

Abstract
In recent years, attention has been given to the interaction between the emotional state of the animal and its ability to learn and remember. Studies into the neural mechanisms underlying these interactions have focused on stress-induced synaptic plasticity impairments in the hippocampus. However, other brain areas, including the amygdala and the prefrontal cortex (PFC), have been implicated in relation to stress-mediated effects on memory. The present study examined whether stress, which impairs hippocampal long-term potentiation (LTP), also affects LTP of the basolateral amygdala (BLA)–PFC pathway in vivo. We first confirmed that the stress protocol we used, i.e., the elevated platform stress, was effective in blocking LTP in the CA1 area of the hippocampus. We then characterized activity and established the ability to induce LTP at the BLA–PFC pathway. Finally, we examined the effects of an exposure to the elevated platform stress on the ability to induce LTP in this pathway. The results indicate that, at the same time when LTP is blocked in the hippocampus, it is also inhibited in the BLA–medial PFC pathway. These results call for a shift from a focused attention on the effects of stress on plasticity in the hippocampus to a system level approach that emphasizes the possible modification of interactions between relevant brain areas after an exposure to a stressful experience.
Keywords: stress, amygdala, prefrontal cortex, long-term potentiation, plasticity, rat
Introduction
In recent years, attention has been given to the interaction between the emotional state of the animal and its ability to learn and remember. Studies into the neural mechanisms underlying these interactions have focused on stress-induced synaptic plasticity impairments in the hippocampus. Exposure to stress can enhance neuronal cell death in the hippocampus and have deleterious effects on broad aspects of cognition and memory processes (for review, see McEwen and Sapolsky, 1995; Kim and Yoon, 1998). Animal studies demonstrated stress-induced deficits in a variety of hippocampal-dependent cognitive tasks, including water maze (Bodnoff et al., 1995; de Quervain et al., 1998), radial maze (Luine et al., 1994), and object recognition tasks (Baker and Kim, 2002). Similarly, stress was found to suppress the ability to induce long-term potentiation (LTP) in the hippocampus (Foy et al., 1987; Diamond and Rose, 1994; Pavlides et al., 1996; Garcia et al., 1997; Xu et al., 1997, 1998; Akirav and Richter-Levin, 1999; Wang et al., 2000).
However, this focus on the effects of stress on hippocampal functioning and plasticity is somewhat misleading. Other brain areas, including the amygdala and the prefrontal cortex (PFC), have been suggested to mediate some aspects of the response to stress and, in particular, to contribute to the interplay between emotions and memory formation.
The amygdala has a pivotal role in mediating stress-related effects on behavior and modulating hippocampal function. Manipulations of the basolateral amygdala (BLA) (stimulation or lesions) modulate hippocampal LTP (Ikegaya et al., 1994, 1995, 1996; Akirav and Richter-Levin, 1999). More specifically, it was demonstrated that, similar to stress, spaced activation of the amygdala inhibits LTP induction in the hippocampus (Akirav and Richter-Levin, 1999, 2002).
Likewise, the medial prefrontal cortex (mPFC) is a key component of the neural circuitry mediating responses to stressful situations. It is known to modulate neuroendocrine function during stress (Meaney and Aitken, 1985; McEwen et al., 1986). It is selectively activated by psychological and social stressors (Thierry et al., 1976). It is involved in working memory and in attentional functions (Williams and Goldman-Rakic, 1995). The mPFC is also a site in which synaptic plasticity can occur and, particularly, can be induced by activating hippocampal projections to the mPFC (Jay et al., 1995; Burette et al., 1997; Takita et al., 1999).
The mPFC and the amygdala have reciprocal anatomical interconnections (Krettek and Price, 1977; Porrino et al., 1981; McDonald, 1987, 1991, 1996; Cassell et al., 1989, 1991; Amaral and Insausti, 1992). Data indicate that the mPFC may influence the activity of the amygdala: for example, lesioning the PFC reduces extinction of cued fear conditioning, an amygdala-dependent task (Morgan and LeDoux, 1995). In a recent study, Garcia et al., (1999) demonstrated that PFC neurons reduce their spontaneous activity in the presence of a tone previously paired with a footshock (Garcia et al., 1999; Herry et al., 1999), indicating that the activity of PFC neurons is inhibited after fear conditioning, a task mediated by the amygdala. How the amygdala may influence activity and plasticity in the PFC and, particularly, how is this influence affected by stress is yet to be established.
