

Abstract
Several lines of evidence indicate that alterations in axonal transport play a critical role in Alzheimer's disease (AD) neuropathology, but the molecular mechanisms that control this process are not understood fully. Recent work indicates that presenilin 1 (PS1) interacts with glycogen synthase kinase 3β (GSK3β). In vivo, GSK3β phosphorylates kinesin light chains (KLC) and causes the release of kinesin-I from membrane-bound organelles (MBOs), leading to a reduction in kinesin-I driven motility (Morfini et al., 2002b). To characterize a potential role for PS1 in the regulation of kinesin-based axonal transport, we used PS1-/- and PS1 knock-inM146V (KIM146V) mice and cultured cells. We show that relative levels of GSK3β activity were increased in cells either in the presence of mutant PS1 or in the absence of PS1 (PS1-/-). Concomitant with increased GSK3β activity, relative levels of KLC phosphorylation were increased, and the amount of kinesin-I bound to MBOs was reduced. Consistent with a deficit in kinesin-I-mediated fast axonal transport, densities of synaptophysin- and syntaxin-I-containing vesicles and mitochondria were reduced in neuritic processes of KIM146V hippocampal neurons. Similarly, we found reduced levels of PS1, amyloid precursor protein, and synaptophysin in sciatic nerves of KIM146V mice. Thus PS1 appears to modulate GSK3β activity and the release of kinesin-I from MBOs at sites of vesicle delivery and membrane insertion. These findings suggest that mutations in PS1 may compromise neuronal function by affecting GSK-3 activity and kinesin-I-based motility.
Keywords: Alzheimer's disease, presenilin, GSK3β, kinesin, axonal transport, growth cones
Introduction
Most early-onset familial Alzheimer's disease (FAD) cases are associated with mutations in presenilin 1 (PS1) and presenilin 2 (PS2; Levy-Lahad et al., 1995; Sherrington et al., 1995). Presenilins are integral membrane proteins with eight transmembrane domains and a hydrophilic loop between transmembrane domains 6 and 7 (Doan et al., 1996; Li and Greenwald, 1998). In neurons the presenilins localize in the endoplasmic reticulum (ER), intermediate compartment, nuclear membranes, and growth cones (Busciglio et al., 1997; Capell et al., 1997; Lah et al., 1997; Annaert et al., 1999; Pigino et al., 2001). Presenilins undergo endoproteolytic cleavage, generating stable N- and C-terminal fragments (NTF and CTF) that interact with other proteins to form a macromolecular complex containing the γ-secretase activity that is responsible for the regulated intramembrane proteolysis of the amyloid precursor protein (APP), Notch 1, ErbB-4, and E-cadherin (Ebinu and Yankner, 2002; Marambaud et al., 2002).
Glycogen synthase kinase 3β (GSK3β) is a component of the WNT signaling pathway with a role in cell proliferation, differentiation, adhesion, microtubule dynamics, apoptosis, and fast axonal transport (Frame and Cohen, 2001; Morfini et al., 2002b). GSK3β is inactivated by phosphorylation at serine 9 (Ser9) in its N terminus (Woodgett, 1994; Frame and Cohen, 2001; Frame et al., 2001). The phosphorylation and dephosphorylation of Ser9 operate as a primary switch to regulate GSK3β kinase activity. GSK3β can inhibit itself by autophosphorylation (Wang et al., 1994) or be inhibited by other kinases that phosphorylate Ser9 (Woodgett, 1994). In vivo substrates for GSK3β include PS1, β-catenin, glycogen synthase, tau, and kinesin-I light chains (Frame and Cohen, 2001; Morfini et al., 2002b).
GSK3β interacts with PS1 heterodimeric complexes and coimmunoprecipitates with PS1 in cell lines (Takashima et al., 1998; Tesco and Tanzi, 2000). GSK3β binds to the hydrophilic loop of PS1 (Takashima et al., 1998), phosphorylates PS1 at Ser397 within the loop domain, and regulates the degradation of PS1 CTF (Kirschenbaum et al., 2001). Previous results indicate that FAD-linked PS1 mutations affect GSK3β kinase activity in transfected cell lines (Takashima et al., 1998; Weihl et al., 1999). These observations suggest the possibility of a molecular and functional interaction between PS1 and GSK3β that is deregulated by PS1 mutations.
Recently, Morfini et al. (2002b) showed that phosphorylation of kinesin-I light chain (KLC) by GSK3β promotes the release of kinesin-I from membrane-bound organelles (MBOs), leading to a reduction in fast anterograde axonal transport. Retrograde transport is not affected, and anterograde transport of vesicles is reduced, but not abolished, indicating that GSK3β regulates anterograde trafficking of MBOs associated with specific kinesin isoforms (Morfini et al., 2002b). These results suggest that the temporal and spatial regulation of GSK3β in neurons is an important element in the control of neuronal membrane trafficking. Relevant pathways for this regulation may involve proteins that interact with microtubules (MTs), that bind to membranes or membrane proteins, or that are components of signaling pathways (Ratner et al., 1998), all of which are characteristics of PS1. Taken together, the above data raise the possibility that interactions between PS1 and GSK3β may play a role in the regulation of vesicle transport and/or delivery. To test this possibility, we examined the effects on GSK3β activity of either eliminating PS1 or introducing the human PS1 gene containing the M146V mutation, which causes a very aggressive early-onset form of FAD. PS1-/- and PS1 knock-inM146V (KIM146V) mice and cultured cells were used to characterize the effect of PS1 and PS1 mutations on the regulation of kinesin-based transport in neurons. We show that PS1 mutations increase GSK3β activity and kinesin-I phosphorylation, leading to reduced levels of kinesin-I bound to MBOs. These observations suggest that PS1 mutations may compromise neuronal function by deregulating GSK3β activity and kinesin-I phosphorylation, leading to impaired transport and/or targeting of MBOs essential for neuronal function such as synaptic vesicles and mitochondria.
Materials and Methods
Cell culture. Neuronal cultures were prepared from PS1 wild-type (WT) or PS1 KI M146V (Guo et al., 1999) mouse embryos at day 16 of gestational age. After dissection the cortical or hippocampal tissue was incubated in 0.25% trypsin in Hank's for 20 min at 37°C, followed by dissociation and plating of the cell suspension in culture dishes or glass coverslips covered with poly-d-lysine (500 μg/ml), at a density of 16,000 cells/cm 2 for immunocytochemistry or 53,000 cell/cm 2 for biochemical analysis. The cultures initially were plated in DMEM plus 10% iron-supplemented calf serum (HyClone, Logan, UT) for 2 hr and then switched to defined media consisting of Neurobasal media plus N2 and B27 supplements (Life Technologies, Grand Island, NY). NT2 cells were cultured as described (Pleasure et al., 1992). Fibroblast cultures were generated from E16 PS1 WT, KO, and KI M146V embryos as described (Taccioli et al., 1998).
Antibodies. The following antibodies (Ab) were used: PSN2, a monoclonal Ab that recognizes the NTF of PS1 (Pigino et al., 2001); 2025, a polyclonal Ab raised against a synthetic peptide corresponding to PS1 amino acid residues 1–20 (Pigino et al., 2001); AD3L, a polyclonal Ab that recognizes the PS1 C terminus (AD3L) (Pigino et al., 2001); anti-β-tubulin isotype III (clone SDL 310; Sigma, St. Louis, MO); anti-β-tubulin (clone DM1B; Sigma); anti-GSK3β monoclonal Ab (BD Transduction Laboratories, San Diego, CA); anti-GSK3β polyclonal Ab (334–348; Calbiochem, San Diego, CA); anti-GSK3β phosphorylated at serine 9 (GSK3β-Pser9; Cell Signaling, Boston, MA), which recognizes the inactive form of GSK3β; anti-cytochrome c (clone 6H2.B4; PharMingen, San Diego, CA); H2, a monoclonal Ab that recognizes kinesin-I heavy chain (Pfister et al., 1989); 63–90, a monoclonal Ab that preferentially recognizes dephosphorylated kinesin-I light chains (Stenoien and Brady, 1997); kinesin-I ALL, a monoclonal Ab that recognizes KLC irrespective of its phosphorylation state (Stenoien and Brady, 1997); anti-synaptophysin (SY38) and anti-APP (Alz-90; both from Chemicon, Temecula, CA); anti-SNAP25 (Alomone Labs, Jerusalem, Israel); and anti-syntaxin-I (BD Transduction Laboratories).
Immunofluorescence. Immunocytochemical staining was performed as described (Pigino et al., 2001). Briefly, cells were fixed for 30 min at 37°C in 4% paraformaldehyde/0.12 m sucrose in PBS and permeabilized with 0.2% Triton X-100 in PBS for 10 min. Then the cultures were blocked for 1 hr in 5% normal goat serum in PBS and incubated overnight at 4°C in a humid chamber with the primary Ab, followed by incubation with the appropriate secondary Abs conjugated with Alexa fluoro-red and fluoro-green (Molecular Probes, Eugene, OR). The fluorescence was visualized with a Zeiss LSM 510 confocal microscope or an Olympus IX-70 inverted microscope.
