

Abstract
The suprachiasmatic nucleus (SCN) circadian clock exhibits a recurrent series of dynamic cellular states, characterized by the ability of exogenous signals to activate defined kinases that alter clock time. To explore potential relationships between kinase activation by exogenous signals and endogenous control mechanisms, we examined clock-controlled protein kinase G (PKG) regulation in the mammalian SCN. Signaling via the cGMP-PKG pathway is required for light- or glutamate (GLU)-induced phase advance in late night. Spontaneous cGMP-PKG activation occurred at the end of subjective night in free-running SCN in vitro. Phasing of the SCN rhythm in vitro was delayed by ∼3 hr after treatment with guanylyl cyclase (GC) inhibitors, PKG inhibition, or antisense oligodeoxynucleotide (αODN) specific for PKG, but not PKA inhibitor or mismatched ODN. This sensitivity to GC-PKG inhibition was limited to the same 2 hr time window demarcated by clock-controlled activation of cGMP-PKG. Inhibition of the cGMP-PKG pathway at this time caused delays in the phasing of four endogenous rhythms: wheel-running activity, neuronal activity, cGMP, and Per1. Timing of the cGMP-PKG-necessary window in both rat and mouse depended on clock phase, established by the antecedent light/dark cycle rather than solar time. Because behavioral, neurophysiological, biochemical, and molecular rhythms showed the same temporal sensitivities and qualitative responses, we predict that clock-regulated GC-cGMP-PKG activation may provide a necessary cue as to clock state at the end of the nocturnal domain. Because sensitivity to phase advance by light-GLU-activated GC-cGMP-PKG occurs in juxtaposition, these signals may induce a premature shift to this PKG-necessary clock state.
Keywords: protein kinase G (PKG), suprachiasmatic nucleus (SCN), circadian, Per1, cGMP, oligodeoxynucleotide (ODN)
Introduction
Circadian timekeeping, even in complex nervous systems, is a property of cells (Michel et al., 1993; Welsh et al., 1995). Ordered, clock-regulated changes in cellular processes partition clock functions into a series of specific time domains that define time of day (Gillette and Mitchell, 2002). The coordinated function of a core group of clock genes and their protein products, articulated by a highly conserved transcriptional-translational feedback loop, provides a framework for molecular definitions of time domains (Okamura et al., 2002).
Regulation of clock elements is mediated by complex cellular processes, including post-translational modifications of proteins via phosphorylation (Garceau et al., 1997; Merrow et al., 1997; Kloss et al., 2001; Lee et al., 2001; Lin et al., 2001; Ripperger and Schibler, 2001; Okamura et al., 2002). Hyperphosphorylation is characteristic of the Drosophila clock elements, PERIOD (dPER) (Kloss et al., 2001) and TIMELESS (dTIM) (Price et al., 1995), as well as the Neurospora clock protein, FREQUENCY (FRQ) (Garceau et al., 1997), during restricted parts of their circadian cycles and is accomplished by specific kinases. dPER is phosphorylated and destabilized by DOUBLETIME (DBL). The effects of light on Drosophila (Naidoo et al., 1999) and Neurospora (Yang et al., 2001) clock elements are also phosphorylation-dependent.
The type of molecular control of circadian timing exhibited by Drosophila and Neurospora is conserved in the mammalian clock in the suprachiasmatic nucleus (SCN) (Okamura et al., 2002). Transcripts of the mammalian homologs (Per1, Per2, Per3) of dPer oscillate in the SCN (Albrecht et al., 1997; Shearman et al., 1997; Shigeyoshi et al., 1997; Sun et al., 1997; Tei et al., 1997). Transcription of mPer1 is induced by light at night, much like frq (Crosthwaite et al., 1997), whereas the pattern of expression of mPer2 is reminiscent of dPer (Zeng et al., 1994; Albrecht et al., 1997; Okamura et al., 2002). Kinase-driven phosphorylation events, whose mediators include casein kinase Iϵ and glycogen synthase kinase orthologs of dDBL and dSHAGGY, respectively (Kloss et al., 1998; Lowrey et al., 2000; Martinek et al., 2001), also regulate putative mammalian clock elements. Additionally, circadian changes in the phosphorylation states of mPER1, mPER2, CLOCK, and BMAL have been demonstrated (Lee et al., 2001).
Complementing the molecular genetic studies, experimental manipulation of kinases alters rhythms in diverse circadian systems (Eskin et al., 1984; Prosser and Gillette, 1989; Prosser et al., 1989; Liu and Gillette, 1996; Krucher et al., 1997; Liu et al., 1997; Comolli and Hastings, 1999). Clock sensitivity to cGMP-dependent pathways has emerged as a conserved feature of the nocturnal domain (Eskin et al., 1984; Prosser and Gillette, 1989). Phase advance of the mammalian clock stimulated by light (Weber et al., 1995; Mathur et al., 1996) or glutamate (GLU) (Ding et al., 1998) requires activation of a guanylyl cyclase (GC)-cGMP-protein kinase G (PKG) signaling cascade during late night. If phase resetting occurs immediately (Best et al., 1999), light-GLU would shift clock state ahead to the end of subjective night. Thus, we hypothesized that light-GLU may prematurely activate cellular events that are activated endogenously by clock-controlled processes at a later time, concurrent with the waning of sensitivity to phase resetting by these stimuli.
We tested this hypothesis by evaluating the suprachiasmatic nucleus (SCN) of rat and mouse for evidence of endogenous activation of cGMP-PKG and clock sensitivity to inhibiting this regulatory node. Herein, we report discovery of a temporal window of spontaneous upregulation in cGMP levels and PKG activity in the SCN at the end of subjective night. Inhibition of the PKG pathway during this sensitive period delays behavioral (wheel-running), physiological (SCN neuronal activity), biochemical (cGMP), and molecular (Per1) oscillations. The magnitudes of the phase delays suggest dynamic altering of clock state, back to the point of maximal sensitivity to phase advance induced by light-GLU activation of GC-cGMP-PKG.
Materials and Methods
Animals and brain slice treatments. Seven- to 10-week-old LE-BluGill rats (University of Illinois) were used for the in vitro studies. Use of this inbred strain greatly reduces interexperimental variation common to outbred animals and is important for achieving high statistical significance with small sample sizes. Rats were provided food and water ad libitum and were housed under a 12 hr light/dark (LD) schedule. Brain slices were prepared between zeitgeber time 1 (ZT 1) and ZT 10, where ZT 0 is defined as the time of the onset of the light phase in the LD cycle. Animals from environments with lights on from either 7:00 A.M. to 7:00 P.M. or 2:00 P.M. to 2:00 A.M. were used to facilitate sampling and to demonstrate phase control of the rhythms investigated. Brains were quickly removed, blocked, and 500 μm coronal hypothalamic slices containing the paired SCN were prepared. Slices were studied for up to 3 d in vitro with continuous perfusion of Earle's Essential Balanced Salt Solution (EBSS) (Invitrogen, Gaithersburg, MD), supplemented with 24.6 mm glucose, 26.2 mm NaHCO3, and 2.5 mg/l gentamycin, and saturated with 95% O2 and 5% CO2 at 37°C, pH 7.4. Under these constant conditions, the SCN generates stable, near 24 hr oscillations in neuronal activity (Prosser and Gillette, 1989) with a characteristic peak around mid-day (approximately circadian time 7, CT 7) (Gillette and Prosser, 1988); measurement of time-of-peak provides an accurate assessment of circadian phase (Gillette et al., 1995).
After equilibration in the brain slice chamber for at least 2 hr, treatments were applied to the brain slice by stopping the perfusion and replacing the medium bathing the slice with EBSS containing one of the following reagents: specific PKG inhibitor, KT5823 (1.0 μm); specific PKA inhibitor, KT5720 (100 nm); GC inhibitors, LY83583 (2.0 μm) and ODQ (20 nm); or PKG-specific oligodeoxynucleotides (ODN) (10 μm).
