

Abstract
γ-Secretase cleavage is the final enzymatic step generating β-amyloid via intramembranous cleavage of the amyloid precursor protein (APP). Presenilin (PS), initially identified as a gene in which mutations account for the vast majority of early-onset autosomal dominant Alzheimer's disease, is a major component of γ-secretase. Enzymatic activity also depends on nicastrin, Aph-1, and Pen-2. We propose a model in which γ-secretase components assemble, interact with substrates initially at a docking site, and then cleave and release substrates. To test this model, we developed a novel morphological technique on the basis of advanced fluorescence microscopy methods, fluorescence lifetime imaging microscopy (FLIM). FLIM allows us to examine protein–protein “proximity” in intact cells. We show that, although the strongest colocalization of APP and PS1 is in the perinuclear area, the strongest interactions detected by FLIM are at or near the cell surface. We also found that APP–PS1 interactions occur even whenγ-secretase inhibitors or “dominant-negative” PS1 mutations are used to block γ-secretase activity. Finally, using nicastrin RNA interference, we demonstrate that nicastrin is critical for APP association with PS1. We interpret these results to suggest that there is a noncatalytic docking site closely associated with PS1–γ-secretase.
Keywords: APP, nicastrin, presenilin, γ-secretase, docking site, FLIM, spatial paradox
Introduction
Alzheimer's disease (AD) is characterized by the accumulation in the brain of senile plaques, composed primarily of the 40–42 amino acid peptide β-amyloid (Aβ). The generation of Aβ depends on cleavage of amyloid precursor protein (APP) by β- and γ-secretases. It is now recognized that presenilin 1 (PS1), a gene known to cause early-onset autosomal dominant Alzheimer's disease, is critical for the γ-secretase activity (De Strooper et al., 1998). Recent studies suggest that at least three other components, nicastrin (Nct), Aph-1, and Pen-2, are also necessary components of the γ-secretase complex (Yu et al., 2000b; Francis et al., 2002; Goutte et al., 2002).
PS1 has been suggested to contain the active site of the γ-secretase. Mutations of either of two critical aspartate residues in PS1 prevent heterodimer formation of presenilin and block γ-secretase cleavage of APP (Wolfe et al., 1999; Yu et al., 2000a). Moreover, photoaffinity cross-linking of γ-secretase transition state analogs resulted in specific tagging of PS1 (and PS2) heterodimers (Esler et al., 2000; Li et al., 2000; Seiffert et al., 2000); this is consistent with the idea that presenilins contain the proteolytic active site. In addition, familial AD autosomal dominant mutations in PS1 or PS2 enhance Aβ42 generation (Selkoe, 2001), also supporting the hypothesis that PS1 is a critical component of γ-secretase.
Where APP and γ-secretase–PS1 interact in cells has been a matter of debate, because PS1 and APP overlap to the greatest extent in the endoplasmic reticulum (ER)–Golgi, but γ-cleavage of substrates seems to occur at or near the cell surface (Haass and DeStrooper, 1999; Cupers et al., 2001), leading to the so-called “spatial paradox.” The evidence that PS1 and APP do interact comes from coimmunoprecipitation experiments, in which APP C-terminal fragments (CTFs) can be coprecipitated with PS1 (Xia et al., 1997, 2000). Similarly, isolation of γ-secretase activity using an active site aspartyl protease inhibitor affinity column and a detergent solubilized in vitro preparation yielded PS1 and nicastrin but, surprisingly, also APP C-terminal fragments (Esler et al., 2002). Additionally, in a solubilized cell-free system, aspartyl protease transition state analogs displayed linear noncompetitive inhibition of γ-secretase activity (Tian et al., 2002). These observations led to the hypothesis that γ-secretase has a “docking” site for APP in addition to a catalytic site and that PS1 might contribute to this as well.
