

Abstract
Loss of sensory function leads to atrophy or death within the developing CNS, yet little is known about the physiology of remaining synapses. After bilateral deafening, gramicidin-perforated-patch recordings were obtained from gerbil inferior colliculus neurons in a brain slice preparation. Afferent-evoked IPSPs had a diminished ability to block current-evoked action potentials in deafened neurons. This change could be attributed, in part, to a loss of potassium-dependent chloride transport function, with little change in K-Cl cotransporter expression. Treatments that suppressed chloride cotransport (bumetanide, cesium, and genistein) had little or no effect on neurons from deafened animals. These same treatments depolarized the EIPSC of control neurons. Semiquantitative RT-PCR and immunohistochemical staining indicated no change in the expression of chloride cotransporter mRNA or protein after deafness. Therefore, profound hearing loss leads rapidly to the disruption of chloride homeostasis, which is likely attributable to the dysfunction of the potassium-dependent chloride cotransport mechanism, rather than a downregulation of its expression. This results in inhibitory synapses that are less able to block excitatory events.
Keywords: inferior colliculus, gerbil, auditory, inhibition, development, KCC2, plasticity
Introduction
The function of central synapses after blindness, deafness, or comparable forms of deafferentation is mostly unknown. However, in vivo results suggest that synaptic strength is adversely affected (Sherman and Spear, 1982; Klinke et al., 1999). An understanding of these changes seems particularly important in light of the compensatory changes that accompany peripheral injury (Pallas et al., 1999; Gold and Knudsen, 2000; Syka, 2002) and the potential to restore stimulation with prosthetic devices (Klinke et al., 1999; Kral et al., 2002). One crucial question is whether the balance between inhibitory and excitatory synaptic drive is altered by hearing loss. For example, some in vivo studies show an apparent decrease of inhibition in the adult inferior colliculus (IC) or cortex after partial hearing loss (Kitzes and Semple, 1985; Rajan, 1998; Mossop et al., 2000). Until recently, there was no direct evidence that inhibitory synapses themselves undergo a change in function after deafness (Vale and Sanes, 2000, 2002), and the molecular loci of these changes remain unexplored. The goal of the present study was to measure inhibitory synaptic strength after deafness and determine whether any loss of strength was attributable to a decline in chloride cotransporter expression or loss of transport function.
Synaptic inhibition mediated by GABAA or glycine receptors involves activation of a Cl- conductance (Bormann et al., 1987). Intracellular chloride ([Cl-]i) is regulated primarily by two cation-chloride cotransporter family members: an Na-K-2Cl cotransporter (NKCC1) leads to cytoplasmic accumulation of chloride, and a K-Cl cotransporter (KCC2) extrudes chloride (Delpire et al., 1994; Payne et al., 1996, 2003; Payne, 1997). During early development, [Cl-]i is relatively high because of NKCC1 activity (Plotkin et al., 1997; Clayton et al., 1998; Kanaka et al., 2001). As KCC2 expression increases, [Cl-]i drops below the electrochemical equilibrium (Lu et al., 1999; DeFazio et al., 2000; Hübner et al., 2001), leading to a transition from inhibitory synapse-evoked depolarizations to hyperpolarizations (Wang et al., 1994; Owens et al., 1996; Ehrlich et al., 1999; Kakazu et al., 1999; Rivera et al., 1999).
Two recent studies demonstrate that synaptic or electrical activity can influence chloride homeostatic mechanisms. GABAA receptor activation facilitates KCC2 expression and the appearance of GABAA-mediated hyperpolarizations in hippocampal cultures (Ganguly et al., 2001). In contrast, axonal injury to motor neurons decreases KCC2 expression, resulting in GABAA-mediated depolarizing responses (Nabekura et al., 2002). There is some indication that epilepsy is also associated with improper chloride balance (Cohen et al., 2002).
Because deafness leads to a depolarization of the inhibitory synaptic reversal potential (Kotak and Sanes, 1996; Vale and Sanes, 2000, 2002), we now examine both the expression and functional status of chloride transport mechanisms in control and deafened gerbils. In control neurons, chloride transport could be blocked by three different pharmacological treatments, whereas the weak inhibitory synapses from deaf animals were unaffected. RT-PCR and immunohistochemical analyses both showed that chloride cotransporters were well expressed in deaf animals, leading to the conclusion that chloride extrusion mechanisms are functionally disrupted. These results suggest that normal auditory activity regulates inhibitory synaptic strength through the functional status of a chloride cotransporter.
Materials and Methods
Cochlear ablation. All of the protocols were reviewed and approved by the New York University Institutional Animal Care and Use Committee. Both cochleas were removed in postnatal day 7 (P7) gerbils (Meriones unguiculatus), using a method described previously (Sanes et al., 1992; Vale and Sanes, 2000). Briefly, P7 pups were anesthetized with hypothermia until respiration ceased and animals did not respond to nociceptive stimuli. A small hole was made in the cochlear wall, and the contents were removed with a forceps. A piece of Gelfoam was placed in the cavity, and the wound was closed. After surgery, animals were warmed on a heating pad and returned to the litter when respiration and motor activity recovered. Successful ablations were confirmed before each brain slice experiment. Experiments were performed 1-7 d after surgery.
Brain slice preparation. Control and bilaterally ablated P8-P14 gerbils were anesthetized with chloral hydrate (350 mg/kg). After decapitation, coronal slices of 300 μm containing the rostral IC were obtained as described previously (Vale and Sanes, 2000). The tissue was cut in cold oxygenated artificial CSF (ACSF) containing (in mm): 123 NaCl, 4 KCl, 1.2 KH2PO4, 1.3 MgSO4, 28 NaHCO3, 15 glucose, 2.4 CaCl2, and 0.4 l-ascorbic acid, pH 7.3 when oxygenated with 95% O2-5% CO2. After 2 hr, slices containing the rostral IC were placed in a recording chamber and superfused with oxygenated ACSF (7 ml/min) at room temperature (22-24°C).
Electrophysiology. Gramicidin-perforated-patch recordings in voltage- and current-clamp mode (PC-501A; Warner Instruments, Hamden, CT) were obtained as described previously (Rhee et al., 1994; Vale and Sanes, 2000, 2002). Recording electrodes were fabricated from borosilicate glass microcapillaries (outer diameter, 1.5 mm), and the tip resistance was 5-10 MΩ. Gramicidin (Sigma, St. Louis, MO) was used as the membrane-perforating agent to allow recording without influencing the [Cl-]i. The internal pipette solution contained (in mM): 132.5 KCl, 0.6 EGTA, 10 HEPES, 2 MgCl2, 2 ATP, 0.3 GTP, and 5 N-(2,6-dimethylphenylcarbamoylmethyl)triethylammonium bromide (QX-314), pH 7.2. The integrity of the perforation was verified by adding QX-314 (Alamone, Jerusalem, Israel) to the intracellular pipette solution. The presence of depolarization-evoked breakaway action potentials (APs) was taken as indication of the integrity of the gramicidin perforation. The progress of perforation was evaluated by monitoring the decrease in membrane resistance. After the membrane resistance had stabilized, data were obtained. In experiments to test for intracellular potassium-dependent chloride transport, KCl was replaced in the internal pipette solution with equimolar cesium gluconate.
Extracellular stimuli (200 μsec pulses) were delivered to the ipsilateral lateral lemniscal pathway (LL) through paired Teflon-insulated platinum electrodes driven by isolated biphasic stimulators (Intronics Instruments, Ontario, Canada). Data were collected using a Macintosh PPC running a custom-designed Igor macro (WaveMetrics, Lake Oswego, OR) called Slice (Kotak et al., 2001). Stimuli were delivered, and data were sampled via an ITC-18 computer interface (Instrutech, Port Washington, NY). Analyses of peak PSC amplitude and reversal potential were performed off-line using a second Igor macro called Slice Analysis.