We undertook the present series of experiments (1) to assess whether LTP can be induced in the BLA–PFC pathway and (2) to further explore whether LTP in this pathway is modified by stress.
Materials and Methods
Electrophysiology. Male Sprague Dawley rats (280–380 gm) were anesthetized (with 40% urethane, 5% chloral hydrate in saline, and 0.5 ml/100 gm, i.p.) and placed in a stereotaxic frame with body temperature maintained at 37 ± 0.5°C. The procedures were performed in a strict accordance with the University of Haifa regulations and the guidelines laid down by the National Institute of Health (NIH publication number 8023). In brief, small holes were drilled in the skull to allow insertion of electrodes in the brain. A recording microelectrode (glass, tip diameter of 2–5 μm, filled with 2 m NaCl, resistance of 1–4 MΩ) was slowly lowered into the prelimbic area of the PFC (3.0–3.3 mm anterior to bregma; 0.7–1.0 mm lateral; 2.8–3.4 mm below the pial surface). A bipolar 125 μm stimulating electrode was implanted in the BLA (3.3 mm anterior to bregma; 5.0–5.3 mm lateral; 6.7 mm below the pial surface).
In the CA1 experiment, the recording electrode was placed in the stratum radiatum (4.2 mm posterior; 2.5–2.8 mm lateral to bregma), and the stimulating electrode was positioned to activate the Schaffer collateral–commissural projection (3.1 mm posterior; 0.3–0.5 mm lateral to bregma).
Evoked responses were digitized (10 kHz) and analyzed using the Cambridge Electronic Design (Cambridge, UK) 1401+ and its Spike 2 software.
Offline measurements were made of the amplitude of EPSP using averages of five successive responses to a given stimulation intensity applied at 0.1 Hz. Test stimuli (monopolar pulses, 100 μsec duration) were delivered at 0.1 Hz. After positioning the electrodes, the rat was left for 30 min before commencing the experiment.
Stress protocol. Behavioral stress protocol was based on Xu et al. (1998). Animals were placed on an elevated platform (12 × 12 cm) for 30 min in a brightly lit room. The animal showed behavioral “freezing,” i.e., immobility for up to 10 min, defecation, and urination. After the procedure, rats were immediately anesthetized and taken for electrophysiological testing.
LTP induction. LTP was induced by applying theta burst stimulation (TBS) to the BLA (three sets of 10 trains; each train consisted of 10 pulses at 100 Hz; intertrain interval, 200 msec; interset interval, 1 min). Field potentials were recorded from the mPFC at 15, 30, 60, and 90 min after the TBS to the BLA. LTP was measured as an increase in EPSP amplitude. Potentiation was measured as a percentage change from baseline and was analyzed using ANOVA.
Pharmacological blockade of LTP. The competitive NMDA receptor antagonist (±)-3-(2-carboxypiperazin-4-yl) propyl-1-phosphonic acid [(±)-CPP], (Tocris, Bristol, UK) was dissolved in 0.9% saline and injected (10 mg/kg, i.p.) 45 min before the application of TBS to the BLA.
Histology. Histological verification of both the recording and stimulating electrodes location was performed on all of the rats.
After electrophysiological testing, marking lesions were made by passing anodal currents (10 mA for 3 sec, 10 mA for 2 min) to the metal bipolar stimulating and recording electrode, respectively. Brains were removed, postfixed over 3 nights in formaldehyde (10%), and sectioned (120 μm) on a sledge microtome. The sections were mounted on gelatin-coated slides, stained in cresyl violet, dehydrated, and coverslipped. The electrode tract and lesion locations were then identifiable under a light microscope.
Statistical analysis. The results are expressed as means ± SEM. For statistical analysis, overall mixed ANOVA, one-way ANOVA, and t test were used as indicated.
