

Abstract
An important milestone in brain development is the transition of neuroprogenitor cells to postmitotic neurons. We report that the bZIP transcription factor ATF5 plays a major regulatory role in this process. In developing brain ATF5 expression is high within ventricular zones containing neural stem and progenitor cells and is undetectable in postmitotic neurons. In attached clonal neurosphere cultures ATF5 is expressed by neural stem/progenitor cells and is undetectable in tau-positive neurons. In PC12 cell cultures nerve growth factor (NGF) dramatically downregulates endogenous ATF5 protein and transcripts, whereas exogenous ATF5 suppresses NGF-promoted neurite outgrowth. Such inhibition requires the repression of CRE sites. In contrast, loss of function conferred by dominant-negative ATF5 accelerates NGF-promoted neuritogenesis. Exogenous ATF5 also suppresses, and dominant-negative ATF5 and a small-interfering RNA targeted to ATF5 promote, neurogenesis by cultured nestin-positive telencephalic cells. These findings indicate that ATF5 blocks the differentiation of neuroprogenitor cells into neurons and must be downregulated to permit this process to occur.
Keywords: ATF5, neural progenitor cells, NGF, neuron, differentiation, ventricular zone
Introduction
A key step in formation of the nervous system is the decision of proliferating neural progenitor cells to exit the cell cycle and undergo neuronal differentiation. Despite major advances in identification and characterization of such progenitor cells (for review, see Placzek and Furley, 1996; Gage, 2000; Kintner, 2002; Schuurmans and Guillemot, 2002), the mechanisms that govern this decision are understood only partially. One system with potential to address this issue is the PC12 line of pheochromocytoma cells (Greene and Tischler, 1976; Burstein and Greene, 1978). In the presence of the neurotrophic factor nerve growth factor (NGF) proliferating neuroblast-like PC12 cells acquire, by means of a transcription-dependent mechanism, a neuronal phenotype characterized by formation of axons, upregulation of a number of neuronal markers, and transition to a postmitotic state. To identify genes responsible for this neuronal differentiation, we used serial analysis of gene expression (SAGE) to provide a comprehensive profile and comparison of transcripts present in PC12 cells before and after 9 d of treatment with NGF (Angelastro et al., 2000). Of the ∼22,000 unique transcripts detected in the cells, ∼4% underwent a sixfold or greater increase or decrease in expression after NGF exposure. Among the identified genes with the greatest change in expression was ATF5, a member of the activating transcription factor/cAMP response element binding protein (ATF/CREB) family. In response to NGF, ATF5 transcripts, which were among the most highly expressed in the cells before treatment, fell by 25-fold in relative expression.
Relatively few studies have been performed to characterize ATF5 (also known as ATFX and ATF-7) and its biological functions (Nishizawa and Nagata, 1992; Pati et al., 1999; Peters et al., 2001; Persengiev et al., 2002). ATF5 is a bZIP transcription factor that forms homodimers, which, at least in vitro, bind the cAMP response element (CRE). In addition, ATF5 represses cAMP-induced transcription in intact cells (Pati et al., 1999; Peters et al., 2001) and has been shown to inhibit apoptosis (Persengiev et al., 2002). This raised the possibility that ATF5 might interfere with the activity of transcription factors such as CREB that appear to promote neuronal differentiation via CRE-mediated gene activation (Finkbeiner et al., 1997; Dawson and Ginty, 2002; Lonze et al., 2002). These properties, along with its downregulation by NGF, thus suggested that ATF5 might be a negative regulator of neuronal differentiation. The findings reported here indicate that ATF5 blocks the transition of neural progenitor cells into neurons and must be downregulated to permit the occurrence of neuronal differentiation.
Materials and Methods
Reagents. Cell culture media RPMI 1640 and DMEM and molecular biology reagents, Taq platinum DNA polymerase, SuperScript II reverse transcriptase, and Lipofectamine 2000 were obtained from Invitrogen (San Diego, CA). Donor horse and fetal bovine sera were from JRH Biosciences (Lenexa, KS). The Marathon cDNA amplification library kit was from Clontech (Palo Alto, CA), and PCR primers were obtained from Integrated DNA Technologies (Coralville, IA) or Invitrogen. Anti-FLAG M2 antibody was from Sigma (St. Louis, MO).
Cell culture. PC12 cells were grown on collagen-coated dishes as previously described (Greene et al., 1998) with or without human recombinant NGF (a generous gift from Genentech, South San Francisco, CA). Dissociated cultures of telencephalic cells were prepared from E14--E15 Sprague Dawley rats. Telencephalic cells were trypsinized (0.05% in 0.53 mm EDTA; Invitrogen) for 30 min (Li et al., 1998), and dissociated cells were centrifuged and resuspended in DMEM containing 5% FBS, 10 ng/ml epidermal growth factor (EGF), and 20 ng/ml basic fibroblast growth factor (bFGF); they were plated on 24-well dishes coated with poly-l-lysine at 3–5 × 105 cells per well (Laywell et al., 1999). The presence of bFGF promotes proliferation of the progenitor cells but does not interfere with their differentiation into neurons (Ghosh and Greenberg, 1995).
Adherent clonal neurosphere cultures were prepared from newborn mouse subependymal zone cells as previously described (Kukekov et al., 1997, 1999). The cell suspension used to generate neurospheres was filtered through sterile gauze and visually verified to contain only single cells.
Cloning of full-length rATF5 and plasmid constructs. SAGE tag CAT-GAGAACCTAGTC was found in rat expressed sequence tag (EST) UI-R-G0-ur-g-10-0-UI (GenBank/European Bioinformatics Institute accession number AI576016), which in turn showed high homology with the 3′-end of murine ATF5. To clone the open reading frame of rat ATF5, we used PCR antisense primer 5′-CTTGGTTTCTCAGTTGCAC-3′ (derived from the sequence of the above EST) for 5′-RACE (rapid amplification of cDNA ends) PCR, using the Clontech Marathon kit according to the manufacturer's protocol. The first-strand cDNA PCR template was prepared from 5 μg of PC12 cell total RNA by reverse transcription with Superscript II. The products of the 5′-RACE PCR included the second of two potential Kozak start sites. Cloning the rATF5 open reading frame that included the first potential start site was achieved with sense PCR primer 5′-TGCACCTGTGCCTCAGCCATGTC-3′. This sequence was obtained from an EST sequence (GenBank/European Bioinformatics Institute accession number AW917099) that overlapped with the 5′-end of the 5′-RACE PCR product described above. Both potential rATF5 forms were FLAG-tagged by PCR with sense primers 5′-CTCGAGAACCATGGACTACAAGGACGATGATGAC-AAAGGATCACTCCTGGCGACCCT-3′ and 5′-CTCGAGAAGCAT-GGACTACAAGGACGATGATGACAAAGGAGCATCCCTACTCAA-GAA-3′ and with 5′-GAATTCTCGAGCTTGGTTTCTCAGTTGCAC-3′ as the antisense primer for both ATF5s, respectively. NTAzip-ATF5 was constructed by overlapping PCR, using FLAG-tagged ATF5 (potential start site 2 form) as the template. PCR product 1 was produced with 5′-CTCGAGAAGCATGGACTACAAGGACGATGATGACAAAGGAGC-ATCCCTACTCAAGAA-3′and 5′-TTCTTCTGCTTCTTTTTCTAGTAG-TTCTTCGTTTTCTCTTGCTAGTTCTTCTGCTCTTTGTTCGAGGGT-GCTGGCAGGACTAGGATA-3′ as primers, and PCR product 2 was made with 5′-GCAAGAGAAAACGAAGAACTACTAGAAAAAGAAGCA-GAAGAACTAGAACAAGAAATGCAGAGCTAGAGGGCGAGTGCCAA-GGG-3′ and 5′ GAATTCTCGAGCTTGGTTTCTCAGTTGCAC-3′ as primers. Products 1 and 2 were mixed, and the product (FL-NTAzip-ATF5) was PCR-amplified with 5′-CTCGAGAAGCATGGACTACAAGGACG-ATGATGACAAAGGAGCATCCCTACTCAAGAA-3′ and 5′-GAATTC-TCGAGCTTGGTTTCTCAGTTGCAC-3′. To generate NTAzip-ATF5, we removed the activation domain from FL-NTAzip-ATF5 by PCR, using primers sense 5′-GAATTCAACCATGGACTACAAGGACGATGATGA-CAAAATGGCATCTATGACTGGAGGACAACAAATGGGAAGAGACC-CAGACCTCGAACAAAGAGCAGAA-3′ and antisense 5′-GAATTC-TCGAGCTTGGTTTCTCAGTTGCAC-3′. NTAzip-ATF5 was N-terminal FLAG-tagged with a predicted open reading frame of MDYKDDDD-KMASMTGGQQMGRDPDLEQRAEELARENEELLEKEAEELEQENAE-LEGECQGLEARNRELRERAESVEREIQYVKDLLIEVYKARSQRTRSA, where the DNA binding motif was replaced with an amphipathic acidic α-helical sequence as marked in bold (Moll et al., 2000). All PCR products were subcloned into the topoisomerase II (Topo II) pCR 2.1 vector and were sequenced to verify identity. After confirmation, all full-length constructs were subcloned into the EcoRI sites of the pCMS-enhanced green fluorescence protein (eGFP) vector.
Retrovirus plasmids were constructed by blunt ligation of eGFP into the XhoI site of QCX (kindly provided by Dr. Jan Kitajewski, Columbia University) (Julius et al., 2000). Subsequently, full-length FLAG-ATF5 was blunt-ligated into the BsiWI site of QCX-eGFP to form the bicistronic Q vector construct (QC-FLAG-ATF5-eGFP) for retrovirus production.