The strength and duration of inhibition was evaluated using current-clamp gramicidin-perforated recordings in the presence of 5 mm kynurenic acid (KYN) (Fluka BioChemika, Ronkonkoma, NY). In these recordings, KCl was used in the internal pipette solution, and QX-314 was retained to confirm the integrity of the perforation. The ability of inhibitory afferents to inhibit evoked action potentials was tested by injecting a short depolarizing current pulse (25-50 msec) that evoked an AP in the postsynaptic neuron, and simultaneously the LL afferents were stimulated. This process was repeated 10 times. The number of times that the LL-evoked IPSP suppressed the current-evoked AP was used to calculate the percentage of inhibition. The time during which the LL-evoked IPSP was able to block the AP was also evaluated by stimulating the LL afferents with latencies from -200 to 0 msec before the current-evoked AP (at 10 msec intervals). Each stimulus was delivered twice at the same latency. The absence of an action potential in two consecutive trials with the same latency was considered to be inhibition.
When only two groups were compared, a Student's t test was used to assess whether the differences were significant. When multiple comparisons were made, a one-way ANOVA was followed by pairwise comparisons (Tukey-Kramer honest significant difference test) to assess significant differences. All of the values are expressed as mean ± SEM, with the number of observations in parentheses. EIPSC values were calculated from linear fits of the current-voltage curves plotting IPSC amplitude versus membrane holding potential.
RT-PCR to determine NKCC1 and KCC2 expression. Slices of 600 μm were obtained from the ICs of P9 and P14 control and BCA animals as described for brain slice preparation, using RNase-free medium. The central nucleus of the IC was dissected under the microscope, and the tissue was immediately frozen in dry ice and kept at -80°C. RNA was isolated from two slices per experimental sample using TRIzol reagent (Invitrogen, San Diego, CA) and treated with RNase-free DNase and phenol extracted twice. RT-PCR assays were performed using the OneSTEP RT-PCR kit (Qiagen, Hilden, Germany). In all of the cases, successful amplification was dependent on reverse transcription demonstrating amplification of mRNA templates. Reactions contained test mRNA (∼5 μl), RNAsin (0.5 μl), and 0.8 μm each primer. Reactions were mixed on ice and cycled as follows: reverse transcription for 30 min at 50°C, heat inactivation for 15 min at 90°C, and 30 PCR cycles (1 min at 94°C, 2 min at 58°C, and 1 min at 72°C). Aliquots (10 μl) were removed at several time points during an extended 5 min incubation at 72°C to monitor exponential amplification. Samples were analyzed by ethidium bromide staining after separation in a 2% agarose-Tris-borate-EDTA gel. Primer pairs were chosen to reside in different exons and within regions of sequence identity between different species, based on the following GenBank entries (accession number in parentheses): human NKCC1 cDNA (U30246), mouse NKCC1 cDNA (U13174), rat NKCC1 cDNA (AF 051561), rat KCC2 cDNA (U55816), and mouse genomic KCC2 sequences (AJ011033), conceptually translated in comparison with the rat cDNA.
Two different primer pairs were used for both NKCC1 (NK5-NK6 and NK7-NK8) and KCC2 (KCC21-KCC22 and KCC21-KCC23). Sizes of amplified DNA fragments were as predicted. The primers used were the following: GPDH1, catcaacgaccccttcattgacctc; GPDH2, atacttggcag gtttctccaggcg; KCC21, cgcagccaccatgctcaacaac; KCC22, acaccaaagatgt tctgcaggcacg; KCC23, ggcccagagacctggaaatcatgtagta; NK5, gaatggagtgg gaagcaaaggctc; NK6, tcctttgggtatggctgactgagg; NK7, ggtggctttttgatgatg gaggtttgac; and NK8, aaggtaaggacgctctgatgattccc.
The intensity of each PCR band was measured with a fixed-size sampling area, using the image analysis program Quantity One (Bio-Rad Laboratories, Hercules, CA). Values were expressed as the ratio of mean intensity for each cotransporter band divided by the glyceraldehyde 3-phosphate dehydrogenase (GAPDH) band for each experimental condition.
Immunohistochemical localization of KCC2 protein. Immunohistochemical localization of KCC2 protein was performed with an affinity-purified rabbit anti-KCC2 polyclonal antibody (Upstate Biotechnology, Lake Placid, NY). P14 control and BCA gerbils were deeply anesthetized with sodium pentobarbital (80 mg/kg). Their vasculature was immediately flushed with 0.9% saline, followed by 4% paraformaldehyde in PBS. Brains were removed, postfixed in 4% paraformaldehyde for 2 hr, frozen in 2-methyl butane on dry ice, embedded in mounting media, and stored at -80°C. A Leica (Nussloch, Germany) cryostat produced 25 μm frozen coronal sections that were mounted on slides.
Immunoperoxidase labeling was performed as described previously (Williams et al., 1999). IC sections from control and BCA animals were washed three times in PBS and then placed in 2% goat serum (GS)-PBS containing 0.2% Triton X-100 for 2 hr at room temperature. Sections were incubated at 4°C for 48 hr with anti-KCC2 primary antibody diluted 1:100 in GS-PBS. After three PBS washes, sections were incubated for 2 hr at room temperature with a biotin-conjugated goat anti-rabbit IgG (1:200 dilution; Vector Laboratories, Burlingame, CA). Sections were washed three times in PBS and incubated for 3 hr in an avidin-horseradish peroxidase solution prepared from an ABC kit (Vector Laboratories). After three washes in PBS, the horseradish peroxidase reaction was performed using diaminobenzidine (DAB) (0.015% in PBS; Sigma) and 0.001% hydrogen peroxide. Sections were washed three times in PBS, taken through an ethanol-xylene dehydration series, and cover-slipped. Photographs of DAB-stained sections were taken with a Leica microscope. Controls for the immunohistochemistry experiments were obtained by incubating one of each four consecutive slides without primary antibody. None of the slides that were run without primary antibody showed staining.
To quantify the immunohistochemical staining pattern, a second set of stains were performed with a biotinylated secondary antibody anti-rabbit IgG (Vector Laboratories) and streptavidin (Alexa Fluor 488 conjugate; Molecular Probes, Eugene, OR). The sections were then examined with a confocal microscope (Leica TCS SP2; 40× objective), and intensity measures were acquired (Leica PowerScan software). An area of 94 μm 2 was imaged in the central nucleus from control and BCA tissue sections, and the mean pixel intensity above background was obtained.
Results
Inhibitory synaptic strength was reduced after complete deafness
Previous reports have demonstrated a deafness-induced change in EIPSC and inhibitory conductance (Vale and Sanes, 2000, 2002). To examine the effective strength of inhibitory synapses after deafness, we tested the ability of IPSPs to block current-evoked APs in control and BCA animals. As shown in Figure 1A, LL-evoked IPSPs and current-evoked APs were first elicited independently. The LL-evoked IPSPs were then presented concurrently or at a fixed latency to the current-evoked action potentials. In control neurons, the evoked IPSPs suppressed APs in 97 ± 2% (n = 15) of the trials (Fig. 1B). In BCA neurons, the IPSPs were able to block APs in only 43 ± 11% (n = 14) of the trials. Thus, the ability of LL-evoked IPSPs to inhibit current-evoked APs was reduced considerably in BCA neurons compared with controls (p < 0.0001; t = -4.859; df = 27). The functional duration of inhibition was also tested by evoking APs at fixed latencies to the IPSPs (see Materials and Methods). Figure 1C shows the mean duration of inhibition in control and BCA animals. The duration of inhibition was significantly longer in control neurons (81 ± 13 msec; n = 10) than in BCA neurons (27 ± 10 msec; n = 13; p < 0.005; t =-3.245; df = 21). Therefore, both measures of synaptic strength indicate that inhibitory connections are less able to suppress suprathreshold excitatory events after deafness.
