

Abstract
Experience-dependent plasticity can be induced in the barrel cortex by removing all but one whisker, leading to potentiation of the neuronal response to the spared whisker. To determine whether this form of potentiation depends on synaptic plasticity, we studied long-term potentiation (LTP) and sensory-evoked potentials in the barrel cortex of α-calcium/calmodulin-dependent protein kinase II (αCaMKII)T286A mutant mice. We studied three different forms of LTP induction: theta-burst stimulation, spike pairing, and postsynaptic depolarization paired with low-frequency presynaptic stimulation. None of these protocols produced LTP in αCaMKIIT286A mutant mice, although all three were successful in wild-type mice. To study synaptic plasticity in vivo, we measured sensory-evoked potentials in the barrel cortex and found that single-whisker experience selectively potentiated synaptic responses evoked by sensory stimulation of the spared whisker in wild types but not in αCaMKIIT286A mice. These results demonstrate that αCaMKII autophosphorylation is required for synaptic plasticity in the neocortex, whether induced by a variety of LTP induction paradigms or by manipulation of sensory experience, thereby strengthening the case that the two forms of plasticity are related.
Keywords: barrels, whiskers, LTP, mouse, synapse, plasticity
Introduction
Sensory experience can have a profound effect on neurons in the neocortex. In the auditory cortex, experience alters the width of receptive field tuning (Kilgard et al., 2001), whereas in the visual cortex, cells are able to alter their occular-dominance and orientation tuning during a critical period of development (Blakemore and Cooper, 1970; Hubel and Wiesel, 1970). In the somatosensory cortex, cells are able to alter their responses to stimulation of specific digits or whiskers, both during development and into adulthood (Simons and Land, 1987; Wall, 1988; Glazewski and Fox, 1996). These experience-dependent changes have been measured by observing spike firing rate but probably rely on the underlying synaptic plasticity of the cortical excitatory neurons. Evidence to support this idea comes from experiments showing that α-calcium/calmodulin-dependent protein kinase II (αCaMKII) and αCaMKII autophosphorylation is required for experience-dependent plasticity (Glazewski et al., 1996, 2000; Gordon et al., 1996; Taha et al., 2002) as well as synaptic plasticity (Silva et al., 1992; Giese et al., 1998).
However, studies showing that αCaMKII autophosphorylation is required for experience-dependent plasticity in the neocortex are thus far only based on recordings from sensory-evoked extracellular action potentials (Glazewski et al., 2000; Taha et al., 2002). Theoretically, αCaMKII autophosphorylation could affect spike activity by altering membrane potential, spike threshold, or accommodation, or affect coupling of synaptic potentials with somatic potentials by altering dendritic voltage-gated channels, all without directly affecting synaptic plasticity. Similarly, evidence that αCaMKII autophosphorylation plays a role in synaptic plasticity is based on recordings in the Schaeffer collateral–CA1 pathway in the hippocampus (Giese et al., 1998), and it is not known whether plasticity mechanisms are identical between neocortical pathways and the Schaeffer collateral–CA1 pathway. Indeed, even hippocampal pathways differ in their requirement for CaMKII autophosphorylation (Errington et al., 2002).
Therefore, to test whether neocortical synaptic plasticity underlies neocortical experience-dependent plasticity, we undertook two types of experiments in the barrel cortex. First, we tested the idea that long-term potentiation (LTP) in the barrel cortex might depend on αCaMKII autophosphorylation, using αCaMKIIT286A point mutant mice (Giese et al., 1998). We studied three different LTP induction paradigms, because it was not clear which paradigm might be most relevant to the natural plasticity induction processes in the barrel cortex. We studied the pathway from layer IV to layer II/III of the near neighboring barrel because this was the most direct pathway that could be involved in the in vivo plasticity, although not the only one (Fox, 2002). Second, we used current source density analysis to measure changes in sensory-evoked dendritic currents associated with experience-dependent plasticity in the barrel cortex. In this way, we were able to measure changes more closely related to sensory-evoked synaptic potentials than was able to be achieved previously with spike recording. Third, we tested whether experience-dependent plasticity was dependent on αCaMKII autophosphorylation in the cortex. The results demonstrate that both LTP and experience-dependent plasticity require αCaMKII autophosphorylation in the barrel cortex and suggest that synaptic plasticity at excitatory pathways plays a major role in the expression of experience-dependent plasticity.
Materials and Methods
Subjects. For Western blot analysis, subjects were three wild-type and three mutant mice. For LTP studies, subjects were male and female mice that were 4–6 weeks of age. A group of 30 wild types and 28 mutants yielded 46 wild-type slices of barrel cortex and 62 slices from the mutants. All animals were studied blind to the genotype.
For current source density studies, subjects were male and female mice, 3–5 months of age. A group of 35 animals was used, including 10 deprived and 7 normally reared homozygotes and 11 deprived and 7 normally reared wild types. All recordings were done blind to the genotype. Animals were genotyped by PCR as described previously (Giese et al., 1998). The animals studied were the F3 and F4 generations of mice in which the original generation was of a hybrid 129/Sv × C57BL/6 background. Only heterozygotes were bred (cousin matings).
Western blot analysis. Brain tissues from the somatosensory cortex and hippocampus were isolated from adult mice. Protein extracts were prepared using 2% SDS, and the protein concentrations were determined with the BCA assay (Pierce, Rockford, IL). Ten micrograms of each sample were separated on 4–15% SDS-PAGE (Bio-Rad, Hercules, CA) and transferred onto polyvinylidene difluoride membranes (Bio-Rad). Antibodies against αCaMKII (monoclonal antibody 6G9; Chemicon, Temecula, CA), β-actin (Clone AC-15; Sigma, St. Louis, MO), and synaptotagmin (rabbit; Sigma) were used. After incubation with a peroxidase-conjugated antibody (Pierce), immunoreactive bands were visualized using a chemiluminescent reagent (SuperSignal West Pico Chemiluminescent Substrate; Pierce) and analyzed by Quantity One (Bio-Rad). After visualization, membranes were washed in stripping buffer (Restore Western Blot Stripping Buffer; Bio-Rad) and reused.
