

Abstract
AMPA receptor-binding protein (ABP) is a multi-postsynaptic density-95/discs large/zona occludens (PDZ) protein that binds to the glutamate receptor 2/3 (GluR2/3) subunits of the AMPA receptor and is implicated in receptor membrane anchorage. A palmitoylated form of ABP localizes to spine heads, whereas a nonpalmitoylated form is found in intracellular clusters. Here, we investigate intracellular cluster formation by ABP and the ability of ABP to associate with GluR2 while in these clusters. We show that ABP interacts with intracellular membranes via the ABP linker I (LI)-set II (SII) subdomain, a region consisting of ABP linker 1 and PDZ4, -5, and -6. This suggests that cluster formation results from LI-SII ABP association with the membrane of a vesicular structure. We present evidence that ABP can self-associate at intracellular membrane surfaces via interactions involving SII. ABP in such membrane clusters can bind and retain GluR2 that has trafficked endocytotically from the plasma membrane. Phosphorylation of GluR2 at serine 880, proximal to the ABP binding site, has been implicated by others in the release of ABP from GluR2 and the mobilization of AMPA receptors for trafficking. We show that binding of GluR2 to ABP blocks phosphorylation of serine 880. This suggests that ABP can stabilize its own association with GluR2. We discuss a model in which ABP can form a protein scaffold at a vesicular membrane that is capable of binding GluR2, leading to formation of an intracellular AMPA receptor pool. Receptors in such a pool may contribute to receptor endocytotic and exocytotic trafficking and recycling.
Keywords: AMPA receptors, ABP, GRIP, trafficking, endocytosis, PDZ domain, phosphorylation
Introduction
AMPA receptors (AMPARs) provide the major fast excitatory currents in the CNS (for review, see Dingledine et al., 1999). Changes in AMPAR synaptic abundance contribute to long-lasting changes in the strength of excitatory synapses, including long-term potentiation (LTP) and long-term depression (LTD). AMPAR synaptic levels are controlled by interactions with receptor-binding proteins (for review, see Barry and Ziff, 2002; Malinow and Malenka, 2002). Two such factors, AMPA receptor-binding protein (ABP) (Srivastava et al., 1998) and the closely related glutamate receptor (GluR)-interacting protein (GRIP) (Dong et al., 1997), are multi-postsynaptic density-95/discs large/zona occludens (PDZ) proteins that bind GluR2 and GluR3 and are implicated in receptor tethering and transport. Although the precise contributions of ABP to AMPAR trafficking are not yet established, recent studies suggest a complex role in which ABP occupies both synaptic and intracellular locations. Immunoelectron microscopy has revealed the presence of ABP proximal to the postsynaptic membrane of hippocampal pyramidal neurons (Srivastava et al., 1998) where it may anchor GluR2 at the synapse (Osten et al., 2000). A synaptic function was also indicated by the detection by immunocytochemistry of synaptophysin-positive ABP puncta that colocalize with GluR2/3 in pyramidal neurons of cortex (Burette et al., 2001). An ABP isoform, pABP-L [palmitoylated ABP-long (seven PDZ) form], has been identified (deSouza et al., 2002), and this isoform may fulfill a synaptic AMPAR-tethering function. pABP-L is targeted by palmitoylation of its N-terminal leader peptide to the plasma membrane at heads of spines where it colocalizes with exogenous, cell surface GluR2. A similarly palmitoylated form of GRIP has been described previously (Yamazaki et al., 2001). The existence of a second, nonsynaptic form of ABP was suggested by the detection in proximal dendrites and somata of cortical pyramidal neurons of ABP-immunopositive puncta that do not colocalize with synaptophysin (Burette et al., 2001). This form of ABP may correspond to a second isoform, ABP-L, which is neither palmitoylated nor found in spines, but colocalizes with GluR2 at intracellular membranes (deSouza et al., 2002). The release of AM-PARs from anchorage by ABP-GRIP at the synaptic plasma membrane may be one step in LTD (Matsuda et al., 2000; Kim et al., 2001). Similarly, the release of AMPARs from intracellular ABP-GRIP tethers may contribute to the dedepression of synapses that have undergone LTD (Daw et al., 2000; Braithwaite et al., 2002).
Here, we employ exogenous expression of ABP mutants to analyze the mechanism of intracellular targeting of ABP, and the capacity of intracellular ABP to associate with GluR2. We identify a subdomain of ABP-L, linker I (LI)-set II (SII), that targets ABP to intracellular membranes. We show that ABP-L can capture exogenous GluR2 intracellularly after GluR2 is endocytosed from the plasma membrane. ABP-L can also block GluR2 phosphorylation and thereby suppress a process that may be required to mobilize AMPARs for transport. Thus ABP-L, acting through its LI-SII region, may stabilize an intracellular pool of AMPA receptors. Retention of AMPARs in such an intracellular pool may contribute to LTD, whereas regulated release of AMPARs from this pool may reverse LTD during synaptic dedepression.
Materials and Methods
Recombinant DNA. To make green fluorescent protein (GFP)-tagged full-length ABP, we amplified fragments by PCR, inserted them into the pEGFP-N1 vector at the EcoRI and SalI sites, and placed GFP at the C terminus. To make other GFP-tagged ABP fragments, ABP fragments were inserted into pEGFP-C2 at the EcoRI and SalI sites, placing GFP at the N terminus of the fragments. These fragments include the first set of PDZ (SI), SI together with linker I (SI-LI), LI together with the second set of PDZ (LI-SII) and mutant forms (see Fig. 1), the second set of PDZ (SII), and SII together with linker II (LII-SII). To insert the ABP (full length)-GFP and the GFP-ABP fragments into Sindbis virus vectors, we amplified the inserts including ABP or ABP fragments plus GFP, using the pEGFP-N1-ABP and pEGFP-C2-ABP fragments as the templates by PCR and subcloned the products into the vector pSinRep5.
Figure 1.

Schematic representation of isoforms and fragments of ABP tagged with GFP. The structures of pABP-L, ABP-L, and ABP-S are shown, with GFP tags at the carboxyl terminus. ABP subdomains including the N-terminal leader, PDZ sets I and II, and linkers I and II are indicated. The N-terminal 18 aa exon of pABP-L, shown as a small shaded box, is palmitoylated (zigzag line), whereas that of ABP-L (empty box) lacks the palmitoylation site. ABP-S is a splice variant that lacks the N-terminal leader, a portion of linker II, and PDZ7. Fragments of ABP, including SI-LI, LI-SII, and SII-LII, were tagged with GFP at their N termini.
Heterologous cell culture and transfection. Heterologous cells including HeLa cells and human embryonic kidney 293T (HEK293T) cells were maintained in DMEM (Invitrogen, San Diego, CA). HeLa cells and HEK293T cells were transfected with Superfect (Qiagen, Hilden, Germany) according to the manufacturer's instructions.
Culture and Sindbis virus infection of hippocampal primary neurons. Hippocampal primary neurons were prepared from E18 Sprague Dawley rat tissues as described previously (Osten et al., 2000), plated at a density of 80,000 per well in six-well dishes, and maintained in Neurobasal medium with B27 for ∼3 weeks before they were used for Sindbis virus infection. All of the pseudovirions were generated as described in the Sindbis Expression System manual (Invitrogen). Briefly, BHK cells were electroporated with RNA transcribed in vitro from pSinRep5 vectors encoding different forms of ABP and from the helper plasmid DH26S. The medium containing pseudovirions was collected and used for infection as described previously (Osten et al., 2000). Hippocampal neurons were infected at ∼21 d in vitro (DIV) with 20 μl of α-MEM virus stock diluted in 500 μl of Neurobasal-B27 medium per well of a six-well dish.
Acid-stripping assay. Twenty-four hours after infection, cells were incubated with the primary monoclonal antibody against Myc tag (see below) for 15 min at 37°C and then washed with cold 1× PBS once. Cells were then treated with cold acid-stripping buffer (0.5 m NaCl, 0.2N acitic acid in PBS) for 3 min, followed by washing five times with cold 1× PBS.
