depolarize all of the synaptosomes in the preparation; however, a wide range of response amplitudes are observed (cf. Nichols and Mollard, 1996). When Ca2+ responses were first evoked with Aβ1-42 and followed by subsequent depolarization, the K+-evoked Ca2+ responses were correspondingly attenuated (Fig. 7C) in a manner somewhat dependent on the amplitude of the initial Aβ1-42-evoked response (Fig. 7D). In this case, even the largest K+-evoked Ca2+ responses were substantially smaller after initial Aβ1-42 stimulation in comparison with typical Ca2+ responses evoked by K+ alone (compare Fig. 7A). These results suggest that the effect of nerve activity on presynaptic responses to Aβ is strictly dependent on the extent of the increase in Ca2+ level in response to presynaptic depolarization. In contrast, the presence of Aβ can attenuate presynaptic Ca2+ increases in response to nerve activity. To some degree, these effects may have resulted from prior activation of voltage-gated Ca2+ channels by either K+ depolarization or Aβ-induced depolarization via nAChRs, with the contribution of voltage-gated Ca2+ channels to the evoked Ca2+ signals varying widely across synaptosomes (Nichols and Mollard, 1996).
Figure 7.

Ca2+ responses in isolated hippocampal nerve endings to Aβ after prior depolarization by KCl compared with Ca2+ responses to depolarization by KCl after Aβ. The averaged responses in A (n = 26) and C (n = 18) are means ± SEM. Individual peak responses to the second agent in B (n = 71) and D (n = 16) are correlated to the initial peak responses of the first agent, with each response to the second agent normalized to the response to the first agent as a percentage.
Discussion
Aβ accumulates in plaques near dystrophic neurites and nerve endings (Brendza et al., 2003). Moreover, synaptic loss is strongly correlated with severity of dementia (Terry et al., 1991; Sze et al., 1997). As yet, effects of Aβ on the presynaptic nerve terminal have not been defined clearly. The present study undertook to explore the possibility that soluble Aβ peptide may modulate presynaptic Ca2+ levels, evaluating first effects on nicotine-induced responses, as an extension of previous work demonstrating an inhibitory interaction between Aβ and nAChRs, and later direct effects of Aβ itself. The observed sustained increases in presynaptic Ca2+ in response to soluble Aβ1-42 raise the question as to whether such an effect would ultimately be toxic. Interestingly, prior depolarization attenuated the effect of Aβ, indicating that the degree of nerve activity and/or relative density of voltage-gated Ca2+ channels on the nerve endings may also play a role in modulating the action of soluble Aβ at presynaptic sites, in particular its toxicity.
Several previous reports have demonstrated an action of soluble Aβ peptides on nicotinic receptors (Dineley et al., 2001; Liu et al., 2001; Pettit et al., 2001). In particular, Aβ was found to block nicotine-stimulated increases in spontaneous neurotransmitter release from cultured hippocampal neurons (Liu et al., 2001), as the first clear indication of an action of Aβ on presynaptic nAChRs. In contrast, a recent report demonstrates a direct activation of nAChR-mediated currents in an oocyte expression system by Aβ (Dineley et al., 2002). Our work confirms and extends these previous findings, showing specifically that Aβ can directly induce increases in presynaptic Ca2+. At picomolar Aβ concentration, the resultant Ca2+ responses appeared to largely involve α7-containing nAChRs, owing to the sensitivity of the Ca2+ responses to nicotinic antagonist α-bungarotoxin. Interestingly, however, prior treatment with Aβ inhibited subsequent action of high nanomolar nicotine, a typical maximal concentration for presynaptic nAChR-evoked responses (Nayak et al., 2001), by occlusion. That relatively high concentrations of nicotine could overcome, to some degree, the occlusion effect of Aβ, in the same synaptosomes, also confirms the involvement of nAChRs. Under the conditions used, these experiments cannot distinguish whether the action of Aβ is competitive or noncompetitive at presynaptic nAChRs, however, but do indicate significant overlap with the α-bungarotoxin site in the case of α7-nAChRs. Previous results indicated that Aβ may be noncompetitive for acetylcholine (Liu et al., 2001), which is consistent with findings of at least two binding sites for Aβ on α7-nAChRs in ligand-binding studies (Wang et al., 2000b), and the results presented here do not exclude this possibility. Further study will be needed to determine how Aβ specifically interacts with presynaptic nAChRs.