Our current studies test these hypotheses in an intact cell system. Using two morphologically based assays of protein “proximity” [fluorescence resonance energy transfer (FRET) and fluorescence lifetime imaging microscopy (FLIM)], we now demonstrate that PS1 and APP are tightly associated in distal subcellular compartments in an intact cell. Genetic and pharmacologic manipulations that block γ-secretase catalytic activity do not prevent the association of APP with PS1, consistent with the presence of an APP docking site on the PS1–γ-secretase complex. In contrast, disrupting the γ-secretase complex by using RNA interference (RNAi) for nicastrin precludes APP–PS1 interactions, suggesting that the absence of nicastrin impairs docking of APP to the presumably disrupted PS1–γ-secretase complex.
Materials and Methods
Cell lines and drug treatments. Chinese hamster ovary (CHO) cells that were stably transfected with APP770 and either wild-type PS1 (wt PS1) or aspartate mutant PS1 (D257A PS1) were grown in OPTI-MEM (Invitrogen, Carlsbad, CA) with 5% fetal bovine serum and appropriate antibiotics (Puromycin or Zeocin, respectively) (Berezovska et al., 2000). To inhibit γ-secretase activity (to modulate or block the active site), cells were incubated for 4 hr with either a small dipeptide analog γ-secretase inhibitor, N-[N-(3,5-difluorophenacetyl)-L-alanyl]-(S)-phenylglycine t-butyl ester (DAPT) (500 nm) (Dovey et al., 2001), or a (hydroxyethyl) urea peptidomimetic WPE-III-31C (1 μm) (Esler et al., 2002) before immunostaining. Human embryonic kidney (HEK) 293 cells were used for the RNAi experiments.
Immunocytochemistry and antibodies. Cells were fixed and immunostained 24 hr after transfection as described previously (Jack et al., 2001). We used a polyclonal C8 antibody (Selkoe et al., 1988) to detect the C terminus of APP. A monoclonal antibody to the loop region of PS1 (Chemicon, Temecula, CA) was used to identify the γ-secretase complex. PS1 N-terminal X81 antibody was a gift from D. Selkoe (Brigham and Women's Hospital, Boston, MA); PS1 C-terminal antibody was purchased from R & D Systems (Minneapolis, MN). For FRET–FLIM studies, we chose cyanine 3 (Cy3)-labeled goat anti-rabbit as the acceptor fluorophore and FITC-labeled goat anti-mouse as the donor fluorophore.
FLIM and FRET assays. FLIM relies on the observation that fluorescence lifetimes (the time of fluorophore emission after brief excitation, measured in picoseconds) are shorter in the presence of a FRET acceptor. A mode-locked Ti-sapphire laser (Spectra-Physics, Fremont, CA) sends a femtosecond pulse every 12 nsec to excite the fluorophore. Images were acquired using a Bio-Rad (Hercules, CA) Radiance 2000 multiphoton microscope. We used a high-speed Hamamatsu (Bridgewater, NJ) detector and hardware and/or software from Becker and Hickl (Berlin, Germany) to measure fluorescence lifetimes on a pixel-by-pixel basis (Bacskai et al., 2003). Donor fluorophore (FITC) lifetimes were fit to two exponential decay curves to calculate the fraction of fluorophores within each pixel that either interact or do not interact with an acceptor. These lifetimes were then mapped by pseudocolor on a pixel-by-pixel basis over the entire image.
Validation of the FLIM assay. The following controls were used to establish and validate the FLIM assay. (1) As a negative control, FITC life-time was measured in the absence of the acceptor. The cells were double stained with primary antibodies, followed by secondary antibodies, including FITC-labeled and nonfluorescent-labeled antibodies (no acceptor fluorophore). (2) As a positive control, FITC lifetime was measured in the presence of an acceptor. For this, the cells were immunostained for presenilin, using a standard monoclonal anti-loop domain antibody. This antibody was visualized with a FITC-labeled goat anti-mouse antibody, followed by reacting with a donkey anti-goat antibody labeled with Cy3 to bring FITC and Cy3 into a close proximity (Fig. 1 A) (Kinoshita et al., 2001). (3) We then used our positive control (Cy3 anti-FITC) to select an area within the cell (one-half of the cell) in which the Cy3 acceptor is destroyed by photobleaching by scanning the area for 30 sec with 568 nm light at 100% laser power. The resulting intensity image shows an increase in FITC intensity because the FITC is “dequenched” in the area in which the Cy3 was photobleached (Fig. 1 B). In contrast, the FITC fluorescence lifetime (FLIM image) within the area in which the acceptor was photobleached reverts to a longer lifetime (blue), identical to the lifetime of FITC in the absence of an acceptor (as in the negative control) (Fig. 1 A). This control shows a close correlation between the FLIM and photobleaching FRET assays. (4) Another negative control that we used was counterstaining of the cells with an antibody predicted to colocalize with PS1 but not interact with PS1. PS1 is expressed in the ER. Double immunostaining shows that PS1 colocalizes with an ER resident protein, BiP (binding protein). However, despite colocalization, the FITC (PS1) lifetime is the same as in the negative control, suggesting that BiP (Cy3 labeled) does not act as an acceptor. This control demonstrates that a change in the fluorescence lifetime, indicating FRET, is a more sensitive measure of proximity than simple colocalization.