Preparation of brain and sciatic nerve homogenates. Adult brains from PS1 WT and KI M146V mice were removed, homogenized in 1 ml of radio immunoprecipitation assay (RIPA) buffer (Pigino et al., 2001), and processed for Western blot analysis. Sciatic nerves were dissected from adult mice as described (Kasa et al., 2001). Briefly, the sciatic nerves were exposed and removed with surgical scissors. Segments 2 cm in length were excised, placed in ice-cold Hank's for 1 min, and processed for SDS-PAGE and Western blot.
Western blot and immunoprecipitation. Changes in protein expression level and association of PS1 and GSK3β with vesicular fractions were analyzed by Western blot as described (Busciglio et al., 2002; Grace and Busciglio, 2003). Immunoprecipitation experiments were performed and controlled as described (Pigino et al., 2001).
Preparation of MBO-enriched fractions and subcellular fractionation. MBOs were obtained as described (Tsai et al., 2000; Morfini et al., 2002b). Briefly, the cells were washed twice with warm PHEM buffer [containing (in mm): 60 PIPES, 25 HEPES, 10 EGTA, and 2 MgCl2, pH 7.4] and extracted for 4 min at 37°C with 0.05% Triton X-100 in PHEM supplemented with 1 mm GTP, 10 μm Taxol, and a mixture of protease inhibitors (Complete; Roche Bioscience, Palo Alto, CA). Then the cells were fixed for immunofluorescence or harvested for Western blot analysis.
Subcellular fractions were collected as described (Morfini et al., 2001b, 2002b). Brains from adult mice were homogenized in 5 ml of homogenization buffer [HB; containing (in mm): 300 sucrose, 10 HEPES, pH 7.4, 5 EDTA, supplemented with protease inhibitors]. Homogenates were spun at 12,500 × g to eliminate cell debris, nuclei, and mitochondria. Supernatants were centrifuged at 39,800 × g for 40 min to obtain a vesicle (V0) pellet. Then the supernatant (S0) was recentrifuged for 40 min at 120,000 × g to obtain a V1 pellet, and this supernatant (S1) was centrifuged for 2 hr at 260,000 × g to obtain the V2 pellet. The remaining supernatant was called cytosol. All vesicle pellets were resuspended in homogenization buffer. Equal amounts of protein from each fraction were processed for Western blot. Growth cone-enriched fractions were prepared as described (Pfenninger et al., 1983; Morfini et al., 2002b). For phosphorylation assays the resuspended V1 vesicle fractions were incubated with 1 mm ATP for 20 min at 37°C (Morfini et al., 2002b). For dephosphorylation experiments identical blotted membranes were blocked first with 5% albumin at room temperature (RT) and then were incubated in dephosphorylation buffer (10 mm HEPES, pH 8, 10 mm MgCl2) with or without calf intestine alkaline phosphatase (200 U/ml; Calbiochem) for 1 hr at RT.
Transfection. Hippocampal neurons and NT2 neuronal cells were transfected as previously described with the expression vectors PS1 wild-type (WT) and PS1 bearing mutations M146V, I143T, and D9, respectively (Pigino et al., 2001). To control transfection efficiency, we used an enhanced green fluorescent protein (EGFP) vector (Clontech, Palo Alto, CA). All constructs were cloned into pcDNA3 (Invitrogen, Carlsbad, CA), and the identity and integrity of each clone were confirmed by sequencing.
Cell viability and caspase activity assays. Neuronal viability was assessed by a propidium iodide exclusion assay as previously described (Busciglio and Yankner, 1995; Grace et al., 2002; Grace and Busciglio, 2003). The activity of caspases 2, 3, 6, 8, and 9 was quantified in triplicate cultures by the Apotarget Caspase Colorimetric Assay kit, following the vendor's procedure (Biosource, Camarillo, CA).
Image analysis. Image analysis was performed as described (Pigino et al., 2001; Grace et al., 2002; Grace and Busciglio, 2003). For synaptic vesicle and mitochondrial density quantification the images of immunostained neurons were visualized with an Olympus IX-70 inverted microscope. Images were captured with a CCD camera (Spot, Diagnostic Instruments, Sterling Heights, MI) driven by Spot image acquisition software and analyzed with NIH Image software. The identity of cultures was coded to avoid experimental bias. To determine the ratio of immunofluorescence (IF) intensity in different subcellular compartments, we obtained confocal images with a Zeiss LSM 510 confocal microscope, and ratio image analysis was performed with the Metamorph/Metafluor software. Ratio analysis was performed in identical volume areas, in the same cell, in at least 30 individual cells per condition.
Statistical analysis. All experiments were repeated at least three times, using different brain specimens or cultures derived from at least three different embryos. Each individual experiment was performed in quadruplicate. In most cases the data were analyzed by ANOVA, followed by post hoc Student–Newman–Keuls test to make all possible comparisons. Data were expressed as the mean ± SEM, and significance was assessed at p < 0.05.
Results
Endogenous PS1 coimmunoprecipitates with GSK3β and colocalizes with active GSK3β in growth cones
To characterize molecular interactions between PS1 and GSK3β in primary neurons, we first analyzed the expression and localization of endogenous PS1 and GSK3β in cultured hippocampal neurons. Western blot analysis revealed that PS1 and GSK3β expression increased steadily during neuronal development (Fig. 1A). Both proteins showed a marked enrichment in a MBO fraction with neuronal maturation (Fig. 1A). Presence in this cellular fraction and resistance to mild permeabilization are consistent with their association with membrane organelles and cytoskeletal elements (Pigino et al., 2001; Morfini et al., 2002b). We further analyzed the association of PS1 and GSK3β with MBOs by subcellular fractionation. Three vesicle fractions and a soluble fraction were obtained by differential centrifugation from mouse brain homogenates (see Materials and Methods). As previously reported, GSK3β was highly enriched in fractions V1 and V2 (Morfini et al., 2002b), whereas PS1 was enriched in fractions V0 and V1 (Fig. 1B). A significant amount of PS1 and GSK3β colocalized in V1 (100,000 × g microsomal pellet), a subpopulation of MBOs, and both were enriched in growth cone preparations (Fig. 1B). Consistent with previous studies (Morfini et al., 2002b), kinesin-I was concentrated in these same fractions also. GSK3β and PS1 coimmunoprecipitated from cell lysates of doubly transfected CosPS1/GSK3β cells (Fig. 1C). Similarly, endogenous GSK3β and PS1 coimmunoprecipitated from mouse brain homogenates (Fig. 1C). Collectively, these results indicate that the cellular distribution of both proteins overlaps in specific vesicular compartments, and the results provide evidence for a molecular interaction between PS1 and GSK3β in nerve tissue.
Figure 1.
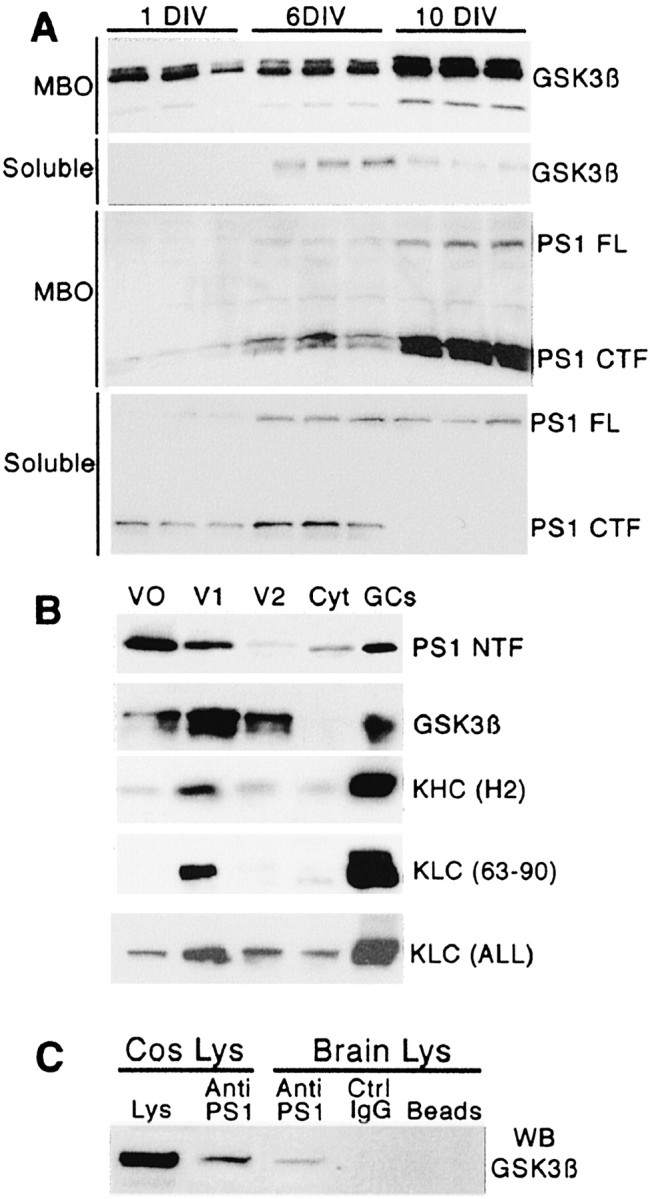
Endogenous PS1 and GSK3β exhibit similar expression profiles during neuronal development, partially colocalize in the same vesicular compartment, and coimmunoprecipitate from brain homogenates. A, Western blot analysis of MBO and soluble fractions during development of hippocampal neurons in culture at 1, 3, and 10 d in vitro (DIV). The figure shows samples of three different homogenates obtained at each time point. Note that the expression pattern is similar for PS1 and GSK3β. Both proteins are highly enriched in the MBO fraction, particularly at 10 DIV. PS1 FL, Full-length PS1; PS1 CTF, C-terminus fragment of PS1. The antibodies include anti-GSK3β (334–348), 1:1000, and anti-PS1 (AD3L), 1:2000. B, PS1 and GSK3β colocalize on a subpopulation of MBOs and are enriched in a growth cone fraction. The Western blots show the localization of PS1, GSK3β, and kinesin-I heavy (KHC) and light (KLC) chains in three vesicle fractions (V0, V1, and V2) and supernatant (Cyt). Significant amounts of PS1 and GSK3β colocalize in V1 where kinesin-I is enriched. PS1 and GSK3β are highly enriched in growth cone fractions (GCs). PS1 NTF, N-terminus fragment of PS1. The antibodies include anti-PS1 (PSN2), 1:1000; anti-KHC (H2), 1:2000; anti-KLC (63–90), 1:2000; and anti-KLC ALL, 1:2000. C, Coimmunoprecipitation of PS1 and GSK3β from Cos cell lysates cotransfected with PS1 and GSK3β expression vectors (Cos Lys) and mouse brain homogenates (Brain Lys). A sample of transfected Cos cell lysates was included as a positive control for GSK3β staining (Lys). The Cos cell lysate that was immunoprecipitated with anti-PS1 antibody (Anti-PS1) shows immunoreactivity with anti-GSK3β in the Western blot. Similarly, anti-PS1 immunoprecipitate from mouse brain homogenate shows immunoreactivity for GSK3β. No GSK3β immunoreactivity is observed in immunoprecipitates for nonimmune mouse IgG (Ctrl IgG) or protein A-Sepharose beads alone (Beads). Immunoprecipitations were performed with anti-PS1 antibody PSN2. Western blot was revealed with anti-GSK3β (Calbiochem).