The CRE-luciferase reporter plasmid was constructed by annealing synthetic oligo 5′-TCGAGTCATGGTAAAAATGACGTCATGGTAAT-TATCATGGTAAAAATGACGTCATGGTAATTATCATGGTAAAAA-TGACGTCATGGTAATTA-3′ to 5′-AGCTTAATTACCATGACGTC-ATTTTTACCATGATAATTACCATGACGTCATTTTTACCATGATAAT-TACCATGACGTCATTTTTACCATGAC-3′ to form a double-stranded DNA (Peters et al., 2001). The annealed DNA was ligated into the XhoI and HindIII sites of the GL3 plasmid.
VP16-CREB (kind gift from Dr. Eric Kandel, Columbia University, New York, NY) was subcloned into the EcoRI and XbaI sites of the pCMS-eGFP vector.
ATF5 antiserum. The CTRGDRKQKKRDQNK peptide corresponding to ATF5 DNA binding domain I (plus an N-terminal cysteine for conjugation to keyhole limpet hemocyanin) was used as the antigen for production of rabbit antiserum (kindly performed by Upstate Biotechnology, Lake Placid, NY).
Western blot analysis. Cultured cells and adult mouse cortex were harvested in Laemmli sample buffer. The protein concentrations were measured by the Bradford assay (Bio-Rad, Hercules, CA); cell proteins were resolved by SDS-PAGE on a 12% gel, and the separated proteins were transferred electrophoretically from the gel to Hybond P membrane (Amersham Biosciences, Arlington Heights, IL) (Towbin et al., 1979). The membranes were blocked for 1 hr in PBS containing 5% milk/1% BSA and were immunolabeled overnight with ATF5 antipeptide antiserum at 1:1000 in PBS containing 5% milk/1% BSA. For detection the blots were washed and probed with goat anti-rabbit horseradish peroxidase (HRP)-conjugated antibody (Pierce, Rockford, IL) and were visualized on film via an enhanced chemiluminescence detection kit (ECL; Amersham Biosciences). For the PC12 cell NGF time course, to normalize for protein loading, the blots were stripped of immunocomplexes as described by Amersham Biosciences and reprobed with ERK1 C-16 antibody (Santa Cruz Biotechnology, Santa Cruz, CA) and goat anti-rabbit HRP-conjugated antibody, followed by ECL film visualization. Densitometry was performed with NIH Image 1.62 software.
Immunohistochemistry. For PC12 cells fluorescence immunohistochemistry was performed as previously described (Angelastro et al., 2001). For dissociated telencephalic cultures the cells were fixed with 4% paraformaldehyde/2% sucrose in PBS for 15 min. After three washes in PBS the cells were blocked in 10% nonimmune goat serum and 0.3% Triton X-100 for 1 hr. The cultures were immunolabeled separately with the following combinations: (1) rabbit anti-GFP (1:1000 dilution; Clontech) and mouse anti-nestin (1:500; rat-401 from the Developmental Studies Hybridoma Bank antibody collection, University of Iowa, Iowa City, IA), (2) rabbit anti-GFP (1:1000 dilution) and mouse TUJ1 (1:2000 dilution; Covance, Princeton, NJ), (3) mouse GFP (1:500; Sigma) and rabbit anti-neurofilament 160 kDa (1:200; kindly provided by Dr. Ronald Liem, Columbia University, New York, NY), or (4) mouse GFP (1:500) and rabbit anti-GFAP (1:500; Dako, Carpinteria, CA) antibody in 10% nonimmune goat serum and 0.3% Triton X-100 for 1 hr, followed by secondary labeling with goat FITC-conjugated anti-rabbit or rhodamine-conjugated anti-mouse antibodies (Alexa) at 1:5000.
For immunolabeling the embryos were fixed in 4% paraformaldehyde in 0.1 m phosphate buffer overnight, subsequently were cryoprotected in 30% sucrose, and then were frozen in O.C.T. compound (Tissue Tek, Miles, Elkhart, IN). Cryosectioned (14 μm) embryos were blocked for 1 hr in 10% nonimmune goat serum and 0.3% Triton X-100. Then the sections were incubated with ATF5 antiserum (1:500) and TUJ1 antibody (1:2000) in 2.5% nonimmune goat serum and 0.3% Triton X-100 overnight. The sections subsequently were incubated for 1 hr with goat FITC-conjugated anti-rabbit and rhodamine-conjugated anti-mouse antibodies in 10% nonimmune goat serum and 0.3% Triton X-100.
For adherent neurospheres the cells were fixed with 4% paraformaldehyde in PBS/2% sucrose for 10 min at room temperature and then permeabilized for 5 min with 0.5% Triton X-100 in ice-cold PBS/2% sucrose. After being blocked with 25% goat or bovine serum in PBS for 20 min, the cultures were incubated with primary antibodies (diluted in 25% serum in PBS for 30 min at room temperature), followed by three washes with PBS, and then were incubated with the appropriate secondary goat anti-rabbit or anti-mouse antibodies conjugated with FITC (Alexa Fluor 488, Molecular Probes, Eugene, OR; A 11001) or Texas Red-X (Molecular Probes, T 6391) for 30 min at room temperature at 1:200. Then the cultures were incubated with the second set of primary and secondary antibodies as above. Immunochemical reagents were anti-AC133/2 antibody (Miltenyi Biotech, Auburn, CA), anti-neurofilament 160 (clone NN18, Sigma), anti-β tubulin isotype III (clone SDL.3D10, Sigma), and goat anti-tau antiserum (clone C-17, Santa Cruz Biotechnology), all diluted according to the manufacturers' recommendations.
Confocal microscopy was performed on either a Zeiss LSM 410 confocal laser-scanning microscope (embryonic brain sections) or on a Bio-Rad Confocal Microscope System 1024ES (neurosphere cultures). Images were obtained under conditions that were identical for both fluorochromes. Confocal images of xy and yz planes confirmed colocalization in brain sections.
In situ hybridization. Nonradioactive in situ hybridization of sections was performed as previously described (Mendelsohn et al., 1999). The antisense ATF5 probe was synthesized by using T3 RNA polymerase and the pCMS-eGFP-ATF5 construct digested with NheI as the template. The corresponding sense probe was synthesized by using T7 RNA polymerase and the pCMS-eGFP-ATF5 construct digested with NotI as the template.
Transient transfections. For PC12 cells the transfection was performed with 0.5 μg of plasmid/well and 6 μl/well of Lipofectamine 2000 for 9 hr, after which time the cells were re-fed with fresh culture medium and then handled as described. For telencephalic cells the transfection was performed with 2.0 μg of plasmid/well and 2 μl/well of Lipofectamine 2000 for 7 hr, followed by an exchange of medium. For transfection of ATF5 small-interfering RNA (siRNA) (AAN19; AAG UCA GCU GCU CUC AGG UAC), 6.67 μg/well of pCMS-eGFP vector was mixed with 80 pmol/well of siRNA in 100 μl of DMEM. An equal amount of DMEM premixed with 1 μl of Lipofectamine 2000/well was added to and mixed with the vector and siRNA. After 30 min the final mixture was added to one-sixth of the volume containing the cells, and the cells were re-fed with fresh culture medium after 7 hr of transfection. For the control the telencephalic cells were transfected with pCMS-eGFP vector alone.
Retrovirus production and infection of telencephalic cells. Nonreplicating retrovirus was made by transfecting subconfluent GP2 293 cells (growing in DMEM plus 10% FBS) with 5 μg of QCX-eGFP or pLeGFP and 5 μg of pVSV-G for the production of empty eGFP retrovirus (as described by Clontech). Likewise, GP2 293 cells were transfected with 5 μgofQC-FLAG-ATF5-eGFP or pLeGFP-FLAG–NTAzip-ATF5 and 5 μg of pVSV-G to make the bicistronic FLAG-ATF5-eGFP or fusion eGFP-FLAG–NTAzip-ATF5 retroviruses, respectively. After 48 hr the medium was collected, and the virus was concentrated by centrifugation at 50,000 × g at 4°C. The final titer was ∼1 × 106 virus particles/ml. The telencephalic cells were infected with 5–10 μl of retrovirus 1 d after plating, and the cells were fixed 7 d after infection.
Scoring of neuronal differentiation. Transfected cells were detected by positive immunostaining for eGFP. Costaining with anti-FLAG established that the GFP-positive PC12 cells also expressed ATF5 constructs. NGF-treated PC12 cells (transfected unless otherwise noted) were scored for processes of length greater than two cell diameters (∼20 μm) (Greene et al., 1998). Transfected telencephalic neurons were scored for the presence of processes with lengths greater than two cell diameters (∼20 μm) and for costaining with TUJ1, nestin, or neurofilament protein (NF-M) antisera/antibodies.
CRE-luciferase reporter assay. PC12 cells were cotransfected with 1 μg of pCMS-eGFP (empty or containing FLAG-tagged ATF5 or FLAG-tagged NTAzip-ATF5) and with 0.2 μg of pGl3-CRE-luciferase reporter and 1 μgof LacZ plasmid/well plus 2 μl/well of Lipofectamine 2000 24 hr before harvesting. The cells were treated with NGF for a total of 1 hr to 3 d. Luciferase levels were assayed by the Promega (Madison, WI) Luciferase System with reporter lysis buffer as described by the manufacturer. The level of LacZ activity was measured as described by Sambrook et al. (1989).
Statistical analyses. Multiple comparisons among the data from different plasmid transfections and retrovirus infections were achieved by Tukey's one-way ANOVA test; comparisons for pairs of data were conducted with Student's t distribution test.