Figure 1.

A, Color-coded images of the negative (FITC only) and positive (Cy3 anti-FITC) controls. The rainbow scale shows fluorescence lifetime as color; if molecules are closer to each other, donor fluorescence (FITC) lifetime is shorter and the color will be closer to red. The graphs show lifetime distribution collected for every pixel of the images; positive control shows a shift to the left. B, The Cy3 acceptor in one-half of the cell (positive control) was destroyed by photobleaching (outlined area), leading to dequenching of the FITC fluorescence intensity and a shift to a longer lifetime.
Photobleach dequenching FRET measurements were made using a Bio-Rad1024 confocal microscope mounted on a Nikon (Tokyo, Japan) Eclipse TE300 inverted microscope. A krypton–argon laser was used to excite FITC and Cy3 (excitation wavelengths, 488 and 568 nm, respectively). FRET was measured using a method developed for laser scanning confocal microscopy (McLean et al., 2000; Kinoshita et al., 2001). The amount of FRET was calculated as a percentage increase in donor (FITC) fluorescence intensity after photobleaching the acceptor (Cy3) in a small part of the cell (∼8 × 8 μm). This ratio was then normalized to the percentage change in FITC fluorescence intensity in a nonbleached area of the cell. The percentage change in fluorescence intensity was compared with the null hypothesis value of 1 by one-group t tests.
Nicastrin RNAi. We used a 21-mer double-stranded RNAi (Dharmacon Research, Lafayette, CO) directed against Nct (Edbauer et al., 2002). Cells grown in 60 mm dishes were transfected with 20 μl of 20 μm Nct RNAi using Effectene reagents (Qiagen, Valencia, CA) according to the instructions of the manufacturer. The cells were harvested 72 hr after transfection, lysed in a buffer containing 1% Triton X-100, and resolved by electrophoresis on a 10–20% Tris-glycine gel (Invitrogen, Carlsbad, CA). HEK 293 cells showed the strongest suppression of nicastrin expression (H4 and CHO cells were also tested) and were used for the FLIM analysis.
Results
To analyze the proximity between different molecules in intact cells, we used two complementary assays: a photobleach dequenching FRET assay that we used for confocal microscopy (Kinoshita et al., 2001) and a FLIM technique performed on a multiphoton microscope (Bacskai et al., 2003).
FLIM reveals a close association of PS1 and APP in distal subcellular compartments
Confocal microscopic analysis of cells double stained with anti-PS1 loop (Chemicon) and C-terminal APP (C8) (Selkoe et al., 1988) antibodies reveals extensive perinuclear colocalization of APP and PS1, as well as lower signal in distal cell compartments (Fig. 2). This method shows subcellular compartment colocalization but not necessarily a close intermolecular interaction. We used FLIM to determine where in the cell APP and PS1 could be found in the closest proximity by analyzing the lifetime of the donor fluorophore.
Figure 2.

A, B, Confocal microscope images of the cells that were double immunostained with FITC-labeled PS1 (A), and Cy3-labeled APP (B) antibodies demonstrate predominantly perinuclear localization of the proteins. Scale bar, 10 μm.