Analysis of PS1 and GSK3β localization in hippocampal neurons by IF and confocal microscopy indicated that PS1 and GSK3β were present in the cell body, neuritic shaft, and growth cones of cultured neurons (Fig. 2A,B). Both proteins were present after mild detergent extraction, which selectively retains membrane-associated proteins, consistent with subcellular fractionation studies (Fig. 2A,B, arrows). At higher magnification GSK3β and PS1 showed extensive colocalization in the central area of the growth cone as well as in individual filopodia of extracted cells (Fig. 2C–E, arrows). Ratio image analysis showed maximum overlap between PS1 and GSK3β in growth cones, strongly suggesting localization of both antigens in the same subcellular structures (Fig. 2F, white pseudocolor). Thus membrane-associated PS1 and GSK3β colocalize and are enriched in the growth cone, a neuronal compartment in which active membrane delivery and insertion take place during neurite extension.
Figure 2.

Endogenous membrane-associated PS1 and GSK3β colocalize in cultured neurons. A, B, Double-immunofluorescence staining of a 2 DIV hippocampal neuron stained with anti-GSK3β (A) and anti-PS1 (B) antibodies. Cultures were extracted before fixation to retain selectively the membrane-associated proteins (see Materials and Methods). PS1 and GSK3β are enriched in growth cones of developing neuritic processes (arrows). The antibodies include anti-PS1 (PSN2), 1:100, and anti-total GSK3β (334–348), 1:400. Scale bar, 20 μm. C–E, Confocal images showing colocalization of PS1 and GSK3β in the growth cone central region and filopodia (yellow in merged image E; arrows). F, The degree of colocalization between membrane-associated GSK3β and PS1 in the confocal image pair from C, D was assessed by ratio image analysis and was pseudocolored according to the degree of colocalization of both fluorescent signals. White denotes the areas of highest immunoreactivity overlap.
FAD-linked PS1 mutations increase GSK3β activity, KLC phosphorylation, and release of kinesin-I from MBOs
To evaluate the possibility of a role for PS1 in the regulation of GSK3β activity, we analyzed fibroblast cultures derived from PS1 WT, PS1-/- (Wong et al., 1997), and PS1 KIM146V (Guo et al., 1999) mice. In PS1 KIM146V mice the endogenous PS1 gene was replaced by a human PS1 gene bearing the M146V mutation, which causes a very aggressive early-onset form of FAD. Western blot analysis of PS1 WT, KIM146V, and PS1-/- fibroblasts showed similar levels of PS1 expression in PS1 WT and KIM146V cells (Fig. 3A), because expression of PS1 KIM146V is driven by the mouse PS1 endogenous promoter. As expected, no PS1 immunoreactivity was detected in PS1-/- cells (Fig. 3A). A marked increase in GSK3β activity was observed in KIM146V and PS1-/- cells, as reflected by the significant reduction in GSK3β-Pser9 immunoreactivity (Fig. 3A). In contrast, no significant changes were detected in the level of total GSK3β, kinesin-I heavy chain (KHC), and tubulin (Fig. 3A). Similarly, expression of FAD-linked PS1 mutations M146V, I143T, and deletion of exon 9 (D9) in NT2 human neuronal cells significantly increased the activation of GSK3β, indicated by a reduction in GSK3β-Pser9 immunoreactivity, with no changes in total GSK3β level (Fig. 3B). None of the mutations that were analyzed caused neuronal death under the conditions of the assay, as assessed by a propidium iodide exclusion assay (Fig. 3C). This rules out the possibility that GSK3β activation was secondary to a neurodegenerative process triggered by the expression of mutant forms of PS1.
Figure 3.

Increased activation of GSK3β in PS1 -/- and PS1 KI M146V fibroblasts and neuronal cells expressing PS1 mutations M146V, I143T, and D9. A, Protein expression analysis of PS1 wild-type (WT), PS1 -/- (KO), and PS1 KI M146V (KI) fibroblasts. The figure shows samples of two different WT, KO, and KI cultures. Reduced levels of GSK3β-Pser9 (inactive form) were detected in PS1 KO and KI as compared with WT fibroblasts. No changes in the expression levels of total GSK3β, kinesin-I heavy chain (KHC), and tubulin were observed. Note the absence of PS1 in KO fibroblast and similar levels of PS1 expression in WT and KI fibroblasts. B, NT2 neuronal cells were transfected with PS1 WT and PS1 mutant M143T, M146V, and D9 constructs. No significant changes were detected in total GSK3β (GSK3β total) and tubulin levels (data not shown). The histogram represents the amount of GSK3β-Pser9 expressed as a percentage of the level in control cells (transfected with GFP). Values are the mean ± SE; n = 3 independent experiments. *p < 0.02 relative to GFP by Student's t test. Transfection and quantitative Western blot analysis were performed as described in Materials and Methods. C, Expression of PS1 mutations does not affect cell viability. Sister cultures to the ones used for the experiment shown in B were used to assess cell viability. The results showed no changes in cell survival associated with the expression of PS1 mutations. Viability was assessed by using a propidium iodide exclusion assay as described in Materials and Methods. At least 200 cells were scored per culture in triplicate cultures. The histogram represents the mean ± SE.
To examine the effect of PS1 mutations on kinesin-I phosphorylation, we used a phosphorylation-sensitive Ab, 63–90, against KLC N-terminal (Stenoien and Brady, 1997). This Ab preferentially recognizes dephosphorylated epitopes in KLCs (Morfini et al., 2001a, 2002b). Western blot analysis with Abs 63–90 (KLC) and H2 (KHC) of V1 vesicle fractions incubated with or without ATP showed an increase in the apparent molecular weight of KLCs after incubation with ATP (Fig. 4A). This shift is attributable to the phosphorylation of KLCs by vesicle-associated kinase(s) and correlates with the detachment of kinesin-I from membranes (Morfini et al., 2002b). Immunoreactivity with 63–90, but not H2, antibody is diminished significantly after incubation of the vesicles with ATP. Dephosphorylation with alkaline phosphatase restores 63–90 immnunoreactivity, indicating that KLCs levels are not affected and consistent with observations that 63–90 Ab immunoreactivity is sensitive to phosphorylation (Fig. 4A). Western blot of the same fractions with anti-KLC ALL Ab, which recognizes KLC irrespective of its phosphorylation state (Stenoien and Brady, 1997), confirmed that similar levels of KLC were present in all samples (Fig. 4A). Western blot analysis of brain homogenates revealed a significant reduction in the immunoreactivity of KLCs detected by Ab 63–90 in KIM146V as compared with WT brains (Fig. 4B), but no changes in protein levels of KHC, tubulin, and GSK3β were observed (Fig. 4B). Analysis of PS1 WT and KIM146V cortical culture homogenates showed a 49 ± 2% reduction (SEM; p < 0.01 by Student's t test) in 63–90 immunoreactivity of 63–90 for KIM146V cortical neurons (Fig. 4C), with no changes in levels of KLC, KHC, and tubulin (Fig. 4C). A similar result was observed in fibroblast from PS1-/- and KIM146V mice (Fig. 4D) and with NT2 cell-expressing mutations M146V, I143T, and D9 (data not shown). These experiments demonstrate increased phosphorylation of KLCs in brain tissue and neurons expressing PS1 mutations concurrent with the increased level of GSK3β kinase activity. To establish the role of kinesin-I phosphorylation in the release of kinesin-I from cargo vesicles, we performed a kinesin release assay (Morfini et al., 2002b). KLC and KHC were released from V1 vesicles (pellet) and were found in the supernatant after incubation with ATP while the vesicle markers synaptophysin, syntaxin-I, and APP remained in the pellet fraction (Fig. 4E). Thus kinesin-I is released from synaptophysin-, syntaxin-I-, and APP-containing vesicles after phosphorylation by V1-associated kinase(s). Importantly, 63–90 immunoreactivity decreases after kinesin is phosphorylated and detached from vesicles, similarly as observed with PS1 mutant cells and tissues (Fig. 4).