Single-unit recordings of SCN neuronal activity. The method used for extracellular recording from the ensemble of SCN neurons has been described in detail previously and validated thoroughly (Prosser and Gillette, 1989). A glass microelectrode filled with 5 M NaCl was lowered into the SCN until the signal from a single cell was encountered. Cells, identified as electrical signals exceeding twice the background level, were observed for stability and counted for 4 min using LabView software. After sampling throughout the rostrocaudal extent of the slice, the electrode was repositioned within the SCN, and the procedure was repeated. Firing rates of individual neurons (n = 45-212, depending on the duration of the experiment) recorded during a single experiment were grouped into 2 hr running averages using 15 min lags. Time-of-peak was determined by visual inspection of the plot for the symmetrically highest point. Phase shifts were determined by comparing the mean time-of-peak from treatment groups to vehicle-treated controls. Effects of pharmacological interventions were evaluated using ANOVA and Student-Neuman-Keuls post hoc analysis or by Student's t test.
Wheel-running activity and assessment of circadian rhythmicity in vivo. Five-week-old B6129PF1/J mice (Jackson Laboratory, Bar Harbor, ME) were surgically implanted with a 26 gauge guide cannula (Plastics One, Inc., Roanoke, VA) aimed at the right SCN (anteroposterior -0.3; mediolateral -0.1; dorsoventral -1.9 from bregma). After surgery, animals were individually housed in cages equipped with a running wheel. Each rotation of the wheel moved a magnet past a hermetically sealed reed switch, closing the circuit. This information was transmitted to a computer equipped with Clocklab Acquisition software (Actimetrics, Inc., Evanston, IL), where wheel-running activity was stored in 6 min bins. Animals were housed on a 12 hr light/dark schedule for at least 10 d after surgery and were released then to constant darkness. After at least 10 d of free-running, animals were removed under dim (<1 lux) red light, gently restrained, and a 33 gauge cannula (Plastics One, Inc.) attached to a Hamilton syringe was inserted into the guide cannula. We injected 300 nl of either 100 μm KT5823 or 0.9% saline over a period of 30 sec. Mice were then returned to their home cage for at least 10 d before receiving an additional injection. Mice were randomized so that half received saline first, followed by KT5823 10 d later, whereas the other half received KT5823 first. Because of differences in the endogenous periods (τ)of the outbred mice, injections took place at different times of day but at the same circadian time. After the final round of injections, mice were killed, and 300 nl of 0.1% thionin was injected into the guide cannula to mark the injection site. Brains were removed, fixed in 4% paraformaldehyde, and sliced for verification of the cannula placement. Only data from mice in which the injection was directly into the SCN and for whom paired data were available were used. Phase shifts were calculated using Clocklab Analysis software (Actimetrics, Inc., Evanston, IL) by comparing the predicted onset of activity to the actual observed onset.
PKG activity assay. The PKG activity assay in SCN reduced slices has been previously validated (Liu et al., 1997) and was determined by in vitro phosphorylation of the PKG specific substrate RKRSRAE (Glass and Krebs, 1982). Reduced slices, containing only the SCN and the underlying optic chiasm, were prepared at ZT 5 and maintained under constant conditions in the brain slice chamber. The SCN in reduced slices maintains a normal circadian rhythm of neuronal activity (Gillette and Reppert, 1987). CT was reckoned from the animal's previous LD cycle, because the SCN from these inbred rats maintains a stable 24 hr rhythm for up to 3 d in vitro (Prosser and Gillette, 1989). Reduced slices were collected at CT 11, 18, and 23, and frozen until assay. A 20 μl aliquot of assay buffer (50 mm HEPES, 10 mm MgCl2, 50 μm IBMX, 1 μm EGTA, and 10 μm DTT, pH 7.4) containing protease inhibitor cocktail (Sigma, St. Louis, MO) and 1.0 μm of the specific PKA inhibitor, KT5720 (Calbiochem, La Jolla, CA), and RKRSRAE (1.0 μg/ml; Peninsula Laboratories, Belmont, CA), was added to each reduced slice. Background was established by addition of KT5823 (1.0 μm; Calbiochem) to half of the samples. We added 5 μm ATP and 1 μCi 32P-ATP to initiate the reaction. The reaction was stopped after 2 min at 37°C by acidification. Reaction mixtures were spotted and dried on P81 filter paper (Millipore, Bedford, MA), washed three times with 0.5% phosphoric acid, and once with ethanol. Dried samples were counted in a liquid scintillation counter. Specific PKG activity was calculated by subtracting activity from background samples. Differences among groups were evaluated statistically using ANOVA, with Student-Neuman-Keuls post hoc analysis.
cGMP measurements. Reduced slices were prepared as for PKG activity assays. Treatments were applied to the bath, as described above. Samples (two reduced SCN slices per tube) were collected at appropriate intervals before and after treatments. cGMP was assayed using a scintillation proximity assay (SPA) (Amersham Biosciences, Arlington Heights, IL) according to the acetylation protocol provided. Peak levels were determined by ANOVA with Student-Newman-Keuls post hoc analysis. Differences in time-of-peak caused by pharmacological interventions were determined by Student's t test. Sample size was based on rigorous statistical testing of the hypothesis.
In situ hybridization. In situ hybridization was performed as previously described (Tischkau et al., 1999, 2000). After treatment, brain slices containing the SCN were fixed overnight in 4% paraformaldehyde. Slices were transferred to 0.1 m PBS with 20% sucrose and maintained at 4°C until sectioning. 10 μm sections were cut at -17°C on a cryostat. The template for the mPer1 probe was kindly provided by Dr. U. Schibler (Balsalobre et al., 1998) and corresponded to nucleotides 660-780 of the published sequence (Sun et al., 1997). Digoxygenin-labeled probes were generated by in vitro transcription. Probe hybridization was visualized using an alkaline phosphatase-labeled anti-digoxygenin antibody (1:100; Roche Products, Hertforshire, UK). Analysis of mPer1-positive cells was made in a midcaudal section of the SCN by an individual blind to the experimental design and identity of the samples.
Results
Endogenous oscillations of cGMP levels and PKG activity
Spontaneous oscillations in cGMP levels and PKG activity were observed in rat SCN maintained under constant conditions (Fig. 1). To establish phase control of the circadian cycle, animals from two different lighting schedules (lights on from 2:00 P.M. to 2:00 A.M. (n = 4) or lights on from 7:00 A.M. to 7:00 P.M.) (n = 3) were used. Results from both lighting schedules were identical; thus, data were pooled. Levels of cGMP were low (∼ 9 fmol/reduced slice) during subjective day and throughout most of subjective night (Fig. 1A). However, a significant rise in endogenous cGMP occurred at the end of subjective night. Levels of cGMP were 44% higher (p < 0.05) at CT 22 (12.22 ± 0.54 fmol/reduced slice; n = 7) compared with CT 20 (8.45 ± 0.36 fmol/reduced slice; n = 7); by CT 24/0, cGMP levels (16.70 ± 0.88 fmol/reduced slice; n = 7) were 74% higher than at CT 20 (p < 0.01). cGMP returned to basal levels by CT 2 (8.48 ± 0.38 fmol/reduced slice; n = 7).
Figure 1.