12-O-Tetradecanoylphorbol-13-acetate treatment. Cells were treated with 12-O-tetradecanoylphorbol-13-acetate (TPA) 24 hr after infection with 100 ng/ml TPA (Sigma, St. Louis, MO) for 15 min at 37°C, followed by fixation and permeabilization (see below).
Antibodies and immunostaining. For live-cell staining of MycGluR2, cells were incubated with a monoclonal antibody against the Myc epitope tag (9E10; 4 μg/ml; Santa Cruz Biotechnology, Santa Cruz, CA) for 15 min at 37°C and washed two times with 1× PBS, followed by the acid-stripping procedure and fixation and permeabilization. For expression of GFP-tagged constructs, cells were washed with cold 1× PBS three times, fixed with 4% paraformaldehyde, and then directly visualized using a Nikon (Melville, NY) PCM2000 confocal microscope. For Flag-tagged or Myc-tagged constructs, cells were permeabilized with 0.2% Triton X-100 and incubated with the anti-Flag M2 antibody (0.4 μg/ml; Eastman Kodak, Rochester, NY), or Myc epitope tag antibody (9E10; 1 μg/ml) or the polyclonal Myc antibody (A-14; 1 μg/ml), or anti-phospho-GluR2 polyclonal serum at 1:1000 dilution (Perez et al., 2001) for 45 min at 37°C. For singly infected neurons, cells were stained with rhodamine red-conjugated donkey anti-mouse secondary antibody at 1:300 dilution for 45 min at 37°C. For doubly infected neurons, in the case of cells treated with acid-stripping buffer, we used rhodamine red-conjugated donkey anti-mouse IgG and Cy5-conjugated donkey anti-rabbit secondary antibodies. In cells treated with TPA, rhodamine red-conjugated donkey anti-rabbit IgG and Cy5-conjugated donkey anti-mouse IgG secondary antibodies were used. All of the secondary antibodies were purchased from Jackson ImmunoResearch (West Grove, PA).
Subcellular fractionation. Cortical neurons were infected at ∼21 DIV with 100 μl of virus stock diluted in 2 ml of Neurobasal-B27 medium per 100 mm dish. Twenty-four hours after infection, cortical neurons were rinsed with cold PBS and 10 mm HEPES and then incubated with 10 mm HEPES for 5 min on ice. Cells were collected and homogenized four times with a Dounce homogenizer. Cell lysates were then spun down at 2500 rpm for 10 min, and 1 ml of supernatant was loaded on the top of 11 ml sucrose gradients (0.12-1.2 m sucrose in 10 mm HEPES). Samples were centrifuged for 4 hr at 23,000 rpm. One milliliter of each fraction (total, 12 fractions) was collected from the top to the bottom of the gradient and transferred to Eppendorf tubes. Each fraction was then mixed with 4 ml of 150 mm NaCl and 10 mm HEPES and centrifuged at 47,000 rpm for 2.5 hr at 4°C. Pellets were solubilized in SDS sample buffer and subjected to SDS-PAGE, and proteins were then transferred onto nitrocellulose for analysis by Western blotting.
Western blotting. Nitrocellulose membranes were probed with α-GFP rabbit polyclonal antibody [0.25 μg/ml; gift of Dr. P. Silver (Harvard University School of Medical, Boston, MA)] in 5% nonfat milk for ABP constructs tagged with GFP. Proteins were detected using SuperSignal West Pico Luminol/Enhancer solution (Pierce, Rockford, IL).
Coimmunoprecipitation. HEK293T cells grown on 10 cm dishes were transfected with plasmids encoding MycGluR2 alone; or MycGluR2 and pABP-L-GFP, ABP-L-GFP, or GFP-LI-SII. Forty-eight hours after transfection, cell lysates were coimmunoprecipitated as described previously (deSouza et al., 2002). Briefly, cells were solubilized with Triton X-100 and immunoprecipitated with monoclonal antibody against Myc (9E10; 1 μg/ml; Santa Cruz Biotechnology). The proteins from the immunoprecipitate (IP) were separated on 8% SDS-PAGE and transferred onto nitrocellulose. The membranes were probed with anti-GFP serum raised in rabbit against His6 GFP (1:5000). As controls, cell lysates were also analyzed by doing Western blots with antibody against Myc or against GFP to check the levels of protein expression and immunoprecipitation.
Results
LI and SII both contribute to intracellular clustering of ABP
The structural organization of the ABP splice variants, pABP-L and ABP-L, including the N-terminal leader, the two sets of PDZ domains, SI and SII, and the two linker regions, LI and LII, is shown in Figure 1. Also shown is the six PDZ form, ABP-S (ABP-short), which lacks the leader, a portion of linker II, and the seventh PDZ. We compared the subcellular localizations of these variants of ABP after tagging with GFP to facilitate visualization and the simultaneous detection of other proteins by immunofluorescence. GFP was added at the C terminus of wild-type ABPs, because the N termini contribute directly to wild-type ABP localization (see below). Constructs were expressed using Sindbis virus vectors in cultured hippocampal neurons (15-21 DIV). ABPS-GFP (six PDZ form; no leader) formed internal clusters in the cell body and dendrites (Fig. 2aA). ABP-L-GFP (seven PDZ form) was, for the most part, localized in similar clusters, but it also distributed significantly proximal to the surface of dendrites (Fig. 2aB). In contrast, pABP-L-GFP (palmitoylated, seven PDZ form) was concentrated along the plasma membrane of dendrites and spines (Fig. 2aC). This suggested that the N-terminal leader directed ABP to the plasma membrane, and that plasma membrane targeting was enhanced by palmitoylation (deSouza et al., 2002).
Figure 2.
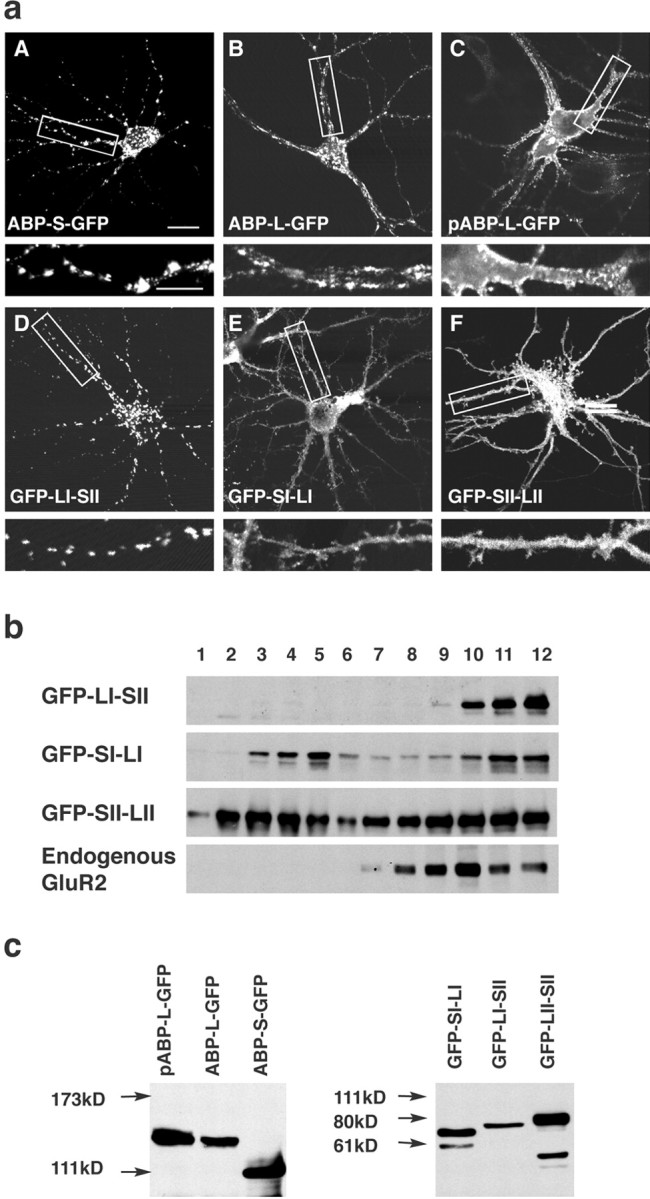
The LI-SII ABP fragment forms internal clusters in hippocampal neurons. a, Hippocampal neurons were infected with Sindbis viruses expressing different full-length isoforms of ABPs or fragments of ABP, all tagged with GFP as indicated. ABPs or fragments of ABP were detected by GFP fluorescence with confocal microscopy. ABP-S-GFP was seen in internal clusters in the cell body and dendrites (A). ABP-L-GFP was for the most part localized in clusters similar to those of ABP-S-GFP, but also distributed to a significant extent at the surface of dendrites (B). pABP-L-GFP was located at the membrane of the cell body and dendrites, as well as in spines (C), and GFP-LI-SII was found extensively in internal clusters (D), whereas GFP-SI-LI and GFP-SII-LII were directed to the cell body, dendrites, and spines (E, F). The bottom panels are enlargements of the areas indicated in the top panels. Scale bars: top panels, 20 μm; bottom panels, 10 μm. b, Sucrose gradient fractionation of lysates of cortical neurons expressing different fragments of ABP. Cortical neurons were infected with viruses that express ABP fragments. GFP-LI-SII and endogenous GluR2 sedimented rapidly, in fractions 9-12, whereas GFP-SI-LI and GFP-SII-LII were widely distributed and present in both slowly and rapidly sedimenting fractions, fractions 2-5 and 9-12, respectively. c, HEK293T cells were transfected with cDNAs encoding different forms of ABP and ABP fragments, all tagged with GFP. After 48 hr of transfection, cell lysates were separated on SDS gel, and proteins were visualized by Western blot with anti-GFP antibody. All of the expressed protein bands displayed the anticipated mobility.