Figure 4.

Enhanced KLC phosphorylation in PS1 KI M146V hippocampal neurons and mouse brain. A, The 63–90 epitope on kinesin-I light chains (KLC) is sensitive to phosphorylation. V1 vesicle fractions were incubated without (Control) and with ATP. The samples were separated in an 8% gel and transferred; immunoblots were prepared with the H2 (kinesin-I heavy chain) and 63–90 antibodies. Notice the increase in relative molecular weight of KLCs after incubation with ATP. This shift is caused by the phosphorylation of KLCs by vesicle-associated kinase(s) and correlates with the detachment of kinesin-I from membranes (Morfini et al., 2002b). Immunoreactivity with 63–90, but not H2, antibody is diminished after the incubation of vesicles with ATP. When transfer membranes are dephosphorylated with alkaline phosphatase (+AP), 63–90 immnunoreactivity is recovered, indicating that the epitope for the Ab 63–90 is sensitive to phosphorylation. The same fractions were separated in a 4–20% gel, and Western blot was performed with the anti-KLC ALL antibody, which recognizes KLC irrespective of its phosphorylation state. Equal amounts of KLC were observed in all samples. B, C, Western blot analysis of WT and PS1 KI M146V total brain homogenates and cortical neuronal cultures. Samples from two WT and two KI brains and cortical cultures are shown. Note the reduction in KLCs staining with Ab 63–90 in PS1 KI M146V brain and cortical culture samples, indicating increased phosphorylation of KLCs in PS1 KI brain and cortical culture homogenates. Similar protein levels of KHC that have been detected with H2 antibody, which recognizes KHC, rule out changes in kinesin-I expression in PS1 KI mouse brain tissue. Similar levels of tubulin and GSK3β rule out general changes in protein expression. Similar levels of KLC immunoreactivity with KLC (ALL) antibody also are observed in cortical cultures. D, Western blot analysis of WT, PS1 KI M146V, and PS1 KO fibroblasts. Note the reduction in Ab 63–90 immunoreactivity. E, Kinesin-I release assay. V1 vesicles were incubated with 1 mm ATP for 30 min at 37°C and centrifuged at 120,000 × g; pellet (P) and supernatant (S) fractions were analyzed by Western blot. After incubation with ATP (+) the KLC and KHC are released from the vesicle fraction (P) and found in the supernatant while the vesicle markers synaptophysin (Syn) syntaxin-I (Synt) and APP remain in the vesicle fraction (P). Omission of ATP (-) results in the complete recovery of kinesin-I with the pellet fraction. Note the reduction in Ab 63–90 immunoreactivity after kinesin-I light chain phosphorylation.
Perfusion of active GSK3β into squid axoplasm drastically reduces anterograde transport of certain MBOs without affecting retrograde transport. This inhibition in anterograde transport is associated with a release of kinesin-I from MBOs, caused by increased phosphorylation of KLC by GSK3β (Morfini et al., 2002b). To determine the effect of increased GSK3β activity and kinesin-I phosphorylation on kinesin-I binding to MBOs in cells expressing PS1 mutants or lacking PS1, we analyzed the association of kinesin-I with MBOs fractions obtained from PS1 WT, KIM146V, and KO fibroblasts. Immunoblots showed a marked reduction in the levels of KLC and KHC associated with MBO-enriched fractions from KIM146V and KO as compared with WT fibroblasts (Fig. 5A). There were also significant reductions in levels of both KHC and KLC in MBO-enriched fractions prepared from NT2 neuronal cell cultures expressing M146V, I143T, and D9 mutations as compared with PS1 WT (Fig. 5B). Thus there is a clear reduction in the amount of kinesin-I associated with MBOs in cells lacking PS1 or expressing PS1 mutations. These results indicate that a lack of PS1 and PS1 mutations results in increased GSK3β activity, KLC phosphorylation, and release of kinesin-I from MBOs.
Figure 5.
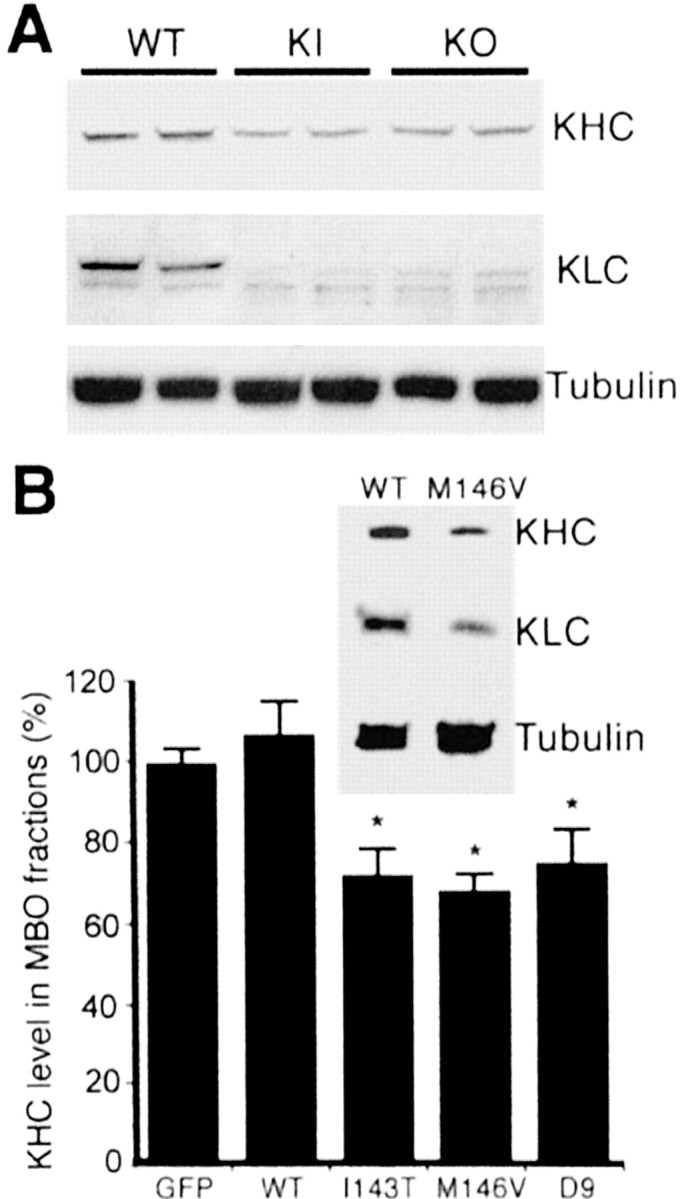
PS1 mutations enhance the release of kinesin-I from MBO-enriched fractions. A, Western blot analysis of KHC and KLC in MBO-enriched fractions from PS1 WT, PS1 KO, and PS1 KI M146V fibroblasts. KHC and KLC were detected with antibodies H2 and 63–90, respectively. Samples of two different cultures were assayed. Note the reduction in KHC and KLC in PS1 KI and KO as compared with WT fibroblasts. Similar tubulin levels were observed in all samples. These results suggest a reduced amount of kinesin-I associated with MBOs in KI and KO fibroblasts. B, Western blot shows reduced levels of both kinesin-I heavy and light chains (KHC and KLC) in MBO fractions prepared from NT2 neuronal cell cultures expressing the PS1 KI M146V mutation. Similar tubulin levels were detected in PS1 WT and M146V. KHC and KLC were detected with antibodies H2 and 63–90, respectively. The histogram shows the result of the quantification of KHC levels in MBO-enriched fractions prepared from transfected cultures expressing PS1 WT and PS1 mutants M143T, M146V, and D9. The amount of KHC in MBO-enriched fractions was expressed as a percentage of the level in control cells (transfected with GFP). Values are the mean ± SE; n = 3 independent experiments. *p < 0.01 relative to GFP by Student's t test. KHCs were detected with antibody H2; similar results were obtained for KLCs (data not shown). Tubulin levels did not change significantly among samples. MBO fractions, transfection, and quantitative Western blot analysis were performed as described in Materials and Methods.
