

Abstract
Active CREB (cAMP responsive element-binding protein) transcription factor is crucial for neuronal survival. Several members of the CREM/ICER (cAMP responsive element modulator/inducible cAMP early repressor) protein family may act as endogenous CREB antagonists. However, their involvement in a process of programmed cell death remains unexplored. Here we report that ICER may play such a role in neuronal apoptosis because it is upregulated in apoptotic neurons in vitro, and overexpression of ICER, delivered in adenoviral vector, evokes programmed cell death of three different kinds of cultured neurons, namely those derived from hippocampal dentate gyrus, cerebral cortex, and superior cervical ganglion. Reporter gene assay with a promoter containing a CREB-responsive sequence revealed a decrease in both basal and induced CRE-dependent gene expression in neurons overexpressing ICER. Finally, the level of expression of the anti-apoptotic protein Bcl-2, a well known CREB target, was markedly diminished in ICER-treated neurons. We suggest that the naturally occurring CREB functional antagonist ICER may have a specific function in programmed cell death of neurons, probably by silencing the expression of anti-apoptotic genes.
Keywords: ICER, CREB, neuronal apoptosis, gene expression, Bcl-2, adenoviral vectors
Introduction
ICER (inducible cAMP early repressor) is a collective name for a group of proteins produced from the CREM/ICER gene by use of an internal promoter within an intron of the CREM (cAMP responsive element modulator) gene (Molina et al., 1993; Stehle et al., 1993). It was first described as the major CREM transcript in the pineal gland during day–night fluctuations of CREM expression (Stehle et al., 1993). The ICER, which contains only a DNA-binding domain, functions as a repressor of transcription of several CRE (cAMP responsive regulatory element)-containing genes (Molina et al., 1993; Stehle et al., 1993; Tinti et al., 1996; Lamas et al., 1997). Studies on pineal gland showed ICER to serve as a negative regulator of cAMP-dependent, phospho-CREB (cAMP regulatory element-binding protein)-driven expression of genes involved in circadian rhythm (Maronde et al., 1999; Pfeffer et al., 2000). Induced expression of ICER was also reported in several other physiological conditions (for review, see Stehle et al., 2001). All of these findings suggest that ICER is involved in mechanisms responsible for returning stimulated cells to the steady-state level (Stehle et al., 2001). However, increased levels of ICER mRNA were also observed in the cortex and the hippocampus after treatment with kainate, NMDA, and dizocilpine maleate (MK-801) (Nedivi et al., 1993; Konopka et al., 1998; Storvik et al., 2000) at doses that provoke neuronal apoptosis (Filipkowski et al., 1994; Hetman et al., 1997), suggesting that it might be worthwhile to consider a possible role for ICER in neuronal cell death, in addition to its putative physiological functions (Stehle et al., 1993; Konopka et al., 1998).
A pro-apoptotic role of ICER in neurons could be of special importance, especially because CREB appears to have antiapoptotic activity in cultured neurons deprived of trophic support (Bonni et al., 1999; Riccio et al., 1999). Moreover, studies on CREB null mutant embryos showed that CREB is necessary for the survival of peripheral neurons at the time of their neurotrophin dependency (Lonze et al., 2002). Furthermore, recent findings link decreased CREB activity to neurotoxicity and neurodegeneration (for review, see Dawson and Ginty, 2002). However, our knowledge about CREB downstream effectors as well as interactions between CREB/ATF (activating transcription factor)/CREM proteins during apoptosis still remains very limited (Walton and Dragunow, 2000; Lonze et al., 2002).
Because no functional data on ICER and neuronal apoptosis have been reported, in this study we tested directly whether ICER may be involved in neuronal cell death in culture. We found that indeed adenovirally delivered ICER produces neuronal apoptosis in three different culture models. Furthermore, l-glutamate-evoked apoptosis of dentate gyrus (DG) neurons and serum deprivation-induced death of cortical neurons are both accompanied by increased ICER expression. Moreover, ICER inhibited CRE-dependent transcription along with diminishing levels of Bcl-2. Thus, our results suggest that ICER, a natural CREB antagonist, may have a specific function in programmed cell death of neurons, perhaps by silencing expression of CRE-dependent genes, including anti-apoptotic ones (Mayr and Montminy, 2001) that are crucial for proper metabolism of the neuronal cell.
Materials and Methods
Adenoviral vectors. Recombinant adenoviruses were constructed by homologous recombination of pRSV-ICER plasmid (containing the ICER IIγcDNA; kindly provided by Dr. P. Sassone-Corsi, Institut de Genetique et de Biologie Moleculaire et Cellulaire, Illkirch-Strasbourg, France) and adenoviral genomic DNA as described earlier (Revah et al., 1996). The recombinant Ad-β-galactosidase (βgal) has been described previously (Stratford-Perricaudet et al., 1992). Viral stocks were prepared according to the procedure described by Revah et al. (1996). Viruses were titered by the end-point dilution method (Lowenstein et al., 1996). Other gene constructs either were previously described (Impey et al., 1998) or commercially available.
Cell culture and adenoviral vector treatment. Primary cultures of DG cells were obtained from 4-d-old Wistar rat pups as described previously (Figiel and Kaczmarek, 1997; Jaworski et al., 2000). Cortical neurons were prepared from newborn Wistar rats (Xia et al., 1996; Hetman et al., 1999). Primary cultures of superior cervical ganglion (SCG) neurons were obtained from 1-d-old Wistar rats (Le Gal La Salle et al., 1993).
All three types of neuronal cell cultures were exposed to adenoviral vectors 3–4 d after plating. DG cells were incubated overnight with 50 pfu per cell [multiplicities of infection (MOI) 50] suspended in a culture medium. The next day, the cultures were washed several times with DMEM (Sigma, St. Louis, MO) and placed in a new culture medium. In case of cortical cultures, culture medium was replaced with Basal Medium Eagle (BME; BioWhittaker, Walkersville, MD) and saved. Adenoviral vectors (MOI, 100) were added to the cultures for 1 hr. Afterward, virus-containing BME was removed, and saved culture medium was added back to the infected cultures. SCG neurons were infected exactly the same way as cortical neurons with the exception that instead of BME, replacement culture medium was changed to L15CO2 medium (Invitrogen Life Technologies, Carlsbad, CA). For quantification of apoptosis, the cultures were fixed as follows: DG, 6 and 24 hr; cortical neurons, 24, 48, and 72 hr; SCG cells, 24, 48, and 72 hr. For other applications the cells were treated as indicated below.
Serum deprivation. Serum deprivation was performed with cortical neurons at 6 d in vitro as described previously (Hetman et al., 1999). Briefly, cells were washed twice with serum-free BME and incubated in serum-free BME supplemented with 35 mm glucose, 1 mm l-glutamine, 100 U/ml penicillin, 0.1 mg/ml streptomycin, and 2.5 mm cytosine arabinoside (all from Sigma) in the presence of the NMDA receptor antagonist MK-801 (10 mm; Sigma). Control cells were washed similarly and then incubated for matched time points in serum-containing conditioned medium.