Results
An exposure to the elevated platform stress blocks LTP in the CA1 area of the hippocampus
Tetanization in vivo of the commissural projection induced long-lasting LTP of the CA1 pyramidal cells that persisted for >90 min. ANOVA analysis revealed a significant difference between the nonstressed (n = 5) and the stressed (n = 5) groups in EPSP amplitude potentiation at all recording time points after tetanization [+1 min, F(1,8) = 25.48, p < 0.005 (nonstressed, 61.3 ± 8.9% increase of the EPSP amplitude when compared with stressed, 3 ± 10.82%); +15 min, F(1,8) = 32.34, p < 0.001 (nonstressed, 58.6 ± 7.7% when compared with stressed, 2 ± 11.06%); +30 min, F(1,8) = 17.69, p < 0.005 (nonstressed, 55.74 ± 9.6% when compared with stressed, -3 ± 12.6%); +60 min, F(1,8) = 24.44, p < 0.001 (nonstressed, 60 ± 10.9 when compared with stressed, -3.4 ± 8.9%); +90 min, F(1,8) = 22.0, p < 0.05 (nonstressed, 64 ± 14% when compared with stressed, -2.18 ± 8.5%)]. This finding is consistent with previous data reporting blockade of CA1 and dentate gurus LTP after exposure to stress (Foy et al., 1987; Shors et al., 1989).
Theta burst stimulation induces LTP in the amygdala–PFC pathway: dependence on NMDA receptor activation
The locations of the recording and stimulating sites in the PFC and BLA are shown in Figure 1A. Amygdala stimulation elicited EPSP in the mPFC that consisted of a negative potential peaking from 15 to 24 msec (Fig. 1). The shape, amplitude, and latency were highly reproducible and remained stable over time.
Figure 1.
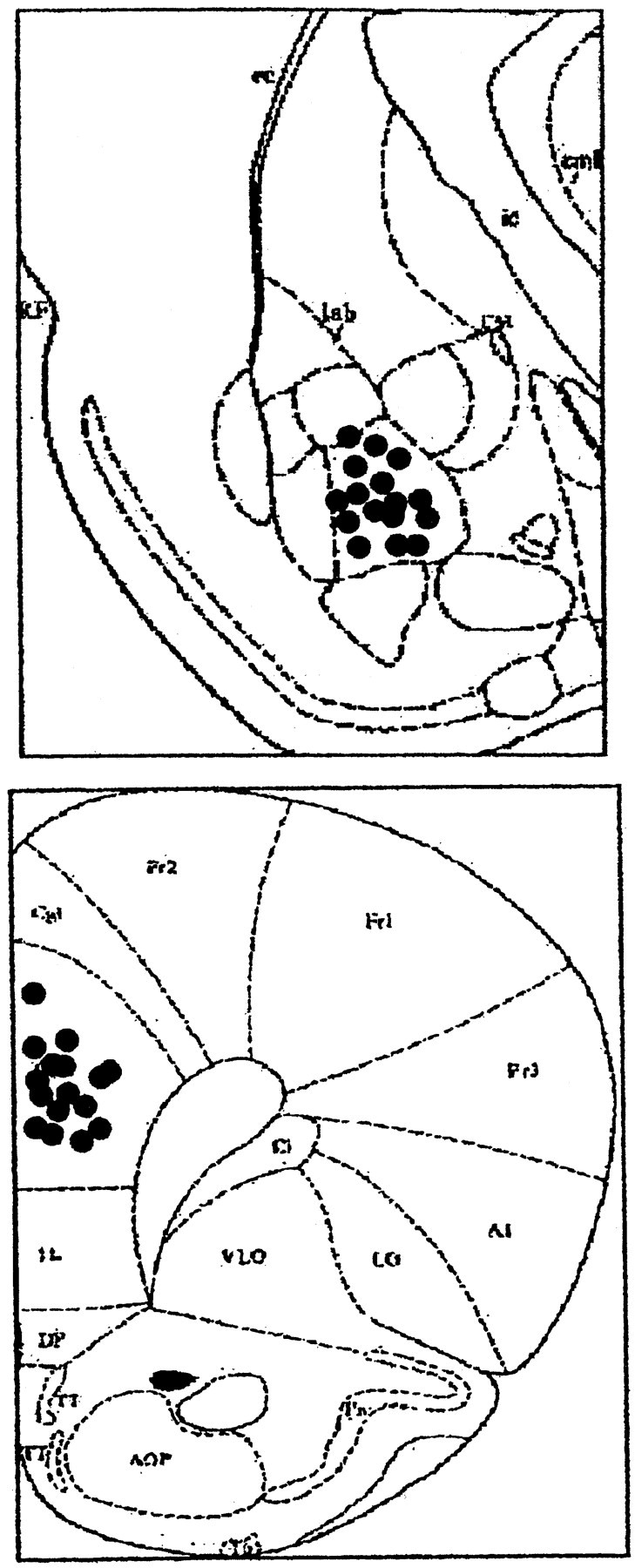
Top, The stimulating electrode in the BLA. Bottom, Schematic diagram representing the location of the recording electrode in the prelimbic area. Solid black circles indicate the locations.