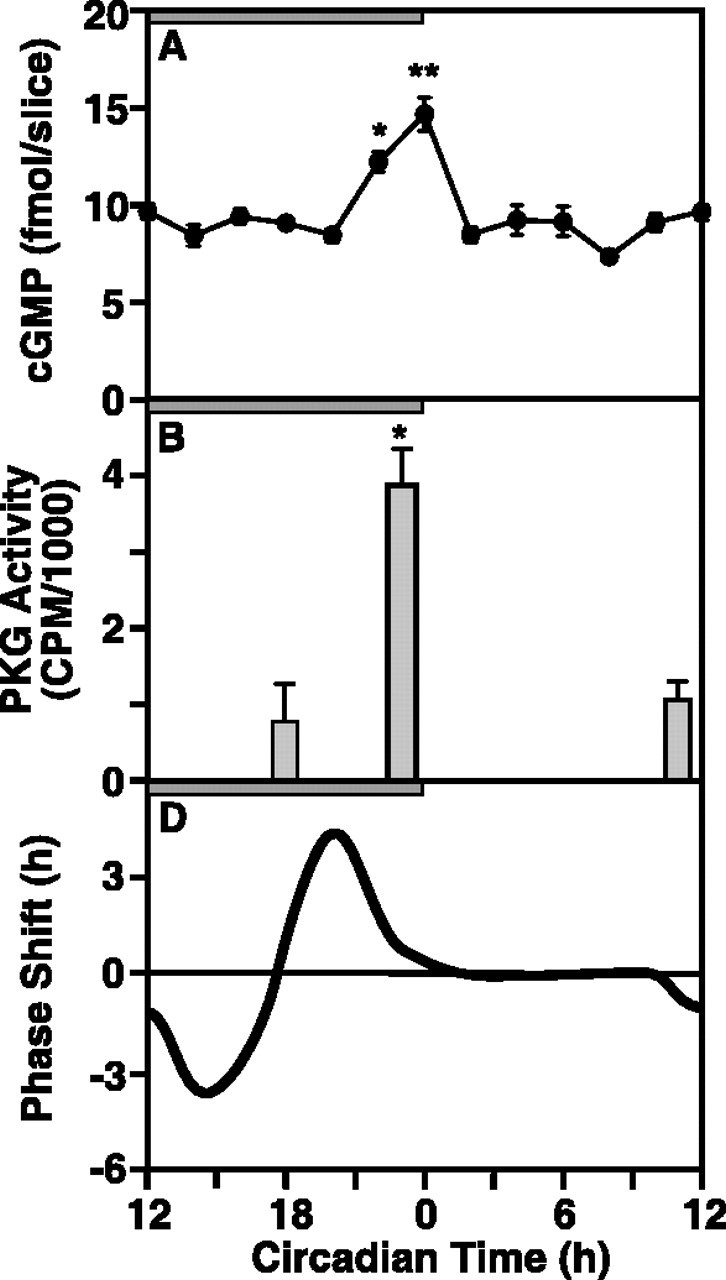
Relationships between oscillations in cGMP levels and PKG activity and the phase response curve for GLU, which relates the time of GLU stimulation to the resultant effect on clock phase observed in rat SCN maintained in vitro. The horizontal gray bar indicates nighttime in the donor colony; slices were maintained in constant light. A, cGMP levels undergo a significant oscillation. Whereas cGMP levels were low during subjective day and most of subjective night, cGMP was significantly higher (ANOVA) at CT 22 (p < 0.05) and CT 24 (p < 0.01) compared with all other times. Data are presented as mean ± SEM for seven experiments. Two rat SCN were pooled within each experiment for every time point. B, In vitro phosphorylation of the specific PKG substrate RKRSRAE reflects the endogenous PKG activity of rat SCN obtained at CT 18, 23, and 11. PKG activity in samples containing 1.0 μm KT5823 was subtracted from activity in uninhibited samples to yield specific PKG activity. PKG specific activity (CPM per reduced slice) was significantly higher at CT 23 compared with CT 11 and 18 (ANOVA; p < 0.05). Data are means ± SEM for four experiments. C, The phase response curve for the effect of GLU on the timing of the oscillation in rat SCN neuronal activity, showing maximum sensitivity to phase advance at CT 19-20 (redrawn from Ding et al., 1994). *Statistical significance, as determined by ANOVA and the Student—Neuman-Keuls post hoc test, indicated by *p < 0.05, **p < 0.01, throughout, except Figure 5.
The rise in cGMP levels was paralleled by a rise in PKG-specific activity (Fig. 1B). Endogenous PKG activity in rat SCN reduced slices was significantly higher (p < 0.05) at the end of the night (CT 23; 3775 ± 362 cpm/reduced slice; n = 4) compared with the end of subjective day (CT 11; 828 ± 167 cpm/reduced slice; n = 4) or the middle of subjective night (CT 18; 1374 ± 348 cpm/reduced slice; n = 4). Interestingly, cGMP levels and PKG activity increase just as sensitivity of the SCN to the cGMP-mediated, GLU-induced phase advance wanes at the end of night (Fig. 1C).
Inhibition of cGMP production or PKG activation at the end of subjective night delays the SCN neuronal activity rhythm
To determine whether this endogenous activation of PKG affects timekeeping, the isoquinoline inhibitor of PKG, KT5823 (1.0 μm) (Kase et al., 1987; Gadbois et al., 1992; Ding et al., 1998), was applied to rat SCN slices for 1 hr at various points in the circadian cycle. The concentration of the PKG inhibitor used was based on a dose-response curve for concentrations ranging between 0.05 and 1.0 μm, with a half-maximal effect at ∼0.2 μm and a plateau above 1.0 μm (data not shown). In control slices, the electrical activity of the ensemble of SCN neurons peaked near CT 7 (CT 6.88 ± 0.11; n = 8) on the first and second days in vitro (Fig. 2A,E). Inhibition of PKG had no effect on the timing of the rhythm when applied at four different time points during subjective day (Fig. 2B,E) or at three points from early to midsubjective night (Fig. 2C,E). However, application of KT5823 at CT 23, the time corresponding to the endogenous increase in PKG activity in the SCN, caused a 2.96 ± 0.24 hr (n = 4; p < 0.01) phase delay in the timing of the electrical activity rhythm (Fig. 2D,E). The phase response curve revealed a very narrow window of sensitivity (Fig. 2E); only treatments between CT 22 and 24 induced significant phase delays. Maximal phase delays were observed after treatment with KT5823 at CT 22 (3.46 ± 0.24 hr; n = 6; p < 0.01).
Figure 2.