Although α7-containing nAChRs seem to be largely involved in the presynaptic actions of Aβ at picomolar concentration, the results indicate that the action of Aβ when present at high nanomolar concentration on presynaptic Ca2+ may be largely at non-α7-containing nAChRs, owing to a more pronounced sensitivity of the responses to the nicotinic antagonist dihydro-β-erythroidine. The results demonstrating that relatively high concentration of Aβ can overcome the occlusion by nicotine of the action of picomolar levels of Aβ are, in contrast, not inconsistent with the possibility that Aβ may be activating a pathway in the nerve endings that is independent of nAChRs. Other indications of direct stimulatory effects of Aβ that are most likely independent of nicotinic receptors have also been noted. For example, Aβ peptide was found to evoke a nonselective inward ion current in rat cortical neurons (Furukawa et al., 1994). The Aβ peptide-induced current was proposed to have resulted from the formation of cation channels, reported previously, wherein Aβ was used at particularly elevated concentrations (micromolar) (Lin et al., 2001). Here, however, Aβ induced increases in presynaptic Ca2+ in a reversible manner that was insensitive to Zn2+, ruling out formation of channels.
An important question is whether Aβ-induced alterations in presynaptic Ca2+ affect the release of neurotransmitter. In one study, Aβ was actually found to have an inhibitory effect on K+-evoked acetylcholine release from hippocampal slices (Kar et al., 1996, 1998), although the time frame of these measurements was rather extended. In view of the sustained increases in presynaptic Ca2+ in response to Aβ that were observed here, it may be that neurotransmitter release evoked subsequent to Aβ treatment would be inhibited perhaps as a consequence of a longer-term depressive and/or toxic effect on the nerve terminal. In contrast, it would be predicted that neurotransmitter release would be initially evoked by Aβ as a result of the increased presynaptic Ca2+. Previous observations of an inhibitory effect of Aβ peptide on nicotine-stimulated increases in spontaneous neurotransmitter release in cultured hippocampal neurons did not, however, note a direct effect of Aβ alone (Liu et al., 2001). Effects, or lack thereof, of Aβ on exocytosis in individual nerve terminals from brain would require imaging using, for example, amphipathic fluorescent dyes such as FM1-43 (Rondé and Nichols, 1998).
Another issue is whether sustained presynaptic Ca2+ in response to Aβ will activate certain intracellular signaling pathways, such as protein kinases. Previous observations using hippocampal slices demonstrated extracellular signal-regulated kinase (ERK) activation in response to Aβ (Dineley et al., 2001). Whether Aβ activates protein kinase pathways, such as ERK, in the presynaptic nerve terminal is currently under investigation.
Despite several lines of evidence, including our own, indicating the possibility of a direct interaction between Aβ and nAChRs, such an interaction remains to be proven. It remains possible that Aβ interacts with a protein that associates with nAChRs and perhaps other signaling molecules. In contrast, nicotinic antagonists were able to inhibit Aβ-evoked presynaptic Ca2+ increases, indicating, as noted previously, some overlap of Aβ interaction with the ligand binding site(s). Sorting out the actual presynaptic targets for Aβ will entail a series of detailed molecular studies using, for example, preparations containing nAChRs modified by site-directed mutagenesis, especially in view of differences noted in the structural components of nAChRs that are essential for agonist binding as compared with antagonist binding (Arias, 1997).
Although speculative at present, our results might suggest that as Aβ begins to accumulate near synaptic sites, nicotine-mediated presynaptic regulation will be initially disrupted, although in a manner dependent on nerve activity-coupled presynaptic Ca2+ changes. Nicotinic receptors, particularly presynaptic nAChRs, have been implicated in long-term potentiation in the hippocampus (Fujii et al., 1999; Matsuyama et al., 2000) as well as the ventral tegmental area (Mansvelder and McGehee, 2000), and, thus, Aβ disruption of signaling via these receptors could have consequences for cognitive function. As Aβ further accumulates, toxic effects on nerve terminals may arise. For example, prolonged Aβ has been shown to alter nerve terminal mitochondrial function (Mattson et al., 1998). A key question is to what extent such toxicity might be a consequence of sustained presynaptic Ca2+ as compared with Aβ gaining entry into the nerve terminal (Kienlen-Campard et al., 2002; Nagele et al., 2002). It is likely that disruption of neuronal function at multiple levels via several pathways ultimately underlies the pathology arising over the course of Alzheimer's disease.
Footnotes
This work was supported in part by the Smokeless Tobacco Research Council. We thank Drs. Michael White and Robert Moreland for helpful comments on this manuscript. We thank Brett Brown for technical assistance.