After establishing that BLA stimulation induces an excitatory field potential in the mPFC, we assessed whether BLA–mPFC is amenable to LTP.
Overall, four groups of rats were tested for LTP: low-frequency control; LTP group, receiving TBS to the BLA; animals in which theta burst stimulation was applied in the presence of the competitive NMDA receptor antagonist CPP; and animals that were exposed to the elevated platform stress. To verify that the baseline response was not different between the different groups, an overall mixed ANOVA [groups × time (4 × 8)] for comparison between the groups before TBS did not reveal a significant difference in EPSP amplitude at -30 min, -15, or -1 min, indicating a similar baseline in all groups (F(3,23) < 1; NS). The stimulation intensity used to elicit a baseline response was not different between the different groups [F(3,23) < 1; NS).
Using overall mixed ANOVA [groups × time (4 × 8)] for post-TBS comparison, we found a significant difference in EPSP amplitude levels between the groups (F(3,23) = 2.84; p = 0.0001), which was further analyzed.
TBS to the BLA induced a robust and long-lasting LTP of the EPSP amplitude in the mPFC, reflecting the potentiation of the BLA–mPFC pathway (n = 8) (Fig. 2A).
Figure 2.

TBS induces LTP in the mPFC. This LTP is blocked by the NMDA receptor antagonist CPP. The increase in EPSP amplitude (LTP) was measured as a percentage of baseline value immediately before TBS to the BLA. Delivering TBS to the BLA induced a robust and long-lasting increase of the amplitude of the evoked field potential in the mPFC, reflecting the potentiation of the amygdala–PFC pathway. This group of LTP was significantly different from the low-frequency stimulation (Low Freq) controls at all the time points after TBS (F(5,7) = 5.149; p < 0.05). The level of potentiation in the LFS group was not significantly different from 100% at any time point. The injection of the competitive NMDA receptor antagonist (10 mg/kg) CPP 45 min before TBS significantly inhibited the induction of LTP; no increase of the EPSP amplitude was observed in the CPP-treated rats at any time point [t test for difference from baseline (100%); t(4) < 1; n = 5; NS]. Top left corner, Representative field potentials in the mPFC evoked during BLA stimulation immediately before and 90 min after TBS. The baseline (thin line) and the potentiated response are superimposed and are averages of 20 evoked responses each. Calibration: 0.2 mV, 10 msec.
Potentiation was followed here for up to 90 min but could be followed for up to 3 hr without decrement (data not shown).
Potentiation levels in the LTP group after TBS to the BLA were significantly different from 100% at all times after TBS [t test for difference from baseline (100%): +1 min, t(7) = 3.039, p < 0.01 (76 ± 13.3%); +15 min, t(7) = 2.971, p < 0.05 (76.6 ± 11.9%); +30 min, t(7) = 3.162, p < 0.05 (72.4 ± 11.3); +60 min, t(7) = 3.75, p < 0.05 (85.9 ± 8.9%); +90 min, t(7) = 3.53, p < 0.05 (89.9 ± 12%)]. Low frequency stimulation controls (n = 5) showed no significant potentiation at any time point [t test for difference from baseline (100%); t(4) < 1; NS) (Fig. 2), and this group was significantly different from the LTP group (F(1,9) = 24.47; p < 0.01). The peak latencies of the EPSP were not changed after TBS.
The application of CPP did not modify the baseline signal before TBS application (data not shown). However, the NMDA receptor antagonist CPP blocked the induction of LTP in the BLA–mPFC pathway at all time points tested (t test for difference from baseline (100%); t(4) < 1; n = 5; NS) (Fig. 2), indicating that, similar to CA1, LTP in the BLA–mPFC pathway is an NMDA-dependent process.