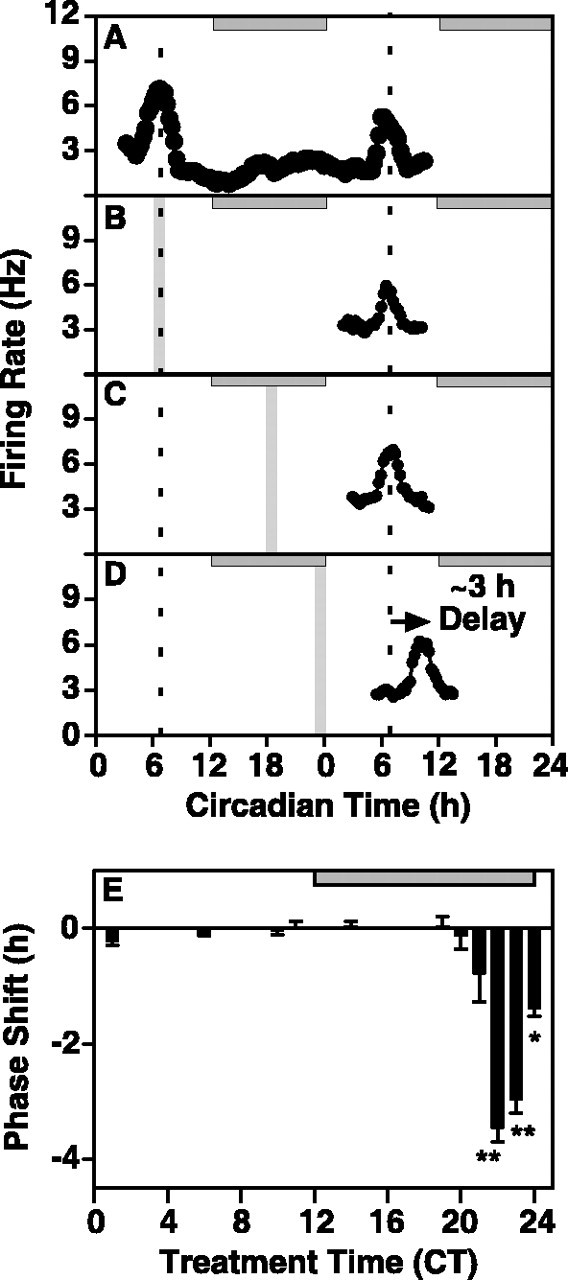
Inhibition of PKG causes a significant phase delay in the electrical activity rhythm of the rat SCN in vitro. The horizontal gray bar indicates nighttime in the donor colony; slices were maintained in constant light. Vertical bars indicate the time and duration of treatment. A, A representative single-unit recording from the ensemble of SCN neurons shows an endogenous electrical activity rhythm with a peak at ∼CT 7 on both day one and day two in vitro. Mean time-of-peak for control experiments (n = 8) was CT 6.88 ± 0.11. B, A 1 hr bath application of the specific PKG inhibitor, KT5823 (1.0 μm), at CT 6 had no effect on the time-of-peak of the SCN electrical activity rhythm on the subsequent day. Data shown are from a representative experiment. The average time-of-peak was CT 6.99 ± 0.23 (n = 3), which is not significantly different from control (Student's t test). C, A 1 hr bath application of KT5823 (1.0 μm) at CT 18 had no effect on the time-of-peak of the SCN electrical activity rhythm on the subsequent day. Data shown are from a representative experiment. The average time-of-peak was CT 6.90 ± 0.45 (n = 3), which was not significantly different from control (Student's t test). D, A 1 hr bath application of KT5823 (1.0 μm) at CT 23 induced a significant phase delay in the SCN electrical activity rhythm (p < 0.01; Student's t test). Data shown are from a representative experiment. The mean time-of-peak was delayed by 3 hr, shifting from CT 6.88 ± 0.11 (n = 7) to CT 9.83 ± 0.24 (n = 4). E, The effects of PKG inhibition are temporally restricted to a narrow window of sensitivity during the late night/predawn period. KT5823 was applied to the bath for 1 hr at 11 different points on the circadian cycle (CT 2, CT 6, CT 10, CT 11, CT 14, and CT 18-23). Each bar represents the mean ± SEM of 3-6 experiments. Sensitivity to phase delay induced by KT5823 was restricted to the end of subjective night. Significant phase delays were observed when the inhibitor was applied at CT 22 (-3.46 ± 0.24; n = 6; p < 0.01), CT 23 (-2.96 ± 0.24; n = 4; p < 0.01), and CT 24 (-1.38 ± 0.14; n = 3; p < 0.05).
To determine if the observed phase delays were specific for inhibition of the cGMP-PKG signaling pathway, several other experiments were performed. An additional isoquinoline inhibitor, KT5720 (100 nm), similar in structure to KT5823 but having higher affinity for PKA (Kase et al., 1987; Gadbois et al., 1992), had no effect on the electrical activity rhythm of rat SCN slices when applied at CT 23 (mean time-of peak CT 6.68 ± 0.33; n = 3) (Fig. 3), which indicates that phase delays associated with KT5823 are not caused by inhibition of PKA. Similar to the effects of PKG inhibition, GC inhibitors induced phase delays in SCN electrical activity rhythms when applied to rat SCN slices at CT 22; LY83583 (2 μm) induced a 3.2 ± 0.32 hr (n = 3; p < 0.01) (Fig. 3) delay, whereas ODQ (20 nm) treatment caused a delay of 3.88 ± 0.45 hr (n = 3; p < 0.01) (Fig. 3). Furthermore, 4 hr treatment of antisense oligonucleotides (αODN) generated against the first 18 nucleotides of PKG caused a 3.12 ± 0.25 hr (n = 4; p < 0.01) phase delay of rat SCN slices (Fig. 4C,D). Notably, the magnitude of this delay was not different than the effects observed with KT5823. PKG αODN bearing three mismatched nucleotides had no effect on the timing of the electrical activity rhythm (time-of-peak = 6.75 ± 0.14; n = 3) (Fig. 4B,D). Scrambled PKG ODN and PKG αODN bearing 1 mismatched nucleotide also had no effect (data not shown). Collectively, these data indicate a specific role for PKG in mediating clock timing at the end of subjective night.
Figure 3.

Sensitivity to phase delay at the end of subjective night is selective for inhibition of the GC-cGMP-PKG-dependent pathway. Similar to the effects of KT5823 (replotted from Fig. 2), the GC inhibitors LY83583 and ODQ caused phase delays of rat SCN electrical activity rhythms when applied at CT 22. LY83583 (2 μm) induced a 3.2 ± 0.32 hr (n = 3; p < 0.01) delay, whereas the delay induced by ODQ (20 nm) was 3.88 ± 0.45 hr (n = 3; p < 0.01). The PKA inhibitor KT5720 (100 nm) had no effect on the electrical activity rhythm when applied at CT 23 (mean time-of-peak, CT 6.68 ± 0.33; n = 3). Duration of each treatment was 1 hr.
Figure 4.
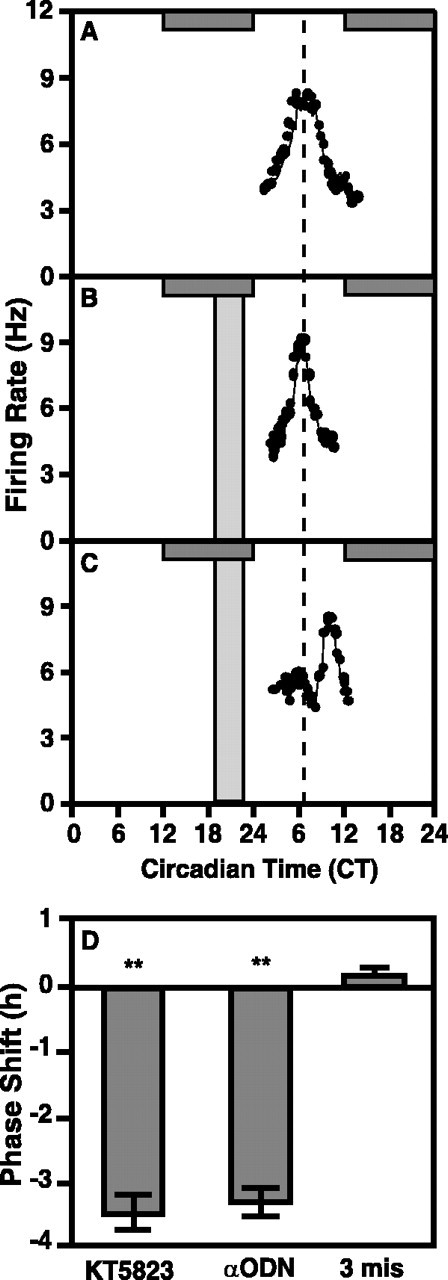
Inhibition of PKG using αODN causes a significant phase delay in the electrical activity rhythm of the rat SCN in vitro. The horizontal gray bar indicates nighttime in the donor colony; slices were maintained in constant light. Vertical bars indicate the time and duration of treatment. A, A representative single unit recording from the ensemble of SCN neurons shows an endogenous electrical activity rhythm with a peak at approximately CT 7 on both days one and two in vitro. Mean time-of-peak for control experiments (n = 8) was CT 6.88 ± 0.11. B, A 4 hr bath application of the PKG αODN (10 μm) bearing three mismatched nucleotides from CT 18-23 had no effect on the time-of-peak of the SCN electrical activity rhythm on the subsequent 2 d. Data shown are from a representative experiment. The average time-of-peak was CT 6.75 ± 0.25 (n = 3), which is not significantly different from control (Student's t test). C, A 4 hr bath application of PKG αODN (10 μm) from CT 19-23 induced a significant phase delay in the SCN electrical activity rhythm (p < 0.01; Student's t test). Data shown are from a representative experiment. The mean time-of-peak was delayed by 3.12 hr, shifting from CT 6.88 ± 0.11 (n = 7) to CT 9.00 ± 0.54 (n = 4). D, Summary of PKG ODN effects. αODN treatments caused phase delays equal in magnitude to those observed after treatment with KT5823, whereas a three base mismatch ODN did not alter phase (replotted from Fig. 2).