Figure 1.
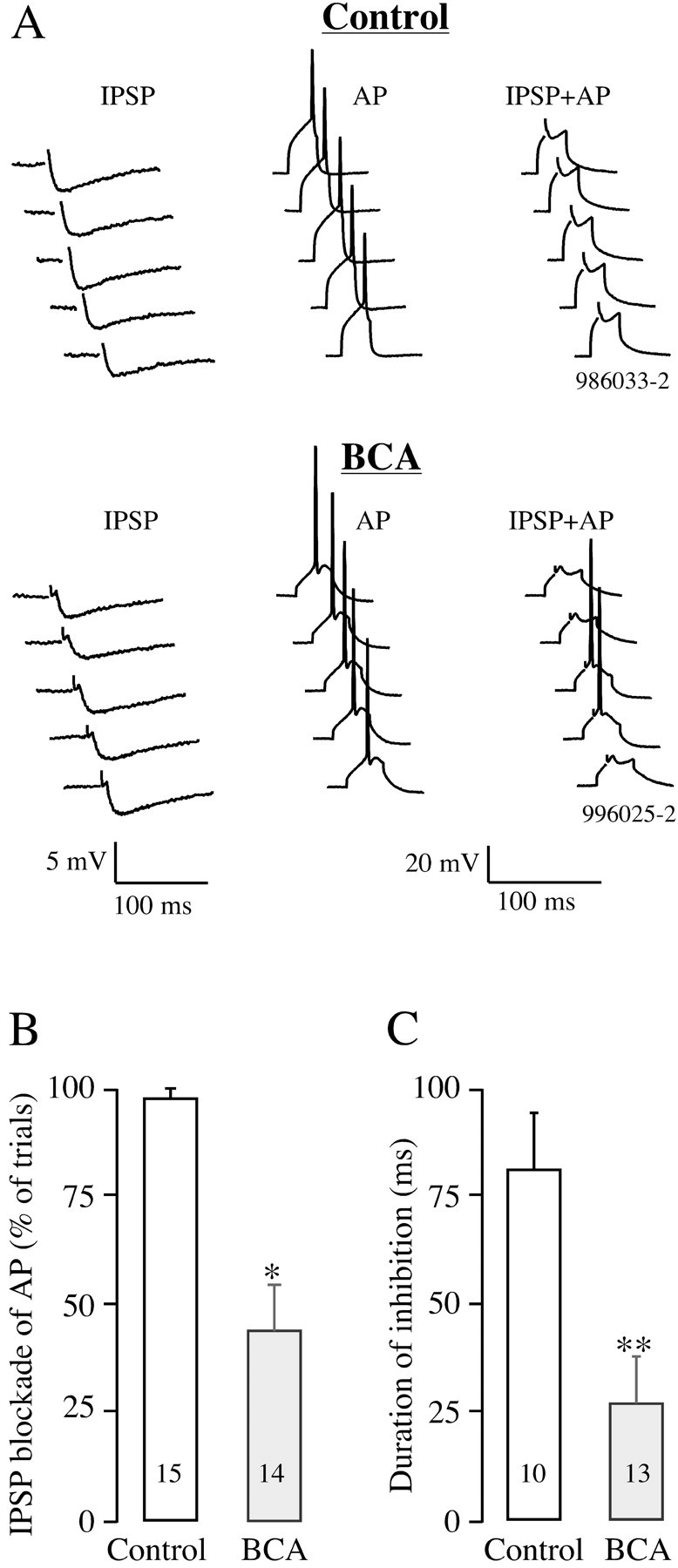
An evaluation of inhibitory synaptic strength in neurons from control and BCA animals. A, Traces show examples of LL-evoked IPSPs, current-evoked action potentials, and the simultaneous presentation of IPSPs with action potentials (from left to right) in control and BCA animals. The number of times that the LL-evoked IPSP inhibited the AP in 10 consecutive trials was counted and used to calculate the percentage of inhibition. B, Mean IPSP ability to inhibit APs is significantly lower in BCA neurons compared with controls. n values are in bars (*p < 0.0001). C, Duration of inhibition was significantly shorter in BCA neurons compared with controls (measurement described in Materials and Methods). n values are in bars (**p < 0.005). Error bars indicate SEM.
An additional set of experiments were performed in whole-cell current-clamp mode to assess neuron excitability. Although resting membrane potential did not differ significantly between groups (control, -53.8 ± 0.8 mV, n = 16; BCA, -52.3 ± 0.8 mV, n = 14), it was possible that action potential threshold changed after deafness. To evaluate this possibility, the action potential threshold was subtracted from resting membrane potential as an indicator of neuron excitability. Action potential threshold was 16 ± 3 mV above rest (n = 15) for control neurons and 18 ± 2 mV above rest (n = 16) for BCA neurons, and there was not a significant difference between groups (p = 0.417; t = -0.824; df = 29).
Blockade of chloride transporters had a smaller effect on deafened neurons
If chloride homeostasis was compromised in deaf animals, then one would expect the loop diuretic bumetanide (Russell, 2000) to have a relatively modest effect on BCA neuron synaptic physiology. In control neurons, 100 μm bumetanide depolarized the EIPSC in all of the neurons tested (12 ± 2 mV; n = 21). In contrast, bumetanide produced only a small depolarization in BCA neurons (4 ± 2 mV; n = 13). As shown in Table 1, the bumetanide-induced change was significantly smaller in BCA neurons compared with controls (p < 0.03; t = 2.375; df = 32). This experiment indicates that bumetanide-sensitive cotransporters, such as KCC2 and NKCC1, participate in the [Cl-]i regulation of control neurons but are less active after deafness. Because bumetanide at 20 μm is a selective antagonist of NKCC (Gillen et al., 1996; Payne, 1997), we examined whether this concentration would cause a negative shift in the EIPSC of BCA neurons. Although the average shift of -5 mV (n = 6) suggested such a trend, there was not a significant difference (p > 0.3; t = 1.07; df = 10), indicating that NKCC does not contribute significantly to Cl- regulation in IC neurons at the ages studied.
Table 1.
Effect of bumetanide on EIPSC
|
|
KYN |
100 μm Bumetanide |
20 μm Bumetanide |
|---|---|---|---|
| Control (n = 21) | −73.1 ± 2.1 | −60.7 ± 2.9* | |
| BCA (n = 13) | −54.0 ± 2.5 | −49.6 ± 1.8 | |
| BCA (n = 6)
|
−55.3 ± 3.3
|
|
−60.3 ± 3.3
|
p < 0.005 versus EIPSC in KYN.
Potassium-dependent chloride extrusion was decreased in deafened neurons
To examine the functional role of KCC2 in control and BCA neurons, we tested the effect of decreasing intracellular K+ concentration ([K+]i)on EIPSC. Because activation of KCC2 requires the simultaneous presence of K+ and Cl-, we examined the effect of a Cs+-containing pipette solution using the gramicidinperforated-patch configuration. Figure 2A shows IPSCs from control and BCA neurons under both recording conditions. The control neuron recorded with Cs+ exhibited a more depolarized EIPSC than that recorded with K+. In contrast, the BCA neuron recorded with Cs+ displayed a similar EIPSC to that recorded with K+. For control neurons, the average EIPSC obtained with K+ was -74 ± 1.8 mV (n = 45), and that obtained with Cs+ was -49.5 ± 2.8 mV (n = 19). In BCA neurons, the average EIPSC obtained with K+ was -49.7 ± 2.2 mV (n = 33), and that obtained with Cs+ was -42.7 ± 2.9 mV (n = 18). An ANOVA indicated that there was a main effect (p < 0.0001; F = 41.943; df = 3), and pairwise comparisons showed that there was a large effect of Cs on EIPSC in control neurons (p < 0.001; t = 7.601; df = 62), but not in BCA neurons (p > 0.05; t = 1.815; df = 49). Control and BCA neurons recorded with Cs+-containing pipette solution did not differ significantly from each another (p > 0.1; t = 1.671; df = 35). Figure 2B summarizes these observations, which indicate that chloride regulation depends on [K+]i in control neurons, but that this mechanism is nearly absent after deafness. The effect of cesium on EIPSC in BCA neurons is similar to that previously reported in immature auditory neurons (Kakazu et al., 1999).