Acute stress blocks LTP in the BLA–mPFC pathway
After establishing the effectiveness of the elevated platform in impairing hippocampal LTP, we further examined whether similar stress exposure will alter LTP in the BLA–mPFC pathway.
Thirty minutes of exposure to stress significantly inhibited the ability of TBS to induce LTP in the mPFC for all the time points tested (t test for difference from baseline (100%); t(7) < 1; NS for all time points) (Fig. 3). Hence, similar to the hippocampus, exposure to inescapable stress attenuates BLA–mPFC LTP.
Figure 3.

Behavioral stress impairs amygdala–PFC LTP. Thirty minutes of exposure to stress significantly inhibited the ability of TBS to induce LTP in the PFC for all of the time points tested [t test for difference from baseline (100%); t(7) < 1; NS for all time points].
Discussion
In the present study, we used in vivo field potential recording to evaluate the effects of stress on plasticity in the excitatory pathway emerging from the BLA to the mPFC. Previous studies have established that the amygdala modulates PFC activity. For example, stimulation of the BLA alters neuronal firing in the mPFC, with latencies consistent with monosynaptic and polysynaptic pathways (Perez-Jaranay and Vives, 1991). Similar to LTP in the CA1, LTP in the BLA–mPFC pathway was found to be NMDA dependent because the selective NMDA receptor competitive antagonist CPP completely blocked its induction.
The data presented here demonstrate for the first time that exposure to inescapable stress effectively blocks the induction of LTP in the mPFC in vivo. The effects of this stressor on BLA–mPFC LTP were similar to its effects on CA1 LTP and compatible with those observed in the hippocampus with other models of stress (Foy et al., 1987; Shors et al., 1989; Diamond and Rose, 1994; Kim et al., 1996; Xu et al., 1997; Mesches et al., 1999). Importantly, although most studies so far have focused on the effects of stress on hippocampal LTP, these findings indicate that, when exposed to stress, LTP is inhibited not only in the hippocampus but also in the mPFC.
To improve future chances of survival, stressful experiences are presumably important events from which to learn. Assuming that LTP-like plasticity is indicative of memory formation processes, it is somewhat surprising that stressful events suppress LTP in two brain areas central to memory formation. It is thus possible that, under emotional conditions, e.g., fear conditioning, it may be essential to “block” the high-order behavior mediated by the hippocampus and PFC and to allow more automatic responses that are dependent on subcortical areas such as the amygdala. It has been suggested that, under these conditions, plasticity in the amygdala will be enhanced (for review, see Diamond et al., 2001). Preliminary data suggests that indeed this is the case (Yaniv et al., 2003), but additional research is required to elaborate this possibility.
The above-presented results should serve to promote a shift from a focused attention on the effects of stress on plasticity in the hippocampus to a system level approach that emphasizes the possible modification of interactions between relevant brain areas after an exposure to a stressful experience.
Footnotes
This study was supported by The National Institute for Psychobiology in Israel founded by The Charles E. Smith Family.
Correspondence should be addressed to Dr. Mouna Maroun, Brain and Behavior Center, Haifa University, Haifa 31905, Israel. E-mail: mmaroun@psy.haifa.ac.il.