Although palmitoylation targets pABP-L to the plasma membrane (deSouza et al., 2002), the subdomain(s) that directs ABP to intracellular clusters is not known. This intracellular targeting domain is likely to be present in ABP-S, because ABP-S forms internal clusters when expressed in neurons (Fig. 2aA). To identify such a domain, we determined the subcellular locations of a series of ABP-S fragments. These fragments included SI-LI, LI-SII, and SII-LII, all of which were tagged with GFP at their N termini (Fig. 1). GFP-LI-SII formed intracellular clusters in the cell body and dendritic shafts, but did not enter spines or associate with the plasma membrane (Fig. 2aD). The construct, Flag-LI-SII, in which the epitope tag is not expected to have a steric effect, was similarly localized (see below). GFP-SI-LI and GFP-SII-LII, in contrast, were distributed diffusely in the cell body, dendrites, and spines (Fig. 2aE,F), as were the corresponding constructs with GFP at their C termini (data not shown). As an additional test of membrane association, we analyzed the sucrose gradient sedimentation of membrane fractions from cortical neurons infected with viruses expressing subfragments of ABP. After fractionation on 0.12-1.2 m sucrose gradients, Western blotting revealed that GFP-LI-SII sedimented rapidly, and was confined to fractions 9-12 (Fig. 2b, fractions 9-12), indicating that this fragment associated with heavier, large membranous and vesicular structures. In agreement with this, endogenous GluR2 was also found exclusively in the rapidly sedimenting fractions. In contrast, GFP-SI-LI and GFP-SII-LII, which do not cluster, were widely distributed through the gradient, with a large proportion sedimenting slowly (Fig. 2a, fractions 2-5). This suggested that the latter fragments are either soluble or associated with smaller vesicles. This experiment correlates clustering with membrane sedimentation. We confirmed by transfection of HEK293T cells the expression of products with the anticipated mobility (Fig. 2c). Together, these results identify LI-SII as an intracellular targeting domain.
Lateral association of SII with LI-SII facilitates the formation of clusters
We showed previously that ABP can self-associate through interactions that involve SII (Srivastava et al., 1998). To determine whether such interactions can contribute to ABP membrane association, we assayed the ability of LI-SII to cluster SII by determining whether LI-SII could draw SII into membrane clusters. When expressed individually in HeLa cells, GFP-LI-SII was clustered intracellularly (Fig. 3A), whereas GFP-SII was distributed diffusely in the cytoplasm (B). When the fragments were tagged with Flag, similar morphologies were seen (Fig. 3C,D), making it unlikely that such results were caused by a steric influence of GFP. Interestingly, when GFP-SII was coexpressed with Flag-LI-SII, it acquired a clustered distribution that colocalized with Flag-LI-SII (Fig. 3E-G). This reveals an interaction of GFP-SII with Flag-LI-SII that draws GFP-SII into clusters. Given previous results that show co-IP of ABP fragments containing SII (Srivastava et al., 1998), these results suggest that one molecule of ABP can interact via SII with a second molecule of ABP that is membrane associated. Such an interaction between membrane-bound ABPs could associate molecules of ABP with one another within a sub-region of a larger membrane surface to form a scaffold.
Figure 3.

Protein-protein interaction contributes to the membrane tethering of LI-SII. HeLa cells were transfected with a cDNA encoding GFP-LI-SII (A), GFP-SII (B), Flag-LI-SII (C), or Flag-SII (D), or cotransfected with GFP-SII and Flag-LI-SII (E-G). After 24 hr of transfection, GFP-tagged protein in A, B, and F were visualized by GFP fluorescence with confocal microscopy. The cells in C, D, and E were stained with anti-Flag antibody. G, Merged image of E and F. When coexpressed with Flag-LI-SII, GFP-SII colocalized with Flag-LI-SII in clusters and at the plasma membrane. Scale bar, 20 μm.
The LI-SII fragment clusters MycGluR2
LI-SII contains PDZ5, the binding site for GluR2 (Srivastava et al., 1998). To determine whether LI-SII can cluster GluR2, we coexpressed GFP-LI-SII with MycGluR2 in hippocampal neurons. The intracellular distribution of MycGluR2 expressed on its own was diffuse within the cell body, dendrites, and spines (Fig. 4a). However, when coexpressed with LI-SII, MycGluR2 redistributed to the intracellular GFP-LI-SII clusters (Fig. 4bA-C). This demonstrated that the LI-SII subdomain can direct the intracellular localization of MycGluR2. In contrast, GFP-SII colocalized with MycGluR2 diffusely in the cell body (data not shown) and in dendrites and spines, rather than in clusters (Fig. 4cA-C). Similarly, SI, SI-LI, and SII-LII (all tagged with GFP), which are fragments that also do not form clusters by themselves, also failed to induce clusters of MycGluR2 (data not shown). Two PDZ binding site mutants of MycGluR2, MycGluR2-SVKE and MycGluR2-Δ10 (Osten et al., 2000), which do not bind ABP in vitro, were not clustered by LI-SII (Fig. 4cD-F) (data not shown). However, MycGluR2Δ31-40, a mutant that fails to bind N-ethylmaleimide-sensitive factor (NSF) but still binds ABP, did associate with LI-SII clusters (Fig. 4cG-I). We conclude that binding of MycGluR2 to a PDZ domain of LI-SII is necessary for intracellular clustering of MycGluR2, but the interaction with NSF is not required. Binding of MycGluR2 to ABP (tagged with GFP), and to GFP-LI-SII, was confirmed by coexpression, immune precipitation, and Western blotting (Fig. 4d, lanes 1-4). Controls for immunoprecipitation (lanes 5-8) and expression of tagged proteins (lanes 9-16) are also shown.
Figure 4.