Reduced APP and synaptophysin immunoreactivity in nerves and cultured neurons from PS1 KIM146V mice
Synaptic vesicle precursors are transported anterogradely by kinesin motors along the axons, and disruption of kinesin-based vesicle motility alters their transport, leading to reduced synaptic density (Ferreira et al., 1992; Yonekawa et al., 1998; Zhao et al., 2001). To analyze the effect of PS1 mutations on synaptic vesicle trafficking, we examined the density of synaptophysin- and syntaxin-I-containing vesicles in neuritic processes of PS1 WT and KIM146V neurons. Both synaptophysin and syntaxin-I are associated with synaptic vesicle membranes and are transported in synaptic vesicle precursors (Okada et al., 1995; Yonekawa et al., 1998). Immunofluorescence analysis showed a diminished number of synaptophysin- and syntaxin-I-containing vesicles in processes of PS1 KIM146V hippocampal neurons (Fig. 6A). Quanti tative analysis confirmed a significant reduction in the density of synaptophysin-immunoreactive dots in KIM146V as compared with WT hippocampal neurons (Fig. 6B); a similar result was obtained when the density of syntaxin-I-containing vesicles was assessed (data not shown). WT and KIM146V neurons exhibited similar viability and caspase activity levels, ruling out a potential effect of neuronal cell death on vesicle transport (Fig. 6C,D). A comparable reduction in synaptophysin immunoreactivity was detected in transfected hippocampal neurons expressing M146V, I143T, and D9 mutations, but not PS1 WT or GFP (Fig. 6E). Thus there is a reduction in the number of synaptic vesicle markers in neurites of primary neurons and neuronal cell lines expressing PS1 mutations.
Figure 6.

Reduced density of synaptic vesicles in hippocampal neurons expressing PS1 mutations. A, Immunofluorescence with anti-synaptophysin (Syn) and anti syntaxin-I (Synt) antibodies of PS1 WT and KI hippocampal neurons at 4 d in culture. Note the reduced number of vesicle clusters (arrows) in KI processes. The gray scale in the images was inverted for clarity. Scale bar, 20 μm. B, Reduced density of synaptophysin-containing vesicles in PS1 KI M146V hippocampal neurons. WT and PS1 KI M146V hippocampal cultures were fixed and double-immunostained with anti-tubulin class III and anti-synaptophysin antibodies. The density of synaptophysin-immunoreactive vesicles was assessed as described (Grace et al., 2002). The numbers represent the mean ± SE; n = 4 independent experiments. *p < 0.05 relative to WT by Student's t test. C, WT and PS1 KI M146V hippocampal neurons exhibit similar viability. Neuronal viability was assessed at 4 DIV by a propidium iodide exclusion assay. No significant differences in the number of viable neurons were detected between WT and PS1 KI M146V cultures. At least 200 neurons were scored per culture in triplicate cultures. The numbers represent the mean ± SE. D, WT and PS1 KI M146V hippocampal neurons exhibit similar caspase activities. No significant differences in caspase activity were observed between WT and PS1 KI M146V cultures. The activity of caspases 2, 3, 6, 8, and 9 was measured in triplicate cultures. Caspase activity in PS1 KI M146V cultures was expressed as a percentage after the activity of each caspase in WT cultures had been normalized as 100. E, A significant reduction in synaptophysin-immunoreactive vesicles was observed in hippocampal neurons expressing PS1 mutations. Transfected neurons were identified by cotransfection with a GFP expression vector (Pigino et al., 2001). Images of transfected neurons were captured at a final magnification of 400×, and the number of synaptophysin-immunoreactive dots was scored. At least 30 cells were analyzed per experimental condition. The numbers represent the mean ± SE; n = 3 independent experiments. *p < 0.02 relative to control (GFP) by Student's t test.
We also examined synaptic protein levels in PS1 KIM146V axons in vivo. The protein levels of PS1, synaptophysin, and APP were reduced markedly in sciatic nerves of KIM146V mice (Fig. 7). In contrast, the levels of tubulin and SNAP25, another synaptic protein, were similar in WT and KIM146V sciatic nerves (Fig. 7). GSK3β-Pser9 and 63–90 immunoreactivity levels were reduced in both sciatic nerves and spinal cord samples. No significant changes were observed in PS1, APP, synaptophysin, SNAP25, or tubulin expression in the KIM146V spinal cord segments, where the cell bodies of neurons projecting their axons into the sciatic nerve are located (Fig. 7), suggesting that there was no decrease in PS1, APP, or synaptophysin protein expression in KIM146V neurons. These results are consistent with a deficit in transport of a population of vesicle precursors and/or specific synaptic proteins associated with PS1 mutations in vitro and in vivo.
Figure 7.

Reduced levels of synaptic proteins in sciatic nerves of PS1 KI M146V mice. Sciatic nerves and spinal cord segments from two WT and two KI animals were dissected and homogenized. Protein (15 mg) was loaded in each lane. Western blots were developed with the indicated antibodies. Note the significant reduction in PS1, APP, and synaptophysin (Syn) in sciatic nerves, but not in the spinal cord, of PS1 KI mice. A reduction in GSK3β-Pser9 and 63–90 immunoreactivity is evident in both sciatic nerve and spinal cord samples from KI mice. No significant changes between PS1 WT and KI samples were detected in the level of total GSK3β, KHC, SNAP25, or tubulin.
Reduced density of mitochondria in neurites of PS1 KIM146V hippocampal neurons
Mitochondria are transported anterogradely by kinesin motors, and defects in kinesin-I transport have been shown to reduce mitochondrial movement along axonal processes (Tanaka et al., 1998). We examined mitochondrial density in cell bodies and neurites of PS1 WT and KIM146V hippocampal neurons by using an antibody against the mitochondrial marker cytochrome c. The result was a significant decrease in mitochondrial density in the neurites, but not in the cell bodies, of KI neurons (Fig. 8), indicating that PS1 mutations also significantly reduce mitochondrial transport into neuritic processes.
Figure 8.

Reduced mitochondrial density in neuritic processes of PS1 KI M146V hippocampal neurons. A–D, Immunofluorescence of PS1 WT and KI M146V (KI) hippocampal neurons stained with an antibody against cytochrome c. Merged images of phase contrast and fluorescence are shown. The gray scale in the images was inverted for clarity. A, B, Representative WT and KI hippocampal neurons fixed at 2 DIV. Note the presence of mitochondria in cell bodies and neuritic processes. C, D, Higher magnification images of neuritic processes. Note the presence of mitochondria along the processes. Scale bars: (in B) A, B, 10 μm; (in D) C, D, 5 μm. E, Assessment of mitochondrial density in neuritic processes and cell bodies of PS1 WT and KI hippocampal neurons. A significant decrease in mitochondrial density was observed in neuronal processes, but not in cell bodies, of KI neurons. PS1 WT and KI hippocampal neurons were fixed at day 2 in culture. To visualize mitochondria, we immunostained the cultures with a monoclonal antibody specific for cytochrome c (1:500), an electron-transporting mitochondrial resident protein. Mitochondrial density in neurites and cell bodies was assessed by image analysis. The images were captured at a final magnification of 400 ×, and the numbers of mitochondria were scored in the cell body and neuritic processes of randomly selected neurons as described in Materials and Methods. At least 20 cells were analyzed per experimental condition. The numbers represent the mean ± SE; n = 3 independent experiments. *p < 0.01 relative to WT by Student's t test.
Discussion
These experiments provide strong evidence for molecular and functional interactions between PS1 and GSK3β. Based on results with PS1 null and mutant models, PS1 appears to modulate the activity of GSK3β in neurons. The absence of both PS1 and PS1 mutations associated with FAD increases GSK3β kinase activity, leading to increased KLC phosphorylation and release of kinesin-I from MBOs. Consistent with increased GSK3β kinase activity, hyperphosphorylation of tau also is associated with PS1 mutations (Takashima et al., 1998; Pigino et al., 2001) and is a hallmark of Alzheimer's disease (AD). There is a precedent for proteins that bind GSK3β and limit its activity. Previous studies indicate that regulation of GSK3β activity can be mediated by proteins that bind GSK3β. For example, GPB/FRAT bind GSK3β in vivo and inhibit its kinase activity (Yost et al., 1998; Freemantle et al., 2002). Moreover, mutations in FRAT that lead to the deregulation of GSK3 have been associated with pathogenesis in leukemia (Yuan et al., 1999; Saitoh et al., 2001).