Electrodes and solutions for in vitro recording. In different experiments, field recording, current clamp, and voltage clamp were used. In all cases, the stimulating electrode was placed accurately within the wall of a layer IV barrel under visual guidance using an Olympus Optical (Tokyo, Japan) BH2 video microscope and a transilluminated slice. For extracellular recording, the recording electrode was positioned in the middle of layers II/III on the nearside of the adjacent barrel column. For intracellular recording, cells were chosen within that same area and patched under visual guidance using a 40× water immersion objective, differential interference contrast optics, and infrared illumination.
Slices were produced by conventional means. The slices were maintained in a submersion chamber continually perfused (2–3 ml/min) with artificial CSF containing (in mm): 119 NaCl, 3.5 KCl, 1 NaH2PO4, 2 CaCl2, 1 MgSO4, 26 NaHCO3, and 10 glucose. The solution was bubbled with 5% CO2–95% O2 and kept at room temperature (21–24°C). The extracellular recording electrode was a carbon fiber microelectrode with the tip etched to 25–40 μm. The intracellular electrodes were 7–10 MΩ and filled with (in mm): 110 K-gluconate, 10 KCl, 2 MgCl2, 2 Na2ATP, 0.03 Na2GTP, 10 HEPES, pH 7.3, and 270 mOsm.
In vitro recording and LTP induction protocols. Whole-cell recordings of synaptic responses in layer II/III pyramidal neurons were taken at the postbreak in resting membrane potential (mean ± SD, 69 ± 4 mV) for current-clamp experiments and at -65 mV for the voltage-clamp experiments. Series resistance was monitored, and the recording was discarded if it changed by >20% during the course of the experiment. Monosynaptic components of the EPSPs or EPSCs were monitored and found to have reversal potentials close to 0 mV for wild types (mean ± SD, 1.8 ± 12.3 mV) and T286A mutants (4.8 ± 9 mV), which were not different (t(19) = 0.38; p > 0.5).
Theta-burst stimulation consisted of groups of four 200 μsec current pulses at 100 Hz repeated at 5 Hz for 10 cycles as described previously (Kirkwood et al., 1993). Field EPSPs were measured in responses to low-frequency (1 per 20 sec) stimulation, low-pass filtered at 10 kHz, and recorded using custom programs generated with LabVIEW software (National Instruments, Austin TX).
Two pairing protocols were used. In both cases, the presynaptic stimulation rate was increased to 2 Hz. With depolarization pairing, current was injected to move the cell to a positive potential (∼0 mV) for 1 min. With spike pairing, a brief 10 msec current pulse was injected, sufficient to produce a postsynaptic spike at the end of the naturally evoked EPSP. The interval between presynaptic and postsynaptic spikes was therefore controlled at 10 msec, and the condition was repeated 120 times.
Whisker deprivation. To induce plasticity, the D1 vibrissa was spared for 18 d, whereas all surrounding vibrissas were deprived every second day. Deprived vibrissas were allowed to regrow for 8–10 d before the recording session. The deprivation technique used was found to not affect vibrissa innervation (Li et al., 1995) and has been described in detail previously (Glazewski et al., 1998).
Anesthesia and surgery. Anesthesia was induced with fluothane (Zeneca, Cheshire, UK) and maintained with intraperitoneal injections of urethane (1.5 gm/kg whole body weight; Sigma). Depth of anesthesia was monitored by testing hindlimb reflexes and monitoring the EEG. Animals were reinjected with 10% of the initial dose of urethane if the hindlimb reflex was brisk. The skull overlying the barrel cortex was thinned by careful drilling. A small hole just large enough for the electrode to pass through was made in the skull before each electrode penetration using a hypodermic needle. All experiments complied with the United Kingdom Animals (Scientific Procedures) Act of 1986.
Electrodes, stimulus, and recording. Glass-insulated carbon fiber microelectrodes were used to record from the cortex (Armstrong-James et al., 1980). Vibrissas were stimulated at 0.1 Hz using a computer-controlled piezoelectric stimulator driven by a voltage source (DS-2; Digitimer, Welwyn Garden, UK). All recordings were made from layers II–V. Usually one electrode penetration was made per animal. The neurons were sampled every 50 μm through the depth of the penetration (-50 to 700 μm). Ten waveforms were averaged for each recording site. Principal and intact (D1) whiskers and one surround-deprived whisker were stimulated at each depth. After advancing the electrode 50 μm, a 10 min pause enabled the recording to stabilize. The stimulus intensity was normalized by adjusting it to 50–60% of the maximal principal whisker response within layer IV (300 μm below the pia) and was kept constant afterward throughout the penetration. The test pulse was generated using Spike2 software (Cambridge Electronic Design, Cambridge, UK). Field potentials [sensory-evoked potentials (SEPs)] were recorded using the Neurolog system (Digitimer) and filtered between 0.1 and 1.5 kHz without a 50 Hz notch filter. Waveforms were amplified 500 times and digitized. Waveforms were monitored, acquired, and averaged using in-house programs written in LabVIEW software (National Instruments).
Current source density analysis. Current source density (CSD) was calculated using methods described previously (Mitzdorf and Singer, 1978; Mitzdorf, 1985) from averaged field potentials as follows:
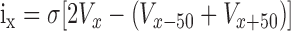 |
where ix and Vx are the current and voltage at depth x μm below the pial surface, Vx - 50 is the depth 50 μm above depth x, and σ is the volume conductance. Gray-scale images of the CSD profiles were made using NIH Image as described previously (Aizenman et al., 1996). The maximum and minimum of the scale were determined by the largest peak current recorded in this series of experiments and were symmetrical at ∼0. The maximum layer II/III sink resulting from “spared” whisker stimulation was identified for each penetration and the spared- and principal-whisker SEPs analyzed for that depth.