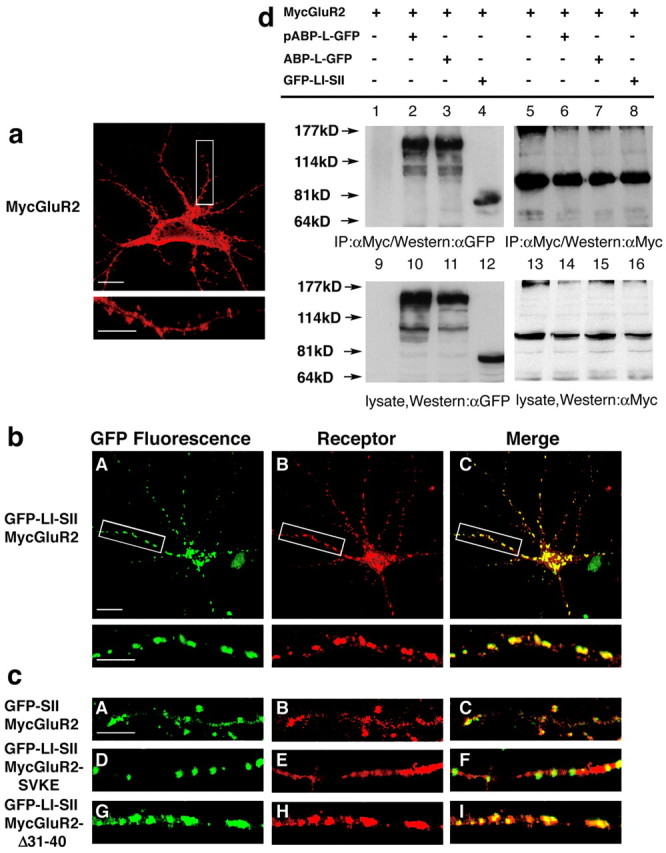
The LI-SII fragment colocalizes and clusters with MycGluR2. a, Hippocampal neurons were infected with a Sindbis virus expressing MycGluR2. After 24 hr of infection, MycGluR2 was detected with the anti-Myc antibody and was distributed diffusely in the cell body, dendrites, and spines. The bottom image is an enlargement of the boxed area in the top image. Scale bars: top image, 20 μm; bottom image, 10 μm. b, Hippocampal neurons coexpressing GFP-LI-SII with MycGluR2. GFP-LI-SII not only formed clusters of its own (A) but also induced MycGluR2 clusters (B). The merged image (C) revealed coclusters of GFP-LI-SII and MycGluR2. The bottom images are enlargements of the boxed areas in the top images. Scale bars: top image, 20 μm; bottom image, 10 μm. c, Control coinfections of hippocampal neurons. GFP-SII and MycGluR2 colocalized in dendrites and spines (A-C). GFP-LI-SII formed clusters, but it did not induce MycGluR2-SVKE clusters (D-F). GFP-LI-SII coclusters with MycGluR2-Δ31-40 (G-I). Clustering requires LI and the interaction of SII with the GluR2 C terminus, but not the NSF binding region of the GluR2 C terminus, residues 31-40. Scale bar, 10 μm. d, HEK293T cells were transfected with the cDNAs encoding MycGluR2 alone or in conjunction with pABP-L-GFP, ABP-L-GFP, and GFP-LI-SII. After 48 hr of transfection, cells lysates were immunoprecipitated with a monoclonal antibody against Myc and blotted with anti-GFP (αGFP) serum (lanes 1-4) and anti-Myc (αMyc) (lanes 5-8). Lysates were also probed with anti-GFP serum (lanes 9-12) or anti-Myc (lanes 13-16), respectively, to confirm the comparable expression levels of all of the proteins.
Trafficking of endocytosed MycGluR2 to LI-SII and ABP-L
The synaptic expression of AMPA receptors is a dynamic process in which exocytotic delivery of AMPA receptors to synapses and endocytic internalization contribute to hippocampal LTP and LTD, respectively (Luscher et al., 2000; Lu et al., 2001). Both processes could involve AM-PARs that are tethered by ABP at internal membranes. Such tethering could generate a receptor pool that serves as a donor of mobile receptors in the case of LTP (or dedepression of LTD) and as the recipient of endocytosed receptors in the case of LTD. To determine whether receptors endocytosed from the plasma membrane can traffic to intracellular clusters of ABP or LI-SII, we analyzed the endocytic targeting of MycGluR2 in the presence or absence of different exogenously expressed ABP forms. We expressed MycGluR2 on its own and also coexpressed MycGluR2 together with pABP-L-GFP, ABP-L-GFP, or GFP-LI-SII in hippocampal neurons. As a control, MycGluR2-SVKE was coexpressed with GFP-LI-SII. Cell surface receptors were labeled by exposure of living neurons to monoclonal antibody against the extracellular Myc epitope. Cells were incubated at 37°C for 15 min to allow internalization of receptor-antibody complexes. The remaining surface antibody was removed by acid stripping, and internalized receptors were visualized by staining with a secondary antibody against mouse IgG. Total MycGluR2 was visualized by incubating cells with a polyclonal antibody against the Myc epitope, followed by incubation with a secondary antibody against rabbit IgG. Internalized MycGluR2 appeared intracellularly proximal to the plasma membrane and within spines, when MycGluR2 was expressed by itself (Fig. 5aA). When the palmitoylated form of ABP, pABP-L-GFP, was coexpressed with MycGluR2, pABP-L-GFP was localized on the plasma membrane (Fig. 5bA), whereas the distribution of internalized MycGluR2 was similar to that seen in control cells not expressing pABP-L-GFP (Fig. 5, compare bB, aA). This suggests that pABP-L-GFP did not affect the localization of internalized MycGluR2. However, when expressed with the nonpalmitoylated ABP-L-GFP, internalized MycGluR2 colocalized partially with intracellular ABP-L-GFP clusters (Fig. 5bD-F). The intracellular colocalization was also seen upon co-expression with GFP-LI-SII (Fig. 5bG-I). In contrast, internalized MycGluR2-SVKE did not colocalize with GFP-LI-SII (Fig. 5bJ-L). This was also evident in higher magnifications of dendrites in which essentially no overlap was seen between internalized MycGluR2-SVKE and GFP-LI-SII (Fig. 5c), confirming the dependence on PDZ interaction of internalized receptor colocalization with ABP. From comparison of B and E in Figure 5b, it is evident that internalized MycGluR2 is copious in the cell body when ABP-L is coexpressed and virtually absent from the cell body when pABP-L is expressed. This is consistent with the internalized receptor trafficking to the location of the particular ABP form. These data suggest that, after endocytosis of GluR2, such as during LTD, AMPA receptors transported from the plasma membrane can bind to ABP-L at an intracellular membrane. Such binding may incorporate internalized receptors into intracellular pools.
Figure 5.