Copyright © 2003 Society for Neuroscience 0270-6474/03/234406-04$15.00/0
References
- Akirav I, Richter-Levin G ( 1999) Biphasic modulation of hippocampal plasticity by behavioral stress and basolateral amygdala stimulation in the rat. J Neurosci 19: 10530–10535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akirav I, Richter-Levin G ( 2002) Mechanisms of amygdala modulation of hippocampal plasticity. J Neurosci 22: 9912–9921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaral DG, Insausti R ( 1992) Retrograde transport of d-[3H]-aspartate injected into the monkey amygdaloid complex. Exp Brain Res 88: 375–388. [DOI] [PubMed] [Google Scholar]
- Baker KB, Kim JJ ( 2002) Effects of stress and hippocampal NMDA receptor antagonism on recognition memory in rats. Learn Mem 9: 58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodnoff SR, Humphreys AG, Lehman JC, Diamond DM, Rose GM, Meaney MJ ( 1995) Enduring effects of chronic corticosterone treatment on spatial learning, synaptic plasticity, and hippocampal neuropathology in young and mid-aged rats. J Neurosci 15: 61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burette F, Jay TM, Laroche S ( 1997) Reversal of LTP in the hippocampal afferent fiber system to the prefrontal cortex in vivo with low-frequency patterns of stimulation that do not produce LTD. J Neurophysiol 78: 1155–1160. [DOI] [PubMed] [Google Scholar]
- Cassell MD, Chittick CA, Siegel MA, Wright DJ ( 1989) Collateralization of the amygdaloid projections of the rat prelimbic and infralimbic cortices. J Comp Neurol 279: 235–248. [DOI] [PubMed] [Google Scholar]
- de Quervain DJ, Roozendaal B, McGaugh JL ( 1998) Stress and glucocorticoids impair retrieval of long-term spatial memory. Nature 394: 787–790. [DOI] [PubMed] [Google Scholar]
- Diamond DM, Rose GM ( 1994) Stress impairs LTP and hippocampal-dependent memory. Ann NY Acad Sci 746: 411–414. [DOI] [PubMed] [Google Scholar]
- Diamond DM, Park CR, Puls MJ, Rose GM ( 2001) Differential effects of stress on hippocampal and amygdaloid LTP. In: Neuronal mechanisms of memory formation (Holscher C, ed), pp 379–403. Cambridge, UK: Cambridge UP.
- Foy MR, Stanton ME, Levine S, Thompson RF ( 1987) Behavioral stress impairs long-term potentiation in rodent hippocampus. Behav Neural Biol 48: 138–149. [DOI] [PubMed] [Google Scholar]
- Garcia R, Musleh W, Tocco G, Thompson RF, Baudry M ( 1997) Time-dependent blockade of STP and LTP in hippocampal slices following acute stress in mice. Neurosci Lett 233: 41–44. [DOI] [PubMed] [Google Scholar]
- Garcia R, Vouimba RM, Baudry M, Thompson RF ( 1999) The amygdala modulates prefrontal cortex activity relative to conditioned fear. Nature 402: 294–296. [DOI] [PubMed] [Google Scholar]
- Herry C, Vouimba RM, Garcia R ( 1999) Plasticity in the mediodorsal thalamo-prefrontal cortical transmission in behaving mice. J Neurophysiol 82: 2827–2832. [DOI] [PubMed] [Google Scholar]
- Ikegaya Y, Saito H, Abe K ( 1994) Attenuated hippocampal long-term potentiation in basolateral amygdala-lesioned rats. Brain Res 656: 157–164. [DOI] [PubMed] [Google Scholar]
- Ikegaya Y, Saito H, Abe K ( 1995) High-frequency stimulation of the basolateral amygdala facilitates the induction of long-term potentiation in the dentate gyrus in vivo. Neurosci Res 22: 203–207. [DOI] [PubMed] [Google Scholar]
- Ikegaya Y, Saito H, Abe K ( 1996) The basomedial and basolateral amygdaloid nuclei contribute to the induction of long-term potentiation in the dentate gyrus in vivo. Eur J Neurosci 8: 1833–1839. [DOI] [PubMed] [Google Scholar]
- Jay TM, Burette F, Laroche S ( 1995) NMDA receptor-dependent long-term potentiation in the hippocampal afferent fiber system to the prefrontal cortex in the rat. Eur J Neurosci 7: 247–250. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Yoon KS ( 1998) Stress: metaplastic effects in the hippocampus. Trends Neurosci 21: 505–509. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Foy MR, Thompson RF ( 1996) Behavioral stress modifies hippocampal plasticity through N-methyl-d-aspartate receptor activation. Proc Natl Acad Sci USA 93: 4750–4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krettek JE, Price JL ( 1977) The cortical projections of the mediodorsal nucleus and adjacent thalamic nuclei in the rat. J Comp Neurol 171: 157–191. [DOI] [PubMed] [Google Scholar]