Figure 2.

The effect of K+ or Cs+ in the recording pipette on EIPSC in neurons from control and BCA animals. A, Representative traces from control and BCA animals at different holding potentials are shown for recordings with K+- and Cs+-containing pipettes. B, For control neurons, the mean EIPSC was significantly more depolarized when the internal pipette solution contained Cs+. However, for BCA neurons, there was not a significant difference between K+- and Cs+-containing pipettes. n values are in bars (*p < 0.0001 vs K+). Error bars indicate SEM.
Phosphorylation-dependent chloride extrusion was decreased in deafened neurons
KCC2 has a C-terminal protein tyrosine kinase (PTK) consensus site, and outward transport of chloride requires PTK activity (Payne, 1997; Kelsch et al., 2001). If PTK-dependent chloride transport is compromised in deaf animals, then a membrane-permeable inhibitor of PTK that reduces KCC2-mediated transport (genistein) should have larger effect on control neurons, compared with BCA neurons (Akiyama et al., 1987; Kelsch et al., 2001).
Figure 3A shows representative IPSCs from control and BCA neurons, and demonstrates the effect of genistein exposure. In the control neuron, a 10 min perfusion of 50 μm genistein caused a significant depolarization of the EIPSC, whereas the same treatment had no effect on the BCA neuron. As shown in Figure 3B, the average EIPSC depolarization for control neurons was 15 ± 4 mV (n = 10), and this was a significant effect (p < 0.006; t = 3.165; df = 18). In contrast, genistein led to an average EIPSC depolarization of only 2 ± 1mV(n = 6) in BCA neurons, and this was not a significant change (p > 0.7; t = 0.273; df = 10). The average genistein-induced EIPSC depolarization was significantly smaller for BCA neurons compared with controls (p = 0.040; t = 2.266; df = 14). Increasing the genistein concentration to 100 μm and perfusion of genistein for 30 min did not heighten the effect on EIPSC in BCA neurons (data not shown).
Figure 3.

The effect of genistein on EIPSC in neurons from control and BCA animals. A, Representative traces from control and BCA animals at different holding potentials are shown for recordings obtained in ACSF and after exposure to 50 μm genistein. KYN was present throughout these recordings. B, The mean EIPSC for control neurons was significantly depolarized in the presence of genistein. However, for BCA neurons, there was not a significant difference. n values are in bars (*p = 0.005 vs EIPSC in ACSF). Error bars indicate SEM.
Chloride cotransporter expression in control and deafened animals
The EIPSC depolarization observed in BCA animals could be caused by reduced expression of an outward chloride cotransporter, such as KCC2, or increased expression of an inward chloride cotransporter, such as NKCC1. To examine this hypothesis, the level of mRNAs encoding both chloride cotransporters in the ICs of control and BCA animals was measured by RT-PCR. In the absence of published DNA sequences for KCC2 and NKCC1 in gerbil, primers were designed in regions of rodent (and human) sequence identity, assuming cross-species sequence identity in those same regions. Indeed, NKCC1, KCC2, and GAPDH sequences were readily amplified after reverse transcription of gerbil mRNA (Fig. 4A). This was true using two different primer pairs for both NKCC1 (either NK5-NK6 or NK7-NK8) and KCC2 (either KCC21-KCC22 or KCC21-KCC23). The gerbil template yielded bands identical in size to their rat counterparts (Fig. 4A), as expected in case of rodent sequence identity.
Figure 4.

Chloride cotransporter mRNA expression in the ICs from control and BCA animals. A, Amplification of NKCC1, KCC2, and GAPDH sequences in gerbil and rat yielded bands of identical size. Primers used were specific for either NKCC1 (top) or KCC2 (bottom), and two different primer sets were used for each of the genes (described in Materials and Methods). GAPDH mRNA was amplified from the same samples as an input control. The number of PCR cycles was 30. B, NKCC1 mRNA expression in control (Con) and BCA gerbils. RNA was isolated from gerbils at days 9 (P9) and 14 (P14) and was subjected to RT-PCR. Sizes of amplified DNA fragments are indicated on the left, and the number of PCR cycles is indicated on the right. C, KCC2 mRNA expression in control and BCA gerbils. RNA was isolated from gerbils at P9 and P14 and was subjected to RT-PCR. Sizes of amplified DNA fragments are indicated on the left, and number of PCR cycles is indicated on the right.
The levels of mRNA encoding either NKCC1 (Fig. 4B) or KCC2 (C) were unchanged in BCA animals compared with controls. This was true at both survival ages examined, P9 and P14. Expression levels of GAPDH showed that mRNA input levels from P9 and P14 samples taken from control or BCA animals were nearly identical (Fig. 3B,C, lanes marked GAPDH). Moreover, the exponential increase in DNA content with cycle number suggested amplification was semiquantitative, satisfying a condition to reliably measure differences in mRNA levels. The ratio of cotransporter band intensity to GAPDH was assessed after 30 cycles for each animal, and this ratio did not exhibit a significant difference among groups for any of the four primers (KCC21-KCC22, p > 0.9, F = 0.103, df = 3; KCC21-KCC23, p > 0.88, F = 0.226, df = 3; NK5-NK6, p > 0.9, F = 0.053, df = 3; and NK7-NK8, p > 0.9, F = 0.007, df = 3). Nor were significant differences observed after 22 or 25-26 cycles. These results indicate that expression of the mRNAs encoding two major chloride cotransporters in the IC is not affected by deafening.
KCC2 protein is similarly localized in control and deafened animals
To determine whether the EIPSC depolarization observed in deaf animals was attributable to reduced translation of the KCC2 protein, the immunohistochemical staining pattern was assessed. Figure 5 shows images of a control section (top), KCC2 staining in the gerbil IC of control (middle), and BCA (bottom) inferior colliculus. At the level of light microscopy, the anti-KCC2 antibody appeared to label the plasma membrane of neuronal somata and neuropil throughout the gerbil IC of both control and BCA animals. There was no apparent difference in the distribution or intensity of staining between control and experimental animals. In confirmation of the staining pattern obtained for the anti-KCC2 antibody in gerbil IC, the immunohistochemical pattern was also observed for adult rat and gerbil cerebellum (data not shown), and this conformed to the pattern described previously (Williams et al., 1999). These results suggest that protein translation of KCC2 cannot account for the deafness-induced change in chloride homeostasis.
Figure 5.