Histology. After every experiment, the animal was deeply anesthetized with urethane and perfused through the heart with 100 mm PBS, followed by PBS containing 4% paraformaldehyde and increasing concentrations of sucrose. The brain was removed and the cortex was flattened, as described previously (Strominger and Woolsey, 1987), and left overnight in 20% sucrose in buffered solution of formaldehyde. Sections of 40 μm were cut tangentially to the surface of flattened cortex and the tissue reacted for cytochrome oxidase (Wong-Riley, 1979). Recording locations were identified from focal lesions (1 μA, 10 sec; tip negative) made at the end of each penetration within the cytochrome oxidase-stained sections of the barrel field.
Statistical analysis. For the in vitro experiments, individual time-course recordings were normalized and averaged within groups as a series of 10 min epochs. ANOVAs were used to test for effects of and interactions between genotype, time, and tetanus protocol. Post hoc repeated-measures t tests were used to find the source of effects and interaction terms.
For the in vivo experiments, the ratio of spared and principal whisker response was calculated for each penetration. Population averages were calculated for spared whisker response, principal whisker response, and the ratio of the two. The averages of these populations were analyzed by ANOVA and, where applicable, post hoc t tests were used.
Results
αCaMKII protein levels in the somatosensory cortex of αCaMKIIT286A mice
Recent studies have shown that αCaMKII levels are reduced in the visual cortex of αCaMKIIT286A point mutants (Taha et al., 2002). Therefore, we measured αCaMKII protein levels in the somatosensory cortex and hippocampus of the same animals. The expression of αCaMKII in mutants and wild types was normalized to the expression of β-actin in each animal to minimize errors in protein loading and transfer (Fig. 1). In somatosensory cortex, αCaMKII expression was significantly reduced by 25% in the mutants (F(1,4) = 13.7; p < 0.05). In contrast, the levels of αCaMKII in the hippocampus of the same mutant mice were not different from those in wild types (F(1,4) = 0.002; p = 0.96). Therefore, the results show that αCaMKII levels are slightly reduced in a region-specific manner in the cortex of αCaMKIIT286A point mutant mice.
Figure 1.

Immunoblot analysis of αCaMKII expression in the somatosensory cortex (Sm Cx) and hippocampus (Hi) in αCaMKII T286A mutants (T286A; n = 3) and wild-type littermates (wt; n = 3). The samples from three mutants (t286A-1, t286A-2, and t286A-3) and three wild-type littermates (wt-1, wt-2, and wt-3) are shown. A, The immunoblots were used first for detecting αCaMKII expression, and, after stripping of the blots, β-actin expression was determined as a standard for normalization. B, Expression in the somatosensory cortex was quantified and plotted in reference to expression in wild-type littermates (white bars). There was only a trend to suggest that αCaMKII expression was reduced in the mutants (F(1,4) = 6.29; p = 0.066), whereas αCaMKII expression normalized to β-actin expression was significantly reduced by 25% (F(1,4) = 13.7; p < 0.05) C, Expression in the hippocampus was quantified and plotted in reference to expression in wild-type littermates (white bars). The αCaMKII expression in the mutants was significantly reduced by 17% (F(1,4) = 9.09; p < 0.05), but this reduction could not be confirmed when the αCaMKII expression was normalized to β-actin expression (F(1,4) = 0.002; p = 0.96). Error bars depict SEMs.
αCaMKII autophosphorylation is required for theta-burst-induced cortical LTP
To study the role of αCaMKII autophosphorylation in cortical LTP, we recorded extracellular field potentials in layers II/III of the barrel cortex in slices prepared from the αCaMKIIT286A mice and their wild-type littermates. To compare our results with previous studies, we used a theta-burst stimulation protocol, which has been shown previously to induce NMDA-dependent LTP in visual cortex (Kirkwood et al., 1993). When the stimulating electrodes were placed in the wall of the barrel under visual guidance and the recording electrode in layers II/III of the neighboring barrel (see Materials and Methods), we were able to evoke LTP (of at least 15%) in all slices derived from wild-type mice.
To test whether the method evoked NMDA-dependent LTP in the barrel cortex, we applied 25 μm d-AP-5 to the slice 10 min before theta-burst stimulation. The peak did not increase significantly above baseline after tetanus (Fig. 2A) (average ± SEM, 104.9 ± 2.5%; p > 0.07; n = 10; t test). In contrast, a second tetanus applied to the same slices 1 hr after the washout produced LTP that persisted for at least 1 additional hour (Fig. 2A) (121.3 ± 5.8%; p < 0.02; n = 10; t test). This result shows that theta-burst stimulation produces NMDA-receptor-dependent LTP in the layer IV to II/III pathway between columns in the barrel cortex.
Figure 2.

Dependence of barrel cortex LTP on NMDA receptors and αCaMKII autophosphorylation. A, Time plots of average extracellular field potentials evoked in the middle of layer II/III by stimulation of layer IV in the nearside barrel. Theta-burst tetanization (leftmost arrow) in the presence of 25 μm d-AP-5 did not induce potentiation. However, when the AP-5 was washed out of the bath and a second tetanus was presented (rightmost arrow), LTP was induced and lasted for at least 1 hr. The dashed line indicates the baseline 15 min before the first (100%) and second (104%) tetanus. B, Theta-burst stimulation was applied (arrow) to slices derived from αCaMKII T286A mice (black circles) or their wild-type littermates (white circles). LTP was not induced in the mutants but was induced in the wild-type mice. EPSP peaks are shown for each stimulus once every 20 sec. Error bars are SEs calculated for the preceding 1 min. fEPSP, Field EPSP.