Fluorescence lifetime is influenced by the surrounding microenvironment and is shortened in the immediate vicinity of a FRET acceptor fluorophore. The degree of lifetime shortening is inherently a quantitative measure of proximity, and changes in this quantity reflect alterations in conformation that can be displayed with very high spatial resolution in a pseudocolor-coded image. The fluorescence lifetime is displayed as a color; if molecules are closer together, the donor fluorescence lifetime will be shorter, and the color will be closer to red. We measured changes in the lifetime of FITC (the donor fluorophore) under different experimental conditions. Our negative control (in the absence of an acceptor fluorophore) showed that the lifetime of FITC (conjugated to IgG, hereafter referred to as FITC) alone is ∼2600 ± 110 psec, whereas in the positive control (Cy3 anti-FITC), the FITC lifetime shortens to 1400 ± 59 psec (Fig. 1).
In each experiment, PS1 loop is stained with FITC, and APP (including the APP C-terminal fragments C83 and C99) is stained with Cy3. Although the strongest colocalization occurs in the Golgi and ER, as seen by confocal microscopy (Fig. 2), FLIM analysis shows that APP and PS1 do not interact closely in these compartments (the green–blue staining in the FLIM image) (Fig. 3). Instead, PS1 and APP come into closest spatial proximity (Fig. 3, red pixels) primarily in more distal compartments near the cell surface. To analyze the fluorescence lifetime in each pixel of the imaged cell, a pseudocolored and enlarged FLIM image (each individual pixel, ∼0.04 μm2) was superimposed onto a look-up table with the average lifetimes for each pixel (Fig. 3B). We found that the average fluorescence lifetime was ∼2118 ± 190 psec on the periphery of the cell and 2402 ± 49 psec in the perinuclear (ER–Golgi) compartments (p < 0.05, compared with 2600 ± 110 psec for no interaction control).
Figure 3.
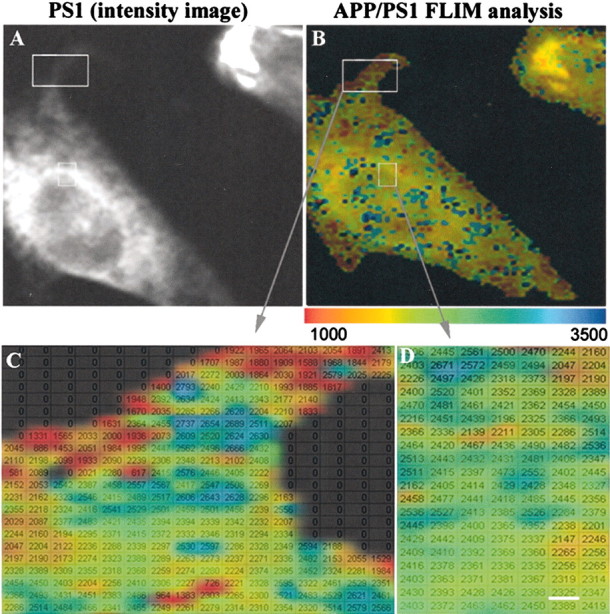
FLIM analysis of the proximity between APP and PS1 molecules within the cell. A, Intensity image shows standard immunostaining pattern for PS1, similar to that shown in Figure 2 A. B, Color-coded FLIM image shows lifetimes, reflecting proximity between PS1 and APP. The cell regions showing closest proximity between PS1 and APP are in the distal compartments near the cell surface. Colorimetric scale shows fluorescence lifetime in picoseconds. C, D, Enlarged boxed areas from B. The FLIM image is superimposed onto a table with calculated average lifetimes for each pixel of the image. Scale bar, 0.2 μm.
Inhibition of γ-secretase function does not prevent association of APP with PS1–γ-secretase
We next asked whether APP and PS1 interactions would still be observed if the γ-secretase function was inhibited. We used the highly potent γ-secretase inhibitor DAPT (Dovey et al., 2001; Micchelli et al., 2002) or a D257A PS1 mutation, which diminishes Aβ and Notch1 processing substantially and appears to be “dominant negative” in terms of γ-secretase function. The shortened lifetime of FITC (PS1) in the presence of Cy3-labeled APP shows that the PS1 loop epitope is in close proximity to the C terminus of APP after each of these manipulations (Table 1). In parallel experiments (sister cultures), we measured Aβ production after DAPT treatment and found a significant decrease in Aβ secretion (data not shown).