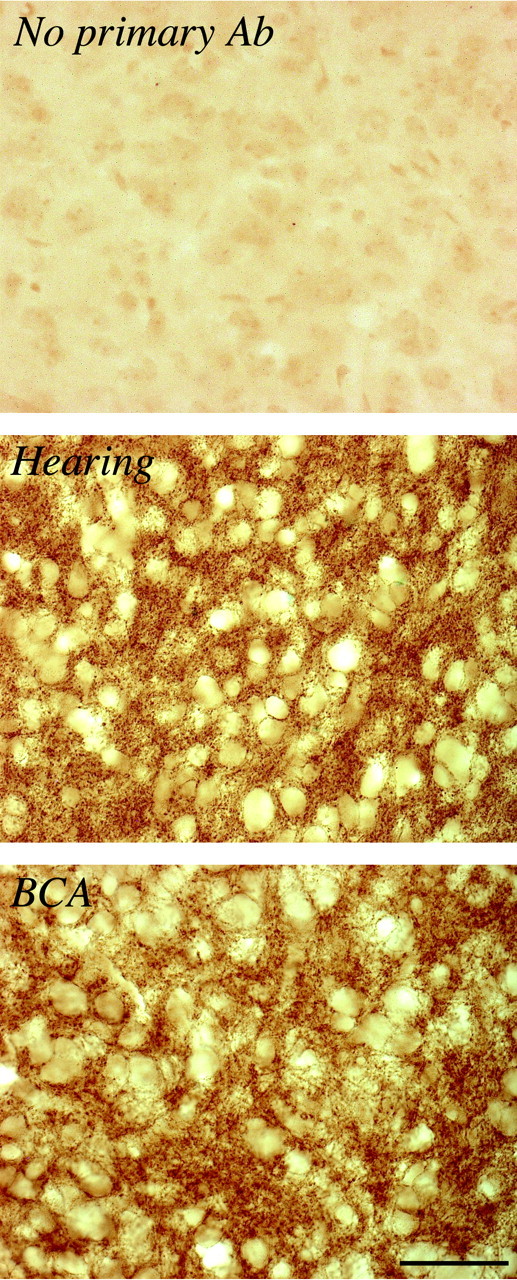
Immunohistochemical staining with anti-KCC2 polyclonal antibody in the ICs of control and BCA animals. Top, Control section in which tissue was not exposed to primary antibody. Middle, KCC2 staining in the IC of control animal. Bottom, KCC2 staining in the IC of BCA animal. Images were obtained at a magnification of 630×. Scale bar, 50 μm.
The immunohistochemical findings were quantified in a separate set of experiments on fluorescently labeled tissue, using a confocal microscope. The mean pixel intensity from control sections (123 ± 7) and from BCA sections (115 ± 7) did not differ significantly (p > 0.3; t = -0.957; df = 18).
Discussion
The proper balance of excitatory and inhibitory drive is an essential feature of all of the neuronal computational circuits. The auditory coding properties that are presumed to underlie sound localization and spectrotemporal discrimination are each sensitive to manipulations that block inhibition (Grothe, 1994; Suga et al., 1997; Carney, 1999; Brand et al., 2002; Klug et al., 2002; Pollak et al., 2002; Syka, 2002). Therefore, regulation of the molecular mechanisms that control inhibitory synaptic strength, such as chloride homeostasis, is a fundamental component of most auditory coding properties. The present study examined the basis for decreased inhibitory synaptic strength after bilateral deafness. Our results show that deafening leads to a functional decrease of chloride transport in the central auditory system without a decrease in the expression of cotransporter mRNA or protein.
BCA decreases inhibitory postsynaptic strength
Many anatomical studies have shown that reduced neural activity or deafferentation lead to postsynaptic cell death, atrophy, and altered metabolism in the central auditory nervous system (Levi-Montalcini, 1949; Webster and Webster, 1979; Parks, 1981; Deitch and Rubel, 1984; Durham and Rubel, 1985; Born and Rubel, 1988; McMullen et al., 1988; Hashisaki and Rubel, 1989; Hyson and Rubel, 1989). However, in vivo electrophysiology studies show that the remaining synapses do function and may undergo physiological alterations. After unilateral deafness, the remaining ear elicits greater-than-normal excitatory responses in the ipsilateral IC (Kitzes and Semple, 1985; McAlpine et al., 1997), and GAD levels decline rapidly (Mossop et al., 2000). In vivo experiments on bilaterally deafened animals suggest that inhibition becomes weaker in that fewer IC neurons are suppressed by auditory nerve stimulation, and GABA release declines (Bledsoe et al., 1995). Partial hearing loss is also associated with decreased inhibitory strength in the auditory cortex (Rajan, 1998). Our current findings suggest that a greater net excitatory drive observed in unilaterally or bilaterally deafened animals may result from decreased inhibition. IPSPs are less reliable at blocking current-evoked action potentials, compared with controls (Fig. 1).
Alteration of intracellular chloride regulators after deafness
In the present study, the depolarization of EIPSC was attributable to a positive shift in ECl-, because the resting membrane potential was unaltered and extracellular Cl- was constant. In control conditions, intracellular chloride is generally lower than expected from passive distribution (Thompson et al., 1988; Thompson and Gahwiler, 1989), and several proteins are involved, including KCC2, NKCC1, the Cl--HCO3- exchanger, and the Na+-dependent Cl--HCO3- exchanger (Payne et al., 2003). Of these, KCC2 is the primary contributor to maintaining low [Cl-]i (Thompson et al., 1988; Payne et al., 1996; Payne, 1997; Plotkin et al., 1997; Kakazu et al., 1999; Lu et al., 1999; Rivera et al., 1999).
In each of three different experimental manipulations, the neurons from deafened animals were less sensitive to blockade of the chloride homeostatic mechanism. First, the loop diuretic bumetanide at 100 μm shifted EIPSC to more positive values in control animals and did not alter EIPSC in BCA neurons (Table 1). Both NKCC1 and KCC2 cotransporters are sensitive to the loop diuretics (Payne, 1997; Isenring et al., 1998; Russell, 2000), and the net depolarization of EIPSC in control neurons suggests that KCC2 activity was attenuated. Second, substitution of Cs+ for K+ in the internal pipette solution depolarized EIPSC by 25 mV in control neurons, but did not modify EIPSC in BCA neurons (Fig. 2). A lack of effect of cesium on intracellular chloride concentration has been reported previously on immature lateral superior olivary neurons (Kakazu et al., 1999). Third, the PTK inhibitor that blocks KCC2 activity, genistein, depolarized EIPSC in control neurons, but did not alter EIPSC in BCA neurons (Fig. 3). These experiments left open the possibility that either KCC2 expression or function had declined.
A second nonexclusive hypothesis was that NKCC activity was upregulated after deafness. NKCC transports Cl- into the cell, particularly in immature neurons (Plotkin et al., 1997; Kakazu et al., 1999). However, 20 μm bumetanide did not significantly affect EIPSC in BCA neurons, suggesting that NKCC activity is low in IC neurons at the ages studied (Gillen et al., 1996; Payne, 1997; Kakazu et al., 2000). Because other chloride transport mechanisms do not maintain low [Cl-]i in the absence of KCC2 (Rivera et al., 2002), we did not assess their function in deafened animals.
Deafness does not affect KCC2 or NKCC1 expression
Our initial hypothesis was that the expression of KCC2 was decreased by deafening. This seemed reasonable, because the manipulation was occurring during early postnatal development, and increased KCC2 expression is commonly observed during this period with an attendant switch to hyperpolarizing response (Ehrlich et al., 1999; Kakazu et al., 1999; Rivera et al., 1999).
Changes in chloride cotransporter expression are observed during normal development and also after injury. For example, a decrease in KCC2 expression occurs after axotomy in vagus motor neurons (Nabekura et al., 2002), and an increase or decrease of KCC2 expression can be elicited in hippocampal cultures by exposure to a GABAA agonist or antagonist, respectively (Ganguly et al., 2001). In fact, depolarizing GABAergic synaptic responses have recently been observed in temporal lobe tissue obtained from human epilepsy patients (Cohen et al., 2002).