X-gal staining of cell cultures. Forty-eight hours after infection, Ad-βgal-infected cell cultures were washed once with PBS and fixed for 5 min in 1% formaldehyde, 0.2% glutaraldehyde in PBS. Then the cells were incubated overnight at 37°C in 0.8 mg/ml X-gal, 4 mm K3Fe(CN)6, 4 mm K4Fe(CN)6, 4 mm MgCl2, 6 H2O in PBS (all from Sigma). After incubation, cultures were washed several times with PBS.
Quantification of apoptosis by nuclear morphological changes. To visualize nuclear morphology, cells were fixed in 4% paraformaldehyde and stained with 2.5 mg/ml DNA dye Hoechst 33258 (bis-benzimide; Sigma). Apoptosis was quantified by scoring the percentage of neurons (as revealed by staining with NeuN antibody) with apoptotic nuclear morphology at the single-cell level after Hoechst staining. Uniformly stained nuclei were scored as healthy, viable neurons. Condensed or fragmented nuclei were scored as apoptotic. Statistical analysis of the data were performed using the nonparametric Kruskal–Wallis ANOVA test.
DNA ladder assay and MTT assay. To examine DNA cleavage, 5 × 106 cortical neurons were plated in a 60 mm dish and infected with Ad-βgal or Ad-ICER (MOI, 100). Seventy-two hours after infection, soluble cytoplasmic DNA was isolated from each plate and loaded to 1.8% agarose gel for DNA ladder analysis, as described (Hetman et al., 1999).
In cortical cultures, neuronal survival was assayed by measuring the conversion of the yellow, water-soluble tetrazolium, MTT (Sigma) to the blue, water-insoluble formazan at 24, 48, 72, and 96 hr after infection with either Ad-βgal or Ad-ICER as described previously (Hetman et al., 1999). Optical blanks, used as controls, were generated by incubating MTT with the conditioned medium. The colorimetric reaction was evaluated spectrophotometrically (A570).
Immunocytochemistry. Immunocytochemical staining was done as described previously (Jaworski et al., 2000). The color reactions were developed using a diaminobenzidine substrate kit (Vector Laboratories, Burlingame, CA). For fluorescence immunocytochemistry, the primary antibody was visualized by incubating the cultures with a biotinconjugated appropriate secondary IgG (Vector Laboratories) diluted 1:500 and then with streptavidin–FITC or streptavidin–Texas Red complex (Vector Laboratories) diluted 1:500. Primary antibodies were as follows: monoclonal anti-MAP2 protein (Sigma) diluted 1:400, monoclonal anti-NeuN (Chemicon, Temecula, CA) diluted 1:500, and polyclonal ICER 1:20,000 (Molina et al., 1993).
RT-PCR analysis. For RT-PCR analysis, total RNA was isolated from 5 × 105 dentate gyrus cells or 5 × 106 cortical cells by use of TRI Reagent (Sigma). The remaining DNA was removed by digestion with DNase I (Roche, Indianapolis, IN). Next, RNA was reverse transcribed by use of Expand Reverse Transcriptase (Roche, Indianapolis, IN) in presence of oligo-dT. The cDNA was then amplified by PCR with a set of oligonucleotides designed to recognize either different isoforms of ICER cDNA (5′ICER: 5′-ACA AGA CCA CTC TGT ATG CA-3′; 3′ICER: 5′-GGG AGA GCA AAT GCT TTT CA-3′) or ICERIIγproduced from adenoviral vector (5′UTRSV: 5′-ACC ATT CAC CAC ATT GGT GT-3′; 3′ICER). The following temperature profile was applied for both reactions: 94°C for 30 sec, 48°C for 30 sec, 72°C for 1 min, followed by 15 min incubation at 72°C. Amplification of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) cDNA was used for verification of the amount of template used for reaction (GAPDH3: 5′-AAC TAC ATG GTC TAC ATG TT-3′; GAPDH4: 5′-GTG GTG CAG GAT GCA TTG CT-3′).
Electrophoretic mobility shift assay. For electrophoretic mobility shift assay (EMSA), 107 cortical neurons were plated in a 100 mm Petri dish. Seventy-two hours after infection with adenoviral vectors, nuclear extracts were isolated and EMSA was done as described previously (Kaminska et al., 1994). A double-stranded 32P-labeled oligonucleotide containing CRE sequence (AGAGATTGCCTGACGTCAGAGAGCTAG) was used as a probe in the binding reactions.
Western blot analysis. For immunoblot analysis, 5 × 106 cortical neurons were plated in a 60 mm Petri dish. Seventy-two hours after infection with adenoviral vectors, total protein extracts were isolated and separated by SDS-PAGE (12%). Next, the proteins were transferred onto Hybond-ECL membrane (Amersham Biosciences, Piscataway, NJ) by semidry electroblotting. Blots were then incubated overnight with 5% nonfat milk in TBS-T (150 mm NaCl, 20 mm Tris, pH 7.4, 0.05% Tween 20) at 4°C. For immunochemical detection, the membranes were incubated at 4°C overnight with the following antibodies: anti-CREM/ICER antibody (Santa Cruz Biotechnology, Santa Cruz, CA; diluted 1:500), ICER (Molina et al., 1993) (diluted 1:1000), CREB (Cell Signaling Technology, Beverly, MA; diluted 1:1000), phospho-CREB (Cell Signaling Technology; diluted 1:1000), GAPDH (Chemicon; diluted 1:2000), or Bcl-2 (BD Transduction Laboratories, Palo Alto, CA; diluted 1:500). The next day, the membranes were washed three times with TBS-T and incubated with the appropriate secondary peroxidase-conjugated antibody (Amersham Biosciences; diluted 1:500) in 5% nonfat milk in TBS-T for 1 hr. After the next three washings, peroxidase was detected with ECL reagent (Amersham Biosciences). For the statistical evaluation of the immunoblots, they were scanned densitometrically, and data were expressed in arbitrary units (AU). For evaluation of the phosphorylated (P)-CREB/CREB ratio, the data in AU were compared for the Ad-ICER versus Ad-β-gal versus control cultures and first nonparametric ANOVA differences among all the groups, and then the Mann–Whitney nonparametric test was applied for all the pairs of comparisons. No difference was found between Ad-β-gal and control cultures. The statistics for the Ad-ICER and Ad-β-gal comparison are shown in Figure 1.
Figure 1.