- Luine V, Villegas M, Martinez C, McEwen BS ( 1994) Stress-dependent impairments of spatial memory. Role of 5-HT. Ann NY Acad Sci 746: 403–404. [DOI] [PubMed] [Google Scholar]
- McDonald AJ ( 1987) Organization of amygdaloid projections to the mediodorsal thalamus and prefrontal cortex: a fluorescence retrograde transport study in the rat. J Comp Neurol 262: 46–58. [DOI] [PubMed] [Google Scholar]
- McDonald AJ ( 1991) Organization of amygdaloid projections to the prefrontal cortex and associated striatum in the rat. Neuroscience 44: 1–14. [DOI] [PubMed] [Google Scholar]
- McDonald AJ ( 1996) Glutamate and aspartate immunoreactive neurons of the rat basolateral amygdala: colocalization of excitatory amino acids and projections to the limbic circuit. J Comp Neurol 365: 367–379. [DOI] [PubMed] [Google Scholar]
- Meaney MJ, Aitken DH ( 1985) [3H]Dexamethasone binding in rat frontal cortex. Brain Res 328: 176–180. [DOI] [PubMed] [Google Scholar]
- Mesches MH, Fleshner M, Heman KL, Rose GM, Diamond DM ( 1999) Exposing rats to a predator blocks primed burst potentiation in the hippocampus in vitro J Neurosci 19: RC18(1–5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen BS, Sapolsky RM ( 1995) Stress and cognitive function. Curr Opin Neurobiol 5: 205–216. [DOI] [PubMed] [Google Scholar]
- McEwen BS, De Kloet ER, Rostene W ( 1986) Adrenal steroid receptors and actions in the nervous system. Physiol Rev 66: 1121–1188. [DOI] [PubMed] [Google Scholar]
- Morgan MA, LeDoux JE ( 1995) Differential contribution of dorsal and ventral medial prefrontal cortex to the acquisition and extinction of conditioned fear in rats. Behav Neurosci 109: 681–688. [DOI] [PubMed] [Google Scholar]
- Pavlides C, Ogawa S, Kimura A, McEwen BS ( 1996) Role of adrenal steroid mineralocorticoid and glucocorticoid receptors in long-term potentiation in the CA1 field of hippocampal slices. Brain Res 738: 229–235. [DOI] [PubMed] [Google Scholar]
- Perez-Jaranay JM, Vives F ( 1991) Electrophysiological study of the response of medial prefrontal cortex neurons to stimulation of the basolateral nucleus of the amygdala in the rat. Brain Res 564: 97–101. [DOI] [PubMed] [Google Scholar]
- Porrino LJ, Crane AM, Goldman-Rakic PS ( 1981) Direct and indirect pathways from the amygdala to the frontal lobe in rhesus monkeys. J Comp Neurol 198: 121–136. [DOI] [PubMed] [Google Scholar]
- Shors TJ, Seib TB, Levine S, Thompson RF ( 1989) Inescapable versus escapable shock modulates long-term potentiation in the rat hippocampus. Science 244: 224–226. [DOI] [PubMed] [Google Scholar]
- Takita M, Izaki Y, Jay TM, Kaneko H, Suzuki SS ( 1999) Induction of stable long-term depression in vivo in the hippocampal-prefrontal cortex pathway. Eur J Neurosci 11: 4145–4148. [DOI] [PubMed] [Google Scholar]
- Thierry AM, Tassin JP, Blanc G, Glowinski J ( 1976) Selective activation of mesocortical DA system by stress. Nature 263: 242–244. [DOI] [PubMed] [Google Scholar]
- Wang J, Akirav I, Richter-Levin G ( 2000) Short-term behavioral and electrophysiological consequences of underwater trauma. Physiol Behav 70: 327–332. [DOI] [PubMed] [Google Scholar]
- Williams GV, Goldman-Rakic PS ( 1995) Modulation of memory fields by dopamine D1 receptors in prefrontal cortex. Nature 376: 572–575. [DOI] [PubMed] [Google Scholar]
- Xu L, Anwyl R, Rowan MJ ( 1997) Behavioral stress facilitates the induction of long-term depression in the hippocampus. Nature 387: 497–500. [DOI] [PubMed] [Google Scholar]
- Xu L, Holscher C, Anwyl R, Rowan MJ ( 1998) Glucocorticoid receptor and protein/RNA synthesis-dependent mechanisms underlie the control of synaptic plasticity by stress. Proc Natl Acad Sci USA 95: 3204–3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaniv D, Vouimba RM, Diamond D, Richter-Levin G ( 2003) Effects of novel versus repeated mild stressful experiences on LTP induced simultaneously in the amygdala and hippocampus in freely behaving rats. Ann NY Acad Sci 985: 556–558. [Google Scholar]