Inhibition of PKG at the end of subjective night delays the wheel-running activity rhythm
To evaluate the importance of PKG in integrated circadian rhythmicity, PKG inhibition studies were performed in vivo using the mouse model. Intra-SCN injection of KT5823 (100 μm) at CT 0 caused a significant phase delay in wheel-running activity onset compared with saline injections (p < 0.01, Student's t test) (Fig. 5). Injection of KT5823 at four time points (n = 26) ranging from 2.5 hr earlier to 2 hr later in the circadian cycle, or injection of vehicle alone, was without significant effect. These data demonstrate that the same temporal window of sensitivity to PKG inhibition is present both in vitro and in vivo. Furthermore, nocturnal PKG sensitivity is not species-dependent: inhibition of PKG at the end of the night delays circadian rhythms in both rats and mice.
Figure 5.

Inhibition of PKG at the end of subjective night induces phase delays in vivo in the mouse. A, B, Representative double-plotted actograms depicting effects of intra-SCN injections at CT 0 of saline (A) (0.3 μl, 0.9% NaCl) or KT5823 (B) (0.3 μl, 100 μm). Each horizontal line indicates 48 hr of data, with the last 24 hr of each line replotted as the first 24 hr of the following line. Treatments are indicated with triangles, and vertical black lines indicate the relative magnitude of running wheel activity. Diagonal lines have been drawn to aid in visualization of the phase shifts. C, A bar graph depicting average phase shifts ± SEM after indicated treatments. Shapes represent individual subject's responses and are paired across treatment conditions. Phase shifts were determined by calculating the difference in hours between two regression lines; one plotted through the 5 d of activity onsets preceding treatment, and the other plotted through the 5 d of activity after a return to stable running-wheel patterns after treatment. There were five animals in each treatment condition. Responses were determined to be significantly different (p < 0.01) by Student's paired t test. D, Representative histology of the intra-SCN injection site. The SCN have been outlined with a dotted line, and arrows point to the location the cannula.
PKG inhibition shifts the phase of the cGMP oscillation
To assess an independent measure of the circadian state during the phase-delay induced by KT5823, cGMP levels were measured in rat SCN reduced slices collected over a 40 hr period. This period extended from 4 hr before to 36 hr after brief application of the inhibitor at CT 22 on the first day in vitro. A significant oscillation of cGMP levels was observed after inhibiting PKG, however, the phase of the rhythm was delayed (Fig. 6). Careful examination of the unperturbed cGMP profile shows that levels were near the daily nadir at CT 18 and CT 20 (Fig. 6A). At CT 22, cGMP was significantly higher (p < 0.05) than at CT 18 or CT 20. In untreated slices, cGMP levels continued to rise after CT 22, peaked at CT 24/0 (p < 0.01), and dropped back to basal levels by CT 2 (Fig. 6A). However, after a 15 min application of KT5823 at CT 22, cGMP levels dropped within 1 hr to the low levels observed at CT 18 and CT 20 in untreated slices. Then, levels of cGMP rose and peaked at CT 4 (p < 0.05; ANOVA), a significant 4 hr phase delay compared with untreated slices (p < 0.05; Student's t test) (Fig. 6B). Levels of cGMP peaked again 24 hr later at CT 4 on the next day, demonstrating both the circadian nature of the cGMP rhythm and stable nature of the phase shift induced by KT5823. KT5823 application at CT 22 also delayed the phase of the endogenous cAMP oscillation by 4 hr (data not shown). However, KT5823 treatment at CT 10 did not affect the temporal profile of the cGMP oscillation (Fig. 6C) or the cAMP oscillation. These data are the first to demonstrate resetting of an SCN biochemical rhythm with similar rate and amplitude as the electrical activity rhythm.
Figure 6.

Inhibition of PKG at the end of subjective night induces phase delays in the rat cGMP oscillation. A, cGMP levels in SCN slices maintained in vitro oscillate with a peak at CT 24. These data are double-plotted for reference of the basal rhythms over 2 d (data from Fig. 1). *p < 0.05 and **p < 0.01 indicate samples that are statistically different compared with CT 20 values. Significance was determined by ANOVA and Student-Neuman-Keuls test. B, A 15 min bath application of KT5823 at CT 22 causes cGMP levels to return to basal (CT 20) levels within 1 hr and causes a significant (p < 0.01; Student's t test) phase delay in the cGMP oscillation. The shift in cGMP persists for 2 d in vitro. Data are means ± SEM for four separate experiments. C, KT5823 had no effect on the cGMP oscillation when applied for 15 min to the bath at CT 10. Individual data points are means ± SEM for four separate experiments.
PKG inhibition shifts the phase of the mPer1 oscillation
To examine the effect of PKG inhibition on the molecular clockwork, the rhythm of Per1 mRNA was assessed aftera1hr KT5823 pulse at CT 22 (Fig. 7). In controls, Per1 mRNA displayed a characteristic oscillation with a peak at CT 4 in rat SCN slices (ANOVA; p < 0.05). After KT5823 treatment, Per1 mRNA levels remained low at CT 4, but rose and peaked at CT 8 (ANOVA; p < 0.05). At CT 4, Per mRNA was significantly higher in controls compared with KT5823-treated slices (Student's t test; p < 0.01). At CT 8 and CT 12, Per1 mRNA was significantly higher in KT5823-treated slices than in controls (Student's t test; p < 0.05). This 4 hr phase delay is similar to the effects on the electrical activity and cGMP rhythms. These data suggest that a PKG-dependent event may be required for the normal timing of transcriptional activation of Per1, so that impeding PKG at this time delays the molecular clockwork.
Figure 7.

Inhibition of PKG at the end of subjective night alters the expression pattern of Per1 mRNA in the rat SCN. A, Representative in situ hybridization histochemistry. In control slices, a significant increase in Per1 mRNA was observed with a peak at CT 4. After KT5823 treatment for 1 hr at CT 22, the endogenous rise in Per1 was delayed by 4 hr; PKG-treated slices showed peak Per1 expression at CT 8. Magnification, 200×. B, Quantitation of A. Per1-positive cells were counted by an experimenter blind to the experimental treatments. Data represent the mean ± SEM of four independent experiments. Statistical analysis (ANOVA with Student-Neuman-Keuls post hoc analysis; p < 0.01) revealed a significant increase in Per1 mRNA at CT 4 and CT 8 in control slices, with a peak at CT 4. In KT5823-treated slices, a significant increase was observed at CT 8 and CT 12 with a peak at CT 8. CT 4 controls had significantly more Per1-positive cells than CT 4-treated slices (p < 0.01; Student's t test). Treated slices had significantly more Per1-positive cells at CT 8 (p < 0.05) and CT 12 (p < 0.01), compared with time-matched controls.
Discussion
Contemporary understanding of circadian time keeping indicates a clock comprised of dynamic temporal domains dominated by the sequential activities of specific molecular and biochemical elements. Temporal domains, which are defined by differential sensitivities to resetting stimuli over a 24 hr cycle (Gillette et al., 1995; Gillette, 1996; Merrow et al., 1997), differ in (1) sensitivities to activation of cell signaling pathways, (2) biochemical and molecular elements, their activities, and subcellular localizations, and (3) transitional sequences, during which one set of elements and processes is replaced with another. Whereas a role for kinases in clock regulation has been established, this study is the first to carefully examine kinase-mediated regulation of a time domain transition.