Table 1.
Aspartate mutations in PS1 or γ-secretase inhibitors do not prevent APP-PS1 FRET
|
Condition |
FRET donor FITC labeled |
FRET acceptor Cy3 labeled |
FITC lifetime (psec) mean ± SD |
|---|---|---|---|
| wt or D257A PS1 (control) | PS1 loop | None | 2600 ± 110 (n = 12) |
| wt PS1 | PS1 loop | APP 770 | 2290 ± 216** (n = 27) |
| wt PS1 | PS1 loop | APP 770 + DAPT | 2416 ± 118* (n = 9) |
| wt PS1 | PS1 loop | APP770 + WPE-111-31C | 1972 ± 264** (n = 30) |
| D257A PS1
|
PS1 loop
|
APP770
|
2255 ± 264** (n = 26)
|
The table shows summary data of the FLIM assay for PS1 loop-APP C-terminus proximity under baseline conditions and in the presence of manipulations to preclude APP γ-secretase cleavage. If there is no interaction, lifetimes on the order of ≈2600 psec are observed. If FRET is detected, a population with a statistically shorter lifetime (approximately < 2400 psec) is observed.
p < 0.05,
p < 0.01, compared with non-FRETing control.
Thus, FLIM analysis revealed that neither Asp mutations nor DAPT treatment prevents interactions between APP and PS1; the fluorescence lifetime was approximately the same as that seen with untreated wild-type PS1 (Table 1).
FRET analysis of the proximity between APP and PS1
To verify that the shortened donor fluorescence lifetimes observed in the FLIM experiments are attributable to FRET between PS1 and APP labels, we confirmed these observations using an alternative strategy to measure FRET: a photobleach dequenching assay.
FRET arises when donor and acceptor molecules are <10 nm apart. Cells stably expressing wt PS1 and APP770 were double labeled with PS1 loop and APP C-terminal antibodies, followed by FITC- and Cy3-labeled secondary antibodies, respectively. A small area within the cell was photobleached at 568 nm (acceptor only), and FITC fluorescence intensity (488 nm) was measured before and after photobleaching. Figure 4A shows the presence of FRET between PS1 and APP CTFs. We observed an 11.6 ± 3% (n = 8; p < 0.05) increase in FITC fluorescence intensity in the bleached area, indicating that the C terminus of APP is in close proximity to the loop region of PS1.
Figure 4.
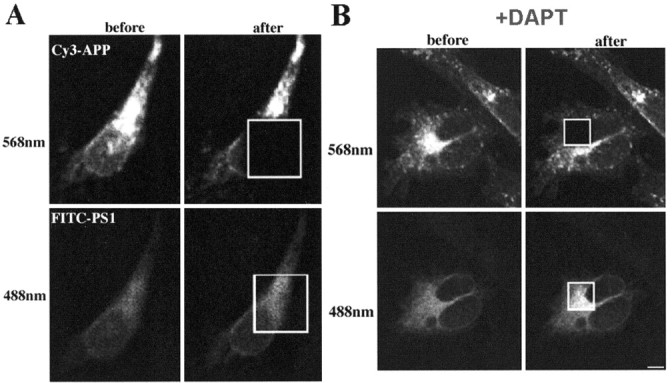
Photobleach dequenching FRET between APP and PS1 demonstrates close proximity between C terminus of APP and loop region of PS1 (A). Cy3-labeled APP (emission, 568 nm) and FITC-labeled PS1 (emission, 488 nm) before and after photobleaching the acceptor (Cy3) in a selected area within the cell. DAPT does not prevent close association of APP with PS1 (B). Scale bar, 10 μm.