Effect of ICER overexpression on CREB function. A, CRE-sequence binding 72 hr after Ad-ICER infection of cortical neurons (electrophoretic mobility shift assay; comp. indicates competition with nonlabeled CREB probe; I, II, and III designate different complexes formed by neuronal nuclear extracts proteins). B, Immunoblotting detection of P-CREB, CREB, CREM, and ICER proteins in Ad-βgalversus Ad-ICER-infected cultures. C, Quantitative analysis of effects of ICER overexpression on P-CREB levels. The immunoblots such as those shown in B were scanned densitometrically and analyzed in such a way that for each lane (n = 10; 5 independent culture experiments) the result of P-CREB expression was divided by the level of CREB immunoreactivity and expressed in arbitrary units (AU). Next, the data were statistically evaluated with nonparametric Kruskal–Wallis ANOVA, followed by Mann–Whitney U test. The difference between P-CREB/CREB ratios in Ad-ICER-versus Ad-β-gal-treated cultures is significant atp=0.008(**). D, Effects of ICER overexpression on firefly luciferase driven by a CRE-containing promoter. Then eurons first were transiently transfected with pCRE-Luc and pRL-TK by use of Lipofectamine2000 and next infected with either Ad-βgal or Ad-ICER. Then, either 10 μm for skolinor vehicle (DMSO) was added to the transfected cells. After 24 hr, lysates from cells were prepared, and luminescence of both firefly and Renilla luciferases was measured with the aid of the Dual-Luciferase Reporter Assay. Results are given as a ratio of luminescence of firefly to Renilla luciferases measured in relative fluorescence units ± SD. E, Immunoblotting detection of Bcl-2 and GAPDH proteins in Ad-ICER- and Ad-βgal-treated cortical neurons; the results of two of three independent experiments are presented. B, C, E, Three days after the plating, the cells were infected with control, β-galactosidase-encoding (Ad-βgal), or ICER-encoding (Ad-ICER) adenoviral vector. Seventy-two hours after infection, the cultures were harvested for protein extraction and immunoblotting analysis.
Transient transfections and reporter gene assay. Cortical neurons, cultured in 24-well dishes (5 × 105 cells per well) were transiently transfected with pCRE-Luc and pRL-TK with the aid of Lipofectamine2000 (Invitrogen Life Technologies, Carlsbad, CA) as described by Ohki et al. (2001) with modifications. Briefly, 3 d after the plating, the cells were exposed to the mixture of plasmid DNA and Lipofectamine2000 (0.6 μg of total DNA per well, with DNA/Lipofectamine2000 ratio 1:2.5) for 2 hr in serum-free culture medium (BME) supplemented with 35 mm glucose, 1 mm glutamine, penicillin (100 U/ml), streptomycin (100 μg/ml) and infected immediately afterward with either Ad-βgal or Ad-ICER. Afterward, the neurons were returned to saved conditioned media and cultured for the next 48 hr before any additional treatment. Next, 10 μm forskolin (Sigma) or vehicle (DMSO) was added to the transfected cells. After next 24 hr, lysates from cells were prepared in Passive Lysis Buffer (Promega, Madison, WI), and luminescence of both firefly and Renilla luciferases in 10 μl of cell lysate were measured by use of Dual-Luciferase Reporter Assay (Promega).
Results
Adenovirally delivered ICER inhibits CRE-dependent transcription
To study ICER function, we developed an adenoviral vector carrying the ICERIIγsequence controlled by the Rous sarcoma virus (RSV) constitutive promoter Ad-ICER. To test whether this experimental tool inhibits CRE-driven transcription in our experimental conditions, we first investigated with an EMSA whether exogenous ICER can bind the CRE sequence. In nuclear extracts prepared from control Ad-βgal-treated cortical neurons, only a slow migrating complex was detected by EMSA (Fig. 1A, I). This band reflected specific binding of the CRE probe as it disappeared in competition experiments with an unlabeled CRE probe (Fig. 1A). On the other hand, in nuclear extracts obtained from Ad-ICER-infected cultures, two additional faster migrating complexes (II, III) were detected (Fig. 1A). Such fast migrating complexes were described previously as containing ICER proteins (Lamas et al., 1997; Trocme et al., 2001). No such binding could be observed in material collected from Ad-βgal-infected cultures (Fig. 1A). By use of immunoblotting technique we did not observe any significant decrease in the CREB or CREM protein levels (Fig. 1B). However, use of an antibody against the phosphorylated form of CREB showed a decrease in its amount in Ad-ICER-treated cultures (Fig. 1B,C).
Next, we checked whether exogenous ICER could inhibit both basal and forskolin-induced CRE-promoter activity. Forskolin is a well known activator of cAMP and thus also CREB function (Gonzalez and Montminy, 1989). The cultures were transiently cotransfected with a plasmid containing firefly luciferase gene under the control of CRE-sequence (pCRE-Luc) as well as plasmid constitutively expressing Renilla luciferase (pRL-TK; for standardization purpose) and next infected with either Ad-βgal or Ad-ICER. Forty-eight hours after the transfection–infection treatment, the cultures were stimulated with 10 μm forskolin for the next 24 hr. The basal level (after vehicle treatment) of CRE-dependent luciferase expression was decreased in Ad-ICER-infected cells almost twofold when compared with Ad-βgal-treated cells (Fig. 1D). Moreover, the levels of CRE-driven reporter expression after for-skolin stimulation were also decreased in ICER-overexpressing cells when compared with cultures infected with control virus (Fig. 1D).
The finding that ICER overexpression resulted in a decrease of CRE-dependent transcription suggested that specific CRE-driven gene(s) might be affected by ICER in neuronal apoptosis. In this context, we studied the expression of a known anti-apoptotic CREB target: Bcl-2 (Wilson et al., 1996; Riccio et al., 1999). We found that the level of Bcl-2 protein, as determined by the immunoblotting 72 hr after infection, in ICER-overexpressing cultures dropped to 62.3 ± 2.5% (n = 6 lines for each virus; three independent culture experiments) of expression level in those treated with Ad-βgal (Fig. 1 E). Thus, our results show that ICER overexpression-evoked apoptosis correlates with an inhibition of CRE-dependent transcription and with a decrease in the amount of the anti-apoptotic protein Bcl-2.
Glutamate-evoked neuronal cell death is accompanied by ICER expression
We have reported previously that high doses of glutamate evoked apoptotic cell death of dentate gyrus neurons cultured in vitro (Figiel and Kaczmarek, 1997) (Fig. 2A). To test the possibility that ICER is related to neuronal cell death, we analyzed ICER mRNA levels in these cultures and found an increase of its expression (Fig. 2B) that peaked at 4 hr after incubation with glutamate and was observed also at 24 hr. On the other hand, glutamate did not change the mRNA level of a housekeeping gene, GAPDH (Fig. 2B).
Figure 2.

Glutamate-evoked neuronal cell death is accompanied by ICER mRNA expression. A, Nuclear morphology of control dentate gyrus cultures (top panel) and after 24 hr exposure to 0.5 mm glutamate (bottom panel) visualized by Hoechst 33258 staining. Arrows indicate fragmented and condensed nuclei. B, Level of ICERII mRNA isoform expression in dentate gyrus cultures treated with 0.5 mm glutamate as verified by RT-PCR. GAPDH mRNA analysis was performed to verify equal reaction and loading.