Because chloride extrusion was clearly diminished in neurons from deaf animals (Figs. 2 and 3, Table 1), we examined cotransporter expression in two ways. Bilateral deafening did not alter the mRNA levels of KCC2 or NKCC1 in IC neurons as assessed by semiquantitative RT-PCR (Fig. 4B,C). Furthermore, immunohistochemical visualization of the KCC2 protein revealed that it was abundant in the ICs of both control and BCA animals.
Brain-derived neurotrophin (BDNF) signaling has been shown to downregulate KCC2 expression by 50% in hippocampal slices, and this is associated with a depolarization of EIPSP (Rivera et al., 2002). Compared with these results, the present findings differ in an interesting way: the EIPSP is depolarized to a similar degree, but there is no indication that KCC2 expression has changed, and certainly not by 50% (Fig. 4). Interestingly, BDNF-dependent expression of KCC2 has also been reported in mice that overexpress the BDNF, but the effect is opposite to that reported in vitro (Aguado et al., 2003). Therefore, the depolarization of EIPSC and loss of inhibitory synaptic strength in deaf animals is attributable to a change in the functional status of potassium-dependent chloride extrusion.
Hypothetical basis for loss of K-Cl cotransport function
Because the present results indicate that KCC2 and NKCC1 expression were unaltered by deafness (Figs. 4 and 5), we propose that the functional status of K-Cl cotransporter(s) was affected. This is somewhat surprising in that published reports on developing systems, as well as models of injury, have associated a depolarized EIPSP with KCC2 expression levels only (Ehrlich et al., 1999; Kakazu et al., 1999; Rivera et al., 1999, 2002; Ganguly et al., 2001; Nabekura et al., 2002). Although there is little information on the regulation of KCC2 in neural tissue, there is a broad literature on the functional modulation of other cation-chloride cotransporter family members. In particular, the normal physiology of KCC1 is closely linked to the cytoplasmic domains; removal of 8 C-terminal or 89 N-terminal amino acids can abolish function without affecting surface expression of the protein in Xenopus oocytes (Casula et al., 2001). In erythrocytes, KCC activation involves a dephosphorylation event, and Src family kinases have been shown to negatively regulate K-Cl cotransporter-activating phosphatases (Jennings and Schulz, 1991; Lauf et al., 1992; Flatman et al., 1996; De Franceschi et al., 1997). In a related manner, potassium-chloride cotransport is regulated by protein kinase G in vascular smooth muscle cells, although mRNA expression is not affected (Adragna et al., 2002).
In contrast to the ubiquitously expressed KCC1, neuron-specific KCC2 has a tyrosine phosphorylation consensus site (Payne, 1997), and a single amino acid substitution is sufficient to reduce transporter activity in oocytes (Strange et al., 2000). Furthermore, KCC2 function can be deactivated by membrane-permeable protein tyrosine kinase inhibitors in cultured hippocampal neurons (Kelsch et al., 2001). It has been suggested that PTK phosphorylation of KCC2 may be part of the overall maturation of GABAergic synapses (Kelsch et al., 2001).
Together, the findings from other systems suggest that deafness leads to an alteration in the KCC2 phosphorylation state, possibly at the tyrosine residue. This is supported by the finding that genistein can depolarize EIPSC in control, but not deaf, neurons (Fig. 3). Auditory-evoked synaptic transmission in the IC could modulate KCC2 phosphorylation via many receptor systems. One viable possibility is that afferent terminals contain and release neurotrophins that elicit postsynaptic KCC2 phosphorylation. Neurotrophin expression is observed in the developing rat IC, and the cognate receptors for BDNF and neurotrophin-3 are expressed at the postsynaptic junctions in gerbil (Hafidi et al., 1996; Hafidi, 1999). Such a modulatory role would complement that long-lasting effect of neurotrophins on KCC2 expression (Rivera et al., 2002; Aguado et al., 2003).
In summary, our results suggest that modulation of KCC2 physiology may help to explain certain CNS pathologies, as well as the physiological set point of normal inhibitory synapses (Golding and Oertel, 1996; Vardi et al., 2000, Kanaka et al., 2001; Ueno et al., 2002). This mechanism differs from the many cases in which KCC2 expression is modified. Although the activity-dependent modulation of KCC2 has yet to be studied, it is known that NMDA receptor activation can stimulate an NKCC cotransporter (Sun and Murali, 1998). Therefore, it may be possible to restore inhibitory synaptic strength by examining how KCC2 depends on ionotropic and metabotropic signaling at the synapse.
Footnotes
This work was supported by National Institutes of Health Grant DC00540 (D.H.S.).
Correspondence should be addressed to Dr. Dan H. Sanes, Center for Neural Science, 4 Washington Place, York University, New York, NY 10003. E-mail: sanes@cns.nyu.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/237516-09$15.00/0
References
- Adragna NC, Zhang J, Di Fulvio M, Lincoln TM, Lauf PK ( 2002) KCl cotransport regulation and protein kinase G in cultured vascular smooth muscle cells. J Membr Biol 187: 157-165. [DOI] [PubMed] [Google Scholar]
- Aguado F, Carmona MA, Pozas E, Aguiló A, Martiínez-Guijarro FJ, Alcantara S, Borrell V, Yuste R, Ibañez CF, Soriano E ( 2003) BDNF regulates spontaneous correlated activity at early developmental stages by increasing synaptogenesis and expression of the K+/Cl- co-transporter KCC2. Development 130: 1267-1280. [DOI] [PubMed] [Google Scholar]
- Akiyama T, Ishida J, Nakagawa S, Ogawara H, Watanabe S, Itoh N, Shibuya M, Fukami Y ( 1987) Genistein, a specific inhibitor of tyrosine-specific protein kinases. J Biol Chem 262: 5592-5595. [PubMed] [Google Scholar]
- Bledsoe SC, Nagase S, Miller JM, Altschuler RA ( 1995) Deafness-induced plasticity in the mature central auditory system. NeuroReport 7: 225-229. [PubMed] [Google Scholar]
- Bormann J, Hamill OP, Sakmann B ( 1987) Mechanism of anion permeation through channels gated by glycine and γ-aminobutyric acid in mouse cultured spinal neurones. J Physiol (Lond) 385: 243-286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Born DE, Rubel EW ( 1988) Afferent influences on brain stem auditory nuclei of the chicken: presynaptic action potentials regulate protein synthesis in nucleus magnocellularis neurons. J Neurosci 8: 901-919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand A, Behrend O, Marquardt T, McAlpine D, Grothe B ( 2002) Precise inhibition is essential for microsecond interaural time difference coding. Nature 417: 543-547. [DOI] [PubMed] [Google Scholar]
- Carney LH ( 1999) Temporal response properties of neurons in the auditory pathway. Curr Opin Neurobiol 9: 442-446. [DOI] [PubMed] [Google Scholar]
- Casula S, Shmukler BE, Wilhelm S, Stuart-Tilley AK, Su W, Chernova MN, Brugnara C, Alper SL ( 2001) A dominant negative mutant of the KCC1 K-Cl cotransporter: both N- and C-terminal cytoplasmic domains are required for K-Cl cotransport activity. J Biol Chem 276: 41870-41878. [DOI] [PubMed] [Google Scholar]