Results
Reciprocal effects of NGF on ATF5 protein expression and neurite outgrowth
Our previous findings revealed that long-term NGF treatment promotes a 25-fold downregulation of ATF5 transcripts in PC12 cells (Angelastro et al., 2000). To determine whether this is reflected at the level of protein expression, we cloned the coding sequence of rat ATF5 (GenBank/European Bioinformatics Institute accession number AY123225) and raised an antiserum against a peptide corresponding to a portion of the deduced sequence of the DNA binding domain. Western immunoblotting with this antiserum detected a single major band in extracts of PC12 cells (Fig. 1A) as well as of HEK-293 cells, primary human neuroblastoma, and mouse brain (data not shown), with an apparent Mr of 20–22 kDa. The nucleotide sequence of rat ATF5 indicates two potential in-frame Kozak start sites, and the apparent Mr of 20–22 kDa indicates preferential use of the second.
Figure 1.
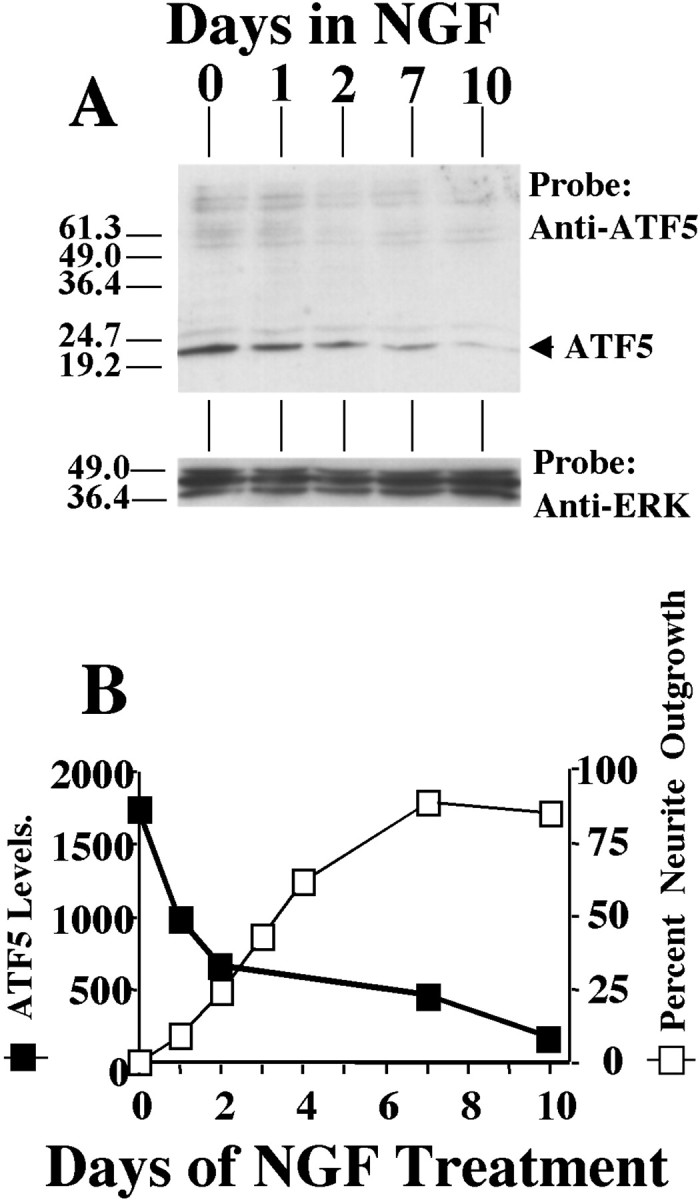
NGF downregulates ATF5 protein in PC12 cells: reciprocal relationship with neurite outgrowth. A, Time course of the effect of NGF treatment on ATF5 protein expression in PC12 cells. Cells were exposed to NGF for the indicated times, and 135 μg of whole cell extracts was subjected to Western immunoblotting with anti-ATF5 and, after stripping, with anti-ERK to normalize for loading. The numbers at left indicate the positions of molecular weight markers (in kDa). Comparable results were achieved in three independent experiments. B, Comparison of the kinetics of NGF-dependent downregulation of ATF5 expression and promotion of neurite outgrowth. The relative levels of ATF5 expression, as determined by densitometry and normalized to levels of ERK protein in the same sample, are reported in arbitrary units. Proportions of cells bearing neurites of length at least twice the diameter of the cell body were determined in the same cultures by scoring at least 200 cells per time point.
A time course of ATF5 protein expression in PC12 cells in response to NGF treatment reveals a drop in levels by 1 d and a progressive fall thereafter, with relatively little detectable expression by 10 d (Fig. 1A,B). Quantification of neurite outgrowth in the same sets of cultures revealed a reciprocal relationship with ATF5 expression (Fig. 1B).
Exogenous ATF5 represses NGF-promoted neurite outgrowth, whereas a dominant-negative ATF5 accelerates initial neuritogenesis
The inverse behaviors of ATF5 expression and neurite outgrowth suggested a possible causal relationship. To test this, we subcloned FLAG-tagged ATF5 into the pCMS-eGFP vector and transfected it into PC12 cells. Two days later NGF was added, and the transfected cells (expressing eGFP and tagged ATF5) were assessed over time for the appearance of neurites. In contrast to cells transfected with empty vector, those expressing exogenous ATF5 showed markedly repressed genesis of neurites over a 5 d time course (Fig. 2A,B).
Figure 2.

Overexpression of ATF5 represses neurite outgrowth in PC12 cells, whereas NTAzip-ATF5 accelerates neuritogenesis. A, Detection and NGF response of PC12 cells expressing exogenous ATF5. PC12 cells were transiently transfected with pCMS-eGFP (a, b) or pCMS-eGFP expressing FLAG-tagged ATF5 (c, d). At 2 d after transfection the cultures were treated with NGF. At 5 d after transfection (i.e., after 3 d of NGF exposure) the cells were fixed and costained with rabbit anti-GFP (a, c) and mouse anti-FLAG antibody (b, d), with detection by FITC (GFP) and rhodamine-conjugated secondary antibody (FLAG-ATF5). Scale bar, 50 μm. B, Quantification of the effects of exogenous ATF5 and of NTAzip-ATF5 on NGF-promoted neurite outgrowth. PC12 cells were transiently transfected with pCMS-eGFP without insert or expressing FLAG-tagged ATF5 or FLAG-tagged NTAzip-ATF5. At 2 d after transfection the cultures were treated with NGF. Cultures were fixed at the indicated times after commencement of NGF exposure and were immunostained with anti-GFP and anti-FLAG as above. Transfected cells (positive for FLAG and/or GFP staining) were assessed for the presence or absence of neurites. The proportions of transfected cells bearing neurites are reported ± SEM; n = 3 cultures (at least 300 transfected cells assessed per culture). Comparable results were achieved in four additional independent experiments, and in all cases (including the data shown) ANOVA analysis indicated p < 0.05 at the 72 hr point of NGF treatment for eGFP versus ATF5. C, NTAzip accelerates NGF-promoted neurite outgrowth. Cultures were transfected, treated, and assessed as in B at 24 hr after NGF exposure. Values represent the mean ± SEM for the results of four independent experiments. In each experiment the data were normalized to the percentage of neurite-bearing cells transfected with pCMS-eGFP. The average percentage of such cells was 10.6 ± 3.7 (NTAzip vs eGFP, p < 0.02; Student's t distribution test).
To assess the possibility that exogenous ATF5 might act at least in part by nonphysiologically sequestering and “squelching” the actions of binding partners, we also prepared a construct encoding an N-terminally truncated form of FLAG-tagged ATF5 possessing an enhanced bZIP domain (NTAzip-ATF5). This was achieved by deleting the N-terminal acidic activation domain and by replacing the DNA binding domain with an amphipathic acidic α-helical sequence containing leucine repeats at each seventh residue. Without activation and DNA binding domains, NTAzip-ATF5 does not interact with DNA or directly affect gene transcription. However, because this protein includes the intact ATF5 leucine zipper, it retains specific interactions with endogenous ATF5 as well as with heterologous binding partners. In addition, the Azip amphipathic acidic α-helical domain should associate tightly with the basic DNA interaction domains of ATF5 binding partners, thereby blocking their functions (Vinson et al., 1993; Krylov et al., 1995; Moitra et al., 1998; Moll et al., 2000). Thus if exogenous ATF5 acts by nonspecific squelching rather than by binding to DNA, NTAzip-ATF5 should have a similar effect. However, in contrast to ATF5, NTAzip-ATF5 did not block NGF-promoted neurite outgrowth (Fig. 2B), thus ruling out a nonspecific action of the former.
In addition to serving as a control for nonspecific squelching, NTAzip-ATF5 acts as a dominant negative for ATF5 and therefore permits evaluation of the consequences of ATF5 loss of function. In the absence of NGF, transfected NTAzip-ATF5 did not stimulate neurite outgrowth (data not shown). However, cells transfected with NTAzip-ATF5 and then exposed to NGF showed a significantly faster (twofold) initial appearance of neurites as compared with controls (Fig. 2C). This reinforces the notion that a physiologic function to ATF5 is to suppress neurite outgrowth and that its downregulation is required for this process to occur. After the first 1–2 d of NGF treatment the effect of NTAzip-ATF5 is much less apparent, presumably because of downregulation of endogenous ATF5.
ATF5 is highly expressed in ventricular zones of developing brain
The suppression of neurite outgrowth by ATF5 in PC12 cells and the potential suitability of this system for modeling the transition of neural progenitor cells to differentiated postmitotic neurons led us to examine the expression of ATF5 in the developing nervous system. In situ hybridization revealed specific expression of ATF5 transcripts in E12–E15 rat embryonic nasal epithelium (see also Hansen et al., 2002), dorsal root and trigeminal ganglia, and brain (data not shown; Fig. 3Aa,Ab). The only signal of comparable strength detected outside the nervous system at these stages was in liver (data not shown). Within E12–E15 rat brain the expression was highest in the ventricular zone (VZ) of the neural epithelium adjacent to the lateral ventricles and the fourth ventricle, sites of intense proliferation of neural cell precursors, and was decreased in overlying structures containing migrating and postmitotic neurons (Fig. 3Aa,Ab).