ICER expression and apoptosis of the dentate gyrus hippocampal neurons
The fact that a pro-apoptotic dose of glutamate caused an increase in expression of ICER mRNA raised a question regarding whether overexpression of ICER is sufficient to induce neuronal cell death. To test such a possibility we used the adenoviral vector Ad-ICER. The ability of adenoviral vectors to transfer genes into DG neurons in culture was verified by infection with Ad-βgal (MOI, 50) (Fig. 3A). Next, the cultures were infected with Ad-ICER, and expression of the transgene after 24 hr was confirmed by RT-PCR. The primers were designed to recognize only the expression of ICER mRNA driven by the adenoviral vector (Fig. 3B). ICER overexpression was accompanied by dramatic changes in morphology of the cells 24 hr after infection (Fig. 3D). In contrast, cultures treated with Ad-βgal did not display any apparent abnormalities (Fig. 3D). The nuclear morphology of neurons infected with either Ad-βgal or Ad-ICER was compared after staining the cell nuclei with Hoechst 33285 dye. Neuronal identity of the cells was revealed by staining for a neuronal marker, NeuN. In noninfected or Ad-βgal-treated cultures at 6 hr (data not shown) as well at 24 hr after infection, the majority of the neurons displayed typical, healthy-looking chromatin structure (Fig. 3C,D). On the other hand, in Ad-ICER-infected cultures >50% of neurons had condensed nuclei 24 hr after treatment (Fig. 3C,D). In contrast, 6 hr after Ad-ICER infection there was no increase in the level of neuronal cell death in comparison with control cultures (data not shown).
Figure 3.
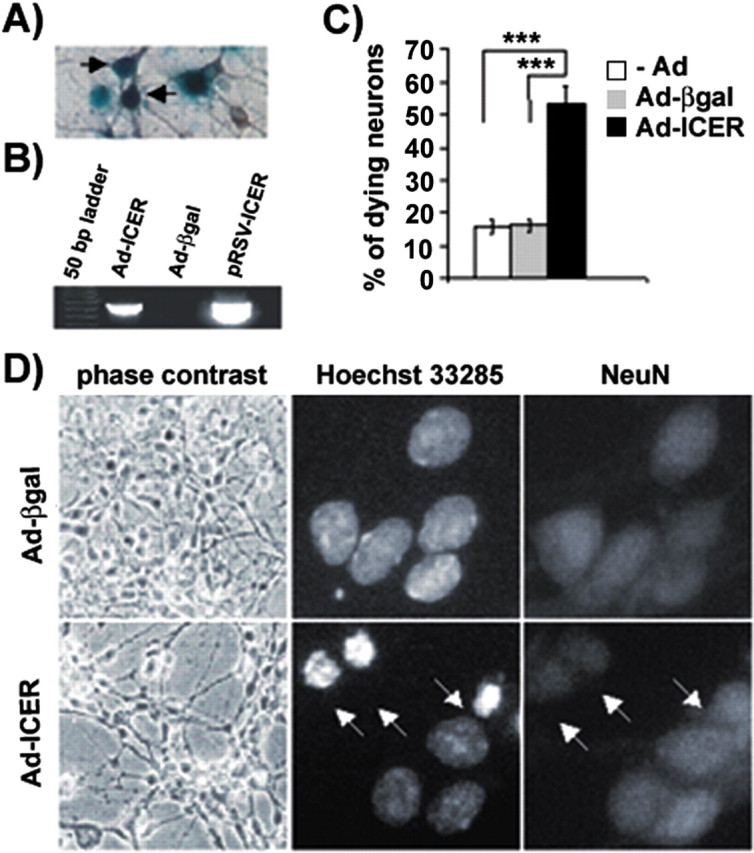
Apoptosis of dentate gyrus neurons induced by adenovirally delivered ICER overexpression. A, β-Galactosidase enzymatic activity in dentate gyrus neurons (note brownish MAP2 immunostaining) infected with an adenoviral vector encoding β-galactosidase (Ad-βgal; arrows). B, RT-PCR analysis of expression of exogenous recombinant adenovirus-delivered ICERIIγgene. RNA was isolated 24 hr after infection (with either Ad-βgal or the adenovirus-carrying ICERIIγgene Ad-ICER). The primers were designed to recognize only the transgenes that were used: Ad-ICER, cDNA from cultures treated with Ad-ICER was used for the reaction; Ad-βgal, cDNA from cultures treated with Ad-βgal; pRSV-ICER, pRSV-ICER plasmid DNA was used as a template for positive control of the reaction. C, Quantitative analysis of neurons with altered chromatin morphology after exposure to Ad-ICER. The cultures were exposed overnight to either Ad-βgal or Ad-ICER (MOI, 50, for both). Cells were fixed 24 hr after infection and stained with Hoechst 33285 and an anti body against NeuN. For each experimental variant, the cells were counted from 24 incidental microscopic fields from four independent cultures grown on 10 mm glass coverslips. Results are shown as an average percentage of neurons with changed chromatin morphology per coverslip ± SEM; ***p < 0.001; Kruskal–Wallis ANOVA. D, Changes of neuronal morphology evoked by Ad-ICER at 24 hr after the infection; arrows point to neuronal (as indicated by the NeuN immunostaining) nuclei with condensed and fragmented chromatin.
ICER expression and apoptosis of the cortical neurons
Dentate gyrus cultures consist of both glial and neuronal cells, and one could reason that the observed Ad-ICER-evoked neuronal cell death might be secondary to the ICER affecting the glia. To extend the observations to predominantly neuronal cultures and to study the phenomenon at the biochemical level, which requires more material than the amount available from the DG cultures, in the following experiments we switched to more homogeneous and abundant cultures of the cerebral cortex neurons. Importantly, it has been reported previously that serum deprivation in the presence of the glutamate receptor antagonist MK-801 evokes apoptotic cell death of cortical neurons cultured in vitro (Hetman et al., 1997) (Fig. 4A). This pro-apoptotic stimulation increased in ICER mRNA expression, evident at 1 hr after serum withdrawal and observed also at 24 hr (Fig. 4B,C). On the other hand, serum deprivation/MK-801 treatment did not change the expression of the mRNA level of a housekeeping gene, GAPDH (Fig. 4B).
Figure 4.

Serum deprivation-evoked neuronal cell death accompanied by ICER mRNA expression. A, Nuclear morphology of control cortical cultures and after 24 hr serum deprivation in the presence of 10 mm MK-801 was visualized by Hoechst 33258 staining. Arrows indicate fragmented and condensed nuclei. B, Levels of ICERII mRNA isoform expression in cortical cultures treated with glutamate were verified by RT-PCR at 1, 3, 6, or 24 hr after the infection. C, Levels of ICER mRNA expression as detected by RT-PCR.