To see whether this form of LTP was also dependent on αCaMKII autophosphorylation, we applied the same protocol to barrel cortical slices prepared from 15 homozygous αCaMKIIT286A mice and 10 wild-type littermates. The mutation prevented LTP, as can be seen in Figure 2B. The response increased to 126.4 ± 6.1% of baseline at 1 hr in wild-type mice but only 104.4 ± 4.5% in the mutants. Analysis revealed a significant main effect of time (F(6,14) = 7.76; p < 0.0005) and genotype (F(1,19) = 14.73; p < 0.002) and an interaction between time and genotype (F(6,14) = 4.41; p < 0.02). Post hoc tests showed that all time points were significantly different between genotypes except for the first two 10 min epochs after tetanization, indicating that LTP induced by theta-burst stimuli rises slowly in the wild types and not at all in the mutants. In five mutants and four wild-type mice, LTP was followed for 1 additional hour. The degree of potentiation was equivalent at 2 hr to that shown at 1 hr in the wild-type mice (140.9 ± 4.9%), whereas no LTP was seen at all in the mutants (91.1 ± 8.2%; data not shown). In conclusion, LTP induced by theta-burst stimulation requires αCaMKII autophosphorylation in layers II/III of the neocortex.
αCaMKII is required for pairing-induced LTP
After pretetanus baseline recording (at the resting membrane potential), LTP was induced by injecting sufficient current to depolarize the cell to ∼0 mV for 1 min while delivering afferent presynaptic stimulation at 2 Hz (depolarization protocol) or by injecting a brief 10 msec current pulse sufficient to induce a postsynaptic action potential directly after the presynaptic stimulus (spike-pairing protocol). Postsynaptic spikes were absent with the depolarization protocol and present with the spike-pairing protocol.
The T286A mutation was again found to prevent LTP, whether induced by spike pairing or the depolarization protocol. Statistical analysis revealed an effect of genotype (F(1,35) = 9.02; p < 0.005) but not of the stimulation protocol (i.e., spike pairing vs depolarization pairing) (F(1,35) = 1.81; p < 0.19) nor any interaction between the two (F(1,35) = 0.29; p = 0.59). We also found an interaction of time and genotype (F(6,30) = 2.49; p < 0.05). As can be seen in Figure 3, this interaction is probably caused by the faster decay of the potentiated state in the αCaMKIIT286A mice compared with wild-type mice. Post hoc tests showed that whereas the wild-type EPSPs remained potentiated at 60 min, the EPSPs decayed back to baseline after 20 min in the mutants (Fig. 3).
Figure 3.

The effect of αCaMKII autophosphorylation on pairing-induced potentiation of cortical EPSPs. Top, Example traces are shown of EPSPs (average of 10 responses) recorded before and after depolarization pairing for cells recorded from a wild-type and αCaMKII T286A homozygote mouse. Immediately after induction, both genotypes show potentiation (0–10 min; middle traces), but only the wild type shows potentiation at 60 min in this case (right traces). Calibration: 2 mV, 50 msec. Bottom, The normalized peak EPSP amplitude is averaged across cells and plotted against time for layer II/III pyramidal cells in response to stimulation of the nearside edge of the layer IV barrel. Averaged normalized EPSPs are represented for 20 cells in the mutants and nine cells in the wild types. Although pairing depolarization with 2 Hz of presynaptic stimulation or pairing a postsynaptic spike 10 msec after the presynaptic stimulus (arrow) induced rapid potentiation in both wild types (white circles) and αCaMKII T286A mutants (black circles), the potentiation decayed in the mutants with in 20 min, where as in the wild types, potentiation was present for at least 1 hr. The dashed line indicates the baseline 15 min before tetanus (100%). Error bars are SE calculated for the preceding 2 min.
Similar results were found in cases in which EPSCs were measured. Layer IV stimulation evoked EPSCs that were recorded in layer II/III cells at a holding potential close to -65 mV. Sustained depolarization to 0 mV for 1 min during 1 Hz presynaptic stimulation produced LTP in wild types but potentiation that decayed to baseline by 40 min in the mutants (Fig. 4). Statistical analysis showed an effect of genotype (F(1,49) = 23.55; p < 0.0001) and time (F(4,46) = 1.73; p < 0.0001) and an interaction between the two factors (F(4,46) = 0.65; p < 0.0001). Post hoc t tests showed that this was because the wild types potentiated at all times after pairing, whereas the αCaMKIIT286A mice showed potentiation at 30 min but not at 40 min after pairing.
Figure 4.

The effect of αCaMKII autophosphorylation on pairing-induced potentiation of cortical EPSCs. Top, Example traces of EPSCs recorded before and 40 min after pairing presynaptic and postsynaptic depolarization for 1 min in a wild-type and an αCaMKII T286A homozygote mouse. Calibration: 100 pA, 10 msec. Bottom, Averaged normalized EPSCs recorded from 31 cells in wild types (white circles) and 31 cells in CaMKII T286A homozygotes (black circles). Pairing presynaptic stimulation at 1 Hz with constant depolarization causes sustained potentiation in wild-type neurons; EPSCs show a slowly decaying level of potentiation that is statistically indistinguishable from baseline at 40 min. The dashed line indicates the baseline 10 min before tetanus (100%). Error bars are SE calculated for the preceding 2–5 min.
These results demonstrate a fundamental deficit in the mechanisms of LTP induction in αCaMKIIT286A mice that is independent of tetanus protocol or whether field EPSPs, EPSPs, or EPSCs are measured (Fig. 5).
Figure 5.

Comparison of the effect of LTP induction protocol and genotype on potentiation. LTP induced by theta-burst potentiation (θ-burst) produced levels of potentiation equivalent to those obtained by spike pairing and depolarization pairing (Depol. pairing) protocols in wild-type mice (white bars). Potentiation was greater in voltage-clamped neurons than in the other cases (141 vs 121–126%). In contrast, none of the four protocols produced LTP in the αCaMKIIT286A mutants. The bars represent averages taken between 50–60 min after tetanus or pairing, except for the EPSCs, which are measured at 30–40 min after pairing. fEPSP, Field EPSP. Error bars represent SEs.