Survival and proper function of neurons depend on the efficient delivery of proteins from the cell body to axonal and dendritic processes. Kinesin-I (Morfini et al., 2002b), PS1, and the active form of GSK3β are enriched and colocalize in growth cones (Figs. 1, 2) where vesicle delivery and membrane insertion are essential for axonal elongation. This distribution is consistent with functional interactions among these proteins that are important in the transport of membrane proteins to neurites. Disruption of these functional interactions would be expected to compromise the efficient delivery of these cargoes. Axons in particular are highly susceptible to transport deficiencies because they lack the elements necessary for protein synthesis. In this context, defects in protein transport have been suggested to play a critical role in AD and other neurodegenerative conditions (de Waegh et al., 1992; Sheetz et al., 1998; Williamson and Cleveland, 1999; Goldstein, 2001; Morfini et al., 2002a), and several studies describe a progressive loss of motor function in AD patients (Sica et al., 1998; Goldman et al., 1999; Waite et al., 2000; Pettersson et al., 2002). A role for PS1 in protein trafficking was suggested initially by the analysis of protein metabolism in PS1-defficient cortical cultures (Naruse et al., 1998). Changes in membrane trafficking in PS1-/- neurons include accumulation of APP and APLP1 (a neuron-specific homolog of APP) C-terminal fragments and a dramatic slowdown in the transit and maturation of TrkB receptors. PS1 mutations also alter intracellular trafficking of β-catenin (Nishimura et al., 1999), leading to suggestions that PS1 is a component of the machinery involved in the selection and sorting of cargo exiting the ER. However, alterations in the delivery of membrane proteins are also consistent with changes in kinesin-based motility in PS1-/- neurons (see below), and these two possibilities are not mutually exclusive.
Impaired synaptic vesicle transport, reduced synaptic density, and altered synaptic function occur in various experimental systems with altered kinesin-I function. Synaptic function and density are affected specifically by kinesin-I mutants in Drosophila (Hurd and Saxton, 1996). Further, mutation in a kinesin-I gene has been associated with spastic paraplegia in humans (Reid et al., 2002).
A number of membrane proteins or MBOs exhibit altered distribution in the PS1 KIM146V and KO cells. Mutations in both PS1 and PS2 reduce levels of secreted APP in the conditioned media of cultured cells (Ancolio et al., 1997; Marambaud et al., 1998), further suggesting that alterations in protein transport are associated with the expression of presenilin mutations. PS1 has been found to undergo fast axonal transport and to accumulate in ligated sciatic nerves (Kasa et al., 2001), and axonal transport of APP is mediated by kinesin-I (Ferreira et al., 1993). Axonal trans port of APP is decreased greatly in a gene-targeted mouse mutant of the neuron-enriched kinesin light chain-1 gene (Kamal et al., 2000). Similarly, Kamal et al. (2001) reported the presence of PS1 in cargo vesicles containing APP, BACE, and kinesin-I. Thus APP and the secretases that generate amyloid β (Aβ) appear to be transported in the same vesicular structures. The decreased density of synaptic vesicles or their precursors observed in hippocampal neurons expressing PS1 mutations (Fig. 6) is consistent with a reduction in their transport. This possibility is favored by a marked decrease in the level of two synaptic proteins, synaptophysin and APP, in sciatic nerves of KIM146V mice. There was also a significant reduction in the levels of APP and synaptophysin in PS1 KIM146V mouse sciatic nerves, but not in the spinal cord, where neuronal cell bodies reside (Fig. 7).
Transport of other membrane proteins or organelles also may be altered by the inhibition of kinesin-I-based transport. Previous reports indicate that mitochondria are transported anterogradely by kinesin-I motors and that disruption of mouse KHC by gene targeting leads to decreased mitochondrial density in neuronal processes (Tanaka et al., 1998). Reduced transport of mitochondria may lead to depletion of ATP in axonal processes and synapses. In this regard, a number of studies have shown impaired energy metabolism in the AD brain (Fiskum et al., 1999; Hirai et al., 2001; Valla et al., 2001). Similarly, mitochondrial density was reduced in neuronal processes, but not cell bodies, of PS1 KIM146V neurons. Finally, a significant reduction was seen in the amounts of kinesin-I bound to membrane structures from neurons and fibroblasts from PS1 KIM146V mice. A model in which misregulation of GSK3β activity by PS1 mutations causes premature release of kinesin-I cargo or impaired transport of MBOs such as mitochondria could account for all of these changes (Morfini et al., 2002b). In the Alzheimer's brain an initial misregulation of transport might lead to a progressive increase in Aβ production and accumulation in the axon and, potentially, to a more generalized disruption and blockage of protein transport. Thus in AD various pathological processes involving PS1, APP, Aβ, and tau may converge to produce a deleterious effect on axonal transport and neuronal homeostasis.
Changes in kinase activities and phosphorylation patterns specifically implicate GSK3β in different pathological conditions (Mandelkow et al., 1992; Eldar-Finkelman et al., 1999; Summers et al., 1999). In particular, a role for GSK3β in AD pathology is suggested by the fact that GSK3β phosphorylates tau to produce AD-like immunoreactivity (Mandelkow et al., 1992; Mulot et al., 1994; Wagner et al., 1996) and that GSK3β expression levels are altered in the Alzheimer's brain (Baum et al., 1996). The discovery that GSK3β plays a role in regulating kinesin-based motility suggests that axonal transport may be a vulnerable step in AD pathogenesis (Morfini et al., 2002a).
Evidence that PS1 mutations produce a misregulation of GSK3β similar to the one observed in PS1-/- cells suggests that PS1 mutations are responsible for a loss of ability to modulate GSK3β. In this regard, all three PS1 mutations that were examined induced similar increases on GSK3β activity. In contrast, the effect of different FAD-linked PS1 mutations on γ-secretase activity appears to differ, promoting either increased Aβ42 production (Borchelt et al., 1996) or reduced Aβ generation (Amtul et al., 2002). Various experimental and clinical phenotypes associated with different PS1 mutations may be the cumulative result of altered PS1 function on multiple cellular pathways, including γ-secretase activity (Ebinu and Yankner, 2002), regulation of intracellular calcium levels (Mattson et al., 1998), regulation of cell cycle protein expression (Janicki et al., 2000), and kinesin-based transport.
In summary, these observations suggest that PS1 affects kinesin-based axonal transport, a phenomenon that correlates with local deregulation of GSK3β activity. Thus PS1 mutations may compromise neuronal function by altering kinesin-based transport mechanisms, including membrane protein transport in the secretory/endocytic pathways and fast anterograde axonal transport. The consequent reductions in efficiency of axonal transport in affected neurons would make them more vulnerable and ultimately lead to neurodegeneration.
Footnotes
This work was supported by grants from the Alzheimer's Association, The Patterson Trust, and the National Institutes of Health (HD38466) to J.B. We are grateful to Dr. Sam Sisodia (University of Chicago) for providing PS1 +/- breeders.