Adenoviral gene transfer of Ad-βgal (MOI, 100) resulted in expression of the transgene in almost 80% of the cortical cells (data not shown). ICERIIγexpression 72 hr after Ad-ICER infection of neurons was documented by RT-PCR (data not shown). Similar to the experiments performed on DG cultures, for the cell death studies the cultures were infected with either Ad-βgal (MOI, 100) or Ad-ICER (MOI, 100). Seventy-two hours after infection, when marked changes in neuronal morphology of Ad-ICER-treated cells were observed under phase-contrast microscope, alterations in chromatin structure were also verified by Hoechst staining. In noninfected cultures or those treated with Ad-βgal, the percentage of dying neurons was very low (Fig. 5A). In contrast, up to 35% of Ad-ICER-infected neurons showed fragmentation and condensation of chromatin (Fig. 5A). Triple staining with Hoechst 33285 and antibodies against NeuN and ICER revealed dying neurons in Ad-ICER-treated cultures that were overexpressing ICER protein (Fig. 5C). Furthermore, a soluble fraction of cytoplasmic DNA isolated from cultures treated with Ad-ICER was found to contain material that was cleaved into oligonucleosomal fragments (DNA laddering), which is one of the hallmarks of apoptosis (Fig. 5B). The cytoplasmic fraction of DNA was absent from the Ad-βgal-infected cultures as well as noninfected neurons (Fig. 5B). To determine cell viability, a colorimetric tetrazolium assay on the basis of mitochondrial conversion of MTT was performed. A significant decrease of neuronal viability in cortical cultures after infection with Ad-ICER was confirmed 96 hr after infection with the MTT test (A570 = 0.283 ± 0.03 SEM) when compared with Ad-βgal-treated cultures (A570 = 0.111 ± 0.03 SEM; p < 0.05; Kruskal–Wallis ANOVA).
Figure 5.

Apoptosis of cortical neurons after ICER overexpression. A, Percentage of cortical apoptotic neurons at 72 hr after infection with Ad-ICER; ***p < 0.001; Kruskal–Wallis ANOVA. B, DNA fragmentation into ladder-like pattern 72 hr after infection of cortical neurons with either Ad-βgal or Ad-ICER. Arrows indicate fragmented, oligonucleosomal DNA. C, Changes in nuclei morphology (Hoechst 33258, left panel) of neurons (NeuN, middle panel) overexpressing ICER (right panel) after Ad-ICER infection of cortical cultures. Arrow points to ICER overexpressing neuron with condensed and fragmented chromatin.
ICER-evoked neuronal apoptosis in cultures derived from superior cervical ganglion
The experiments performed on cultures of DG and cerebral cortex showed that overexpression of ICER in CNS neurons was sufficient to cause their apoptotic death. To extend these observations to the neurons derived from the peripheral nervous system, we investigated the effect of ICER on SCG cultures. These cultures are homogenously neuronal and provide a well characterized model of physiological apoptosis and CREB-dependent survival (Riccio et al., 1999).
Initially, we confirmed that SCG neurons could be efficiently infected with Ad-βgal, the control recombinant adenovirus. Next, the SCG neurons were treated either with ICER-encoding adenoviral vector (MOI, 100) or with control recombinant adenovirus (Ad-βgal; MOI, 100) and fixed 6, 24, 48, and 72 hr after infection. Condensation of chromatin was evident at 72 hr after infection (36.2 ± 3.6% of neurons; p < 0.001; Kruskal–Wallis ANOVA), and to a lesser extent also at 48 hr, only in the Ad-ICER-infected cultures. We did not observe any changes in nuclear morphology of ICER-treated neurons at 6 and 24 hr or in cultures treated with control virus or noninfected even after 72 hr (7.9 ± 1.3 and 5.5 ± 1.1% of dying neurons, respectively).
Discussion
The survival and programmed cell death (apoptosis) of neurons depend on the activity of a number of genes, and therefore transcription factors controlling gene expression play a key role in these events (Dragunow and Preston, 1995; Herdegen et al., 1997; Pettmann and Henderson, 1998; Hughes et al., 1999; Sastry and Rao, 2000; Walton and Dragunow, 2000; Yuan and Yankner, 2000; Zagulska-Szymczak et al., 2001). CREB is one of the transcription factors that has already been studied in the context of neuronal survival, physiological apoptosis, and neurodegeneration (for review, see Finkbeiner, 2000; Walton and Dragunow, 2000). Increased levels of its phosphorylated active form were observed in neurons resistant to brain ischemia, whereas decrease of CREB phosphorylation was observed in neurons vulnerable to this treatment (Walton et al., 1996; Hu et al., 1999; Mabuchi et al., 2001). Furthermore, stimulation of extrasynaptic NMDA receptors, which leads to excitotoxic cell death of hippocampal neurons, resulted in a decrease in CREB phosphorylation, whereas synaptic stimulation increased the amount of phospho-CREB and was able to prevent staurosporine-evoked apoptosis (Hardingham et al., 2002). Additionally, CREB might be involved in protection against the adverse effects of deafferentation (Zirpel et al., 2000). Furthermore, Riccio et al. (1999) demonstrated that transcriptionally active CREB is necessary and sufficient for survival of sympathetic neurons under both normal and NGF-withdrawal conditions. Recently, Mantamadiotis et al. (2002) showed extensive apoptosis and progressive neurodegeneration in mouse brain in conditional CREB knock-out on CREM -/- background. One of the pro-survival targets for CREB seems to be bcl-2 (Riccio et al., 1999; Mabuchi et al., 2001), which was shown to be able to block apoptosis of the sympathetic neurons evoked by neurotrophin starvation (Garcia et al., 1992).
In contrast to growing evidence of the significance of CREB for neuronal survival, involvement of other CRE-binding proteins that belong to the CREM/ICER family in making decisions about neuronal cell fate remains essentially unexplored. In this report, we describe for the first time a novel, pro-apoptotic function of the transcription factor ICER, an endogenous, functional CREB antagonist. We show the increased levels of ICERII mRNA after glutamate stimulation that leads to apoptotic death of dentate gyrus neurons as well as under conditions of trophic deprivation of the cortical neurons in vitro. We also provide evidence that infection of three different types of neurons, namely dentate gyrus, cortical, and sympathetic neurons, with recombinant adenovirus carrying ICERIIγcDNA is capable of provoking robust neuronal cell death. Morphological features, including nuclear fragmentation, together with DNA-laddering, widely accepted methods for visualizing apoptotic changes (Willingham, 1999), have proven the apoptotic character of the neuronal cell death evoked by ICER. We also provide evidence for the competence of exogenous ICER to block CRE-dependent transcription both at the basal state and on activation with forskolin. Additionally our studies on the phosphorylated, i.e., possibly transcriptionally active form of CREB (P-CREB), revealed its downregulation in cortical cultures treated with Ad-ICER. Finally, we show that the level of Bcl-2 anti-apoptotic protein (Vaux et al., 1988, 1992) that was previously described to be regulated via CRE sequence (Wilson et al., 1996; Riccio et al., 1999) was decreased in cultures infected with adenoviral vector coding for ICERIIγ.