Whisker deprivation causes potentiation of sensory-evoked potentials
To test whether SEPs are enhanced in the barrel cortex of wild-type mice after whisker stimulation, we stimulated a single whisker while recording the SEP at a known depth within the cortex (Fig. 6). By averaging SEPs at each depth and measuring them every 50 μm, it was possible to construct a CSD profile of SEPs and currents within an electrode penetration between the cortical surface and layer V. The maximum amplitude of the CSDs decreased as a function of distance from the principal barrel of the stimulated whisker. We therefore compared CSDs in deprived and undeprived mice at similar distances from the barrel of the spared whisker (Fig. 7). The distances were measured from microlesions made at the end of recording in each penetration visualized in the cytochrome oxidase histology of the barrel field. Even without normalizing the currents across animals, it was clear from the CSD profiles that larger currents were evoked by the spared whisker after a period of 18 d of single-whisker experience compared with currents evoked by the same whisker in undeprived animals. We found that currents were greater in all layers after deprivation. The earliest currents after stimulation of the spared whisker occurred in layers II/III, followed by layer IV and V.
Figure 6.

The effect of whisker deprivation on SEPs in the barrel cortex of wild-type (WT) and homozygous αCaMKII T286A mice. Left column, SEPs were recorded in the D2 barrel-column for a rapid deflection of the D2 whisker (dashed line) or the D1 whisker (solid line). In a normal animal, the amplitude of the potential is smaller for the surround receptive-field whisker (D1 in this case) than for the principal whisker (D2 in this case) at most depths. Middle column, Whisker deprivation increases the amplitude of the spared whisker SEPs so that they were at least as great as those evoked by the principal whisker. Right column, Despite a period of single-whisker experience, the spared (D1) whisker (solid line) shows a far smaller response than the principal whisker in homozygous αCaMKII T286A mice (dashed line). Calibration: 3 mV, 50 msec. Note that the potentials at intermediate depths are not shown for clarity (i.e., those at -50, 50, 150 μm, etc.).
Figure 7.

CSD analysis of SEPs in wild-type mice. CSD is plotted for penetrations made in the barrel neighboring D1. Currents were evoked by stimulating the D1 whisker and calculated from field potentials measured every 50 μm. Responses are normalized across all cases and plotted on a gray scale in which the maximum sink is represented by white, the maximum source by black, and zero by mid-gray. Examples are shown at various distances from the D1 barrel penetration as indicated by the insets, in which the white circle represents the location of the penetration. Top row, The earliest sink can be seen in superficial layers and later in layer IV in animals deprived of all but the D1 whisker. The amplitude of the sinks and sources decreases with distance from the D1 barrel. Bottom row, The equivalent CSD analysis in animals that have not undergone whisker deprivation. Cases were taken for identical D1 stimulation at distances from the D1 barrel matched as closely as possible to those shown on the top row. The sinks and sources evoked by D1 stimulation were far weaker in undeprived animals.
αCaMKII autophosphorylation is necessary for potentiation of SEPs
To test whether αCaMKII autophosphorylation was required for potentiation of SEPs, we repeated the deprivation study in homozygous αCaMKIIT286A mice and their wild-type littermates. The CSD profiles indicated a lack of plasticity in the mutants, because they were unchanged between the deprived and undeprived mutant animals (Fig. 8). To quantify the data, we identified the locus of the largest superficial layer II/III sink for each penetration and compared the SEP amplitude for the principal whisker (which was always a deprived whisker for the deprived mice) for each penetration and the spared whisker (Fig. 6).
Figure 8.

Whisker-deprivation plasticity depends on autophosphorylation of αCaMKII as assessed by CSD analysis. CSD is plotted on a gray scale as described in Figure 7. The sinks (white) and sources (black) evoked by stimulating the spared whisker (D1) were weak in both deprived and undeprived αCaMKII T286A mutants. Penetrations were chosen for matched distances from the D1 barrel. The insets show the position of the penetration relative to the D1 barrel (white circle). Top row, Deprived animals. Bottom row, Undeprived animals.
In wild-type mice, the spared-whisker SEP increased to the same amplitude or greater than the principal-whisker SEP. The increase in the ratio of spared-whisker to principal-whisker SEP, from 0.45 to 1.64 (averaged across the barrel neighboring the spared barrel), in the wild-type mice did not occur in the αCaMKIIT286A mice (0.60 in undeprived mice vs 0.74 in deprived mice). Statistical analysis showed that the ratio of the spared-to the principal-whisker response was affected by deprivation (F(1,33) = 2.9; p < 0.0005) and by genotype (F(1,33) = 4.5; p < 0.05), and that there was an interaction between genotype and deprivation (F(1,33) = 1.8; p < 0.005). The interaction was caused by an increase in the spared-whisker-evoked SEPs that was not observed in the αCaMKIIT286A animals (Fig. 9). These findings therefore show that experience-dependent synaptic potentiation normally occurs in the mouse barrel cortex, and that this plasticity requires autophosphorylation of αCaMKII.
Figure 9.

Quantification of SEPs as a function of distance from the principal barrel of the spared whisker. The ratio of the spared to principal whisker sensory-evoked potentials is plotted as a function of distance from the edge of the barrel of the spared whisker. Each point represents a ratio of time-averaged responses for the maximum layer II/III sink in each penetration. The black bar beneath the x-axis indicates the approximate extents of the neighboring barrel. Top left, In undeprived wild-type mice, the ratio decreases with increasing distance from the D1 barrel. The average ratio of the D1 to the principal whisker-evoked potential between 50 and 250 μm from the edge of the D1 barrel is shown by the horizontal dashed line. Top right, After a period of single-whisker experience, the spared whisker evoked a far greater response in the neighboring barrel. Bottom left, Response ratios decreased with distance from the D1 barrel in homozygous αCaMKII T286A mice, just as in wild-type mice. Bottom right, A period of single-whisker experience does not lead to an increase in the ratio of the D1 to principal whisker response in the mutants.