Correspondence should be addressed to Jorge Busciglio, Department of Neuroscience, University of Connecticut Health Center, 263 Farmington Avenue, Farmington, CT 06030. E-mail: busciglio@nso1.uchc.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/234499-10$15.00/0
References
- Amtul Z, Lewis PA, Piper S, Crook R, Baker M, Findlay K, Singleton A, Hogg M, Younkin L, Younkin SG, Hardy J, Hutton M, Boeve BF, Tang-Wai D, Golde TE ( 2002) A presenilin 1 mutation associated with familial frontotemporal dementia inhibits γ-secretase cleavage of APP and Notch. Neurobiol Dis 9: 269–273. [DOI] [PubMed] [Google Scholar]
- Ancolio K, Marambaud P, Dauch P, Checler F ( 1997) α-Secretase-derived product of β-amyloid precursor protein is decreased by presenilin 1 mutations linked to familial Alzheimer's disease. J Neurochem 69: 2494–2499. [DOI] [PubMed] [Google Scholar]
- Annaert WG, Levesque L, Craessaerts K, Dierinck I, Snellings G, Westaway D, George-Hyslop PS, Cordell B, Fraser P, De Strooper B ( 1999) Presenilin 1 controls γ-secretase processing of amyloid precursor protein in pre-Golgi compartments of hippocampal neurons. J Cell Biol 147: 277–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum L, Hansen L, Masliah E, Saitoh T ( 1996) Glycogen synthase kinase 3 alteration in Alzheimer disease is related to neurofibrillary tangle formation. Mol Chem Neuropathol 29: 253–261. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, Prada CM, Kim G, Seekins S, Yager D, Slunt HH, Wang R, Seeger M, Levey AI, Gandy SE, Copeland NG, Jenkins NA, Price DL, Younkin SG, Sisodia SS ( 1996) Familial Alzheimer's disease-linked presenilin 1 variants elevate Aβ1–42/1–40 ratio in vitro and in vivo Neuron 17: 1005–1013. [DOI] [PubMed] [Google Scholar]
- Busciglio J, Yankner BA ( 1995) Apoptosis and increased generation of reactive oxygen species in Down's syndrome neurons in vitro Nature 378: 776–779. [DOI] [PubMed] [Google Scholar]
- Busciglio J, Hartmann H, Lorenzo A, Wong C, Baumann K, Sommer B, Staufenbiel M, Yankner BA ( 1997) Neuronal localization of presenilin-1 and association with amyloid plaques and neurofibrillary tangles in Alzheimer's disease. J Neurosci 17: 5101–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busciglio J, Pelsman A, Wong C, Pigino G, Yuan M, Mori H, Yankner BA ( 2002) Altered metabolism of the amyloid β precursor protein is associated with mitochondrial dysfunction in Down's syndrome. Neuron 33: 677–688. [DOI] [PubMed] [Google Scholar]
- Capell A, Saffrich R, Olivo JC, Meyn L, Walter J, Grunberg J, Mathews P, Nixon R, Dotti C, Haass C ( 1997) Cellular expression and proteolytic processing of presenilin proteins is developmentally regulated during neuronal differentiation. J Neurochem 69: 2432–2440. [DOI] [PubMed] [Google Scholar]
- de Waegh SM, Lee VM, Brady ST ( 1992) Local modulation of neurofilament phosphorylation, axonal caliber, and slow axonal transport by myelinating Schwann cells. Cell 68: 451–463. [DOI] [PubMed] [Google Scholar]
- Doan A, Thinakaran G, Borchelt DR, Slunt HH, Ratovitsky T, Podlisny M, Selkoe DJ, Seeger M, Gandy SE, Price DL, Sisodia SS ( 1996) Protein topology of presenilin 1. Neuron 17: 1023–1030. [DOI] [PubMed] [Google Scholar]
- Ebinu JO, Yankner BA ( 2002) A RIP tide in neuronal signal transduction. Neuron 34: 499–502. [DOI] [PubMed] [Google Scholar]
- Eldar-Finkelman H, Schreyer SA, Shinohara MM, LeBoeuf RC, Krebs EG ( 1999) Increased glycogen synthase kinase-3 activity in diabetes- and obesity-prone C57BL/6J mice. Diabetes 48: 1662–1666. [DOI] [PubMed] [Google Scholar]
- Ferreira A, Niclas J, Vale RD, Banker G, Kosik KS ( 1992) Suppression of kinesin expression in cultured hippocampal neurons using antisense oligonucleotides. J Cell Biol 117: 595–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira A, Caceres A, Kosik KS ( 1993) Intraneuronal compartments of the amyloid precursor protein. J Neurosci 13: 3112–3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiskum G, Murphy AN, Beal MF ( 1999) Mitochondria in neurodegeneration: acute ischemia and chronic neurodegenerative diseases. J Cereb Blood Flow Metab 19: 351–369. [DOI] [PubMed] [Google Scholar]
- Frame S, Cohen P ( 2001) GSK3 takes centre stage more than 20 years after its discovery. Biochem J 359: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frame S, Cohen P, Biondi RM ( 2001) A common phosphate binding site explains the unique substrate specificity of GSK3 and its inactivation by phosphorylation. Mol Cell 7: 1321–1327. [DOI] [PubMed] [Google Scholar]
- Freemantle SJ, Portland HB, Ewings K, Dmitrovsky F, DiPetrillo K, Spinella MJ, Dmitrovsky E ( 2002) Characterization and tissue-specific expression of human GSK-3-binding proteins FRAT1 and FRAT2. Gene 291: 17–27. [DOI] [PubMed] [Google Scholar]
- Goldman WP, Baty JD, Buckles VD, Sahrmann S, Morris JC ( 1999) Motor dysfunction in mildly demented AD individuals without extrapyramidal signs. Neurology 53: 956–962. [DOI] [PubMed] [Google Scholar]
- Goldstein LS ( 2001) Kinesin molecular motors: transport pathways, receptors, and human disease. Proc Natl Acad Sci USA 98: 6999–7003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace E, Busciglio J ( 2003) Aberrant activation of focal adhesion proteins mediates fibrillar Aβ-induced neuronal dystrophy. J Neurosci 23: 493–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace E, Rabiner C, Busciglio J ( 2002) Characterization of neuronal dystrophy induced by fibrillar amyloid β: implications for Alzheimer's disease. Neuroscience 114: 265–273. [DOI] [PubMed] [Google Scholar]
- Guo Q, Fu W, Sopher BL, Miller MW, Ware CB, Martin GM, Mattson MP ( 1999) Increased vulnerability of hippocampal neurons to excitotoxic necrosis in presenilin-1 mutant knock-in mice. Nat Med 5: 101–106. [DOI] [PubMed] [Google Scholar]
- Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, Johnson AB, Kress Y, Vinters HV, Tabaton M, Shimohama S, Cash AD, Siedlak SL, Harris PL, Jones PK, Petersen RB, Perry G, Smith MA ( 2001) Mitochondrial abnormalities in Alzheimer's disease. J Neurosci 21: 3017–3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurd DD, Saxton WM ( 1996) Kinesin mutations cause motor neuron disease phenotypes by disrupting fast axonal transport in Drosophila Genetics 144: 1075–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janicki SM, Stabler SM, Monteiro MJ ( 2000) Familial Alzheimer's disease presenilin-1 mutants potentiate cell cycle arrest. Neurobiol Aging 21: 829–836. [DOI] [PubMed] [Google Scholar]
- Kamal A, Stokin GB, Yang Z, Xia CH, Goldstein LS ( 2000) Axonal transport of amyloid precursor protein is mediated by direct binding to the kinesin light chain subunit of kinesin-I. Neuron 28: 449–459. [DOI] [PubMed] [Google Scholar]
- Kamal A, Almenar-Queralt A, LeBlanc JF, Roberts EA, Goldstein LS ( 2001) Kinesin-mediated axonal transport of a membrane compartment containing β-secretase and presenilin-1 requires APP. Nature 414: 643–648. [DOI] [PubMed] [Google Scholar]
- Kasa P, Papp H, Pakaski M ( 2001) Presenilin-1 and its N-terminal and C-terminal fragments are transported in the sciatic nerve of rat. Brain Res 909: 159–169. [DOI] [PubMed] [Google Scholar]
- Kirschenbaum F, Hsu SC, Cordell B, McCarthy JV ( 2001) Glycogen synthase kinase-3β regulates presenilin 1 C-terminal fragment levels. J Biol Chem 276: 30701–30707. [DOI] [PubMed] [Google Scholar]
- Lah JJ, Heilman CJ, Nash NR, Rees HD, Yi H, Counts SE, Levey AI ( 1997) Light and electron microscopic localization of presenilin-1 in primate brain. J Neurosci 17: 1971–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, Yu CE, Jondro PD, Schmidt SD, Wang K ( 1995) Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science 269: 973–977. [DOI] [PubMed] [Google Scholar]
- Li X, Greenwald I ( 1998) Additional evidence for an eight-transmembrane-domain topology for Caenorhabditis elegans and human presenilins. Proc Natl Acad Sci USA 95: 7109–7114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelkow EM, Drewes G, Biernat J, Gustke N, Van Lint J, Vandenheede JR, Mandelkow E ( 1992) Glycogen synthase kinase-3 and the Alzheimer-like state of microtubule-associated protein tau. FEBS Lett 314: 315–321. [DOI] [PubMed] [Google Scholar]
- Marambaud P, Alves da Costa C, Ancolio K, Checler F ( 1998) Alzheimer's disease-linked mutation of presenilin 2 (N141I-PS2) drastically lowers APPα secretion: control by the proteasome. Biochem Biophys Res Commun 252: 134–138. [DOI] [PubMed] [Google Scholar]
- Marambaud P, Shioi J, Serban G, Georgakopoulos A, Sarner S, Nagy V, Baki L, Wen P, Efthimiopoulos S, Shao Z, Wisniewski T, Robakis NK ( 2002) A presenilin-1/γ-secretase cleavage releases the E-cadherin intracellular domain and regulates disassembly of adherens junctions. EMBO J 21: 1948–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Guo Q, Furukawa K, Pedersen WA ( 1998) Presenilins, the endoplasmic reticulum, and neuronal apoptosis in Alzheimer's disease. J Neurochem 70: 1–14. [DOI] [PubMed] [Google Scholar]