Correspondence should be addressed to Dr. Robert A. Nichols, Department of Pharmacology and Physiology, Drexel University College of Medicine (formerly MCP Hahnemann University), 245 North 15th Street, Philadelphia, PA 19102. E-mail: robert.nichols@drexel.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/236740-08$15.00/0
References
- Arias HR ( 1997) Topology of ligand binding sites on the nicotinic acetylcholine receptor. Brain Res Rev 25: 133-191. [DOI] [PubMed] [Google Scholar]
- Auld DS, Kar S, Quirion R ( 1998) β-Amyloid peptides as direct cholinergic neuromodulators: a missing link? Trends Neurosci 21: 43-49. [DOI] [PubMed] [Google Scholar]
- Brendza RP, O'Brien C, Simmons K, McKeel DW, Bales KR, Paul SM, Olney JW, Sanes JR, Holtzman DM ( 2003) PDAPP; YFP double transgenic mice: a tool to study amyloid-associated changes in axonal, dendritic, and synaptic structures. J Comp Neurol 456: 375-383. [DOI] [PubMed] [Google Scholar]
- Clarke PB, Reuben M ( 1996) Release of [3H]-noradrenaline from rat hippocampal synaptosomes by nicotine: mediation by different nicotinic receptor subtypes from striatal [3H]-dopamine release. Br J Pharmacol 117: 595-606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coggan JS, Paysan J, Conroy WG, Berg DK ( 1997) Direct recording of nicotinic responses in presynaptic nerve terminals. J Neurosci 17: 5798-5806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di íaz-Hernández M, Pintor J, Castro E, Miras-Portugal MT ( 2002) Colocalisation of functional nicotinic and ionotropic nucleotide receptors in isolated cholinergic synaptic terminals. Neuropharmacology 42: 20-33. [DOI] [PubMed] [Google Scholar]
- Dineley KT, Westerman M, Bui D, Bell K, Hsiao Ashe K, Sweatt JD ( 2001) β-Amyloid activates the mitogen-activated protein kinase cascade via hippocampal α7 nicotinic acetylcholine receptors: in vitro and in vivo mechanisms related to Alzheimer's disease. J Neurosci 21: 4125-4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dineley KT, Bell KA, Bui D, Sweatt JD ( 2002) β-Amyloid peptide activates α7 nicotinic acetylcholine receptors expressed in Xenopus oocytes. J Biol Chem 277: 25056-25061. [DOI] [PubMed] [Google Scholar]
- Dunkley PR, Jarvie PE, Heath JW, Kidd GJ, Rostas JAP ( 1986) A rapid method for isolation of synaptosomes on Percoll gradients. Brain Res 372: 115-129. [DOI] [PubMed] [Google Scholar]
- El Khoury J, Hickman SE, Thomas CA, Loike JD, Silverstein SC ( 1998) Microglia, scavenger receptors, and the pathogenesis of Alzheimer's disease. Neurobiol Aging 19: S81-S84. [DOI] [PubMed] [Google Scholar]
- Fabian-Fine R, Skehel P, Errington ML, Davies HA, Sher E, Stewart MG, Fine A ( 2001) Ultrastructural distribution of the α7 nicotinic acetylcholine receptor subunit in rat hippocampus. J Neurosci 21: 7993-8003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii S, Ji Z, Morita N, Sumikawa K ( 1999) Acute and chronic nicotine exposure differentially facilitate the induction of LTP. Brain Res 846: 137-143. [DOI] [PubMed] [Google Scholar]
- Furukawa K, Abe Y, Akaike N ( 1994) Amyloid β protein-induced irreversible current in rat cortical neurones. NeuroReport 5: 2016-2018. [DOI] [PubMed] [Google Scholar]
- Garzon-Rodriguez W, Sepulveda-Becerra M, Milton S, Glabe CG ( 1997) Soluble amyloid Aβ(1-40) exists as a stable dimer at low concentrations. J Biol Chem 272: 21037-21044. [DOI] [PubMed] [Google Scholar]
- Gravina SA, Ho L, Eckman CB, Long KE, Otvos Jr L, Younkin LH, Suzuki N, Younkin SG ( 1995) Amyloid β protein (Aβ) in Alzheimer's disease brain. Biochemical and immunocytochemical analysis with antibodies specific for forms ending at Aβ 40 or Aβ 42(43). J Biol Chem 270: 7013-7016. [DOI] [PubMed] [Google Scholar]
- Gray R, Rajan AS, Radcliffe KA, Yakehiro M, Dani JA ( 1996) Hippocampal synaptic transmission enhanced by low concentrations of nicotine. Nature 383: 713-716. [DOI] [PubMed] [Google Scholar]
- Huang TH, Yang DS, Plaskos NP, Go S, Yip CM, Fraser PE, Chakrabartty A ( 2000) Structural studies of soluble oligomers of the Alzheimer β-amyloid peptide. J Mol Biol 297: 73-87. [DOI] [PubMed] [Google Scholar]