Discussion
The relationship between an artificially induced synaptic plasticity mechanism such as LTP and naturally induced potentiation is not easy to prove. The advantage of studying the relationship in the somatosensory cortex is that the form of potentiation is very similar in the two cases. Sensory-evoked potentials are enhanced in layer II/III of the neighboring cortical column after single-whisker experience, and a similar form of potentiation can be induced by theta-burst stimulation in the IV to II/III pathway between columns in slices (this study) or in vivo (Glazewski et al., 1998). From a conservative viewpoint, we can only say that we have not disproved that the two phenomena are different. However, LTP and experience-dependent potentiation do show a remarkably detailed level of dependence on the same synaptic mechanism, even to the level of requiring the same αCaMKII phosphorylation state.
We found that total αCaMKII levels were lower in the barrel cortex of T286A mutants than wild types. However, the effect of the T286A mutation appears to depend on the inability of αCaMKII to autophosphorylate rather than a reduction in αCaMKII levels. In the visual cortex, the αCaMKIIT286A mice show a 50% decrease in αCaMKII (Taha et al., 2002). A decrease in total αCaMKII of 50% in the somatosensory cortex does cause a significant decrease in cortical plasticity, as observed in the heterozygous αCaMKII knock-out mice (Glazewski et al., 1996). However, the inhibition of plasticity in the T286A point mutant, which has a 25% reduction in αCaMKII, is far more severe than that of the αCaMKII heterozygote, which has a larger reduction in αCaMKII. Therefore, the greater effect on plasticity in the point mutant must be attributable to the dependence of plasticity on autophosphorylation at the T286 site.
Barrel cortex LTP
The results presented here show for the first time that neocortical LTP is dependent on autophosphorylation of αCaMKII. Regardless of whether LTP is induced by theta-burst stimulation, pairing presynaptic and postsynaptic depolarization, by sustained depolarization, or by spike pairing, LTP is absent in αCaMKIIT286A mice. The fact that the same conclusion is reached regardless of the LTP induction protocol used increases confidence that αCaMKII autophosphorylation is a basic requirement for induction of LTP in the neocortex rather than a permissive or marginal factor. The fact that low-frequency pairing of presynaptic activity with forced postsynaptic depolarization fails to induce LTP in the mutants shows that the mutation does not affect LTP by altering the transmission properties of the circuit.
Previous studies using knock-out mice had concluded that αCaMKII is required for hippocampal LTP (Silva et al., 1992). The same point mutation in αCaMKII studied here has also been shown to almost completely block LTP in the Schaeffer collateral–CA1 pathway of the hippocampus (Giese et al., 1998), where the residual potentiation is attributed to an NMDA-independent component. However, LTP at the perforant path-dentate gyrus pathway shows no requirement for αCaMKII autophosphorylation (Errington et al., 2002). The diversity of synaptic plasticity mechanisms within the hippocampus and across other brain regions argues against assuming that plasticity mechanisms discovered in CA1 will necessarily be used in other cortical areas. However, our results suggest that the plasticity exhibited in the layer IV to II/III pathway of the cortex has relatively similar properties to that of CA1, as suggested previously (Kirkwood et al., 1993).
We studied the effects of spike-pairing as a more natural form of LTP induction that is likely to contribute to endogenous plasticity in the cortex (Feldman, 2000). Action potentials are likely to back-propagate in the relatively short apical dendrites of mouse layer II/III pyramidal cells (Golding et al., 2002), are naturally evoked by sensory stimuli (Larkum and Zhu, 2002), are also known to affect spine calcium levels in a nonlinear manner (Koester and Sakmann, 1998; Schiller et al., 1998), and clearly affect the scale and direction of plasticity contingent on the timing of presynaptic and postsynaptic activity (Markram et al., 1997; Feldman, 2000). We confirmed that pairing the postsynaptic spike, so as to occur 10 msec after the presynaptic spike, does produce LTP in neurons of wild-type mice but not in αCaMKIIT286A mutants.
Not all cells in the cortex showed LTP, although all slices from wild types showed potentiation in the extracellular fields. This suggests some heterogeneity of cell type or pathway in the barrel cortex. Glazewski and Fox (1996) previously noted that whisker deprivation only causes a shift in the receptive fields of ∼50% of the cells recorded, which supports this view. However, when the response of a population of layer II/III cells is studied, whether by electrical stimulation of layer IV and extracellular recording in a slice or by whisker stimulation and extracellular recording of the evoked SEPs in the entire animal, the subpopulation of cells that does potentiate tends to dominate the response. Consequently, population recordings reveal potentiation in every animal.
Cortical SEPs
The results presented here show for the first time that sensoryevoked synaptic responses are potentiated after single-whisker experience, and that this property of the barrel cortex is blocked in αCaMKIIT286A mice. The field potentials evoked by whisker stimulation will reflect population activity over a limited distance in the cortex. The most influential factor contributing to the low-frequency component of the field is the synaptic potentials. Therefore, a lack of potentiation in the αCaMKIIT286A mice indicates a loss of sensory-evoked synaptic plasticity.
The synaptic potentials tend to occur synchronously in the barrel cortex, because whisker stimulation can be made with rapid onset (1 msec) and at a discrete moment in time, producing a high level of temporal summation. The small dipoles created by the layer II/III pyramidal cells also summate spatially to some degree, although not as much as in the hippocampus, where the somata are more closely aligned. Both factors ensure a relatively large SEP and strong sinks and sources in the CSD analysis. Somatic and even dendritic action potentials contribute relatively little to the CSD analysis, because they are rapid events that tend not to summate temporally among cells. Whereas SEPs tend to measure several millivolts in amplitude and last tens of milliseconds, spikes are rarely >2–300 μV and last 1 msec. Spikes therefore compose <10% of the voltage at a very brief time point. In addition, spikes are sharply attenuated with distance far more than the lower-frequency components. Finally, any local spikes are filtered out electronically before additional analysis. Consequently, potentiation of the SEPs seen as a result of whisker deprivation is most likely a good reflection of the degree of potentiation of the excitatory synaptic potentials within the cortex.