- Morfini G, Szebenyi G, Richards B, Brady ST ( 2001a) Regulation of kinesin: implications for neuronal development. Dev Neurosci 23: 364–376. [DOI] [PubMed] [Google Scholar]
- Morfini G, Tsai MY, Szebenyi G, Brady ST ( 2001b) Approaches to study interactions between kinesin motors and membranes. Methods Mol Biol 164: 147–162. [DOI] [PubMed] [Google Scholar]
- Morfini G, Pigino G, Beffert U, Busciglio J, Brady ST ( 2002a) Fast axonal transport misregulation and Alzheimer's disease. Neuromolecular Med 2: 89–100. [DOI] [PubMed] [Google Scholar]
- Morfini G, Szebenyi G, Elluru R, Ratner N, Brady ST ( 2002b) Glycogen synthase kinase-3 phosphorylates kinesin light chains and negatively regulates kinesin-based motility. EMBO J 21: 281–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulot SF, Hughes K, Woodgett JR, Anderton BH, Hanger DP ( 1994) PHF-tau from Alzheimer's brain comprises four species on SDS-PAGE that can be mimicked by in vitro phosphorylation of human brain tau by glycogen synthase kinase-3β. FEBS Lett 349: 359–364. [DOI] [PubMed] [Google Scholar]
- Naruse S, Thinakaran G, Luo JJ, Kusiak JW, Tomita T, Iwatsubo T, Qian X, Ginty DD, Price DL, Borchelt DR, Wong PC, Sisodia SS ( 1998) Effects of PS1 deficiency on membrane protein trafficking in neurons. Neuron 21: 1213–1221. [DOI] [PubMed] [Google Scholar]
- Nishimura M, Yu G, Levesque G, Zhang DM, Ruel L, Chen F, Milman P, Holmes E, Liang Y, Kawarai T, Jo E, Supala A, Rogaeva E, Xu DM, Janus C, Levesque L, Bi Q, Duthie M, Rozmahel R, Mattila K, Lannfelt L, West-away D, Mount HT, Woodgett J, St. George-Hyslop P ( 1999) Presenilin mutations associated with Alzheimer disease cause defective intracellular trafficking of β-catenin, a component of the presenilin protein complex. Nat Med 5: 164–169. [DOI] [PubMed] [Google Scholar]
- Okada Y, Yamazaki H, Aizawa Y, Hirokawa N ( 1995) The neuron-specific kinesin superfamily protein KIF1A is a unique monomeric motor for anterograde axonal transport of synaptic vesicle precursor. Cell 81: 769–780. [DOI] [PubMed] [Google Scholar]
- Pettersson AF, Engardt M, Wahlund LO ( 2002) Activity level and balance in subjects with mild Alzheimer's disease. Dement Geriatr Cogn Disord 13: 213–216. [DOI] [PubMed] [Google Scholar]
- Pfenninger KH, Ellis L, Johnson MP, Friedman LB, Somlo S ( 1983) Nerve growth cones isolated from fetal rat brain: subcellular fractionation and characterization. Cell 35: 573–584. [DOI] [PubMed] [Google Scholar]
- Pfister KK, Wagner MC, Stenoien DL, Brady ST, Bloom GS ( 1989) Monoclonal antibodies to kinesin heavy and light chains stain vesicle-like structures, but not microtubules, in cultured cells. J Cell Biol 108: 1453–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pigino G, Pelsman A, Mori H, Busciglio J ( 2001) Presenilin 1 mutations reduce cytoskeletal association, deregulate neurite growth, and potentiate neuronal dystrophy and tau phosphorylation. J Neurosci 21: 834–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleasure SJ, Page C, Lee VM ( 1992) Pure, postmitotic, polarized human neurons derived from NTera 2 cells provide a system for expressing exogenous proteins in terminally differentiated neurons. J Neurosci 12: 1802–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratner N, Bloom GS, Brady ST ( 1998) A role for cyclin-dependent kinase(s) in the modulation of fast anterograde axonal transport: effects defined by olomoucine and the APC tumor suppressor protein. J Neurosci 18: 7717–7726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid E, Kloos M, Ashley-Koch A, Hughes L, Bevan S, Svenson IK, Graham FL, Gaskell PC, Dearlove A, Pericak-Vance MA, Rubinsztein DC, Marchuk DA ( 2002) A kinesin heavy chain (KIF5A) mutation in hereditary spastic paraplegia (SPG10). Am J Hum Genet 71: 1189–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh T, Moriwaki J, Koike J, Takagi A, Miwa T, Shiokawa K, Katoh M ( 2001) Molecular cloning and characterization of FRAT2, encoding a positive regulator of the WNT signaling pathway. Biochem Biophys Res Commun 281: 815–820. [DOI] [PubMed] [Google Scholar]
- Sheetz MP, Pfister KK, Bulinski JC, Cotman CW ( 1998) Mechanisms of trafficking in axons and dendrites: implications for development and neurodegeneration. Prog Neurobiol 55: 577–594. [DOI] [PubMed] [Google Scholar]
- Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K ( 1995) Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature 375: 754–760. [DOI] [PubMed] [Google Scholar]
- Sica RE, Pereyra S, Mangone CA ( 1998) Loss of motor units in Alzheimer's disease. Electromyogr Clin Neurophysiol 38: 475–479. [PubMed] [Google Scholar]
- Stenoien DL, Brady ST ( 1997) Immunochemical analysis of kinesin light chain function. Mol Biol Cell 8: 675–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers SA, Kao AW, Kohn AD, Backus GS, Roth RA, Pessin JE, Birnbaum MJ ( 1999) The role of glycogen synthase kinase 3β in insulin-stimulated glucose metabolism. J Biol Chem 274: 17934–17940. [DOI] [PubMed] [Google Scholar]
- Taccioli GE, Amatucci AG, Beamish HJ, Gell D, Xiang XH, Torres Arzayus MI, Priestley A, Jackson SP, Marshak Rothstein A, Jeggo PA, Herrera VL ( 1998) Targeted disruption of the catalytic subunit of the DNA-PK gene in mice confers severe combined immunodeficiency and radiosensitivity. Immunity 9: 355–366. [DOI] [PubMed] [Google Scholar]
- Takashima A, Murayama M, Murayama O, Kohno T, Honda T, Yasutake K, Nihonmatsu N, Mercken M, Yamaguchi H, Sugihara S, Wolozin B ( 1998) Presenilin 1 associates with glycogen synthase kinase-3β and its substrate tau. Proc Natl Acad Sci USA 95: 9637–9641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Kanai Y, Okada Y, Nonaka S, Takeda S, Harada A, Hirokawa N ( 1998) Targeted disruption of mouse conventional kinesin heavy chain, KIF5B, results in abnormal perinuclear clustering of mitochondria. Cell 93: 1147–1158. [DOI] [PubMed] [Google Scholar]
- Tesco G, Tanzi RE ( 2000) GSK3β forms a tetrameric complex with endogenous PS1-CTF/NTF and β-catenin. Effects of the D257/D385A and FAD-linked mutations. Ann NY Acad Sci 920: 227–232. [DOI] [PubMed] [Google Scholar]
- Tsai MY, Morfini G, Szebenyi G, Brady ST ( 2000) Release of kinesin from vesicles by hsc70 and regulation of fast axonal transport. Mol Biol Cell 11: 2161–2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valla J, Berndt JD, Gonzalez-Lima F ( 2001) Energy hypometabolism in posterior cingulate cortex of Alzheimer's patients: superficial laminar cytochrome oxidase associated with disease duration. J Neurosci 21: 4923–4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner U, Utton M, Gallo JM, Miller CC ( 1996) Cellular phosphorylation of tau by GSK-3β influences tau binding to microtubules and microtubule organization. J Cell Sci 109: 1537–1543. [DOI] [PubMed] [Google Scholar]
- Waite LM, Broe GA, Grayson DA, Creasey H ( 2000) Motor function and disability in the dementias. Int J Geriatr Psychiatry 15: 897–903. [DOI] [PubMed] [Google Scholar]
- Wang QM, Fiol CJ, DePaoli-Roach AA, Roach PJ ( 1994) Glycogen synthase kinase-3β is a dual specificity kinase differentially regulated by tyrosine and serine/threonine phosphorylation. J Biol Chem 269: 14566–14574. [PubMed] [Google Scholar]
- Weihl CC, Ghadge GD, Kennedy SG, Hay N, Miller RJ, Roos RP ( 1999) Mutant presenilin-1 induces apoptosis and downregulates Akt/PKB. J Neurosci 19: 5360–5369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson TL, Cleveland DW ( 1999) Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat Neurosci 2: 50–56. [DOI] [PubMed] [Google Scholar]
- Wong PC, Zheng H, Chen H, Becher MW, Sirinathsinghji DJ, Trumbauer ME, Chen HY, Price DL, Van der Ploeg LH, Sisodia SS ( 1997) Presenilin 1 is required for Notch1 and DII1 expression in the paraxial mesoderm. Nature 387: 288–292. [DOI] [PubMed] [Google Scholar]
- Woodgett JR ( 1994) Regulation and functions of the glycogen synthase kinase-3 subfamily. Semin Cancer Biol 5: 269–275. [PubMed] [Google Scholar]
- Yonekawa Y, Harada A, Okada Y, Funakoshi T, Kanai Y, Takei Y, Terada S, Noda T, Hirokawa N ( 1998) Defect in synaptic vesicle precursor transport and neuronal cell death in KIF1A motor protein-deficient mice. J Cell Biol 141: 431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yost C, Farr 3rd GH, Pierce SB, Ferkey DM, Chen MM, Kimelman D ( 1998) GBP, an inhibitor of GSK-3, is implicated in Xenopus development and oncogenesis. Cell 93: 1031–1041. [DOI] [PubMed] [Google Scholar]
- Yuan H, Mao J, Li L, Wu D ( 1999) Suppression of glycogen synthase kinase activity is not sufficient for leukemia enhancer factor-1 activation. J Biol Chem 274: 30419–30423. [DOI] [PubMed] [Google Scholar]
- Zhao C, Takita J, Tanaka Y, Setou M, Nakagawa T, Takeda S, Yang HW, Terada S, Nakata T, Takei Y, Saito M, Tsuji S, Hayashi Y, Hirokawa N ( 2001) Charcot–Marie–Tooth disease type 2A caused by mutation in a microtubule motor KIF1Bβ. Cell 105: 587–597. [DOI] [PubMed] [Google Scholar]