Treatment with γ-secretase inhibitors (or Asp mutations in PS1) prevents processing of substrates at the γ-secretase site. Therefore, we tested whether FRET would be altered by DAPT treatment. Treatment with DAPT also revealed significant FRET between APP and PS1 (19.8 ± 2%; n = 8; p < 0.05), indicating that the γ-secretase inhibitor does not disrupt association of APP with the PS1 containing the γ-secretase complex (Fig. 4B). This was also true for the D257A PS1 mutation; we observed a 25.9 ± 11% (n = 8; p < 0.05) increase in FITC fluorescence intensity compared with a non-FRET control.
Therefore, both the FLIM and photobleaching assays suggest that APP interacts closely with PS1, even when γ-secretase is inhibited.
These data suggest either that DAPT and the D257A mutation stabilize an APP–PS1 active site complex (acting as noncompetitive inhibitors) or that there is a second binding site on the PS1–γ-secretase complex for APP. We reasoned that an agent that binds to the active site would compete with APP, blocking the APP–PS1 interaction if the first possibility were the case. We used a well characterized transition state analog, WPE-III-31C, that binds to the active site of γ-secretase (Esler et al., 2002). The cells were treated with WPE-III-31C to examine whether APP would still be associated with the PS1 loop region. The data show clearly that, although the active site is occupied, there remains a tight association between APP and PS1 (Table 1). Thus, our data suggest that there is a docking site, distinct from the catalytic site, on the PS1–γ-secretase complex.
The association of WPE-III-31C with the active site suggests its possible use as a histological reagent to identify the subcellular location of the active site and as a FRET reagent to probe the microenvironment near the active site. It has been observed that transition state analogs such as WPE-III-31C bind specifically to the heterodimeric form of PS1 and not to the holoprotein before cleavage. We therefore expect them not to bind to the D257A mutant protein that does not undergo cleavage. We used a biotinylated form of WPE-III-31C (Esler et al., 2002) as a histological reagent. Analysis of wild-type PS1 or the D257A mutant PS1 CHO cells exposed to biotinylated WPE-III-31C showed a striking difference in Cy3-strepavidin distribution. Figure 5 shows little staining of the D257A cells, which did not colocalize with PS1 immunostaining by a monoclonal antibody developed with FITC. In contrast, wt PS1-expressing cells were strongly stained by WPE-III-31C, and this staining colocalized with PS1 immunostaining. Note that there was less WPE-III-31C–Cy3 signal in the proximal perinuclear compartment compared with PS1 immunoreactivity, confirming our observation that some of this PS1 is in a different conformation and is likely not proteolytically active.
Figure 5.
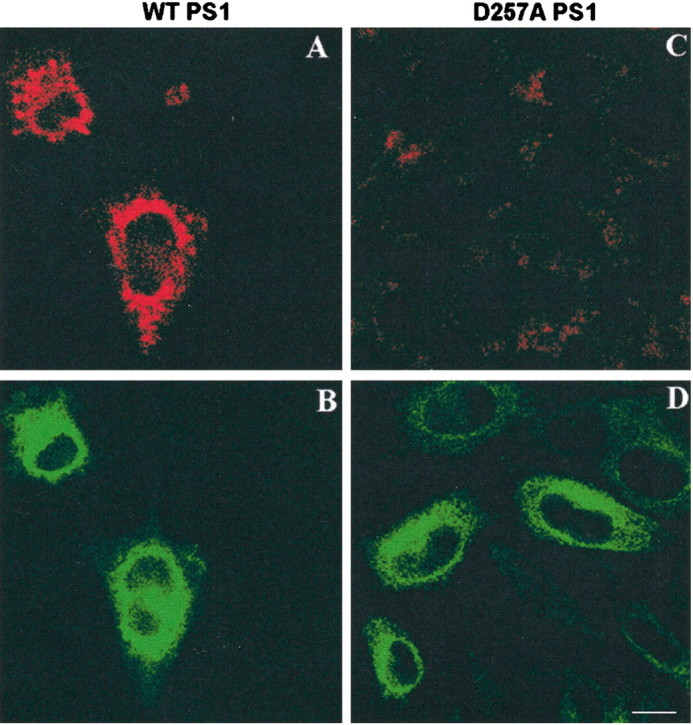
A–D, Confocal microscope images of wtPS1 (A, B) and aspartate mutant (D257A) PS1 (C, D) double stained with biotinylatedγ-secretase inhibitor WPE-III-31C-bi (A, C) and PS1 antibody (B, D). PS1 immunostaining colocalizes with Cy3 streptavidin-labeled WPE-III-31C-bi in the cells expressing wtPS1. WPE-III-31C-bi does not bind to aspartate mutant PS1 holoprotein. Scale bar, 20 μm.