Toward a complete theory of experience-dependent plasticity
The weight of evidence is that experience-dependent plasticity is expressed in the cortex rather than subcortically (Fox, 1994, 2002; Glazewski and Fox, 1996; Wallace et al., 2001). Plasticity may be induced by whisker deprivation setting up a spike-firing pattern in the somatosensory system that induces synaptic plasticity in a particular cortical pathway. Those synaptic plasticity mechanisms then require autophosphorylation of CaMKII. The activity patterns set up by whisker deprivation have been studied recently by Kelly et al. (1999), who showed that removing whiskers surrounding a particular spared whisker leads to an increased firing in the barrel of the spared whisker during natural whisking behavior. The response to single-whisker stimulation is unaffected by this manipulation, but the excitation produced by a single whisker would normally be damped down by lateral inhibition generated by coincident activation of the surround whiskers during whisking. Presumably, a prolonged period of increased firing in the barrel of the spared whisker causes potentiation in the postsynaptic targets of those layer IV cells (Fox, 2000), either by a spike-pairing mechanism (Feldman, 2000) or simply because of the increased firing rate.
We have shown that a variety of firing patterns do lead to synaptic plasticity in the barrel cortex, either spike pairing or theta-burst firing. Theta-burst frequencies are similar to those generated during whisking behavior in the rodent and might be expected to occur with higher-frequency bursts in single-whisker mice than in normal mice. Although synaptic plasticity can occur in the absence of postsynaptic spikes, in vivo it is very likely that postsynaptic spikes will occur during sensory responses, which makes induction of plasticity by spike pairing highly relevant.
The results of this study show that potentiation is dependent on αCaMKII autophosphorylation. Why autophosphorylation is important for potentiation is not entirely clear. Evidence suggests it is not necessary for expression of LTP (Lisman et al., 2002), although it is required for induction. Nevertheless, autophosphorylation does last for ∼1 hr after induction of LTP in the hippocampus and would be expected to prolong the capacity of the kinase to phosphorylate and enable it to translocate to the postsynaptic density (Shen and Meyer, 1999). Both factors would tend to improve the chances of αCaMKII phosphorylating synaptic substrates including AMPA receptors at the serine 831 site of the glutamate receptor type 1 (GluR1) subunit, a process known to increase conductance and therefore produce potentiation (Barria et al., 1997). The fact that LTP is not entirely abolished in mice lacking the GluR1 subunit (Hoffman et al., 2002) implies that at least one other mechanism is involved.
The results of this and other studies in the barrel cortex offer a possibility of a complete description of a plasticity process from circuit behavior through to molecular mechanism. This study makes a connection between the neuronal circuit behavior and synaptic plasticity by showing that both experience-dependent plasticity and LTP critically depend on αCaMKII autophosphorylation in the same pathway.
Footnotes
This work was supported by Medical Research Council Grant G9900931 to K.F. We thank Phil Blanning for genotyping and John Curry for help with the histology.
Correspondence should be addressed to Prof. Kevin Fox, School of Biosciences, Cardiff University, Museum Avenue, Cardiff CF10 3US, UK. E-mail: foxkd@cf.ac.uk.
S. Glazewski's present address: McKay Institute of Communication and Neuroscience, Keele University, Staffordshire ST5 5BG, UK.
P. Pakhotin's present address: School of Biological Sciences, Manchester University, Manchester M13 9PT, UK.
Copyright © 2003 Society for Neuroscience 0270-6474/03/234428-09$15.00/0
N.H., S.G., and P.P. contributed equally to this work.
References
- Aizenman CD, Kirkwood A, Bear MF ( 1996) A current source density analysis of evoked responses in slices of adult rat visual cortex: implications for the regulation of long-term potentiation. Cereb Cortex 6: 751–758. [DOI] [PubMed] [Google Scholar]
- Armstrong-James M, Fox K, Millar J ( 1980) A method for etching the tips of carbon fibre microelectrodes. J Neurosci Methods 2: 431–432. [DOI] [PubMed] [Google Scholar]
- Barria A, Muller D, Derkach V, Griffith LC, Soderling TR ( 1997) Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science 276: 2042–2045. [DOI] [PubMed] [Google Scholar]
- Blakemore C, Cooper GF ( 1970) Development of the brain depends on the visual environment. Nature 228: 477–478. [DOI] [PubMed] [Google Scholar]
- Errington ML, Bradshaw KD, Peters M, Bliss TVP, Giese KP ( 2002) LTP is abolished in area CA1 but survives in the dentate gyrus of alphaCaMKII-T286A mutant mice: an in vivo and in vitro study. Abstr Fed Eur Soc Neurosci 3: 262. [Google Scholar]
- Feldman DE ( 2000) Timing-based LTP and LTD at vertical inputs to layer II/III pyramidal cells in rat barrel cortex. Neuron 27: 45–56. [DOI] [PubMed] [Google Scholar]
- Fox K ( 1994) The cortical component of experience-dependent synaptic plasticity in the barrel cortex. J Neurosci 14: 7665–7679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox K ( 2000) Timing is everything. Neuron 27: 1–3. [DOI] [PubMed] [Google Scholar]
- Fox K ( 2002) Pathways and mechanisms for plasticity in the barrel cortex. Neuroscience 111: 799–814. [DOI] [PubMed] [Google Scholar]
- Giese KP, Fedorov NB, Filipkowski RK, Silva AJ ( 1998) Autophosphorylation at Thr286 of the alpha calcium-calmodulin kinase II in LTP and learning. Science 279: 870–873. [DOI] [PubMed] [Google Scholar]