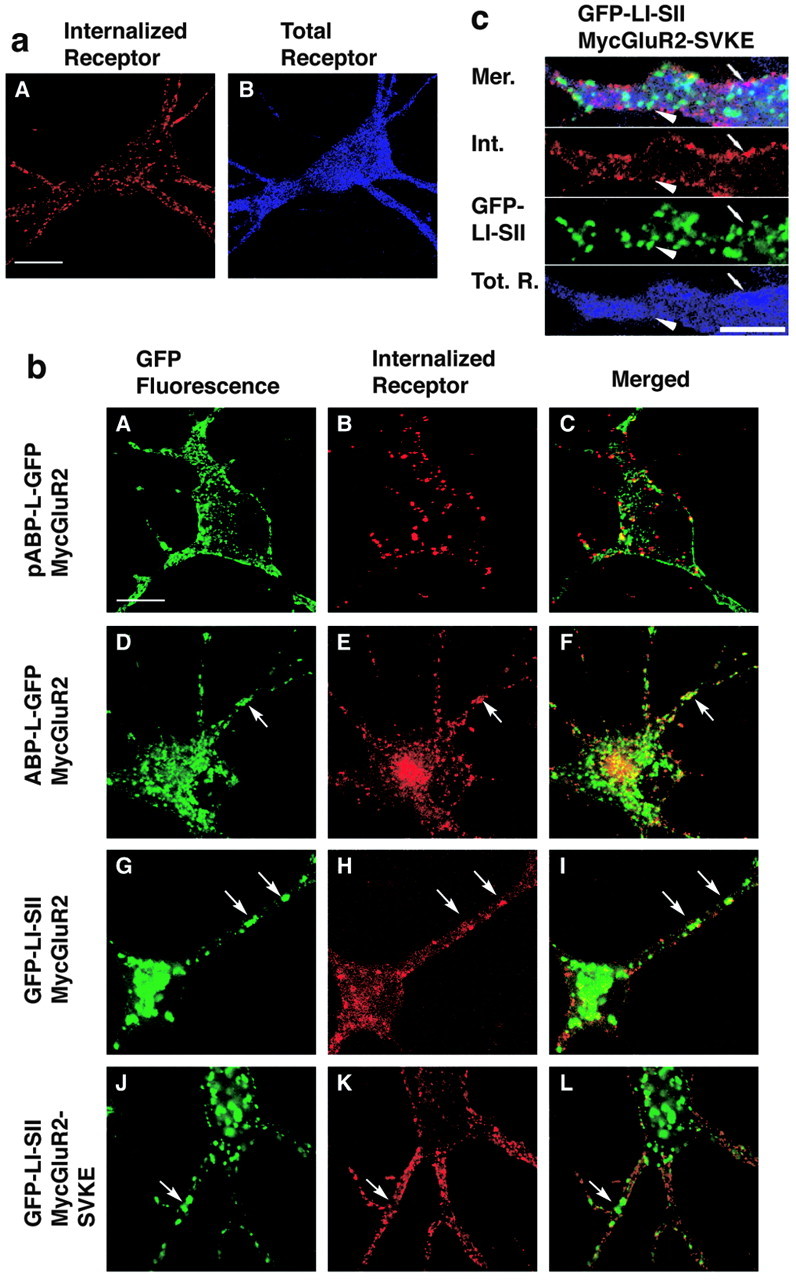
Internalized MycGluR2 colocalizes with pABP-L-GFP, ABP-L-GFP, and GFP-LI-SII. a, Neurons were infected with Sindbis viruses expressing MycGluR2 alone. After 24 hr of infection, living cells were incubated with monoclonal anti-Myc antibody, followed by acid stripping and immunostaining. The total receptor was probed by the polyclonal anti-Myc antibody. Scale bar, 20 μm. b, Neurons coexpressed MycGluR2 with ABP or ABP fragments as indicated. All of the ABPs were seen by GFP fluorescence (A, D, G, J). MycGluR2 and its mutant were detected as red (internalized; B, E, H, K) or blue (total proteins; C, F, I, L). MycGluR2 (B) was detected on the plasma membrane and in spines (A), which does not colocalize exactly with pABP-L-GFP, when coexpressed with pABP-L-GFP. However, it coclustered with ABP-L-GFP (compare D, E) or with GFP-LI-SII (compare G, H), when coexpressed with ABP-L-GFP or GFP-LI-SII, respectively. As a control, Myc GluR2-SVKE was also coexpressed with GFP-LI-SII (J-L). Internalized MycGluR2-SVKE did not cocluster with GFP-LI-SII (compare J, K). Arrows indicate the sites at which MycGluR2 and ABPs colocalized or not. c, Dendritic expression of GFP-LI-SII and MycGluR2-SVKE at higher magnification. Panels are merged (Mer.) images, internalized MycGluR2-SVKE (Int.), GFP-LI-SII, and total receptor (Tot. R.). Note that the GFP-LI-SII clusters (arrowhead) and internalized receptor (small arrow) do not colocalize with each other. Scale bar, 10 μm.
ABP and LI-SII suppress PKC phosphorylation of MycGluR2 at serine 880
Phosphorylation of serine 880 (S880) of GluR2, adjacent to the PDZ binding site, appears to trigger a series of steps leading to GluR2 trafficking (Matsuda et al., 1999, 2000; Chung et al., 2000; Perez et al., 2001). Such a mechanism may contribute to cerebellar LTD at parallel fiber-Purkinje cell synapses (Xia et al., 2000) and in hippocampus (Kim et al., 2001). Notably, Daw et al. (2000) showed that introduction of a GluR2 C-terminal peptide (Pep2-SVKI) into hippocampal neurons causes the AMPAR-mediated EPSC amplitude to increase in a PKC-dependent manner. Disruption, by the peptide, of an interaction between GluR2 and an intracellular tethering protein was proposed to release GluR2, allowing it to be phosphorylated by PKC. In its mobile, phosphorylated form, the receptor may be reinserted into the synaptic membrane, increasing the AMPAR-mediated EPSC amplitude (Daw et al., 2000). The specificity of peptide disruption of tethering suggested that the tethering protein was ABP-GRIP. Because we had observed that ABP could retain GluR2 intracellularly, we analyzed further the regulation of the internal GluR2 pool by the receptor-ABP interaction. Specifically, we determined the effect of tethering GluR2 by ABP on S880 phosphorylation. We first determined whether the binding of pABP-L-GFP or ABP-L-GFP to Myc-GluR2 influences PKC phosphorylation of MycGluR2. We coexpressed MycGluR2 and either pABP-L-GFP or ABP-L-GFP in hippocampal neurons and then treated the cells with TPA for 10 min to activate PKC. Cells were fixed and assayed for S880 phosphorylation with an S880-PO4-specific antibody (α-S880-PO4). Total MycGluR2 was detected with an antibody against the Myc epitope tag. As a control, MycGluR2 was expressed on its own, and the neurons were either treated or not treated with TPA. Without treatment, staining of MycGluR2 with α-S880-PO4 antibody revealed no phosphorylated receptor (Fig. 6aB), whereas staining with anti-Myc antibody revealed that most of the receptor was distributed diffusely in cell body, dendrites, and spines (Fig. 6aA). In contrast, when treated with TPA, phosphorylated MycGluR2 was detected strongly on the plasma membrane (Fig. 6aD). Coexpression of pABP-L-GFP or ABP-L-GFP with MycGluR2 suppressed TPA induction of S880 phosphorylation (Fig. 6bA-D and E-H, respectively) (compare bC,G; aD). Upon TPA treatment, MycGluR2 co-expressed with GFP-LI-SII was primarily composed of unphosphorylated MycGluR2 found in intracellular compartments coclustered with GFP-LI-SII (Fig. 6b, compare I, J). The phosphorylated MycGluR2 was distributed evenly on the plasma membrane. Cells expressing GFP-LI-SII displayed relatively lower α-S880-PO4 staining than controls lacking the ABP fragment (Fig. 6, compare bK, aD). In contrast, MycGluR2-SVKE did not cocluster with LI-SII (Fig. 6bM-P) confirming the PDZ dependence of clustering. More importantly, this mutant, which fails to bind LI-SII, was readily phosphorylated upon TPA treatment. In this case, phosphorylated MycGluR2-SVKE was strongly expressed on the cell surface (Fig. 6b, compare O, C,G,K).
Figure 6.