Despite correlative results obtained in gene expression studies on the brain treated with stimuli provoking neurodegeneration by drugs acting on glutamate receptors (Konopka et al., 1998; Storvik et al., 2000), no direct evidence was previously available on the functional ability of ICER to evoke neuronal cell death. Our results directly indicating such a possibility are not surprising, however. The major biological function of ICER appears to be its antagonism toward CREB, as indicated by studies on a number of genes (Molina et al., 1993; Stehle et al., 1993; Tinti et al., 1996; Lamas et al., 1997). In this report we add the gene encoding Bcl-2 to this list of those that are negatively regulated by ICER. Our data are in a good agreement with a postulated prosurvival activity of CREB and Bcl-2 (Walton et al., 1996; Hu et al., 1999; Walton and Dragunow, 2000; Mabuchi et al., 2001). Directly relevant to our work are also the results obtained with use of dominant negative mutants of CREB in vitro that showed this transcription regulator to be necessary for the pro-survival neuronal response to trophic factors (Bonni et al., 1999; Riccio et al., 1999), as well as studies by Mabuchi et al. (2001) who reported that blockade of CRE-dependent Bcl-2 expression by use of CRE-decoy oligonucleotides resulted in enhanced neuronal cell death within cultures exposed to glutamate. However, to the best of our knowledge, there was no previous evidence that overexpression of naturally occurring CREB functional antagonist(s) may result in neuronal cell death. Some previous reports, however, involved ectopic overexpression of ICER to study regulation of expression of various genes (Zauli et al., 2000; Colgin et al., 2001). Notably, the pro-aptotic effect of ICER has not been shown in either neuronal-like cell lines or in neurons. The discrepancy of these results and ours might be explained by the differences in experimental design, mainly by the fact that in most of these studies ICER was overexpressed for the shorter periods of time that are necessary to observe its severe pro-apoptotic effect. Such a delay of apoptotic effect of inhibition of CRE-dependent transcription was described previously by Riccio et al. (1999) for dominant negative mutants of CREB.
In addition to the reports showing correlation of ICER mRNA expression and neurodegeneration, a physiological role for this protein has also been suggested by studies documenting its elevated expression in conditions with enhanced neuronal activity but with no apparent component of cell death. The activation of ICER gene expression has been reported in the pineal gland during day–night fluctuations (Stehle et al., 1993), in hippocampus, frontal cortex, and cerebellum after electroconvulsive seizures (Fitzgerald et al., 1996), in visual and barrel cortex in response to sensory stimulation (Konopka et al., 1998; Staiger et al., 2000), in amygdala after lithium chloride injection (Spencer and Houpt, 2001), in hypothalamus after osmotic stimulation (Luckman and Cox, 1995), and in adrenal medulla after trans-synaptic stimulation with reserpine (Trocme et al., 2001). An explanation for the differential involvement of ICER in various phenomena could be derived from analysis of the time course of expression. In those reports suggesting physiological roles for ICER, its expression was short lasting, in contrast to our results obtained after glutamate stimulation of DG cultures as well as kainic acid treatment of animals (Konopka et al., 1998; S. Szymczak and J. Jaworski, unpublished observations). Thus, it might be suggested that prolonged ICER activity is detrimental to the neurons. This hypothesis might be supported further by the finding that HIV-1 Tat protein treatment of PC-12 cells resulted in prolonged increased expression of ICER mRNA (Zauli et al., 2000), and exposure of both PC-12 and neurons to high doses of this protein was found to induce programmed cell death (Weeks at al., 1995; Kruman et al., 1998). Furthermore, Zauli et al. (2000) showed that injection of Tat protein into rat striatum caused progressive loss of dopaminergic neurons in substantia nigra.
Our results show a decrease of CREB phosphorylation in Ad-ICER-overexpressing cultures without a significant decrease of the total amount of CREB protein. Most of the current models of initiation of CREB transcriptional activity suggest that CREB binding to its specific DNA sequence precedes CREB phosphorylation as well as further recruitment of transcription coactivators (for review, see Shaywitz and Greenberg, 1999). Our results showing that ICER overexpression diminishes the CREB DNA binding fully support such a notion.
Finally, we note that our approach with ectopic expression of ICER, an endogenous antagonist of CREB reported herein to have a specific biological function, might be useful for studies on ICER in other physiological and pathological conditions. It should also prove to be valuable in probing cellular activities of CREB that have recently been investigated extensively. Notably, adenoviral vectors appear to offer a very convenient tool for such studies. They are devoid of significant side effects in culture, and they have been used previously to deliver also anti-apoptotic genes into the cells (Simons et al., 1999; Kugler et al., 2001).
In conclusion, our data as well as the aforementioned considerations strongly suggest that the mechanism of ICER-evoked apoptosis in neurons is based on its ability to antagonize CREB anti-apoptotic action that involves activation of pro-survival genes. Thus, we postulate that the effects of ICER on neuronal survival might depend on a balance between the active form of CREB on the one hand and ICER on the other.
Footnotes
This research was supported by KBN (Komitet Badan Naukowych, Poland) Grant Z-KBN004/P04/98. Joint studies between the Laboratory of Molecular Neuroscience and Laboratoire de Genetique Moleculaire de la Neurotransmission et des Processus Neurodegeneratifs were a part of a Centre National de la Recherche Scientifique–Polish Academy of Sciences collaborative project. J.J. was supported by scholarships of the French Government, the Polish Foundation of Experimental and Clinical Oncology, the Batory Foundation, the European Molecular Biology Organization, and the Foundation for Polish Science. We express our gratitude to Dr. Sassone-Corsi for providing ICERIIγcDNA, Dr. Molina for antibody against ICER, S. De Gois and Dr. Millecamps for introducing us to SCG culture, and Dr. Nikolaev and K. Radwanska for assistance with statistical analysis. We thank also Drs. M. Sheng, G. Schütz, J. Kuznicki, R. Przewlocki, and J. Platenik for helpful comments.
Correspondence should be addressed to Dr. Jacek Jaworski, Picower Center for Learning and Memory, Massachusetts Institute of Technology, E18-215, 77 Massachusetts Avenue, Cambridge, MA02139. E-mail: jaworski@mit.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/234519-08$15.00/0
J.J. and B.M. contributed equally to the results of this work.