Figure 3.
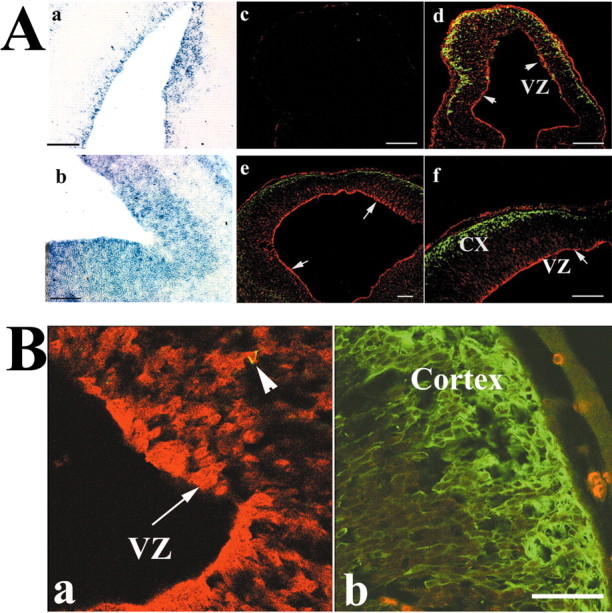
ATF5 is expressed differentially in the ventricular zones of E12–E15 rat brain. Aa, Ab, Expression of ATF5 message in developing rat brain. In situ hybridization was performed by using an ATF5 antisense probe in sagittal sections of E15 rat brain. Shown is the area around the fourth ventricle (a) and the telencephalon (b). There was no positive signal with a control ATF5 sense probe. c–f, Expression of ATF5 protein in coronal sections of E12 (c, d) and E14 (e, f) rat telencephalon. c, Staining with preimmune serum. d–f, Costaining with anti-ATF5 (red) and anti-tubulin β (class III; TUJ1 antibody; green). Arrows indicate staining of ATF5 in the ventricular zone (VZ); CX, cortex. Scale bar, 100 μm. B, High-power confocal images of reciprocal expression of ATF5 (red) and tubulin β (class III) in coronal sections of E14 rat telencephalon. Immunochemical staining was performed as in A. Images showing the ventricular zone (a) and cortex (b) are from the same section and were photographed in the same confocal z-plane section (1.3 μm). Arrowhead shows a migratory cell undergoing a transition from a progenitor to a neuron by exhibiting both ATF5 and tubulinβ (class III) staining. Colocalization was confirmed by yz and xz confocal images. Scale bar, 20 μm.
In view of the pattern of ATF5 transcripts in developing brain, we next examined ATF5 protein expression there by immunohistochemistry. ATF5 protein was expressed strongly in the VZ of E12 and E14 telencephalon and fell to undetectable levels toward the surface of the developing cortex (Fig. 3Ac–Af, B). Double staining with the TUJ1 antibody that recognizes tubulin βIII, a marker for postmitotic neurons (Lee et al., 1990), showed a converse pattern of staining (Fig. 3A,B), thus indicating that ATF5 is highly expressed in proliferating neural progenitor cells and is undetectable in differentiated neurons. A comparable pattern also was observed in E14 rat embryo telencephalon at higher magnification with confocal microscopy (Fig. 3B). At E17 the ATF5 expression remained confined mainly to the VZ in contrast to the large expansion of TUJ1-positive staining in the cortical area (Fig. 4A–F).
Figure 4.

Reciprocal expression of ATF5 and tubulin β (class III) in E17 rat brain. A–C, Expression of ATF5 (red) and tubulin β (class III; green) in the area of the anterior (A–C) and posterior (D–F) lateral ventricles of the E17 rat brain. Immunohistochemical staining was performed as in Figure 3 and as discussed in Materials and Methods. Scale bar, 100 μm.
ATF5 is a marker for neural stem/progenitor cells, but not for mature neurons, in clonal neural progenitor cell cultures
The above findings indicate that ATF5 is highly expressed in proliferating PC12 cells and in VZ progenitor cells, but not in postmitotic neurons. To examine further the correlation between ATF5 expression and neuronal differentiation, we prepared cultures of neural progenitor cells from the neurogenic subventricular zone or hippocampal dentate gyrus of newborn mouse brain. Clones derived from single cell suspensions were expanded and cultured as neurospheres under nonadherent conditions in the presence of EGF, bFGF, and insulin and then were plated onto poly-l-ornithine and laminin with 10% fetal bovine serum to trigger substrate attachment and neurogenesis (Kukekov et al., 1999; Laywell et al., 2000). Cells at the centers of the cultured neurospheres proliferate as stem/progenitor cells, whereas those that migrate to the culture periphery differentiate into neurons and glia (Fig. 5E). ATF5 expression was very high at the three-dimensional core of the cultures. Costaining with antibodies to the AC133 antigen, a marker for hematopoietic and neural stem cells (Yin et al., 1997; Uchida et al., 2000; Bhatia, 2001; Yu et al., 2002), revealed extensive coexpression with ATF5 in this region (Fig. 5A). AC133 antigen localization appeared to be mainly at the cell surface and plasma membrane, whereas ATF5 appeared to be localized mainly to nuclei. ATF5 was also expressed in cells positive for nestin (Fig. 5B), an intermediate filament expressed by neuroectodermal progenitors (Lendahl et al., 1990).
Figure 5.

ATF5 is expressed in neural stem cells and progenitor cells, but not in mature neurons in attached neurosphere cultures. Attached clonal neurosphere cultures were established from the subventricular zone and hippocampal dentate gyrus of newborn mouse brain and were maintained as described in Materials and Methods. Cultures were fixed and costained as follows: A, ATF5 (red) and AC133 (green), a stem cell marker. Thick arrows show examples of nuclear staining; thin arrows show cytoplasmic staining. B, ATF5 (red) and nestin (green), a marker for neural progenitor cells. Arrows indicate nuclear staining. C, D, ATF5 (red) and NF-M(green), a marker for the neuronal lineage. Arrows show nuclear staining in C and cell body in D. E,F, ATF5 (red) and anti-tau (green), a neuronal marker. Comparable results were achieved in 10 independent experiments. Arrows show neurons at the periphery of the cultures; arrowhead shows stem and neural progenitor cells at the center of the culture. Stained cells were examined and photographed by confocal microscopy. Scale bars: A, 20 μm; B–F, 50 μm.
Colocalization experiments also were performed with ATF5 and neuronal markers. The 160 kDa neurofilament protein (NF-M) was detected in cells outgrowing toward the culture periphery. A subpopulation of such cells, which generally appeared to have short neurite-like processes, costained for nuclear ATF5 (Fig. 5C). For such cells the staining of ATF5 and NF-M appeared to be of relatively low intensity, indicating that these were immature neuronal cells in transition with rising levels of NF-M expression and falling levels of ATF5. Another population of cells with more advanced neuronal morphology strongly stained for NF-M but was negative for expression of ATF5 (Fig. 5D). Finally, costaining with antiserum for the neuronal marker tau (Takemura et al., 1991) revealed a set of tau-positive cells at the periphery of the cultures with clear neuronal morphology (Fig. 5E,F). Unlike the progenitor cells in the centers of the cultures that were positive for ATF5 expression and negative for tau, the tau-positive cells in the periphery did not costain for ATF5. Taken together, these observations indicate that ATF5 is expressed in neural stem (AC133+) and progenitor (nestin+) cells, including those committed to the neuronal lineage, and are downregulated in differentiated, postmitotic neurons (tau+).
ATF5 represses, but dominant-negative ATF5 and ATF5 siRNA accelerate, neuronal differentiation of neural progenitor cells
The above described expression pattern of ATF5 raised the possibility that the presence of this protein, as in PC12 cells, may block proliferating neural progenitor cells from undergoing neuronal differentiation. To assess this, we transfected rat E14 telencephalic cell cultures [3 d in vitro (DIV) and containing a mixture of proliferating progenitor cells, postmitotic neurons, and a small number of glial cells (Ghosh and Greenberg, 1995)] with pCMS-eGFP containing no insert, FLAG-ATF5, or FLAG-NTAzip-ATF5; transfected cells (identifiable by eGFP expression) were scored 3 d later for neuronal morphology and expression of nestin and tubulin βIII (Fig. 6A). In contrast with cells transfected with empty vector, few cells transfected with ATF5 exhibited neuronal morphology. In addition, ATF5 greatly repressed expression of the neuronal marker tubulin βIII. On the other hand, ATF5 significantly increased the proportion of cells expressing nestin, a marker for neural progenitor cells. NTAzip-ATF5 did not mimic ATF5, thus (as in the case of PC12 cells) ruling out a potential nonphysiological squelching action of ATF5. As compared with control transfectants, somewhat fewer cells transfected with NTAzip-ATF5 expressed nestin, and there was a tendency for a greater number with neuronal markers.
Figure 6.