- Clayton GH, Owens GC, Wolff JS, Smith RL ( 1998) Ontogeny of cation-Cl- cotransporter expression in rat neocortex. Brain Res Dev Brain Res 109: 281-292. [DOI] [PubMed] [Google Scholar]
- Cohen I, Navarro V, Clemenceau S, Baulac M, Miles R ( 2002) On the origin of interictal activity in human temporal lobe epilepsy in vitro Science 298: 1418-1421. [DOI] [PubMed] [Google Scholar]
- DeFazio RA, Keros S, Quick MW, Hablitz JJ ( 2000) Potassium-coupled chloride cotransport controls intracellular chloride in rat neocortical pyramidal neurons. J Neurosci 20: 8069-8076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Franceschi L, Fumagalli L, Olivieri O, Corrocher R, Lowell CA, Berton G ( 1997) Deficiency of Src family kinases Fgr and Hck results in activation of erythrocyte K/Cl cotransport. J Clin Invest 99: 220-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deitch JS, Rubel EW ( 1984) Afferent influences on brain stem auditory nuclei of the chicken: time course and specificity of dendritic atrophy following deafferentation. J Comp Neurol 229: 66-79. [DOI] [PubMed] [Google Scholar]
- Delpire E, Rauchman MI, Beier DR, Hebert SC, Gullans SR ( 1994) Molecular cloning and chromosome localization of a putative basolateral Na+-K+-2Cl- cotransporter from mouse inner medullary collecting duct (mIMCD-3) cells. J Biol Chem 269: 25677-25683. [PubMed] [Google Scholar]
- Durham D, Rubel EW ( 1985) Afferent influences on brain stem auditory nuclei of the chicken: succinate dehydrogenase activity following cochlea removal. J Comp Neurol 231: 446-456. [DOI] [PubMed] [Google Scholar]
- Ehrlich I, Lohrke S, Friauf E ( 1999) Shift from depolarizing to hyperpolarizing glycine action in rat auditory neurones is due to age-dependent Cl- regulation. J Physiol (Lond) 520: 121-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flatman PW, Adragna NC, Lauf PK ( 1996) Role of protein kinases in regulating sheep erythrocyte K+-Cl- cotransport. Am J Physiol 271: C255-C263. [DOI] [PubMed] [Google Scholar]
- Ganguly K, Schinder AF, Wong ST, Poo M ( 2001) GABA itself promotes the developmental switch of neuronal GABAergic responses from excitation to inhibition. Cell 105: 521-532. [DOI] [PubMed] [Google Scholar]
- Gillen CM, Brill S, Payne JA, Forbush B ( 1996) Molecular cloning and functional expression of the K-Cl cotransporter from rabbit, rat and human. A new member of the cation-chloride cotransporter family. J Biol Chem 271: 16237-16244. [DOI] [PubMed] [Google Scholar]
- Gold JI, Knudsen EI ( 2000) Abnormal auditory experience induces frequency-specific adjustments in unit tuning for binaural localization cues in the optic tectum of juvenile owls. J Neurosci 20: 862-877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golding NL, Oertel D ( 1996) Context-dependent synaptic action of glycinergic and GABAergic inputs in the dorsal cochlear nucleus. J Neurosci 16: 2208-2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grothe B ( 1994) Interaction of excitation and inhibition in processing of pure tone and amplitude-modulated stimuli in the medial superior olive of the mustached bat. J Neurophysiol 71: 706-721. [DOI] [PubMed] [Google Scholar]
- Hafidi A ( 1999) Distribution of BDNF, NT-3 and NT-4 in the developing auditory brainstem. Int J Dev Neurosci 17: 285-294. [DOI] [PubMed] [Google Scholar]
- Hafidi A, Moore T, Sanes DH ( 1996) Regional distribution of neurotrophin receptors in the developing auditory brainstem. J Comp Neurol 367: 454-464. [DOI] [PubMed] [Google Scholar]
- Hashisaki GT, Rubel EW ( 1989) Effects of unilateral cochlea removal on anteroventral cochlear nucleus neurons in developing gerbils. J Comp Neurol 283: 465-473. [DOI] [PubMed] [Google Scholar]
- Hübner CA, Stein V, Hermans-Borgmeyer I, Meyer T, Ballanyi K, Jentsch TJ ( 2001) Disruption of KCC2 reveals an essential role of K-Cl cotransport already in early synaptic inhibition. Neuron 30: 515-524. [DOI] [PubMed] [Google Scholar]
- Hyson RL, Rubel EW ( 1989) Transneuronal regulation of protein synthesis in the brain stem auditory system of the chick requires synaptic activation. J Neurosci 9: 2835-2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isenring P, Jacoby SC, Payne JA, Forbush B ( 1998) Comparison of Na-K-Cl cotransporters. J Biol Chem 273: 11295-11301. [DOI] [PubMed] [Google Scholar]
- Jennings ML, Schulz RK ( 1991) Okadaic acid inhibition of KCl cotransport. Evidence that protein dephosphorylation is necessary for activation of transport by either cell swelling or N-ethylmaleimide. J Gen Physiol 97: 799-817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakazu Y, Akaike N, Komiyama S, Nabekura J ( 1999) Regulation of intracellular chloride by cotransporters in developing lateral superior olive neurons. J Neurosci 19: 2843-2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakazu Y, Uchida S, Takashi N, Akaike N, Nabekura J ( 2000) Reversibility and cation selectivity of the K+-Cl- cotransport in rat central neurons. J Neurophysiol 84: 281-288. [DOI] [PubMed] [Google Scholar]
- Kanaka C, Ohno K, Okabe A, Kuriyama K, Itoh T, Fukuda A, Sato K ( 2001) The differential expression patterns of messenger RNAs encoding K-Cl cotransporters (KCC1, 2) and Na-K-2Cl cotransporter (NKCC1) in the rat nervous system. Neuroscience 104: 933-946. [DOI] [PubMed] [Google Scholar]
- Kelsch W, Hormuzdi S, Straube E, Lewen A, Monyer H, Misgeld U ( 2001) Insulin-like growth factor 1 and a cytosolic tyrosine kinase activate chloride outward transport during maturation of hippocampal neurons. J Neurosci 21: 8339-8347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitzes LM, Semple MN ( 1985) Single-unit responses in the inferior colliculus: effects of neonatal unilateral cochlear ablation. J Neurophysiol 53: 1483-1500. [DOI] [PubMed] [Google Scholar]
- Klinke R, Kral A, Heid S, Tillein J, Hartmann R ( 1999) Recruitment of the auditory cortex in congenitally deaf cats by long-term cochlear electro-stimulation. Science 285: 1729-1733. [DOI] [PubMed] [Google Scholar]
- Klug A, Bauer EE, Hanson JT, Hurley L, Meitzen J, Pollak GD ( 2002) Response selectivity for species-specific calls in the inferior colliculus of Mexican free-tailed bats is generated by inhibition. J Neurophysiol 88: 1941-1954. [DOI] [PubMed] [Google Scholar]
- Kotak VC, Sanes DH ( 1996) Developmental influence of glycinergic transmission: regulation of NMDA receptor-mediated EPSPs. J Neurosci 16: 1836-1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotak VC, DiMattina C, Sanes DH ( 2001) GABAB and Trk receptor signaling mediates long-lasting inhibitory synaptic depression. J Neurophysiol 86: 536-540. [DOI] [PubMed] [Google Scholar]
- Kral A, Hartmann R, Tillein J, Heid S, Klinke R ( 2002) Hearing after congenital deafness: central auditory plasticity and sensory deprivation. Cereb Cortex 12: 797-807. [DOI] [PubMed] [Google Scholar]
- Lauf PK, Bauer J, Adragna NC, Fujise H, Zade-Oppen AM, Ryu KH, Delpire E ( 1992) Erythrocyte K+-Cl- cotransport: properties and regulation. Am J Physiol 263: C917-C932. [DOI] [PubMed] [Google Scholar]