SCN sensitivity to resetting during the second half of subjective night is dominated by cGMP-dependent signaling events. Exogenous PKG stimulation, initiated by light pulses to the eyes (Weber et al., 1995; Mathur et al., 1996) or in the SCN slice by exogenous application of GLU (Ding et al., 1994, 1998) or membrane-soluble analogs of cGMP (Prosser and Gillette, 1989), advances the clock toward a biochemical transitional state in which nighttime processes are replaced with daytime processes. This state change is the biological equivalent of dawn in the natural world. Our studies identify clock-driven changes in the cGMP-PKG system at this transition point. An endogenous rise in cGMP levels and PKG activity in rat SCN maintained in vitro occurs juxtaposed to the termination of sensitivity to phase advances via exogenous cGMP-PKG-activation. This endogenous increase coincides with the appearance of a 2 hr window of sensitivity to phase delay by PKG inhibition at the end of subjective night of the rat SCN. Transient inhibition of this endogenous rise in cGMP or activation of PKG at the end of the night phase delays clock phase by similar degrees or 3.5 hr. This suggests that PKG-mediated processes perform a necessary function at this clock time.
The temporal relationship between phase shifting by exogenous PKG activation and endogenous clock-controlled PKG-activation is striking (Fig. 8). The unperturbed clock naturally progresses through the entire nocturnal domain toward the time of endogenous accumulation of cGMP and consequent activation of PKG that marks the end of night. Directly before reaching the point of cGMP accumulation, the clock is sensitive to phase advance via light-GLU activating cGMP-dependent signal transduction events (Weber et al., 1995; Mathur et al., 1996; Ding et al., 1998). Because of this juxtaposition, we hypothesize that these exogenous stimuli prematurely activate a process that occurs normally in the subsequent temporal domain, caused by the endogenous rise in cGMP. This event would effectively signal a transition from one temporal domain into another. When unperturbed until late night, cGMP accumulation-PKG activation under endogenous clock control would initiate the changes, thereby terminating sensitivity to nocturnal phase resetting agents. These changes would end the nocturnal state and initiate transition into the clock state defined as morning. A similar model may also explain night-to-day transitions in other circadian systems. For example, the window of sensitivity to a depolarizing stimulus applied to basal retinal neurons in the Bulla eye, which mimics light-induced phase shifts, is followed immediately by a period of endogenous depolarization (McMahon et al., 1984). Inhibition of spontaneous depolarization at the dawn transition by a pulse of low-calcium or hyperpolarization phase delays the Bulla pacemaker (Khalsa and Block, 1988, 1990).
Figure 8.

Model of circadian clock regulation by cGMP-PKG at the end of subjective night. The SCN is sensitive to phase resetting by stimuli dependent on cGMP and PKG activation during the last half of subjective night. These stimuli cause phase advances that may be manifested as shifting clock state forward several hours to the end of the night (solid arrow). The domain of endogenous rise in cGMP and PKG activity (gradient gray) is a narrow window of time that is coincident with the waning of sensitivity to phase advance stimulated by light-GLU via cGMP-PKG-dependent mechanisms. Inhibiting PKG activity during this time causes a 3 hr phase delay. This may be a dynamic process that shifts the clock back in time, into the domain of sensitivity to phase resetting by exogenous stimuli that activate the cGMP-PKG pathway (dashed arrow). The tight temporal relationship between exogenous sensitivity and endogenous activation of cGMP-PKG suggests that light-GLU may prematurely stimulate a system poised to respond ∼3.5 hr later to clock-controlled processes. The clock-controlled rise in cGMP levels and concomitant activation of PKG at the end of the night comprise a critical event in this time domain.
The data presented herein are consonant with our hypothetical model. When the endogenous rise in PKG activity is blocked transiently by exposure to KT5823, significant phase delays in electrical activity, cGMP, and Per1 oscillations in the SCN slice from rat are observed. The data suggest that inhibition of the endogenous rise in PKG activity effectively interrupts clock progression. Within 1 hr of PKG inhibition at CT 22, cGMP returns to low levels typically observed between CT 18 and CT 20 (Fig. 6). Accumulation of cGMP begins again, so that it peaks ∼4 hr later. Subsequent peaks in Per1 and spontaneous neuronal activity are delayed similarly, given the limits of our resolution. The immediate drop in cGMP and 4 hr delay in return to the peak cGMP level suggest that PKG activation state feeds back on the system that regulates it, and that the clock mechanism is acutely sensitive to cGMP-PKG state at this point in the night. The 4 hr delay in the cGMP peak and attendant PKG activation could account entirely for the phase delay in firing rate and Per1 oscillations, which are delayed by the same amount but peak later.
Wheel-running activity in mice is also significantly delayed. The magnitude of this behavioral phase delay is, not surprisingly, somewhat smaller than the delays of rhythms in the SCN slice. Phase shifts in vivo are commonly smaller than those observed invitro in brain slices (Ding et al., 1994). This likely reflects the increased complexity of feedback information encountered in the behaving animal as well as limitations incurred by the experimental design. Notably, mice were used for the wheel-running experiments, whereas rats were used for all slice experiments. Thus, differences in the magnitude of the phase shift may also reflect innate differences in the circadian rhythmicity of the two species as well as differences in their abilities to adjust their clock in response to input. For example, phase delays and phase advances in response to light are similar in rats, with peak responses occurring at CT 14 and CT 19-20, respectively. In contrast, mice display large phase delays with maximal sensitivity at CT 14 (G. F. Buchanan and M. U. Gillette, unpublished observations), but relatively small phase advances with a maximal effect at CT 22 (0.5 hr). Unilateral injection of KT5823 into only one of the paired SCN may have impeded total PKG inhibition.
Nevertheless, four independent measures of phase, oscillations in cGMP concentration, mPer1 levels, neuronal activity, and wheel-running activity, are delayed in parallel, suggesting coupling of these processes. The fact that multiple circadian output pathways are affected by inhibition of the endogenous activation of PKG suggests that the normal clock-controlled upregulation of cGMP-PKG is either an input signal or is directly coupled to core elements of the molecular clockwork. The order in which the delayed oscillations peak, from cGMP to Per1 to electrical activity, reflects normal phase relationships driven by the clock rather than acute changes induced by PKG inhibition. Importantly, the amplitude of the delay does not directly reflect the duration of PKG inhibition. Rather, inhibition is rapidly reversed after removal of KT5823 such that sensitivity to GLU stimulation reemerges within 15 min; the amplitude of phase advance inducible by GLU after KT5823 treatment is concordant with resumption of a CT 20-like state (S. A. Tischkau and M. U. Gillette, unpublished observations).
The nature of biochemical events that require activation of PKG at the end of the night is unknown. Changes in the levels, activation states, and/or subcellular localization of temporal regulators at this time may require PKG-mediated phosphorylation before the clock transits from the nighttime domain. A substrate of PKG phosphorylation may be necessary to sustain the rise of cGMP because cGMP levels drop rapidly after PKG inhibition. This demonstrates integrated regulation of the pathway such that inhibition of the kinase immediately feeds back to downregulate its activator, cGMP. Thus, the delay likely represents the time required to restore element or elements downstream from PKG, which may be vulnerable to degradation in an unphosphorylated state, but are required during the clock state at the end of the night.