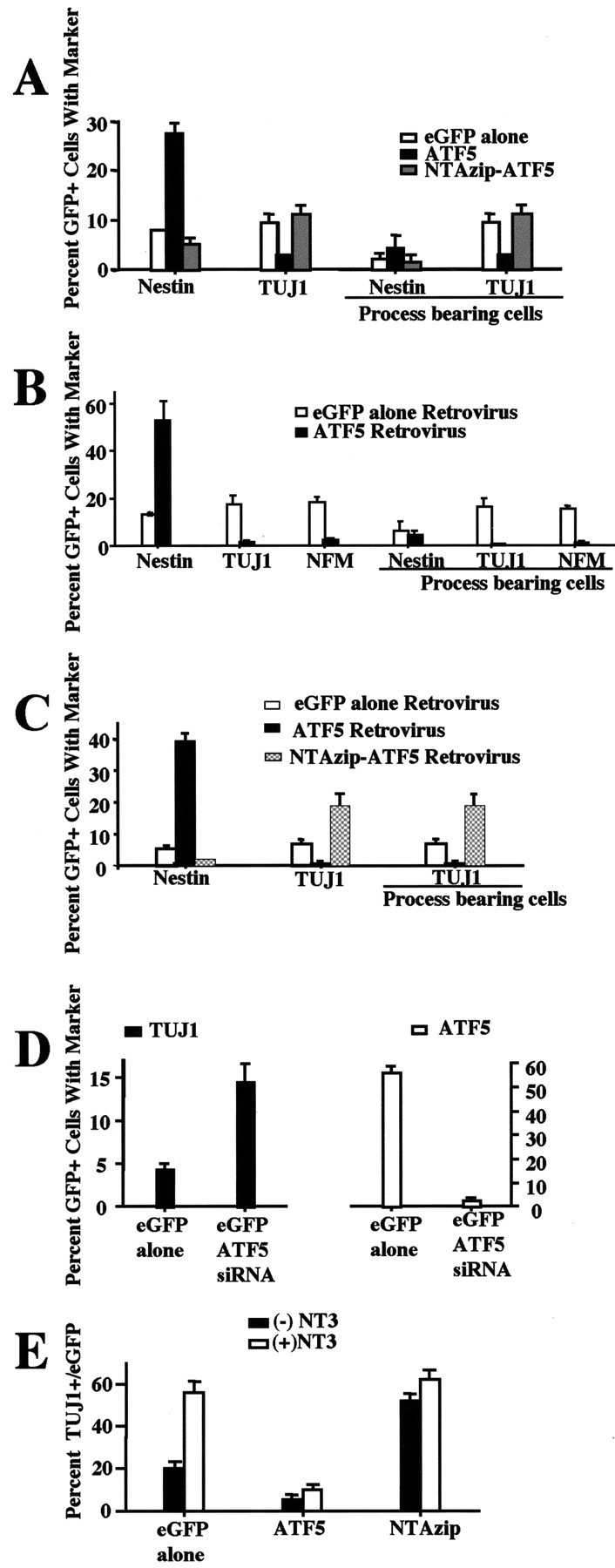
ATF5 represses, and NTAzip-ATF5 promotes, neurite outgrowth and expression of neuronal markers in neural progenitor cells. A, Cultured E14 telencephalic cells were transiently transfected with pCMS-eGFP containing no insert (empty vector), FLAG-ATF5, or NTAzip-ATF5. At 3 d after transfection the cultures were fixed and coimmunostained for GFP and either nestin or tubulinβ (class III; TUJ1 antibody). Transfected cells (GFP +) were assessed for the presence of neurite-like processes as well as for coexpression of the indicated markers. Values represent the mean ± SEM for three cultures in which at least 300 transfected cells were evaluated per culture. Comparable results were achieved in four independent experiments (ANOVA analysis of transfected cells. Total cells: nestin/eGFP alone vs nestin/ATF5, p < 0.001; TUJ1/eGFP alone vs TUJ1/ATF5, p < 0.05; nestin/eGFP alone vs nestin/NTAzip and TUJ1/eGFP alone vs TUJ1/NTAzip, no significant difference. Process-bearing cells: TUJ1/GFP alone vs TUJ1/ATF5, p < 0.05; nestin/eGFP alone vs nestin/NTAzip and TUJ1/eGFP alone vs TUJ1/NTAzip, no significant difference). B, Cultured E14 telencephalic cells were infected with retroviruses expressing eGFP or FLAG-ATF5 and eGFP. At 1 week after infection the cultures were fixed and assessed as in A as well as for NF-M expression. Comparable results were achieved in three independent experiments (ANOVA analysis. Total cells: nestin/eGFP alone vs nestin/ATF5, p < 0.001; TUJ1/GFP alone vs TUJ1/ATF5, p < 0.01; NFM/eGFP vs NFM/ATF5, p < 0.001. Process-bearing cells: TUJ1/GFP alone vs TUJ1/ATF5, p < 0.001; NFM/GFP alone vs NFM/ATF5, p < 0.01; nestin/eGFP alone vs nestin/ATF5, no significant difference. TUJI vs NFM, no significance both with eGFP alone and +ATF5). C, E14 telencephalon cells were infected with retroviruses expressing eGFP, eGFP and FLAG-ATF5, or eGFP-FLAG–NTAzip-ATF5. At 4 d after infection the cultures were fixed and evaluated as in A. Comparable results were achieved in two independent experiments (ANOVA analysis. Nestin/GFP alone vs nestin/ATF5, p < 0.001; total and process-bearing cells: TUJ1/eGFP alone vs TuJ1/ATF5, p < 0.01; TUJ1/GFP alone vs TUJ1/NTAzip, p < 0.05). D, Cultured E14 telencephalic cells were transiently transfected with pCMS-eGFP with or without ATF5 siRNA. At 4 d after transfection the cultures were fixed and coimmunostained either for GFP and TUJ1 antibody or with GFP and ATF5 antiserum. Transfected cells (GFP +) were assessed for the presence of the neuronal marker tubulinβ (class III; TUJ1) or ATF5. Values represent the mean±SEM for six cultures in which atleast 300 transfected cells were evaluated perculture. Comparable results were achieved in three independent experiments (two with E14 telencephalon cells cultured with serum plus EGF and FGF2 and one experiment with only serum) (ANOVA analysis. TUJ1/eGFP alone vs TUJ1/ATF5 siRNA, p < 0.001; ATF5/GFP alone vs ATF5/ATF5 siRNA, p < 0.001). E, ATF5 suppresses NT3-promoted neuronal differentiation. E15 telencephalon cells were infected with retroviruses expressing eGFP, eGFP and FLAG-ATF5, or eGFP-FLAG–NTAzip-ATF5, all ± NT3. At 4 d after infection and maintenance of ± NT3 treatment the cultures were fixed and evaluated as in A for eGFP and TUJ1 expression. Comparable results were achieved in two independent experiments (ANOVA analysis. -NT3/eGFP alone vs +NT3/GFP alone, p < 0.001; -NT3/eGFP alone vs -NT3/ATF5, p < 0.05; +NT3/eGFP alone vs +NT3/ATF5, p < 0.001; -NT3/eGFP alone vs -NT3/NTAzip, p < 0.001; +NT3/GFP alone vs +NT3/NTAzip, no significant difference).
To ensure initial expression only in proliferating cells in our telencephalic cell cultures and to permit transgene delivery at an early point after establishment of the cultures (which was technically unfeasible with our transfection conditions), we constructed and infected them at 1 DIV with retroviral vectors expressing eGFP, eGFP-FLAG–NTAzip-ATF5, or FLAG-ATF5 and eGFP. In this paradigm ATF5 once again suppressed neurite outgrowth and expression of neuronal markers (NF-M and TUJ1) and led to an increase in proportion of nestin-positive cells at either 7 d (Fig. 6B) or 4 d (Fig. 6C) after infection. Moreover, loss of function of endogenous ATF5 promoted by NTAzip-ATF5 significantly enhanced the genesis of neurite-bearing TUJ1-positive cells in cultures assessed at 3 d (data not shown) and 4 d (Fig. 6C) after viral exposure. The dominant-negative construct also promoted a fall in nestin-positive cells, which presumably reflects the increase in neuronal differentiation. The increase in TUJ1-positive cells was greater than can be accounted for by the fall of nestin-positive cells, indicating either that the antibody we used led to an underestimation of the numbers of nestin-positive progenitor cells in the cultures or that at least some neurons were generated from a population of nestin-expressing progenitors. To corroborate our findings that NTAzip-ATF5 accelerates neurogenesis by specifically interfering with the function of endogenous ATF5 rather than via nonspecific actions, we used siRNA to downregulate endogenous ATF5 selectively. After 3 DIV the E14 telencephalic cells were transfected with GFP or with GFP plus ATF5 siRNA. On day 4 after transfection with the siRNA the proportion of transfected cells with detectable endogenous ATF5 fell by 96% as compared with controls (Fig. 6D). Significantly, the reduction of endogenous ATF5 resulted in a 3.4-fold increase in neurogenesis as judged by the appearance of TuJ1 staining (Fig. 6D) and neurite outgrowth (data not shown). In contrast, an irrelevant siRNA synthesized to target downregulation of the protein POSH had no effect on the development of neuronal markers or processes. Taken together, our findings support a model in which ATF5 suppresses the transition between neural progenitor cells and postmitotic neurons and in which loss of or interference with ATF5 function accelerates neuronal differentiation.
The limited degree of neuronal differentiation in the telencephalic cultures appears to occur in response to endogenous factors. To determine whether ATF5 also can regulate CNS neuronal differentiation promoted by a defined trophic agent, we tested the effects of exogenous ATF5 and NTAzip-ATF5 in the presence and absence of NT3, a neurotrophin previously reported to drive telencephalic progenitor cell differentiation into neurons (Ghosh and Greenberg, 1995). As shown in Figure 6E, NT3 nearly tripled the level of neurogenesis in the cultures, and ATF5 suppressed this by five- to sixfold. On the other hand, NTAzip-ATF5, in contrast to its marked promotion of neuronal differentiation in the absence of NT3, had no significant effect on neurogenesis in the presence of NT3. The latter observation would suggest that neuronal differentiation in the cultures is stimulated by NT3 maximally and cannot be promoted further by interfering with endogenous ATF5 activity. Moreover, it appears that NT3 leads to the downregulation of endogenous ATF5 in that none of the neurons formed in its presence exhibited detectable ATF5 immunostaining (data not shown). In conclusion, these findings indicate that, as in the case of NGF, NT3 promotes neurogenesis by a mechanism that can be suppressed by exogenous ATF5 and that includes loss of endogenous ATF5 expression.