- Levi-Montalcini R ( 1949) The development of the acoustico-vestibular centers in the chick embryo in the absence of the afferent root fibers and of descending fiber tracts. J Comp Neurol 91: 209-241. [DOI] [PubMed] [Google Scholar]
- Lu J, Karadhed M, Delpire E ( 1999) Developmental regulation of the neuronal-specific isoform of K-Cl cotranspoter KCC2 in postnatal rat brains. J Neurobiol 39: 558-568. [PubMed] [Google Scholar]
- McAlpine D, Martin RL, Mossop JE, Moore DR ( 1997) Response properties of neurons in the inferior colliculus of the monaurally deafened ferret to acoustic stimulation of the intact ear. J Neurophysiol 78: 767-779. [DOI] [PubMed] [Google Scholar]
- McMullen NT, Goldberger B, Suter CM, Glaser EM ( 1988) Neonatal deafening alters non pyramidal dendrite orientation in auditory cortex: a computer microscopic study in the rabbit. J Comp Neurol 267: 92-106. [DOI] [PubMed] [Google Scholar]
- Mossop JE, Wilson MJ, Caspary DM, Moore DR ( 2000) Down-regulation of inhibition following unilateral deafening. Hear Res 147: 183-187. [DOI] [PubMed] [Google Scholar]
- Nabekura J, Ueno T, Okabe A, Furuta A, Iwaki T, Shimizu-Okabe C, Fukuda A, Akaike N ( 2002) Reduction of KCC2 expression and GABAA receptor-mediated excitation after in vivo axonal injury. J Neurosci 22: 4412-4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens DF, Boyce LH, Davis MB, Kriegstein AR ( 1996) Excitatory GABA responses in embryonic and neonatal cortical slices demonstrated by gramicidin perforated-patch recordings and calcium imaging. J Neurosci 16: 6414-6423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallas SL, Littman T, Moore DR ( 1999) Cross-modal reorganization of callosal connectivity without altering thalamocortical projections. Proc Natl Acad Sci USA 96: 8751-8756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks TN ( 1981) Changes in the length and organization of nucleus laminaris dendrites after unilateral otocyst ablation in chick embryos. J Comp Neurol 202: 47-57. [DOI] [PubMed] [Google Scholar]
- Payne JA ( 1997) Functional characterization of the neuronal-specific K-Cl cotransporter: implications for [K+]o regulation. Am J Physiol 273: C1516-C1525. [DOI] [PubMed] [Google Scholar]
- Payne JA, Stevenson TJ, Donaldson LF ( 1996) Molecular characterization of a putative K-Cl cotransporter in rat brain. J Biol Chem 27: 16245-16252. [DOI] [PubMed] [Google Scholar]
- Payne JA, Rivera C, Voipio J, Kaila K ( 2003) Cation-chloride cotransporters in neuronal communication, development, and trauma. Trends Neurosci 26: 199-206. [DOI] [PubMed] [Google Scholar]
- Plotkin MD, Snyder EY, Hebert SC, Delpire E ( 1997) Expression of the Na-K-2Cl cotransporter is developmentally regulated in postnatal rat brains: a possible mechanism underlying GABA's excitatory role in immature brain. J Neurobiol 33: 781-795. [DOI] [PubMed] [Google Scholar]
- Pollak GD, Burger RM, Park TJ, Klug A, Bauer EE ( 2002) Roles of inhibition for transforming binaural properties in the brainstem auditory system. Hear Res 168: 60-78. [DOI] [PubMed] [Google Scholar]
- Rajan R ( 1998) Receptor organ damage causes loss of cortical surround inhibition without topographic map plasticity. Nat Neurosci 1: 138-143. [DOI] [PubMed] [Google Scholar]
- Rhee JS, Ebihara S, Akaike N ( 1994) Gramicidin perforated patch-clamp technique reveals glycine-gated outward chloride current in dissociated nucleus solitarii neurons of the rat. J Neurophysiol 72: 1103-1108. [DOI] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K ( 1999) The K+/Cl- cotransporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 397: 251-255. [DOI] [PubMed] [Google Scholar]
- Rivera C, Li H, Thomas-Crusells J, Lahtinen H, Viitanen T, Nanobashvili A, Kokaia Z, Airaksinen MS, Voipio J, Kaila K, Saarma M ( 2002) BDNF-induced TrkB activation down-regulates the K+-Cl- cotransporter KCC2 and impairs neuronal Cl- extrusion. J Cell Biol 159: 747-752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell JM ( 2000) Sodium-potassium-chloride cotransport. Physiol Rev 80: 211-276. [DOI] [PubMed] [Google Scholar]
- Sanes DH, Markowitz S, Bernstein J, Wardlow J ( 1992) The influence of inhibitory afferents on the development of postsynaptic dendritic arbors. J Comp Neurol 321: 637-644. [DOI] [PubMed] [Google Scholar]
- Sherman SM, Spear PD ( 1982) Organization of visual pathways in normal and visually deprived cats. Physiol Rev 62: 738-855. [DOI] [PubMed] [Google Scholar]
- Strange K, Singer TD, Morrison R, Delpire E ( 2000) Dependence of KCC2 K-Cl cotransporter activity on a conserved carboxy terminus tyrosine residue. Am J Physiol 279: C860-C867. [DOI] [PubMed] [Google Scholar]
- Suga N, Zhang Y, Yan J ( 1997) Sharpening of frequency tuning by inhibition in the thalamic auditory nucleus of the mustached bat. J Neurophysiol 77: 2098-2114. [DOI] [PubMed] [Google Scholar]
- Sun D, Murali SG ( 1998) Stimulation of Na+-K+-2Cl- cotransporter in neuronal cells by excitatory neurotransmitter glutamate. Am J Physiol 275: C772-C779. [DOI] [PubMed] [Google Scholar]
- Syka J ( 2002) Plastic changes in the central auditory system after hearing loss, restoration of function, and during learning. Physiol Rev 82: 601-636. [DOI] [PubMed] [Google Scholar]
- Thompson SM, Gahwiler BH ( 1989) Activity-dependent disinhibition. II. Effects of extracellular potassium, furosemide, and membrane potential on ECl- in hippocampal CA3 neurons. J Neurophysiol 61: 512-523. [DOI] [PubMed] [Google Scholar]
- Thompson SM, Deisz RA, Prince DA ( 1988) Outward chloride cation cotransport in mammalian cortical neurons. Neurosci Lett 89: 49-54. [DOI] [PubMed] [Google Scholar]
- Ueno T, Okabe A, Akaike N, Fukuda A, Nabekura J ( 2002) Diversity of neuron-specific K+-Cl- cotransporter expression and inhibitory postsynaptic potential depression in rat motoneurons. J Biol Chem 277: 4945-4950. [DOI] [PubMed] [Google Scholar]
- Vale C, Sanes DH ( 2000) Afferent regulation of inhibitory synaptic transmission in the developing auditory midbrain. J Neurosci 20: 1912-1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale C, Sanes DH ( 2002) The effect of bilateral deafness on excitatory and inhibitory synaptic strength in the inferior colliculus. Eur J Neurosci 16: 2394-2404. [DOI] [PubMed] [Google Scholar]
- Vardi N, Zhang L-L, Payne JA, Sterling P ( 2000) Evidence that different cation chloride cotransporters in retinal neurons allow opposite responses to GABA. J Neurosci 20: 7657-7663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Reichling DB, Kyrozis A, MacDermott AB ( 1994) Developmental loss of GABA- and glycine-induced depolarization and Ca2+ transients in embryonic rat dorsal horn neurons in culture. Eur J Neurosci 6: 1275-1280. [DOI] [PubMed] [Google Scholar]
- Webster DB, Webster M ( 1979) Effects of neonatal conductive hearing loss on brain stem auditory nuclei. Ann Otol Rhinol Laryngol 88: 684-688. [DOI] [PubMed] [Google Scholar]
- Williams JR, Sharp JW, Kumari VG, Wilson M, Payne JA ( 1999) The neuron-specific K-Cl cotransporter, KCC2. Antibody development and initial characterization of the protein. J Biol Chem 274: 12656-12664. [DOI] [PubMed] [Google Scholar]