References
- Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME ( 1999) Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science 286: 1358–1362. [DOI] [PubMed] [Google Scholar]
- Colgin MA, Smith RL, Wilcox CL ( 2001) Inducible cyclic AMP early repressor produces reactivation of latent herpes simplex virus type 1 in neurons in vitro. J Virol 75: 2912–2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson TM, Ginty DD ( 2002) CREB family transcription factors inhibit neuronal suicide. Nat Med 8: 450–451. [DOI] [PubMed] [Google Scholar]
- Dragunow M, Preston K ( 1995) The role of inducible transcription factors in apoptotic nerve cell death. Brain Res Rev 21: 1–28. [DOI] [PubMed] [Google Scholar]
- Figiel I, Kaczmarek L ( 1997) Cellular and molecular correlates of glutamate-evoked neuronal programmed cell death in the in vitro culture of rat hippocampal dentate gyrus. Neurochem Int 31: 229–240. [DOI] [PubMed] [Google Scholar]
- Filipkowski RK, Hetman M, Kaminska B, Kaczmarek L ( 1994) DNA fragmentation in rat brain after intraperitoneal administration of kainate. NeuroReport 5: 1538–1540. [DOI] [PubMed] [Google Scholar]
- Finkbeiner S ( 2000) CREB couples neurotrophin signals to survival messages. Neuron 25: 11–14. [DOI] [PubMed] [Google Scholar]
- Fitzgerald LR, Vaidya VA, Terwilliger RZ, Duman RS ( 1996) Electroconvulsive seizure increases the expression of CREM (cyclic AMP response element modulator) and ICER (inducible camp early repressor) in rat brain. J Neurochem 66: 429–432. [DOI] [PubMed] [Google Scholar]
- Garcia I, Martinou I, Tsujimoto Y, Martinou JC ( 1992) Prevention of programmed cell death of sympathetic neurons by the bcl-2 proto-oncogene. Science 258: 302–304. [DOI] [PubMed] [Google Scholar]
- Gonzalez GA, Montminy MR ( 1989) Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell 59: 675–680. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H ( 2002) Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci 5: 405–414. [DOI] [PubMed] [Google Scholar]
- Herdegen T, Skene P, Bahr M ( 1997) The c-Jun transcription factor—bipotential mediator of neuronal death, survival and regeneration. Trends Neurosci 5: 227–231. [DOI] [PubMed] [Google Scholar]
- Hetman M, Danysz W, Kaczmarek L ( 1997) Increased expression of cathepsin D in retrosplenial cortex of MK-801-treated rats. Exp Neurol 147: 229–237. [DOI] [PubMed] [Google Scholar]
- Hetman M, Kanning K, Cavanaugh JE, Xia Z ( 1999) Neuroprotection by brain-derived neurotrophic factor is mediated by extracellular-signal-regulated kinase and phosphatidylinositol-3 kinase. J Biol Chem 274: 22569–22580. [DOI] [PubMed] [Google Scholar]
- Hu BR, Fux CM, Martone ME, Zivin JA, Ellisman MH ( 1999) Persistent phosphorylation of cyclic AMP responsive element binding protein and activating transcription factor 2 transcription factors following transient cerebral ischemia in rat brain. Neuroscience 89: 437–452. [DOI] [PubMed] [Google Scholar]
- Hughes PE, Alexi T, Walton M, Williams CE, Dragunow M, Clark RG, Gluckman PD ( 1999) Activity and injury-dependent expression of inducible transcription factors, growth factors and apoptosis-related genes within the central nervous system. Prog Neurobiol 57: 421–450. [DOI] [PubMed] [Google Scholar]
- Impey S, Obrietan K, Wong ST, Poser S, Yano S, Wayman G, Deloulme JC, Chan G, Storm DR ( 1998) Cross talk between ERK and PKA is required for Ca 2+ stimulation of CREB-dependent transcription and ERK nuclear translocation. Neuron 21: 869–883. [DOI] [PubMed] [Google Scholar]
- Jaworski J, Figiel I, Proszynski T, Kaczmarek L ( 2000) Efficient expression of tetracycline-responsive gene following transfection of dentate gyrus neurons in vitro. J Neurosci Res 60: 754–760. [DOI] [PubMed] [Google Scholar]
- Kaminska B, Filipkowski RK, Zurkowska G, Lason W, Przewlocki R, Kaczmarek L ( 1994) Dynamic changes in composition of the AP-1 transcription factor DNA binding activity in rat brain following kainate induced seizures and cell death. Eur J Neurosci 6: 1558–1566. [DOI] [PubMed] [Google Scholar]
- Konopka D, Szklarczyk AW, Filipkowski RK, Trauzold A, Nowicka D, Hetman M, Kaczmarek L ( 1998) Plasticity- and neurodegeneration-linked CREM/ICER mRNA expression in the rat brain. Neuroscience 86: 499–510. [DOI] [PubMed] [Google Scholar]
- Kruman II, Nath A, Mattson MP ( 1998) HIV-1 protein Tat induces apoptosis of hippocampal neurons by a mechanism involving caspase activation, calcium overload, and oxidative stress. Exp Neurol 154: 276–288. [DOI] [PubMed] [Google Scholar]
- Kugler S, Meyn L, Holzmuller H, Gerhardt E, Isenmann S, Schulz JB, Bahr M ( 2001) Neuron-specific expression of therapeutic proteins: evaluation of different cellular promoters in recombinant adenoviral vectors. Mol Cell Neurosci 17: 78–96. [DOI] [PubMed] [Google Scholar]
- Lamas M, Molina C, Foulkes NS, Jansen E, Sassone-Corsi P ( 1997) Ectopic ICER expression in pituitary corticotroph AtT20 cells: effects on morphology, cell cycle, and hormonal production. Mol Endocrinol 11: 1425–1434. [DOI] [PubMed] [Google Scholar]
- Le Gal La Salle G, Robert JJ, Berrard S, Ridoux V, Stratford-Perricaudet LD, Perricaudet M, Mallet J ( 1993) An adenovirus vector for gene transfer into neurons and glia in the brain. Science 259: 988–990. [DOI] [PubMed] [Google Scholar]
- Lonze BE, Riccio A, Cohen S, Ginty DD ( 2002) Apoptosis, axonal growth defects, and degeneration of peripheral neurons in mice lacking CREB. Neuron 34: 371–385. [DOI] [PubMed] [Google Scholar]
- Lowenstein PR, Shering AF, Bain D, Castro MG, Wilkinson GWG ( 1996) The use of adenovirus vectors to transfer genes to identified target brain cells in vitro. In: Protocols for gene transfer in neuroscience: towards gene therapy of neurological disorders (Lowenstein PR, Enquist LW, eds), pp 93–114. New York: Wiley.
- Luckman SM, Cox HJ ( 1995) Expression of inducible camp early repressor (ICER) in hypothalamic magnocellular neurons. Mol Brain Res 34: 231–238. [DOI] [PubMed] [Google Scholar]
- Mabuchi T, Kitagawa K, Kuwabara K, Takasawa K, Ohtsuki T, Xia Z, Storm D, Yanagihara T, Hori M, Matsumoto M ( 2001) Phosphorylation of cAMP response element-binding protein in hippocampal neurons as a protective response after exposure to glutamate in vitro and ischemia in vivo. J Neurosci 21: 9204–9213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantamadiotis T, Lemberger T, Bleckman SC, Kern H, Kretz O, Martin-Villalba A, Tronche F, Kellendonk C, Gau D, Kapfhammer J, Otto C, Schmid W, Schutz G ( 2002) Disruption of CREB function in brain leads to neurodegeneration. Nat Genet 31: 47–54. [DOI] [PubMed] [Google Scholar]
- Maronde E, Wicht H, Tasken K, Genieser HG, Dehghani F, Olcese J, Korf HW ( 1999) CREB phosphorylation and melatonin biosynthesis in the rat pineal gland: involvement of cyclic AMP dependent protein kinase type II. J Pineal Res 27: 170–182. [DOI] [PubMed] [Google Scholar]
- Mayr B, Montminy M ( 2001) Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol 2: 599–609. [DOI] [PubMed] [Google Scholar]
- Molina CA, Foulkes NS, Lalli E, Sassone-Corsi P ( 1993) Inducible and negative autoregulation of CREM: an alternative promoter directs the expression of ICER, an early response repressor. Cell 75: 875–886. [DOI] [PubMed] [Google Scholar]
- Nedivi E, Hevroni D, Naot D, Israeli D, Citri Y ( 1993) Numerous candidate plasticity-related genes revealed by differential cDNA cloning. Nature 363: 718–722. [DOI] [PubMed] [Google Scholar]
- Ohki EC, Tilkins ML, Ciccarone VC, Price PJ ( 2001) Improving the transfection efficiency of post-mitotic neurons J Neurosci Methods 112: 95–99. [DOI] [PubMed] [Google Scholar]
- Pettmann B, Henderson CE ( 1998) Neuronal cell death. Neuron 20: 633–647. [DOI] [PubMed] [Google Scholar]
- Pfeffer M, Maronde E, Korf HW, Stehle JH ( 2000) Antisense experiments reveal molecular details on mechanisms of ICER suppressing cAMP-inducible genes in rat pinealocytes. J Pineal Res 29: 24–33. [DOI] [PubMed] [Google Scholar]
- Revah F, Horellou P, Vigne E, Le Gal La Salle G, Robert JJ, Perricaudet M, Mallet J ( 1996) Gene transfer into the central and peripheral nervous system using adenoviral vectors. In: Protocols for gene transfer in neuroscience: towards gene therapy of neurological disorders (Lowenstein PR, Enquist LW, eds), pp 81–92. New York: Wiley.