Inhibition of neurite outgrowth by ATF5 involves repression of CRE transactivation
The work of Peters et al. (2001) has established that ATF5 homodimers specifically bind to CRE elements, and there is evidence that CRE plays an important role in neuronal differentiation and maintenance (Finkbeiner et al., 1997). We therefore next determined whether ATF5 regulates CRE activity in neuronal cells and whether this action plays a role in ATF5-mediated suppression of neuronal differentiation. We also wished to determine whether the presence of NGF would affect the capacity of ATF5 to regulate CRE activity. Accordingly, PC12 cells were cotransfected with a CRE-luciferase reporter construct, a lacZ expression construct (for normalization of transfection efficiency), and pCMS-eGFP containing no insert, FLAG-ATF5, or FLAG-NTAzip-ATF5; 1 d later the cells were harvested and assessed for reporter activity. A portion of the cultures were treated with NGF for 2 d before and during the 24 hr after transfection (3 d NGF treatment), whereas others either were unexposed to NGF or were exposed to the factor at the time of transfection (1 d NGF treatment) or during the last hour before harvesting (1 hr NGF treatment). Without or after 1 hr of NGF treatment there was relatively little constitutive CRE transactivation. The effect of exogenous ATF5 was somewhat variable at this time, with suppression of activity in some experiments and not others (Fig. 7A,B), possibly reflecting cell culture conditions. At 1 d with NGF there was a small (50%) but statistically significant increase in CRE activity in comparison with naive cells, and this was reduced to baseline by exogenous ATF5; at day 3 there was a 10-fold increase in CRE reporter activity as compared with untreated cells, and this again was reduced substantially by exogenous ATF5. NTAzip-ATF5 did not reduce CRE activity, thus making it unlikely that ATF5 interferes with CRE transactivation by nonphysiologic interaction with CRE-regulatory proteins. Moreover, neither ATF5 nor NTAzip-ATF5 expression suppressed expression of a SRE reporter (data not shown). In addition to establishing that ATF5 suppresses CRE transactivation in intact neuronal cells, these findings indicate that NGF elevates basal CRE activity and that this occurs at a time when endogenous ATF5 levels have fallen by approximately two-thirds (Fig. 1).
Figure 7.
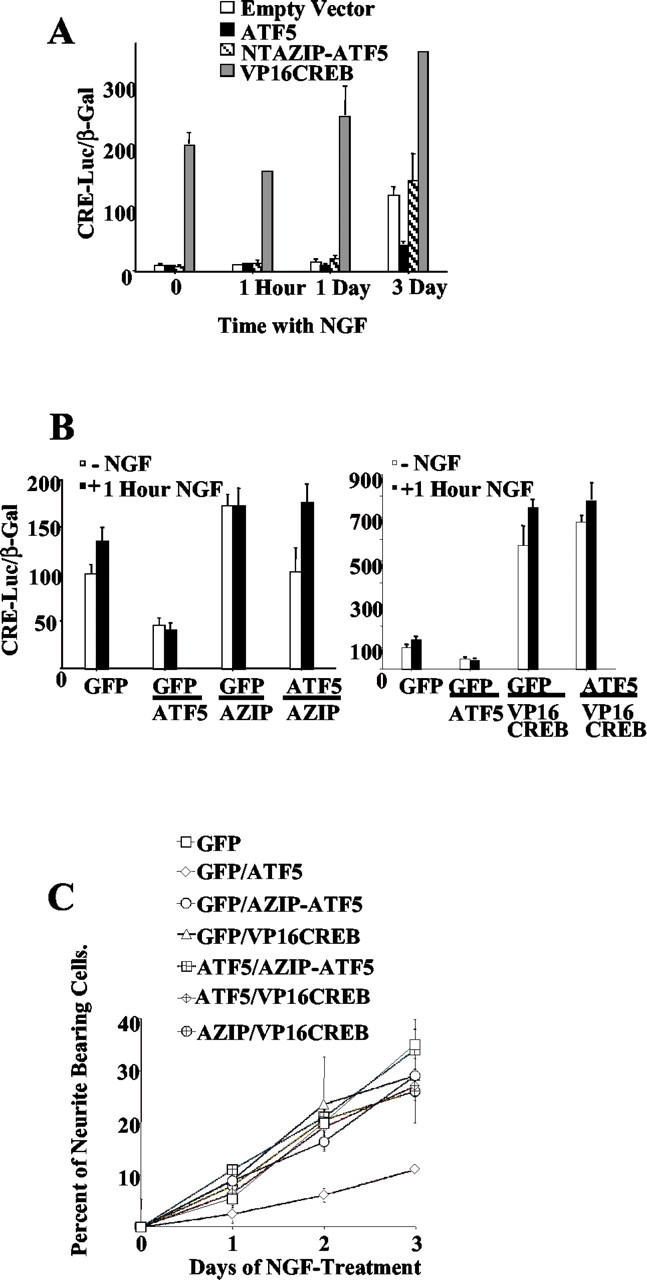
NTAzip-ATF5 and VP16-CREB reverse ATF5-promoted repression of CRE-mediated gene expression and of neurite outgrowth. A, PC12 cells were cotransfected with pGl3-CRE-luciferase, pcDNA-LacZ, and 1 μg/culture of pCMS-eGFP expressing no insert (empty vector), FLAG-ATF5, FLAG-NTAzip-ATF5, or VP16-CREB. The cultures also were exposed to NGF for 2 d before and during the time of transfection (for a total of 3 d), during the time of transfection (1 d), or during the last hour before harvesting. At 1 d after transfection the cells were harvested and assessed for luciferase expression and LacZ activity (β-gal). Values represent mean normalized CRE-luciferase activity (in arbitrary units) ± SEM (n = 3). Comparable results were achieved in three independent experiments [Student's t distribution test. Empty vector (eGFP alone) vs VP16-CREB at all times, p < 0.001; GFP alone vs ATF5, p < 0.033 by day 3]. B, PC12 cells were cotransfected with pGl3-CRE-luciferase, pcDNA-LacZ, and the indicated combinations of pCMS-eGFPexpressing no insert(GFP),FLAG-ATF5(ATF5),FLAG-NTAzip-ATF5(AZIP), or VP16-CREB. The latter vectors were used each at 0.5 μg/culture, and empty vector was added as needed to bring the total DNA level to 1 μg/culture. Cultures were harvested 1 d later for assay of luciferase expression and LacZ activity (β-gal). Where indicated, NGF was added to the medium 1 hr before harvesting. Values represent mean normalized CRE-luciferase activity (in arbitrary units) ± SEM (n = 6), with data pooled from two independent experiments (Student's t distribution test. -NGF eGFP alone vs ATF5, p < 0.003; eGFP alone vs NTAzip, p < 0.0003; eGFP alone vs ATF5/NTAzip, no significant difference; eGFP alone vs VP16-CREB and VP16-CREB/ATF5, p < 0.0001. +NGF eGFP alone vs ATF5, p < 0.0001; eGFP alone vs NTAzip, p < 0.02; eGFP alone vs ATF5/NTAzip, p < 0.02; eGFP alone vs VP16-CREB and VP16-CREB/ATF5, p < 0.0001). C, PC12 cells were cotransfected with the indicated constructs, and NGF was added to the medium 2 d later. Transfected cells (identified for eGFP) were assessed for neurite outgrowth at the indicated times. Values represent the mean ± SEM of results for three cultures in which at least 300 transfected cells were scored per culture. Comparable results were obtained in two independent experiments (ANOVA analysis after 72 hr of NGF-treatment. eGFP alone vs ATF5, p < 0.001; eGFP alone vs NTAzip-ATF5, NTAzip/ATF5, VP16-CREB, or VP16-CREB/ATF5, no significant difference).
If ATF5 suppresses neuronal differentiation by binding to CRE and inhibiting its transactivation, then one would predict that this action should be reversed either by a dominant-negative ATF5 protein without DNA binding or activation sites or by a strong competitive CRE activator. The former characteristics are fulfilled by NTAzip-ATF5, which should form tight heterodimers with ATF5 but does not bind DNA. In support of our hypothesis, coexpression of NTAzip-ATF5 blocked inhibition of CRE reporter activity by ATF5 (Fig. 7B) and reversed ATF5-dependent suppression of NGF-promoted neurite outgrowth (Fig. 7C).
With respect to a competitive CRE activator, we used VP16-CREB, a constitutively active form of the CRE binding protein CREB (Lu et al., 1998; Barco et al., 2002). Cotransfection of pCMS-eGFP–VP16-CREB into PC12 cells produced strong transactivation of the CRE reporter (Fig. 7A), and this essentially was unaffected by the additional cotransfection of FLAG-ATF5 (Fig. 7B). We next assessed whether driving CRE with VP16-CREB would reverse the actions of ATF5 on neurite outgrowth. Transfection of PC12 cells with pCMS-eGFP–VP16-CREB alone did not elicit neurite outgrowth in the absence of NGF and, as in the case of FLAG-NTAzip-ATF5, enhanced the initial rate of neuritogenesis in the presence of NGF (Fig. 7C). Significantly, cotransfection of VP16-CREB along with FLAG-ATF5 reversed the suppression of NGF-stimulated neurite outgrowth achieved with ATF5 alone (Fig. 7C). Taken together, these findings further support a model in which CRE transactivation is required for neuronal differentiation but is blocked reversibly by ATF5.