- Glazewski S, Fox K ( 1996) The time-course of experience-dependent synaptic potentiation and depression in barrel cortex of adolescent rats. J Neurophysiol 75: 1714–1729. [DOI] [PubMed] [Google Scholar]
- Glazewski S, Chen C-H, Silva A, Fox K ( 1996) The requirement for αCAMKII in experience-dependent plasticity of the barrel cortex. Science 272: 421–423. [DOI] [PubMed] [Google Scholar]
- Glazewski S, Herman C, McKenna M, Chapman PF, Fox K ( 1998) Long-term potentiation in vivo in layer II/III of rat barrel cortex. Neuropharmacology 37: 581–592. [DOI] [PubMed] [Google Scholar]
- Glazewski S, Giese KP, Silva A, Fox K ( 2000) The role of the alpha-CaMKII autophosphorylation in neocortical experience-dependent plasticity. Nat Neurosci 3: 911–918. [DOI] [PubMed] [Google Scholar]
- Golding NL, Staff NP, Spruston N ( 2002) Dendritic spikes as a mechanism for cooperative long-term potentiation. Nature 418: 326–331. [DOI] [PubMed] [Google Scholar]
- Gordon JA, Cioffi D, Silva AJ, Stryker MP ( 1996) Deficient plasticity in the primary visual cortex of alpha-calcium/calmodulin-dependent protein kinase II mutant mice. Neuron 17: 491–499. [DOI] [PubMed] [Google Scholar]
- Hoffman DA, Sprengel R, Sakmann B ( 2002) Molecular dissection of hippocampal theta-burst pairing potentiation. Proc Natl Acad Sci USA 99: 7740–7745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubel DH, Wiesel TN ( 1970) The period of susceptibility to the physiological effects of unilateral eye closure in kittens. J Physiol (Lond) 206: 419–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly MK, Carvell GE, Kodger JM, Simons DJ ( 1999) Sensory loss by selected whisker removal produces immediate disinhibition in the somatosensory cortex of behaving rats. J Neurosci 19: 9117–9125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilgard MP, Pandya PK, Vazquez J, Gehi A, Schreiner CE, Merzenich MM ( 2001) Sensory input directs spatial and temporal plasticity in primary auditory cortex. J Neurophysiol 86: 326–338. [DOI] [PubMed] [Google Scholar]
- Kirkwood A, Dudek SM, Gold JT, Aizenman CD, Bear MF ( 1993) Common forms of synaptic plasticity in the hippocampus and neocortex in vitro Science 260: 1518–1522. [DOI] [PubMed] [Google Scholar]
- Koester HJ, Sakmann B ( 1998) Calcium dynamics in single spines during coincident pre- and postsynaptic activity depend on relative timing of back-propagating action potentials and subthreshold excitatory postsynaptic potentials. Proc Natl Acad Sci USA 95: 9596–9601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkum ME, Zhu JJ ( 2002) Signaling of layer 1 and whisker-evoked Ca 2+ and Na + action potentials in distal and terminal dendrites of rat neocortical pyramidal neurons in vitro and in vivo J Neurosci 22: 6991–7005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Glazewski S, Lin X, Elde R, Fox K ( 1995) The effect of vibrissae deprivation on follicle innervation, neuropeptide synthesis in the trigeminal ganglion and S1 barrel field cortical plasticity. J Comp Neurol 357: 465–481. [DOI] [PubMed] [Google Scholar]
- Lisman J, Schulman H, Cline H ( 2002) The molecular basis of alphaCaMKII function in synaptic and behavioural memory. Nat Rev Neurosci 3: 175–190. [DOI] [PubMed] [Google Scholar]
- Markram H, Lubke J, Frotscher M, Sakmann B ( 1997) Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science 275: 213–215. [DOI] [PubMed] [Google Scholar]
- Mitzdorf U ( 1985) Current source-density method and application in cat cerebral cortex: investigation of evoked potentials and EEG phenomena. Physiol Rev 65: 37–65. [DOI] [PubMed] [Google Scholar]
- Mitzdorf U, Singer W ( 1978) Prominent excitatory pathways in the cat visual cortex (A7 and A18): a current source density analysis of electrically evoked potentials. Exp Brain Res 33: 371–394. [DOI] [PubMed] [Google Scholar]
- Schiller J, Schiller Y, Clapham DE ( 1998) NMDA receptors amplify calcium influx into dendritic spines during associative pre- and postsynaptic activation. Nat Neurosci 1: 114–118. [DOI] [PubMed] [Google Scholar]
- Shen K, Meyer T ( 1999) Dynamic control of alphaCaMKII translocation and localization in hippocampal neurons by NMDA receptor stimulation. Science 284: 162–166. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Stevens CF, Tonegawa S, Wang Y ( 1992) Deficient hippocampal long-term potentiation in alpha-calcium-calmodulin kinase II mutant mice. Science 257: 201–206. [DOI] [PubMed] [Google Scholar]
- Simons DJ, Land PW ( 1987) Early experience of tactile stimulation influences organization of somatic sensory cortex. Nature 326: 694–697. [DOI] [PubMed] [Google Scholar]
- Strominger RN, Woolsey TA ( 1987) Templates for locating the whisker area in fresh flattened mouse and rat cortex. J Neurosci Methods 22: 113–118. [DOI] [PubMed] [Google Scholar]
- Taha S, Hanover JL, Silva AJ, Stryker MP ( 2002) Autophosphorylation of αCAMKII is required for ocular dominance plasticity. Neuron 36: 483–491. [DOI] [PubMed] [Google Scholar]
- Wall JT ( 1988) Variable organization in cortical maps of the skin as an indication of the lifelong adaptive capacities of circuits in the mammalian brain. Trends Neurosci 11: 549–557. [DOI] [PubMed] [Google Scholar]
- Wallace H, Liming K, Glazewski S, Fox K ( 2001) The role of cortical activity in experience-dependent potentiation and depression of sensory responses in barrel field. J Neurosci 21: 3881–3894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong-Riley M ( 1979) Changes in the visual system of monocularly sutured or enucleated cats demonstrable with cytochrome oxidase histochemistry. Brain Res 171: 11–28. [DOI] [PubMed] [Google Scholar]