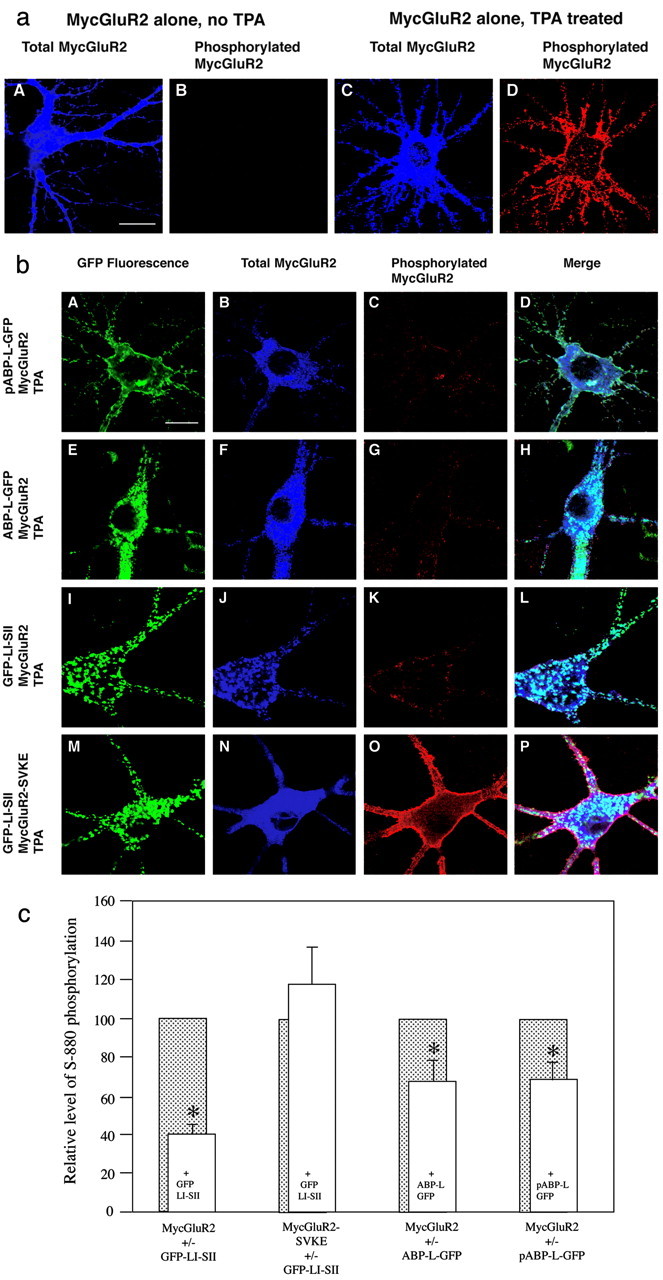
Ser880 phosphorylation of GluR2 is partially inhibited by coexpression of ABP. a, Hippocampal neurons were infected with Sindbis virus expressing MycGluR2 alone. The effect of TPA was assessed by comparing neurons not treated (A, B) or treated (C, D) with TPA. Total MycGluR2 was detected by antibody against Myc epitope, which is shown in blue (A, C), and the phosphorylated MycGluR2 was probed with antibody against the S880-PO4 phosphorylated peptide and is shown in red (B, D). In the absence of TPA, no signal is detected using antibody against the phosphorylated peptide (B). In the presence of TPA, intensive staining was shown using the same antibody (D). Scale bar, 20 μm. b, Hippocampal neurons were coinfected with Sindbis virus expressing pABP-L-GFP (Figure legend continued.) and MycGluR2 (A-D); ABP-L-GFP and MycGluR2 (E-H); GFP-LI-SII and MycGluR2 (I-L); or GFP-LI-SII and MycGluR2-SVKE (M-P). After 24 hr of infection, neurons were treated with TPA to induce PKC. GFP-tagged ABPs were visualized by fluorescence (A, E, I, M). Total (B, F, J, N) and phosphorylated (C, G, K, O) receptors were detected as shown in a. D, H, L, P, Merged images. Coexpression with MycGluR2 of pABP-L-GFP (C), ABP-L-GFP (G), or GFP-LI-SII (K) reduced phosphorylation relative to MycGluR2 expressed on its own (D). GFP-LI-SII coclusters with the total MycGluR2, which is primarily the unphosphorylated MycGluR2 (compare I, J). In contrast, the level of phosphorylated MycGluR2-SVKE in cells coinfected with MycGluR2-SVKE and GFP-LI-SII is higher (compare O with C, G, K). Scale bar, 20 μm. c, Neurons were infected and treated with TPA as shown in b. The coinfections were with viruses expressing MycGluR2 and GFP-LI-SII; MycGluR2-SVKE and GFP-LI-SII; MycGluR2 and ABP-L-GFP; or MycGluR2 and pABP-L-GFP. Cells were scanned with a confocal microscope, images were quantitated, and the ratio of phosphorylated MycGluR2 (red channel) to total MycGluR2 (blue channel) was determined. The phosphorylated MycGluR2 signals (open bars) were normalized to that obtained from cells with single infection with MycGluR2, scanned from the same coverslip (shaded bars). For each group, 10 or more cells were scanned. Whereas the phosphorylation of MycGluR2 was highly significantly reduced in presence of GFP-LI-SII compared with the control (p < 0.0002), the phosphorylation of MycGluR2-SVKE was not significantly changed in the presence of GFP-LI-SII (p > 0.4). The phosphorylation of MycGluR2 was significantly reduced in the presence of ABP-L-GFP or pABP-L-GFP (p < 0.05). Error bar indicates SEM. *p < 0.05.
To confirm the apparent reduction in S880 phosphorylation resulting from tethering of MycGluR2 by ABP, we quantitated the ratio of S880-PO4 MycGluR2 (red image) to total MycGluR2 (blue image) for all of the coinfection groups. We normalized the results by comparing the ratio of S880-PO4 to total MycGluR2 in cells expressing the MycGluR2 and ABP proteins, with the ratio in singly infected cells from the same coverslip expressing only the MycGluR2 subunit. As shown in Figure 6c, both pABP-L-GFP and ABP-L-GFP reduced S880-PO4 of MycGluR2 (p < 0.05, respectively). S880-PO4 of MycGluR2 was even more highly significantly reduced in the neurons expressing GFP-LI-SII (p < 0.0002) (Fig. 6c). In contrast, S880-PO4 of MycGluR2-SVKE was not reduced by LI-SII (p > 0.4) (Fig. 6c). We conclude that ABP can partially inhibit the phosphorylation of MycGluR2 at S880 by PKC through interaction with the PDZ binding site within the C terminus of the receptor. In addition, the LI-SII fragment displayed an increased ability to suppress the phosphorylation of MycGluR2 compared with the wild-type ABP.
Dependence of intracellular clusters on LI and PDZ6
The mechanism of association of LI-SII and ABP-L with internal membranes is not known. ABP and LI-SII could be tethered by binding of the C terminus of an integral membrane protein to an ABP PDZ domain. However, SII, which contains the same set of PDZ domains as LI-SII, is mostly cytosolic in HeLa in contrast to LI-SII, which is membrane associated in these same cells. This suggests that simple integral membrane protein binding to the PDZ domains of ABP is not sufficient for membrane tethering. This also implies a function for LI. We showed that LI-SII can stabilize SII at a membrane (Fig. 3). This reinforces the likelihood that a region of LI is essential for the membrane association of these complexes. To begin to deduce the basis for membrane association, we defined more precisely the amino acids within LI required for intracellular targeting. We expressed in neurons a series of deletions of GFP-LI-SII lacking progressively greater portions of the N-terminal region of LI (Fig. 7a) and localized the mutants by GFP fluorescence using confocal microscopy. GFP-LI-SII formed strong clusters intracellularly (Fig. 7bA). The length of LI is 120 aa. Constructs with partial deletions of LI of up to 80 aa, including GFP-LIΔ20-SII, GFP-LIΔ40-SII, LIΔ60-SII, and GFP-LIΔ80-SII, still formed clusters (Fig. 7bB) (data not shown). GFP-SII, which represents the full truncation of LI from LI-SII, was diffusely distributed (Fig. 7bC), indicating that the C-terminal 40 aa of linker I are essential for forming the internal clusters. To analyze this 40 aa region further, we deleted 100 aa of N-terminal LI. The resulting mutant, GFP-LIΔ100-SII displayed a diffuse distribution (Fig. 7bD). Inspection of the sequence between aa 80 and 100 of LI revealed a region rich in basic amino acids that is highly conserved in GRIP. In ABP, 10 of 20 aa in this stretch are basic. For GRIP, 9 of the corresponding 20 amino acids are basic (Fig. 7c, aa in blue). We also found that deletion of PDZ6 from GFP-LI-SII in mutant GFP-LI-PDZ4,5 (Fig. 7a) led to a highly diffuse distribution, although a few clusters were seen along the dendrites (bE). This demonstrates that clustering also depends on the integrity of SII. Similar structural requirements for clustering were observed in HeLa cells (data not shown). These results demonstrate the residue 80-100 subregion of LI, which contains a stretch of basic amino acids, as well as the presence of PDZ6 are required for intracellular clustering.
Figure 7.