Discussion
Regulation of endogenous ATF5 protein in PC12 cells and neural progenitor cells
In consonance with our past observations of ATF5 transcripts, we found that ATF5 protein is expressed in PC12 cells and drops to nearly undetectable levels during NGF-promoted neuronal differentiation. Similarly, both ATF5 transcripts and protein are highly expressed in neural progenitor cells and absent from postmitotic neurons. The observed fall in ATF5 protein expression most likely reflects the downregulation of ATF5 transcripts. ATF5 has been reported to be a substrate for ubiquitin-conjugating enzymes, including Cdc34 (Pati et al., 1999), and hence is likely to have a relatively rapid turnover that would produce efficient loss of expression after transcriptional downregulation.
Western immunoblotting permitted us to deduce the major cellular form of ATF5 protein. The ATF5 cDNA sequence predicts two potential in-frame methionine start sites that would lead to proteins of ∼30 and 20 kDa. Our observation that the major form of ATF5 in cells is of an apparent Mr of 20–22 kDa indicates favored use of the second site. When a canonical Kozak initiation consensus sequence was included upstream of the first methionine, the larger protein was expressed (data not shown), thus indicating that the 22 kDa form is not formed by cleavage of a 30 kDa precursor.
ATF5 represses neuronal differentiation of neural progenitor cells
The downregulation of ATF5 expression by NGF in PC12 cells, the progressive loss of ATF5 expression that occurs as cells leave the ventricular zone and enter the developing cortex, and the presence of ATF5 in neural stem and progenitor cells, but not in well differentiated neurons in neurosphere cultures, suggested that this factor may play a causal role in regulating neuronal differentiation. In support of this supposition exogenous ATF5 suppressed both neurite outgrowth in PC12 cell cultures and differentiation of cultured neural progenitor cells. Conversely, loss of ATF5 function (evoked by NTAzip, an ATF5 dominant negative) nearly doubled the initial rate of NGF-promoted neuritogenesis by PC12 cells and significantly enhanced neurogenesis in telencephalic cell cultures. In support of this an ATF5 siRNA that effectively reduced endogenous ATF5 levels also promoted a 3.6-fold enhancement of neurogenesis by cultured telencephalic cells.
The effect of exogenous ATF5 does not appear to be limited solely to neurite outgrowth in that virally induced ATF5 expression in proliferating progenitor cells also blocked the appearance of several neuronal markers and led to an increase in the numbers of cells that expressed nestin, a marker for neural progenitor cells. The increase in the numbers of nestin-positive cells induced by exogenous ATF5 appeared to be greater than could be accounted for merely by blocking progenitor cell differentiation. One possible explanation is that nestin-positive cells expressing exogenous ATF5 continued to proliferate rather than leaving the cell cycle and differentiating. However, further studies will be necessary to confirm whether ATF5 plays a role in directly blocking proliferating progenitor cells from acquiring a postmitotic state.
Taken together, our observations with developing rat brain and neurosphere cultures indicate a scenario in which ATF5 is highly expressed in neural stem and progenitor cells and suppresses their differentiation. The action of appropriate neurotrophic factors leads to downregulation of ATF5, thereby permitting differentiation of neural progenitor cells into neurons. Our present findings thus suggest that ATF5 acts in a permissive rather than instructional manner in that it does not appear to play a role in specifying cell fate directly per se but rather acts as a negative suppressor that must be downregulated to permit the transition of neural progenitor cells to neurons. In this role ATF5 would function to prevent stem and progenitor cells from undergoing terminal differentiation until stimulated by appropriate neurotrophic agents.
Further support for the notion that ATF5 acts as a negative permissive regulator rather than as an instructional factor comes from our observations with NTAzip-ATF5. This modified form of ATF5 should act as a dominant negative that prevents interaction of ATF5 with DNA as well as with other potential protein binding partners. This is borne out by the capacity of NTAzip-ATF5 to reverse the effect of ATF5 on CRE reporter activity.
Nevertheless, when expressed in PC12 cells, NTAzip did not promote neurite outgrowth in the absence of NGF. Thus although ATF5 downregulation appears to be necessary for neuronal differentiation, loss of ATF5 activity does not appear to be sufficient to promote this process. Factors such as NGF appear both to downregulate negative permissive agents such as ATF5 and to provide instructional information that actively promotes neuronal differentiation. In the CNS neuroprogenitor cultures that were used here, downregulation and instructional activity were likely to be supplied by endogenously synthesized and released factors such as NT3 and BDNF (Ghosh and Greenberg, 1995).
The expression pattern of NGF during embryogenesis makes it unlikely that this factor is a key regulator of ATF5 expression in developing brain. However, many other potential neurotrophic factors are present there that could fulfill a similar role. For instance, BDNF and NT3 and their cognate receptors TrkB and TrkC are present in rat ventricular progenitor cells at E13 and E15 (Fukumitsu et al., 1998), and BDNF (Ahmed et al., 1995) and NT3 (Ghosh and Greenberg, 1995) promote differentiation of cultured neuronal progenitor cells.
Our experiments were focused on neuronal differentiation and did not establish whether ATF5 also affects glial cell differentiation. However, the localization of ATF5 in brain areas that also give rise to glial progenitor cells, its colocalization with nestin, which is present in progenitor cells for both neurons and glia, and our preliminary observations that ATF5 colocalizes with GFAP in neuroprogenitor cell cultures and that exogenous ATF5 suppresses GFAP expression suggest that ATF5 also may be a negative regulator of astrocyte differentiation.
Although ATF5 expression negatively correlates with neuronal differentiation, this may not be the case universally for differentiation of other cell types. Peters et al. (2001) reported that ATF5 transcripts were elevated markedly when human Caco-2 cells reached confluency and spontaneously differentiated into a brush border-bearing polarized cell layer.
Suppression of neuronal differentiation by ATF5 involves CRE
On the basis of reports that ATF5 homodimers bind CRE, but not C/EBP or AP1, sites (Peters et al., 2001) and that ATF5 represses cAMP-mediated activation of a CRE reporter in JEG3 cells (Pati et al., 1999), we examined the effect of ATF5 on the activity of a CRE reporter in PC12 cells. Our findings confirm that ATF5 suppresses cellular CRE transactivation. As discussed above, it is significant that NTAzip-ATF5 did not mimic the suppressive actions of ATF5 on neurite outgrowth and CRE activity but rather antagonized these effects, thus indicating that ATF5 acts by binding to DNA rather than by nonspecific “squelching” of binding partners.
We observed that basal CRE activity substantially increased by 3 d of NGF treatment. One potential cause for this is the concurrent fall in endogenous ATF5 expression and subsequent loss of ATF5-mediated CRE repression; however, we cannot rule out the possibility that NGF regulates additional proteins that affect CRE activity.
Although NTAzip-ATF5 blocked the inhibitory effects of exogenous ATF5 on its own, it had no or relatively little effect on CRE reporter activity. If, as we propose, CRE-dependent gene activation is suppressed by endogenous ATF5, then it might have been anticipated that basal CRE activation would be elevated in response to NTAzip-ATF5. Because this was not the case, this raises the possibility that one or more factors in addition to ATF5 act to suppress CRE in neural progenitor cells and that these also are downregulated during neuronal differentiation.
To assess whether interference with CRE-mediated gene regulation might account for the mechanism by which ATF5 interferes with neuronal differentiation, we coexpressed it with VP16-CREB, a constitutively active fusion protein that includes the CREB DNA binding domain and transactivation domain of the HSV VP16 protein. VP16-CREB potently activated the CRE reporter, and this effect was not blocked by coexpression of ATF5. Significantly, coexpressed VP16-CREB overrode ATF5-mediated inhibition of neurite outgrowth. This finding supports a model in which neuronal differentiation requires CRE-mediated gene activation and in which such activation is repressed in neural progenitor cells by factors such as ATF5. In this light, it is of interest that PACAP, a potent activator of adenylate cyclase, promotes mitotic exit and neuronal differentiation of cultured cortical neuron precursor cells (Dicicco-Bloom et al., 1998) and that NGF-promoted differentiation of PC12 cells is synergized by cell-permeant cAMP derivatives (Gunning et al., 1981).
In summary, our findings indicate that both positive and negative regulators govern the transition of neural progenitor cells to neurons. On one hand, ATF5 is highly expressed in neural stem and progenitor cells and suppresses their neuronal differentiation, apparently by competing for binding to CREs. On the other hand, neuronal differentiation is accompanied by and appears to require the downregulation of ATF5 expression. This can be accomplished by neurotrophic factors such as NGF and NT3. Although such downregulation may be necessary, it is not sufficient to permit neuronal differentiation. The latter also appears to require instructive signals that may be imparted by neurotrophic factors and/or activators of adenylate cyclase.
Footnotes
This work was supported in part by grants from National Institutes of Health (NIH)–National Institute of Neurological Disorders and Stroke (L.A.G., D.A.S.) and NIH (C.M.) and by the McKnight Brain Institute and University of Florida Shands Cancer Center (T.I., V.K.). G.S. was supported by grants from the Columbia University Summer Research Program for Science Teachers. We thank Drs. James Goldman and Arnold Kriegstein for helpful discussions, Claudine Bitel and Ekatherina Batourina for outstanding technical assistance, and Timothy W. Vaught for superb assistance and performance of confocal microscopy.