Critical elements at the end of subjective night may include activators of transcription. Preliminary studies indicate that light-GLU-induced phase advances cause phosphorylation of the transcription factor CREB in the SCN (Ginty et al., 1993; Ding et al., 1997) and require subsequent activation of transcription at CRE-mediated sites (Tischkau et al., 2003). PKG activation may mediate CREB phosphorylation (Gudi et al., 2000) (Tischkau and Gillette, unpublished results). Transcription of a number of genes, including the immediate early genes c-fos and junB (Guido et al., 1999) and the circadian clock gene, Per1 (Sun et al., 1997; Tei et al., 1997), is initiated near dawn in the SCN, immediately after the endogenous rise in cGMP-PKG. The phase delay in the Per1 oscillation induced by PKG inhibition at the end of subjective night we report suggests involvement of PKG-dependent processes in initiation of endogenous Per1 transcription.
Although the importance of kinases in regulating clock function is well established, our results provide evidence of a direct temporal relationship between specific kinase activity necessary for accomplishing an exogenously stimulated phase shift and for key events within a defined state of the circadian clock. They identify cGMP-PKG-regulated changes as requisite at the end of the nighttime domain. Endogenous processes that activate the cGMP-PKG pathway at the end of night are unknown, but may be predicted to bear some relation to the exogenous activators. The clock-controlled nature of these temporally restricted spheres of kinase influence suggests that PKG may mediate post-translational effects common both to light-induced phase advance and to clock timekeeping. By comparing PKG substrates critical to both processes and cellular mechanisms by which they regulate time domain transitions, insights as to temporal convergence of these dynamic clock states should emerge.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants NS22155, HL67007, and a University Scholar award (M.U.G.), NIH Grant NS10170 (S.A.T.), NIH Grant GM07143 (S.M.A.), and NIH Grant NS11158 (J.W.M.). We thank U. Schibler for mPer1 probe and S. Baker for manuscript preparation.
Correspondence should be addressed to Dr.M.U.Gillette, Department of Cell and Structural Biology, University of Illinois at Urbana-Champaign, B107 Chemical and Life Sciences Laboratory, 601 South Goodwin Avenue, Urbana, IL 60801. E-mail: mgillett@life.uiuc.edu.
S. A. Tischkau's present address: Department of Veterinary Biosciences, University of Illinois at Urbana-Champaign, 3615 VMBSB, 2001 South Lincoln Avenue, Urbana, IL 60801.
E. T. Weber's present address: Department of Biology, Rider University, 2083 Lawrenceville Road, Lawrenceville, NJ 08648.
Copyright © 2003 Society for Neuroscience 0270-6474/03/237543-08$15.00/0
References
- Albrecht U, Sun ZS, Eichele G, Lee CC ( 1997) A differential response of two putative mammalian circadian regulators, mper1 and mper2, to light. Cell 91: 1055-1064. [DOI] [PubMed] [Google Scholar]
- Balsalobre A, Damiola F, Schibler U ( 1998) A serum shock induces circadian gene expression in mammalian tissue culture cells. Cell 93: 929-937. [DOI] [PubMed] [Google Scholar]
- Best JD, Maywood ES, Smith KL, Hastings MH ( 1999) Rapid resetting of the mammalian circadian clock. J Neurosci 19: 828-835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comolli JC, Hastings JW ( 1999) Novel effects on the Gonyaulax circadian system produced by the protein kinase inhibitor staurosporine. J Biol Rhythms 14: 11-19. [DOI] [PubMed] [Google Scholar]
- Crosthwaite SK, Dunlap JC, Loros JJ ( 1997) Neurospora wc-1 and wc-2: transcription, photoresponses, and the origins of circadian rhythmicity. Science 276: 763-769. [DOI] [PubMed] [Google Scholar]
- Ding JM, Chen D, Weber ET, Faiman LE, Rea MA, Gillette MU ( 1994) Resetting the biological clock: mediation of nocturnal circadian shifts by glutamate and NO. Science 266: 1713-1717. [DOI] [PubMed] [Google Scholar]
- Ding JM, Faiman LE, Hurst WJ, Kuriashkina LR, Gillette MU ( 1997) Resetting the biological clock: mediation of nocturnal CREB phosphorylation via light, glutamate, and nitric oxide. J Neurosci 17: 667-675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding JM, Buchanan GF, Tischkau SA, Chen D, Kuriashkina L, Faiman LE, Alster JM, McPherson PS, Campbell KP, Gillette MU ( 1998) A neuronal ryanodine receptor mediates light-induced phase delays of the circadian clock. Nature 394: 381-384. [DOI] [PubMed] [Google Scholar]
- Eskin A, Takahashi JS, Zatz M, Block GD ( 1984) Cyclic guanosine 3′:5′-monophosphate mimics the effects of light on a circadian pacemaker in the eye of Aplysia J Neurosci 4: 2466-2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadbois DM, Crissman HA, Tobey RA, Bradbury EM ( 1992) Multiple kinase arrest points in the G1 phase of nontransformed mammalian cells are absent in transformed cells. Proc Natl Acad Sci USA 89: 8626-8630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garceau NY, Liu Y, Loros JJ, Dunlap JC ( 1997) Alternative initiation of translation and time-specific phosphorylation yield multiple forms of the essential clock protein FREQUENCY. Cell 89: 469-476. [DOI] [PubMed] [Google Scholar]
- Gillette MU ( 1996) Regulation of entrainment pathways by the suprachiasmatic circadian clock: sensitivities to second messengers. Prog Brain Res 111: 121-132. [DOI] [PubMed] [Google Scholar]
- Gillette MU, Prosser RA ( 1988) Circadian rhythm of the rat suprachiasmatic brain slice is rapidly reset by daytime application of cAMP analogs. Brain Res 474: 348-352. [DOI] [PubMed] [Google Scholar]
- Gillette MU, Mitchell JW ( 2002) Signaling in the suprachiasmatic nucleus: selectively responsive and integrative. Cell Tissue Res 309: 99-107. [DOI] [PubMed] [Google Scholar]
- Gillette MU, Reppert SM ( 1987) The hypothalamic suprachiasmatic nuclei: circadian patterns of vasopressin secretion and neuronal activity in vitro. Brain Res Bull 19: 135-139. [DOI] [PubMed] [Google Scholar]
- Gillette MU, Medanic M, McArthur AJ, Liu C, Ding JM, Faiman LE, Weber ET, Tcheng TK, Gallman EA ( 1995) Intrinsic neuronal rhythms in the suprachiasmatic nuclei and their adjustment. Ciba Found Symp 183: 134-144. [DOI] [PubMed] [Google Scholar]
- Ginty DD, Kornhauser JM, Thompson MA, Bading H, Mayo KE, Takahashi JS, Greenberg ME ( 1993) Regulation of CREB phosphorylation in the suprachiasmatic nucleus by light and a circadian clock. Science 260: 238-241. [DOI] [PubMed] [Google Scholar]
- Glass DB, Krebs EG ( 1982) Phosphorylation by guanosine 3′:5′-monophosphate-dependent protein kinase of synthetic peptide analogs of a site phosphorylated in histone H2B. J Biol Chem 257: 1196-1200. [PubMed] [Google Scholar]
- Gudi T, Casteel DE, Vinson C, Boss GR, Pilz RB ( 2000) NO activation of fos promoter elements requires nuclear translocation of G-kinase I and CREB phosphorylation but is independent of MAP kinase activation. Oncogene 19: 6324-6333. [DOI] [PubMed] [Google Scholar]
- Guido ME, de Guido LB, Goguen D, Robertson HA, Rusak B ( 1999) Daily rhythm of spontaneous immediate-early gene expression in the rat suprachiasmatic nucleus. J Biol Rhythms 14: 275-280. [DOI] [PubMed] [Google Scholar]