The LI and the entire set II are required for forming the internal clusters. a, Schematic representation of GFP-tagged ABP fragments. GFP-tagged ABP fragments, GFP-LI-SII and its deletion mutants, GFP-LIΔ80-SII, GFP-LIΔ100-SII, GFP-SII, and GFP-LI-PDZ4,5, were expressed in hippocampal neurons as indicated. The presence of the internal clusters is indicated by a plus (+), whereas the absence of the cluster is labeled as minus (-). b, ABP fragments were expressed in hippocampal neurons as indicated. After 24 hr of transfection, GFP-tagged protein was visualized by fluorescence with confocal microscopy. GFP-LI-SII and GFP-LIΔ80-SII formed internal clusters (A, B), whereas GFP-SII, GFP-LIΔ100-SII, and GFP-LI-PDZ4,5 were diffusely distributed in cell body, dendrites, and spines (C-E). Scale bar, 20 μm. c, Sequence comparison of the distal 40 amino acids of linker I in ABP and GRIP. Linker I of ABP has 120 amino acids, whereas that of GRIP has 135 amino acids. The aa 80-100 region of linker I of ABP is required for membrane association of the ABP membrane binding domain and contains a subregion rich in basic residues (underlined).
Discussion
The mechanisms of AMPA receptor trafficking have received extensive scrutiny because of the role of trafficking in the regulation of synaptic strength (for review, see Barry and Ziff, 2002; Malinow and Malenka, 2002). Channel trafficking involves subunit-specific interactions with cytosolic proteins, and the trafficking pathways taken by particular heteromers are dictated by channel subunit composition. Receptors that contain GluR1 may be inserted de novo into synapses by a mechanism that is dependent on activity. In contrast, receptors containing GluR2/3 enter only those synapses that already contain AMPARs, by an activity-independent mechanism involving receptor replacement (Shi et al., 2001). The presence of GluR1 in a channel has a dominant role in establishing receptor-trafficking properties (Shi et al., 2001). Here, we analyze ABP, a multi-PDZ GluR2- and GluR3-binding protein that has been implicated in receptor membrane anchorage (Srivastava et al., 1998; Daw et al., 2000; Osten et al., 2000; deSouza et al., 2002) and transport (Wyszynski et al., 2002). AMPARs in hippocampus are predominantly heteromers of GluR2 with either GluR1 or GluR3 (Wenthold et al., 1996). Therefore, the contributions of GluR2 and associated proteins to trafficking are likely to be significant in the hippocampus. We studied ABP interaction with GluR2 expressed from Sindbis virus, which assembles predominately into homomeric channels (Osten et al., 2000). Because GluR2 homomeric channels cycle constitutively by an activity-independent mechanism between the plasma membrane and internal receptor pools (Shi et al., 2001), the current studies are most relevant to GluR2/3 activity-independent, constitutive recycling pathways. Such pathways may complement the activity-dependent pathway involving GluR1-containing receptor insertion.
Mechanism of ABP intracellular localization
At least three regions of ABP appear to contribute to ABP subcellular targeting. Palmitoylation at the N terminus directs ABP to the plasma membrane and heads of spines (deSouza et al., 2002), whereas LI and SII both contribute to the targeting of ABP to internal membrane. Previous studies have defined other functions for LI and for SII. SII contains PDZ5, which is the binding site of GluR2 (Srivastava et al., 1998), and PDZ6, which binds to liprin-α (Wyszynski et al., 2002) and to EphB receptor and ephrin B ligands (Torres et al., 1998; Bruckner et al., 1999; Lin et al., 1999). The liprin-α interaction with PDZ6 is implicated in AMPA receptor accumulation at synapses, either through direct recruitment of receptors or through linking the GRIP-ABP complex to microtubule transport motors (Wyszynski et al., 2002). In addition, recent crystallographic and hydrodynamic evidence indicates that PDZ6 of GRIP can form antiparallel dimers and provides a basis for SII-dependent ABP self-interaction (Im et al., 2003). Multimerization of ABP molecules could cluster ABP and thereby sort associated proteins such as AMPA receptors within an intracellular membrane surface. Because membrane association of LI-SII was shown here to depend on PDZ6, which is also the apparent dimerization site (Im et al., 2003), oligomerization may also contribute to membrane binding. The contribution of LI appears to depend on a 20 aa stretch that contains a region rich in basic amino acids that is also highly conserved in GRIP. Basic aa regions of other proteins have been found to function in membrane association through binding to phospholipids (for review, see McLaughlin et al., 2002). Additional studies will be required to establish the role of the basic region of ABP.
Function of ABP in receptor cycling
In contrast to pABP-L, which appears at the plasma membrane, LI-SII displays a vesicle-like intracellular distribution and its sedimentation on sucrose gradients also suggests an association with membranes. Strictly speaking, all of the clusters, both those of the pABP-L as well as those of ABP-L, are intracellular in the sense that ABP is not an integral membrane protein. In a previous report (deSouza et al., 2002), we compared the localizations of pABP-L and ABP-L and showed that the palmitoylated form, pABP-L, lines the plasma membrane of HeLa cells and colocalizes with the surface form of GluR2 in neurons, whereas ABP-L forms clusters in HeLa that lie in the midst of the cytoplasm and localizes with GluR2 that resides in intracellular endomembrane structures. Mutation of the palmitoylation site of pABP-L released ABP to form the intracellular clusters resembling those of ABP-L, a shift consistent with the loss of palmitate, a lipophillic plasma membrane-targeting motif. These observations support the conclusion that the ABP in clusters is associated with membrane rather than protein aggregates. The ability of endocytosed GluR2 to traffic to sites containing intracellular ABP also implies an endosomal location for the intracellular ABP. Confocal images of cells expressing exogenous GRIP have revealed cavities within the membranous structures formed by GRIP, a finding consistent with a vesicular organization of these clusters (B. States and E. Ziff, unpublished observations). Nonetheless, despite extensive efforts to demonstrate the colocalization of ABP and its fragments with intracellular compartment markers, including endocytosed transferrin and the LDL (low-density lipoprotein) receptor, which label endosomes, and LAMP2 (lysosome-associated membrane protein 2), which labels lysosomes, we have not been successful in defining the membrane compartment that is associated with intracellular ABP and GRIP.
Mutations that disrupt the GluR2-ABP interaction increased the relative rate of GluR2 endocytosis (Osten et al., 2000), suggesting a function for ABP in anchoring GluR2 at the plasma membrane. Here, we report that the intracellular form of ABP, ABP-L, and also LI-SII can bind GluR2 that traffics from the plasma membrane. This indicates function for ABP-L in tethering AMPARs intracellularly. The localization of internalized GluR2 reflected the form of ABP that was expressed, with internalized GluR2 accumulating at intracellular locations upon expression of ABP-L but not pABP-L. Other work also supports a receptor-tethering role for ABP-GRIP. A peptide block of the interaction of GluR2 with ABP-GRIP increased AMPAR-mediated EPSCs in CA1 hippocampal neurons, indicating that release of AMPARs from an internal ABP-GRIP tether can stimulate AMPAR exocytosis. The EPSC increase was observed in cells in which LTD had been induced (Daw et al., 2000). This suggested that dedepression of LTD, which involves receptor reinsertion into the synapse via exocytosis, is inhibited by tethering AMPAR intracellularly to ABP-GRIP or a similar factor. Other studies using PDZ binding site mutations also suggest such a mechanism (Braithwaite et al., 2002). However, it remains to be determined whether expression of pABL-L versus ABP-L can influence the rate of exocytosis of AMPA receptors.
Daw et al. (2000) have proposed that two intracellular pools of AMPA receptors are found in proximity to synapses: a constitutively trafficked pool composed of receptors with phosphorylated GluR2 and a regulated pool composed of receptors with unphosphorylated GluR2. In this model, phosphorylation of S880 by PKC releases receptors from an intracellular pool for insertion into the synaptic membrane. The model accounts for the finding that dedepression, which results from outward transport of AMPA receptors released from intracellular tethers, depends on PKC (Daw et al., 2000). We showed that ABP blocks PKC phosphorylation of GluR2, and that the inhibition requires PDZ binding to GluR2. Binding of metabotropic (m)GluR7a to the PDZ domain of PICK1 (protein interacting with C-kinase) reduces mGluR7a phosphorylation by PKCα in vitro (Dev et al., 2000). Thus, PDZ binding may more generally block receptor phosphorylation by masking or physically blocking the phosphorylation site. Our work suggests that GRIP-ABP may act as an insertion clamp through its ability to block S880 phosphorylation of GluR2. Such a clamp could prevent the release of receptors from an intracellular pool and block receptor entry into the proposed constitutive recycling pool. Receptor uncoupling from ABP-GRIP may be required before a kinase is able to phosphorylate S880 of GluR2. Once GluR2 is phosphorylated, its recoupling to ABP-GRIP would be blocked, and the receptor would be released for exocytotic transport.