- Irizarry MC, McNamara M, Fedorchak K, Hsiao K, Hyman BT ( 1997) APPSw transgenic mice develop age-related Aβ deposits and neuropil abnormalities, but no neuronal loss in CA1. J Neuropathol Exp Neurol 56: 965-973. [DOI] [PubMed] [Google Scholar]
- Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y ( 1994) Visualization of Aβ 42(43) and Aβ40 in senile plaques with end-specific Aβ monoclonals: evidence that an initially deposited species is Aβ42(43). Neuron 13: 45-53. [DOI] [PubMed] [Google Scholar]
- Kar S, Seto D, Gaudreau P, Quirion R ( 1996) β-Amyloid-related peptides inhibit potassium-evoked acetylcholine release from rat hippocampal slices. J Neurosci 16: 1034-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar S, Issa AM, Seto D, Auld DS, Collier B, Quirion R ( 1998) Amyloid β-peptide inhibits high-affinity choline uptake and acetylcholine release in rat hippocampal slices. J Neurochem 70: 2179-2187. [DOI] [PubMed] [Google Scholar]
- Kienlen-Campard P, Miolet S, Tasiaux B, Octave JN ( 2002) Intracellular amyloid-β1-42, but not extracellular soluble amyloid-β peptides, induces neuronal apoptosis. J Biol Chem 277: 15661-15670. [DOI] [PubMed] [Google Scholar]
- Kiyosawa A, Katsurabayashi S, Akaike N, Pang ZP, Akaike N ( 2001) Nicotine facilitates glycine release in the rat spinal dorsal horn. J Physiol (Lond) 536: 101-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein WL, Krafft GA, Finch CE ( 2001) Targeting small Aβ oligomers: the solution to an Alzheimer's disease conundrum? Trends Neurosci 24: 219-224. [DOI] [PubMed] [Google Scholar]
- Kotilinek LA, Bacskai B, Westerman M, Kawarabayashi T, Younkin L, Hyman BT, Younkin S, Ashe KH ( 2002) Reversible memory loss in a mouse transgenic model of Alzheimer's disease. J Neurosci 22: 6331-6335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulak JM, McIntosh JM, Yoshikami D, Olivera BM ( 2001) Nicotine-evoked transmitter release from synaptosomes: functional association of specific presynaptic acetylcholine receptors and voltage-gated calcium channels. J Neurochem 77: 1581-1589. [DOI] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL ( 1998) Diffusible, nonfibrillar ligands derived from Aβ1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA 95: 6448-6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Léna C, Changeux J-P ( 1997) Role of Ca2+ ions in nicotinic facilitation of GABA release in mouse thalamus. J Neurosci 17: 576-585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H, Bhatia R, Lal R ( 2001) Amyloid β protein forms ion channels: implications for Alzheimer's disease pathophysiology. FASEB J 15: 2433-2444. [DOI] [PubMed] [Google Scholar]
- Liu Q-S, Kawai H, Berg DK ( 2001) β-Amyloid peptide blocks the response of α7-containing nicotinic receptors on hippocampal neurons. Proc Natl Acad Sci USA 98: 4734-4739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzo A, Yankner BA ( 1994) β-Amyloid neurotoxicity requires fibril formation and is inhibited by Congo red. Proc Natl Acad Sci USA 91: 12, 243-12: 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lue L-F, Kuo Y-M, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J ( 1999) Soluble amyloid β peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol 155: 853-862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lue L-F, Walker DG, Brachova L, Beach TG, Rogers J, Schmidt AM, Stern DM, Yan SD ( 2001) Involvement of microglial receptor for advanced glycation endproducts (RAGE) in Alzheimer's disease: identification of a cellular activation mechanism. Exp Neurol 171: 29-45. [DOI] [PubMed] [Google Scholar]
- Mansvelder HD, McGehee DS ( 2000) Long-term potentiation of excitatory inputs to brain reward areas by nicotine. Neuron 27: 349-357. [DOI] [PubMed] [Google Scholar]
- Marchi M, Risso F, Viola C, Cavazzani P, Raiteri M ( 2002) Direct evidence that release-stimulating α7* nicotinic cholinergic receptors are localized on human and rat brain glutamatergic axon terminals. J Neurochem 80: 1071-1078. [DOI] [PubMed] [Google Scholar]