- Kase H, Iwahashi K, Nakanishi S, Matsuda Y, Yamada K, Takahashi M, Murakata C, Sato A, Kaneko M ( 1987) K-252 compounds, novel and potent inhibitors of protein kinase C and cyclic nucleotide-dependent protein kinases. Biochem Biophys Res Commun 142: 436-440. [DOI] [PubMed] [Google Scholar]
- Khalsa SB, Block GD ( 1988) Calcium channels mediate phase shifts of the Bulla ocular pacemaker. J Comp Physiol [A] 164: 195-206. [DOI] [PubMed] [Google Scholar]
- Khalsa SB, Block GD ( 1990) Calcium in phase control of the Bulla circadian pacemaker. Brain Res 506: 40-45. [DOI] [PubMed] [Google Scholar]
- Kloss B, Price JL, Saez L, Blau J, Rothenfluh A, Wesley CS, Young MW ( 1998) The Drosophila clock gene double-time encodes a protein closely related to human casein kinase I epsilon. Cell 94: 97-107. [DOI] [PubMed] [Google Scholar]
- Kloss B, Rothenfluh A, Young MW, Saez L ( 2001) Phosphorylation of period is influenced by cycling physical associations of double-time, period, and timeless in the Drosophila clock. Neuron 30: 699-706. [DOI] [PubMed] [Google Scholar]
- Krucher NA, Meijer L, Roberts MH ( 1997) The cyclin-dependent kinase (cdk) inhibitors, olomoucine and roscovitine, alter the expression of a molluscan circadian pacemaker. Cell Mol Neurobiol 17: 495-507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Etchegaray JP, Cagampang FR, Loudon AS, Reppert SM ( 2001) Post-translational mechanisms regulate the mammalian circadian clock. Cell 107: 855-867. [DOI] [PubMed] [Google Scholar]
- Lin FJ, Song W, Meyer-Bernstein E, Naidoo N, Sehgal A ( 2001) Photic signaling by cryptochrome in the Drosophila circadian system. Mol Cell Biol 21: 7287-7294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Gillette MU ( 1996) Cholinergic regulation of the suprachiasmatic nucleus circadian rhythm via a muscarinic mechanism at night. J Neurosci 16: 744-751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Ding JM, Faiman LE, Gillette MU ( 1997) Coupling of muscarinic cholinergic receptors and cGMP in nocturnal regulation of the suprachiasmatic circadian clock. J Neurosci 17: 659-666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowrey PL, Shimomura K, Antoch MP, Yamazaki S, Zemenides PD, Ralph MR, Menaker M, Takahashi JS ( 2000) Positional syntenic cloning and functional characterization of the mammalian circadian mutation tau. Science 288: 483-492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinek S, Inonog S, Manoukian AS, Young MW ( 2001) A role for the segment polarity gene shaggy/GSK-3 in the Drosophila circadian clock. Cell 105: 769-779. [DOI] [PubMed] [Google Scholar]
- Mathur A, Golombek DA, Ralph MR ( 1996) cGMP-dependent protein kinase inhibitors block light-induced phase advances of circadian rhythms in vivo. Am J Physiol 270: R1031-R1036. [DOI] [PubMed] [Google Scholar]
- McMahon DG, Wallace SF, Block GD ( 1984) Cellular Analysis of the Bulla ocular circadian pacemaker system II. Neurophysiological basis of circadian rhythmicity. J Comp Physiol [A] 155: 379-385. [Google Scholar]
- Merrow MW, Garceau NY, Dunlap JC ( 1997) Dissection of a circadian oscillation into discrete domains. Proc Natl Acad Sci USA 94: 3877-3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel S, Geusz ME, Zaritsky JJ, Block GD ( 1993) Circadian rhythm in membrane conductance expressed in isolated neurons. Science 259: 239-241. [DOI] [PubMed] [Google Scholar]
- Naidoo N, Song W, Hunter-Ensor M, Sehgal A ( 1999) A role for the proteasome in the light response of the timeless clock protein. Science 285: 1737-1741. [DOI] [PubMed] [Google Scholar]
- Okamura H, Yamaguchi S, Yagita K ( 2002) Molecular machinery of the circadian clock in mammals. Cell Tissue Res 309: 47-56. [DOI] [PubMed] [Google Scholar]
- Price JL, Dembinska ME, Young MW, Rosbash M ( 1995) Suppression of PERIOD protein abundance and circadian cycling by the Drosophila clock mutation timeless. EMBO J 14: 4044-4049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosser RA, Gillette MU ( 1989) The mammalian circadian clock in the suprachiasmatic nuclei is reset in vitro by cAMP. J Neurosci 9: 1073-1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosser RA, McArthur AJ, Gillette MU ( 1989) cGMP induces phase shifts of a mammalian circadian pacemaker at night, in antiphase to cAMP effects. Proc Natl Acad Sci USA 86: 6812-6815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripperger JA, Schibler U ( 2001) Circadian regulation of gene expression in animals. Curr Opin Cell Biol 13: 357-362. [DOI] [PubMed] [Google Scholar]
- Shearman LP, Zylka MJ, Weaver DR, Kolakowski LF Jr, Reppert SM ( 1997) Two period homologs: circadian expression and photic regulation in the suprachiasmatic nuclei. Neuron 19: 1261-1269. [DOI] [PubMed] [Google Scholar]
- Shigeyoshi Y, Taguchi K, Yamamoto S, Takekida S, Yan L, Tei H, Moriya T, Shibata S, Loros JJ, Dunlap JC, Okamura H ( 1997) Light-induced resetting of a mammalian circadian clock is associated with rapid induction of the mPer1 transcript. Cell 91: 1043-1053. [DOI] [PubMed] [Google Scholar]
- Sun ZS, Albrecht U, Zhuchenko O, Bailey J, Eichele G, Lee CC ( 1997) RIGUI, a putative mammalian ortholog of the Drosophila period gene. Cell 90: 1003-1011. [DOI] [PubMed] [Google Scholar]
- Tei H, Okamura H, Shigeyoshi Y, Fukuhara C, Ozawa R, Hirose M, Sakaki Y ( 1997) Circadian oscillation of a mammalian homologue of the Drosophila period gene. Nature 389: 512-516. [DOI] [PubMed] [Google Scholar]
- Tischkau SA, Gallman EA, Buchanan GF, Gillette MU ( 2000) Differential cAMP gating of glutamatergic signaling regulates long-term state changes in the suprachiasmatic circadian clock. J Neurosci 20: 7830-7837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischkau SA, Barnes JA, Lin FJ, Myers EM, Barnes JW, Meyer-Bernstein EL, Hurst WJ, Burgoon PW, Chen D, Sehgal A, Gillette MU ( 1999) Oscillation and light induction of timeless mRNA in the mammalian circadian clock. J Neurosci 19: RC15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischkau SA, Mitchell JW, Tyan SH, Buchanan GF, Gillette MU ( 2003) Ca2+/cAMP response element-binding protein (CREB)-dependent activation of Per1 is required for light-induced signaling in the suprachiasmatic nucleus circadian clock. J Biol Chem 278: 718-723. [DOI] [PubMed] [Google Scholar]
- Weber ET, Gannon RL, Rea MA ( 1995) cGMP-dependent protein kinase inhibitor blocks light-induced phase advances of circadian rhythms in vivo. Neurosci Lett 197: 227-230. [DOI] [PubMed] [Google Scholar]
- Welsh DK, Logothetis DE, Meister M, Reppert SM ( 1995) Individual neurons dissociated from rat suprachiasmatic nucleus express independently phased circadian firing rhythms. Neuron 14: 697-706. [DOI] [PubMed] [Google Scholar]
- Yang Y, Cheng P, Zhi G, Liu Y ( 2001) Identification of a calcium/calmodulin-dependent protein kinase that phosphorylates the Neurospora circadian clock protein FREQUENCY. J Biol Chem 276: 41064-41072. [DOI] [PubMed] [Google Scholar]
- Zeng H, Hardin PE, Rosbash M ( 1994) Constitutive overexpression of the Drosophila period protein inhibits period mRNA cycling. EMBO J 13: 3590-3598. [DOI] [PMC free article] [PubMed] [Google Scholar]