Correspondence should be addressed to James M. Angelastro, Columbia University College of Physicians and Surgeons, 15-401, 630 West 168th Street, New York, NY 10032. E-mail: jma14@columbia.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/234590-11$15.00/0
References
- Ahmed S, Reynolds BA, Weiss S ( 1995) BDNF enhances the differentiation but not the survival of CNS stem cell-derived neuronal precursors. J Neurosci 15: 5765–5778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelastro JM, Klimaschewski L, Tang S, Vitolo OV, Weissman TA, Donlin LT, Shelanski ML, Greene LA ( 2000) Identification of diverse nerve growth factor-regulated genes by serial analysis of gene expression (SAGE) profiling. Proc Natl Acad Sci USA 97: 10424–10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelastro JM, Moon NY, Liu DX, Yang AS, Greene LA, Franke TF ( 2001) Characterization of a novel isoform of caspase-9 that inhibits apoptosis. J Biol Chem 276: 12190–12200. [DOI] [PubMed] [Google Scholar]
- Barco A, Alarcon JM, Kandel ER ( 2002) Expression of constitutively active CREB protein facilitates the late phase of long-term potentiation by enhancing synaptic capture. Cell 108: 689–703. [DOI] [PubMed] [Google Scholar]
- Bhatia M ( 2001) AC133 expression in human stem cells. Leukemia 15: 1685–1688. [DOI] [PubMed] [Google Scholar]
- Burstein DE, Greene LA ( 1978) Evidence for RNA synthesis-dependent and -independent pathways in stimulation of neurite outgrowth by nerve growth factor. Proc Natl Acad Sci USA 75: 6059–6063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson TM, Ginty DD ( 2002) CREB family transcription factors inhibit neuronal suicide. Nat Med 8: 450–451. [DOI] [PubMed] [Google Scholar]
- Dicicco-Bloom E, Lu N, Pintar JE, Zhang J ( 1998) The PACAP ligand/receptor system regulates cerebral cortical neurogenesis. Ann NY Acad Sci 865: 274–289. [DOI] [PubMed] [Google Scholar]
- Finkbeiner S, Tavazoie SF, Maloratsky A, Jacobs KM, Harris KM, Greenberg ME ( 1997) CREB: a major mediator of neuronal neurotrophin responses. Neuron 19: 1031–1047. [DOI] [PubMed] [Google Scholar]
- Fukumitsu H, Furukawa Y, Tsusaka M, Kinukawa H, Nitta A, Nomoto H, Mima T, Furukawa S ( 1998) Simultaneous expression of brain-derived neurotrophic factor and neurotrophin-3 in Cajal–Retzius, subplate, and ventricular progenitor cells during early development stages of the rat cerebral cortex. Neuroscience 84: 115–127. [DOI] [PubMed] [Google Scholar]
- Gage FH ( 2000) Mammalian neural stem cells. Science 287: 1433–1438. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Greenberg ME ( 1995) Distinct roles for bFGF and NT-3 in the regulation of cortical neurogenesis. Neuron 15: 89–103. [DOI] [PubMed] [Google Scholar]
- Greene LA, Tischler AS ( 1976) Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci USA 73: 2424–2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene LA, Farinelli SE, Cunningham ME, Park DS ( 1998) Culture and experimental use of the PC12 rat pheochromocytoma cell line. In: Culturing nerve cells, 2nd Ed (Goslin K, Banker G, eds), pp 161–187. Cambridge, MA: MIT.
- Gunning PW, Landreth GE, Bothwell MA, Shooter EM ( 1981) Differential and synergistic actions of nerve growth factor and cyclic AMP in PC12 cells. J Cell Biol 89: 240–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen MB, Mitchelmore C, Kjaerulff KM, Rasmussen TE, Pedersen KM, Jensen NA ( 2002) Mouse Atf5: molecular cloning of two novel mRNAs, genomic organization, and odorant sensory neuron localization. Genomics 80: 344–350. [DOI] [PubMed] [Google Scholar]
- Julius MA, Yan Q, Zheng Z, Kitajewski J ( 2000) Q vectors, bicistronic retroviral vectors for gene transfer. Biotechniques 28: 702–708. [DOI] [PubMed] [Google Scholar]
- Kintner C ( 2002) Neurogenesis in embryos and in adult neural stem cells. J Neurosci 22: 639–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krylov D, Olive M, Vinson C ( 1995) Extending dimerization interfaces: the bZIP basic region can form a coiled coil. EMBO J 14: 5329–5337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukekov VG, Laywell ED, Thomas LB, Steindler DA ( 1997) A nestin-negative precursor cell from the adult mouse brain gives rise to neurons and glia. Glia 21: 399–407. [DOI] [PubMed] [Google Scholar]
- Kukekov VG, Laywell ED, Suslov O, Davies K, Scheffler B, Thomas LB, O'Brien TF, Kusakabe M, Steindler DA ( 1999) Multipotent stem/progenitor cells with similar properties arise from two neurogenic regions of adult human brain. Exp Neurol 156: 333–344. [DOI] [PubMed] [Google Scholar]
- Laywell ED, Kukekov VG, Steindler DA ( 1999) Multipotent neurospheres can be derived from forebrain subependymal zone and spinal cord of adult mice after protracted postmortem intervals. Exp Neurol 156: 430–433. [DOI] [PubMed] [Google Scholar]
- Laywell ED, Rakic P, Kukekov VG, Holland EC, Steindler DA ( 2000) Identification of a multipotent astrocytic stem cell in the immature and adult mouse brain. Proc Natl Acad Sci USA 97: 13883–13888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MK, Rebhun LI, Frankfurter A ( 1990) Posttranslational modification of class III β-tubulin. Proc Natl Acad Sci USA 87: 7195–7199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lendahl U, Zimmerman LB, McKay RD ( 1990) CNS stem cells express a new class of intermediate filament protein. Cell 60: 585–595. [DOI] [PubMed] [Google Scholar]
- Li W, Cogswell CA, LoTurco JJ ( 1998) Neuronal differentiation of precursors in the neocortical ventricular zone is triggered by BMP. J Neurosci 18: 8853–8862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonze BE, Riccio A, Cohen S, Ginty DD ( 2002) Apoptosis, axonal growth defects, and degeneration of peripheral neurons in mice lacking CREB. Neuron 34: 371–385. [DOI] [PubMed] [Google Scholar]
- Lu R, Yang P, Padmakumar S, Misra V ( 1998) The herpesvirus transactivator VP16 mimics a human basic domain leucine zipper protein, luman, in its interaction with HCF. J Virol 72: 6291–6297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelsohn C, Batourina E, Fung S, Gilbert T, Dodd J ( 1999) Stromal cells mediate retinoid-dependent functions essential for renal development. Development 126: 1139–1148. [DOI] [PubMed] [Google Scholar]
- Moitra J, Mason MM, Olive M, Krylov D, Gavrilova O, Marcus-Samuels B, Feigenbaum L, Lee E, Aoyama T, Eckhaus M, Reitman ML, Vinson C ( 1998) Life without white fat: a transgenic mouse. Genes Dev 12: 3168–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moll JR, Olive M, Vinson C ( 2000) Attractive interhelical electrostatic interactions in the proline- and acidic-rich region (PAR) leucine zipper subfamily preclude heterodimerization with other basic leucine zipper subfamilies. J Biol Chem 275: 34826–34832. [DOI] [PubMed] [Google Scholar]
- Nishizawa M, Nagata S ( 1992) cDNA clones encoding leucine-zipper proteins which interact with G-CSF gene promoter element 1-binding protein. FEBS Lett 299: 36–38. [DOI] [PubMed] [Google Scholar]
- Pati D, Meistrich ML, Plon SE ( 1999) Human Cdc34 and Rad6B ubiquitin-conjugating enzymes target repressors of cyclic AMP-induced transcription for proteolysis. Mol Cell Biol 19: 5001–5013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persengiev SP, Devireddy LR, Green MR ( 2002) Inhibition of apoptosis by ATFx: a novel role for a member of the ATF/CREB family of mammalian bZIP transcription factors. Genes Dev 16: 1806–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters CS, Liang X, Li S, Kannan S, Peng Y, Taub R, Diamond RH ( 2001) ATF-7, a novel bZIP protein, interacts with the PRL-1 protein-tyrosine phosphatase. J Biol Chem 276: 13718–13726. [DOI] [PubMed] [Google Scholar]
- Placzek M, Furley A ( 1996) Patterning cascades in the neural tube. Neural development. Curr Biol 6: 526–529. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T ( 1989) Molecular cloning: a laboratory manual, 2nd Ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory.
- Schuurmans C, Guillemot F ( 2002) Molecular mechanisms underlying cell fate specification in the developing telencephalon. Curr Opin Neurobiol 12: 26–34. [DOI] [PubMed] [Google Scholar]
- Takemura R, Kanai Y, Hirokawa N ( 1991) In situ localization of tau mRNA in developing rat brain. Neuroscience 44: 393–407. [DOI] [PubMed] [Google Scholar]
- Towbin H, Staehelin T, Gordon J ( 1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA 76: 4350–4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida N, Buck DW, He D, Reitsma MJ, Masek M, Phan TV, Tsukamoto AS, Gage FH, Weissman IL ( 2000) Direct isolation of human central nervous system stem cells. Proc Natl Acad Sci USA 97: 14720–14725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinson CR, Hai T, Boyd SM ( 1993) Dimerization specificity of the leucine zipper-containing bZIP motif on DNA binding: prediction and rational design. Genes Dev 7: 1047–1058. [DOI] [PubMed] [Google Scholar]
- Yin AH, Miraglia S, Zanjani ED, Almeida-Porada G, Ogawa M, Leary AG, Olweus J, Kearney J, Buck DW ( 1997) AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood 90: 5002–5012. [PubMed] [Google Scholar]
- Yu Y, Flint A, Dvorin EL, Bischoff J ( 2002) AC133-2, a novel isoform of human AC133 stem cell antigen. J Biol Chem 277: 20711–20716. [DOI] [PubMed] [Google Scholar]