Footnotes
This work was supported by National Institutes of Health Grant AG13620 (E.B.Z.). J.F. and S.D. are associates and E.B.Z. is an investigator of the Howard Hughes Medical Institute. We thank C. Misra, I. Greger, and B. Jordan for critical reading of this manuscript and I. Greger for assistance with the sedimentation assays. We also thank T. Serra for help in preparation of this manuscript.
Correspondence should be addressed to Dr. Edward B. Ziff, Howard Hughes Medical Institute, Department of Biochemistry, New York University School of Medicine, 550 First Avenue, New York, NY 10016. E-mail: edward.ziff@med.nyu.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/237592-10$15.00/0
References
- Barry MF, Ziff EB ( 2002) Receptor trafficking and the plasticity of excitatory synapses. Curr Opin Neurobiol 12: 279-286. [DOI] [PubMed] [Google Scholar]
- Braithwaite SP, Xia H, Malenka RC ( 2002) Differential roles for NSF and GRIP/ABP in AMPA receptor cycling. Proc Natl Acad Sci USA 99: 7096-7101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruckner K, Pablo Labrador J, Scheiffele P, Herb A, Seeburg PH, Klein R ( 1999) EphrinB ligands recruit GRIP family PDZ adaptor proteins into raft membrane microdomains. Neuron 22: 511-524. [DOI] [PubMed] [Google Scholar]
- Burette A, Khatri L, Wyszynski M, Sheng M, Ziff EB, Weinberg RJ ( 2001) Differential cellular and subcellular localization of AMPA receptor-binding protein and glutamate receptor-interacting protein. J Neurosci 21: 495-503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung HJ, Xia J, Scannevin RH, Zhang X, Huganir RL ( 2000) Phosphorylation of the AMPA receptor subunit GluR2 differentially regulates its interaction with PDZ domain-containing proteins. J Neurosci 20: 7258-7267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daw MI, Chittajallu R, Bortolotto ZA, Dev KK, Duprat F, Henley JM, Collingridge GL, Isaac JT ( 2000) PDZ proteins interacting with C-terminal GluR2/3 are involved in a PKC-dependent regulation of AMPA receptors at hippocampal synapses. Neuron 28: 873-886. [DOI] [PubMed] [Google Scholar]
- deSouza S, Fu J, States B, Ziff EB ( 2002) Differential palmitoylation directs targeting of the AMPA receptor-binding protein ABP to spines and intracellular clusters. J Neurosci 22: 3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dev KK, Nakajima Y, Kitano J, Braithwaite SP, Henley JM, Nakanishi S ( 2000) PICK1 interacts with and regulates PKC phosphorylation of mGLUR7. J Neurosci 20: 7252-7257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF ( 1999) The glutamate receptor ion channels. Pharmacol Rev 51: 7-61. [PubMed] [Google Scholar]
- Dong H, O'Brien RJ, Fung ET, Lanahan AA, Worley PF, Huganir RL ( 1997) GRIP: a synaptic PDZ domain-containing protein that interacts with AMPA receptors. Nature 386: 279-284. [DOI] [PubMed] [Google Scholar]
- Im YJ, Park SH, Rho SH, Lee JH, Kang GB, Sheng M, Kim E, Eom SH ( 2003) Crystal structure of GRIP1 PDZ6-peptide complex reveals the structural basis for class II PDZ target recognition and PDZ domain-mediated multimerization. J Biol Chem 278: 8501. [DOI] [PubMed] [Google Scholar]
- Kim CH, Chung HJ, Lee HK, Huganir RL ( 2001) Interaction of the AMPA receptor subunit GluR2/3 with PDZ domains regulates hippocampal long-term depression. Proc Natl Acad Sci USA 98: 11725-11730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin D, Gish GD, Songyang Z, Pawson T ( 1999) The carboxyl terminus of B class ephrins constitutes a PDZ domain binding motif. J Biol Chem 274: 3726-3733. [DOI] [PubMed] [Google Scholar]
- Lu W, Man H, Ju W, Trimble WS, MacDonald JF, Wang YT ( 2001) Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron 29: 243-254. [DOI] [PubMed] [Google Scholar]
- Luscher C, Nicoll RA, Malenka RC, Muller D ( 2000) Synaptic plasticity and dynamic modulation of the postsynaptic membrane. Nat Neurosci 3: 545-550. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC ( 2002) AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci 25: 103-126. [DOI] [PubMed] [Google Scholar]
- Matsuda S, Mikawa S, Hirai H ( 1999) Phosphorylation of serine-880 in GluR2 by protein kinase C prevents its C terminus from binding with glutamate receptor-interacting protein. J Neurochem 73: 1765-1768. [DOI] [PubMed] [Google Scholar]
- Matsuda S, Launey T, Mikawa S, Hirai H ( 2000) Disruption of AMPA receptor GluR2 clusters following long-term depression induction in cerebellar Purkinje neurons. EMBO J 19: 2765-2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin S, Wang J, Gambhir A, Murray D ( 2002) PIP2 and proteins: interactions, organization, and information flow. Annu Rev Biophys Biomol Struct 31: 151-175. [DOI] [PubMed] [Google Scholar]
- Osten P, Khatri L, Perez JL, Kohr G, Giese G, Daly C, Schulz TW, Wensky A, Lee LM, Ziff EB ( 2000) Mutagenesis reveals a role for ABP/GRIP binding to GluR2 in synaptic surface accumulation of the AMPA receptor. Neuron 27: 313-325. [DOI] [PubMed] [Google Scholar]
- Perez JL, Khatri L, Chang C, Srivastava S, Osten P, Ziff EB ( 2001) PICK1 targets activated protein kinase Cα to AMPA receptor clusters in spines of hippocampal neurons and reduces surface levels of the AMPA-type glutamate receptor subunit 2. J Neurosci 21: 5417-5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi S, Hayashi Y, Esteban JA, Malinow R ( 2001) Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons. Cell 105: 331-343. [DOI] [PubMed] [Google Scholar]
- Srivastava S, Osten P, Vilim FS, Khatri L, Inman G, States B, Daly C, DeSouza S, Abagyan R, Valtschanoff JG, Weinberg RJ, Ziff EB ( 1998) Novel anchorage of GluR2/3 to the postsynaptic density by the AMPA receptor-binding protein ABP. Neuron 21: 581-591. [DOI] [PubMed] [Google Scholar]
- Torres R, Firestein BL, Dong H, Staudinger J, Olson EN, Huganir RL, Bredt DS, Gale NW, Yancopoulos GD ( 1998) PDZ proteins bind, cluster, and synaptically colocalize with Eph receptors and their ephrin ligands. Neuron 21: 1453-1463. [DOI] [PubMed] [Google Scholar]
- Wenthold RJ, Petralia RS, Blahos J, Niedzielski AS ( 1996) Evidence for multiple AMPA receptor complexes in hippocampal CA1/CA2 neurons. J Neurosci 16: 1982-1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyszynski M, Kim E, Dunah AW, Passafaro M, Valtschanoff JG, Serra-Pages C, Streuli M, Weinberg R, Sheng M ( 2002) Interaction between GRIP and Liprin-α/SYD2 is required for AMPA receptor targeting. Neuron 34: 39-52. [DOI] [PubMed] [Google Scholar]
- Xia J, Chung HJ, Wihler C, Huganir RL, Linden DJ ( 2000) Cerebellar long-term depression requires PKC-regulated interactions between GluR2/3 and PDZ domain-containing proteins. Neuron 28: 499-510. [DOI] [PubMed] [Google Scholar]
- Yamazaki M, Fukaya M, Abe M, Ikeno K, Kakizaki T, Watanabe M, Sakimura K ( 2001) Differential palmitoylation of two mouse glutamate receptor interacting protein 1 forms with different N-terminal sequences. Neurosci Lett 304: 81-84. [DOI] [PubMed] [Google Scholar]