- Matsuyama S, Matsumoto A, Enomoto T, Nishizaki T ( 2000) Activation of nicotinic acetylcholine receptors induces long-term potentiation in vivo in the intact mouse dentate gyrus. Eur J Neurosci 12: 3741-3747. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Partin J, Begley JG ( 1998) Amyloid β-peptide induces apoptosis-related events in synapses and dendrites. Brain Res 807: 167-176. [DOI] [PubMed] [Google Scholar]
- McGehee DS, Heath MJS, Gelber S, Devay P, Role LW ( 1995) Nicotine enhancement of fast excitatory synaptic transmission in CNS by presynaptic receptors. Science 269: 1692-1696. [DOI] [PubMed] [Google Scholar]
- McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL ( 1999) Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol 46: 860-866. [DOI] [PubMed] [Google Scholar]
- Nagele RG, D'Andrea MR, Anderson WJ, Wang H-Y ( 2002) Intracellular accumulation of β-amyloid1-42 in neurons is facilitated by the α7 nicotinic acetylcholine receptor in Alzheimer's disease. Neuroscience 110: 199-211. [DOI] [PubMed] [Google Scholar]
- Nayak SV, Rondé P, Spier AD, Lummis SCR, Nichols RA ( 2000) Nicotinic receptors co-localize with 5-HT3 serotonin receptors on striatal nerve terminals. Neuropharmacology 39: 2681-2690. [DOI] [PubMed] [Google Scholar]
- Nayak SV, Dougherty JJ, McIntosh JM, Nichols RA ( 2001) Ca2+ changes induced by different presynaptic nicotinic receptors in separate populations of individual striatal nerve terminals. J Neurochem 76: 1860-1870. [DOI] [PubMed] [Google Scholar]
- Nichols RA, Mollard P ( 1996) Direct observation of 5-HT3 receptor-induced increases in calcium levels in individual brain nerve terminals. J Neurochem 67: 581-592. [DOI] [PubMed] [Google Scholar]
- Pettit DL, Shao Z, Yakel J ( 2001) β-Amyloid1-42 peptide directly modulates nicotinic receptors in the rat hippocampal slice. J Neurosci 21: RC120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radcliffe KA, Fisher JL, Gray R, Dani JA ( 1999) Nicotinic modulation of glutamate and GABA synaptic transmission of hippocampal neurons. Ann NY Acad Sci 868: 591-610. [DOI] [PubMed] [Google Scholar]
- Rondé P, Nichols RA ( 1998) High calcium permeability of serotonin 5-HT3 receptors on presynaptic nerve terminals from rat striatum. J Neurochem 70: 1094-1103. [DOI] [PubMed] [Google Scholar]
- Rondé P, Nichols RA ( 2001) Postsynaptic target regulates functional responses induced by 5-HT3 serotonin receptors on axonal varicosities of NG108-15 hybrid neuroblastomal cells. Neuroscience 102: 979-987. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ ( 1998) The cell biology of β-amyloid precursor protein and presenilin in Alzheimer's disease. Trends Cell Biol 8: 447-453. [DOI] [PubMed] [Google Scholar]
- Sze CI, Troncoso JC, Kawas C, Mouton P, Price DL, Martin LJ ( 1997) Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline and Alzheimer disease. J Neuropathol Exp Neurol 56: 933-944. [DOI] [PubMed] [Google Scholar]
- Teplow DB ( 1998) Structural and kinetic features of amyloid β-protein fibrillogenesis. Amyloid Int J Exp Clin Invest 5: 121-142. [DOI] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R ( 1991) Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 30: 572-580. [DOI] [PubMed] [Google Scholar]
- Walter J, Kaether C, Steiner H, Haass C ( 2001) The cell biology of Alzheimer's disease: uncovering the secrets of secretases. Curr Opin Neurobiol 11: 585-590. [DOI] [PubMed] [Google Scholar]
- Wang H-Y, Lee DHS, Davis CB, Shank RP ( 2000a) Amyloid peptide Aβ1-42 binds selectively and with picomolar affinity to α7 nicotinic acetylcholine receptors. J Neurochem 75: 1155-1161. [DOI] [PubMed] [Google Scholar]
- Wang H-Y, Lee DHS, D'Andrea MR, Peterson PA, Shank RP, Reitz AB ( 2000b) β-Amyloid1-42 binds to α7 nicotinic acetylcholie receptor with high affinity. J Biol Chem 275: 5626-5632. [DOI] [PubMed] [Google Scholar]
- Westerman M, Cooper-Blacketer D, Mariash A, Kotilinek L, Kawarabayashi T, Younkin LH, Carlson G, Younkin S, Ashe KH ( 2002) The relationship between Aβ and memory in the Tg2576 mouse model of Alzheimer's disease. J Neurosci 22: 1858-1867. [DOI] [PMC free article] [PubMed] [Google Scholar]