- Riccio A, Ahn S, Davenport CM, Blendy JA, Ginty DD ( 1999) Mediation by a CREB family transcription factor of NGF-dependent survival of sympathetic neurons. Science 286: 2358–2361. [DOI] [PubMed] [Google Scholar]
- Sastry PS, Rao SK ( 2000) Apoptosis and nervous system. J Neurochem 74: 1–20. [DOI] [PubMed] [Google Scholar]
- Shaywitz AJ, Greenberg ME ( 1999) CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem 68: 821–861. [DOI] [PubMed] [Google Scholar]
- Simons M, Beinroth S, Gleichmann M, Liston P, Korneluk RG, MacKenzie AE, Bahr M, Klockgether T, Robertson GS, Weller M, Schulz JB ( 1999) Adenovirus-mediated gene transfer of inhibitors of apoptosis protein delays apoptosis in cerebellar granule neurons. J Neurochem 72: 292–301. [DOI] [PubMed] [Google Scholar]
- Spencer CM, Houpt TA ( 2001) Dynamics of c-fos and ICER mRNA expression in rat forebrain following lithium chloride injection. Mol Brain Res 93: 113–126. [DOI] [PubMed] [Google Scholar]
- Staiger JF, Bisler S, Schleicher A, Gass P, Stehle JH, Zilles K ( 2000) Exploration of a novel environment leads to the expression of inducible transcription factors in barrel-related columns. Neuroscience 99: 7–16. [DOI] [PubMed] [Google Scholar]
- Stehle JH, Foulkes NS, Molina CA, Simonneaux V, Pevet P, Sassone-Corsi P ( 1993) Adrenergic signals direct rhythmic expression of transcriptional repressor CREM in the pineal gland. Nature 365: 314–320. [DOI] [PubMed] [Google Scholar]
- Stehle JH, von Gall C, Korf HW ( 2001) Analysis of cell signalling in the rodent pineal gland deciphers regulators of dynamic transcription in neural/endocrine cells. Eur J Neurosci 14: 1–9. [DOI] [PubMed] [Google Scholar]
- Storvik M, Linden A-M, Kontkanen O, Lakso M, Castren E, Wong G ( 2000) Induction of cAMP response element modulator (CREM) and inducible cAMP early repressor (ICER) expression in rat brain by uncompetitive N-methyl-d-aspartate receptor antagonist. J Pharmacol Exp Ther 294: 52–60. [PubMed] [Google Scholar]
- Stratford-Perricaudet LD, Makeh I, Perricaudet M, Briand P ( 1992) Wide-spread long-term gene transfer to mouse skeletal muscles and heart. J Clin Invest 90: 626–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinti C, Conti B, Cubells JF, Kim KS, Baker H, Joh TH ( 1996) Inducible cAMP early repressor can modulate tyrosine hydroxylase gene expression after stimulation of cAMP synthesis. J Biol Chem 271: 25375–25381. [DOI] [PubMed] [Google Scholar]
- Trocme C, Ravassard P, Sassone-Corsi P, Mallet J, Biguet NF ( 2001) CREM and ICER are differentially implicated in trans-synaptic induction of tyrosine hydroxylase gene expression in adrenal medulla and sympathetic ganglia of rat. J Neurosci Res 65: 91–99. [DOI] [PubMed] [Google Scholar]
- Vaux DL, Cory S, Adams JM ( 1988) Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature 335: 440–442. [DOI] [PubMed] [Google Scholar]
- Vaux DL, Weissman IL, Kim SK ( 1992) Prevention of programmed cell death in Caenorhabditis elegans by human bcl-2. Science 258: 1955–1957. [DOI] [PubMed] [Google Scholar]
- Walton M, Sirimanne E, Williams C, Gluckman P, Dragunow M ( 1996) The role of the cyclic AMP response element binding protein (CREB) in hypoxic-ischemic brain damage and repair. Mol Brain Res 43: 21–29. [DOI] [PubMed] [Google Scholar]
- Walton MR, Dragunow M ( 2000) Is CREB a key to neuronal survival? Trends Neurosci 23: 48–52. [DOI] [PubMed] [Google Scholar]
- Weeks BS, Lieberman DM, Johnson B, Roque E, Green M, Loewenstein P, Oldfield EH, Kleinman HK ( 1995) Neurotoxicity of the human immunodeficiency virus type 1 tat transactivator to PC12 cells requires the Tat amino acid 49–58 basic domain. J Neurosci Res 42: 34–40. [DOI] [PubMed] [Google Scholar]
- Willingham MC ( 1999) Cytochemical methods for the detection of apoptosis. J Histochem Cytochem 47: 1101–1110. [DOI] [PubMed] [Google Scholar]
- Wilson BE, Mochon E, Boxer LM ( 1996) Induction of bcl-2 expression by phosphorylated CREB proteins during B-cell activation and rescue from apoptosis. Mol Cell Biol 16: 5546–5556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z, Dudek H, Miranti CK, Greenberg ME ( 1996) Calcium influx via NMDA receptor induces immediate early gene transcription by MAP kinase/ERK-dependent mechanism. J Neurosci 16: 5425–5436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Yankner BA ( 2000) Apoptosis in the nervous system. Nature 407: 802–809. [DOI] [PubMed] [Google Scholar]
- Zagulska-Szymczak S, Filipkowski R, Kaczmarek L ( 2001) Kainate-induced genes in the hippocampus: lessons from expression patterns. Neurochem Int 8: 485–501. [DOI] [PubMed] [Google Scholar]
- Zauli G, Secchiero P, Rodella L, Gibellini D, Mirandola P, Mazzoni M, Milani D, Dowd DR, Capitani S, Vitale M ( 2000) HIV-1 Tat-mediated inhibition of the tyrosine hydroxylase gene expression in dopaminergic neuronal cells. J Biol Chem 275: 4159–4165. [DOI] [PubMed] [Google Scholar]
- Zirpel L, Janowiak MA, Veltri CA, Parks TN ( 2000) AMPA receptormediated, calcium-dependent CREB phosphorylation in a subpopulation of auditory neurons surviving activity deprivation. J Neurosci 20: 6267–6275. [DOI] [PMC free article] [PubMed] [Google Scholar]