FLIM analysis of the biotinylated transition state analog WPE-III-31C suggests a close association with the PS1 loop region
We reasoned that if WPE-III-31C bound to the active site and if the transmembrane domains (TMs) near the PS1 loop are near the active site, we might be able to detect an interaction between the ligand [WPE-III-31C-biotin (-bi)] and the PS1 loop region using FLIM–FRET. We examined this possibility in wtPS1-expressing cells and found that there was strong FRET, as observed by a shortening in fluorescence lifetimes, between the PS1 loop antibody and WPE-III-31C. Interestingly, neither a PS1 N-terminal antibody nor an APP C-terminal antibody showed statistically significant FRET with WPE-111-31C, despite complete colocalization of the PS1 N-terminal antibody with WPE-111-31C at the light level (Table 2). These data reinforce the specificity and proximity dependence of the FLIM assay and act as an additional negative control for the technique.
Table 2.
PS1 loop shows the strongest FRET with WPE-III-31C-biotin
|
Condition |
FRET donor FITC labeled |
FRET acceptor Cy3 labeled |
FITC lifetime (psec) mean ± SD |
|---|---|---|---|
| wt PS1 | PS1 NT (X81) | None | 2630 ± 82 (n = 9) |
| wt PS1 | PS1 loop | WPE-III-31C-biotin | 1815 ± 162* (n = 15) |
| wt PS1 | PS1 NT (X81) | WPE-III-31C-biotin | 2672 ± 44 (n = 14) |
| wt PS1
|
PS1 CT
|
WPE-III-31C-biotin
|
2220 ± 110* (n = 12)
|
The degree of the fluorescence lifetime shortening indicates that the γ-secretase transition state analog WPE-III-31C is in the closest proximity to the PS1 loop region and far away from PS1 N terminus (n, indicates number of cells). NT, N terminus; CT, C terminus.
p < 0.001 compared with non-FRETing control.
Nicastrin is important for APP–PS1 association
The above experiments show that there is an APP–PS1 interaction even in the presence of pharmacological inhibitors and dominant-negative PS1 mutations. Nicastrin has been reported to be a major component of the γ-secretase complex, and nicastrin RNAi blocks γ-secretase function (Edbauer et al., 2002). We tested whether nicastrin interactions with PS1 and γ-secretase were important for the association of substrates to the γ-secretase complex. We used a nicastrin RNAi knock-down approach, followed by FLIM analysis, to examine APP–PS1 interaction in HEK 293 cells. Three days after treatment with nicastrin small interfering RNA, the amount of immunodetectable nicastrin was minimal as seen by Western blot analysis (Fig. 6A). There was no evidence of enhanced cell death in the treated (compared with mock treated) cells. Nicastrin RNAi-treated cells were then immunostained with APP and PS1 antibodies as described above. The proximity between endogenous APP CTF and PS1 loop was then analyzed by FLIM. Importantly, the same pattern of PS1–APP FLIM occurred using endogenous proteins as we had observed in stable transfected CHO cells. In contrast to our previous studies of D257A PS1 mutations, DAPT and WPE-III-31C, which did not affect the association of APP with PS1, we found that nicastrin RNAi treatment completely disrupts the APP–PS1 assembly (Fig. 6B).
Figure 6.

Nicastrin is important for the association of APP with PS1–γ-secretase complex. A, Nct RNAi leads to a significant inhibition of nicastrin expression in the cells. B, A decrease in the FITC–PS1 lifetime in APP–PS1 double-immunostained cells is observed in mock-treated cells, indicating a close proximity between the two proteins. This association is eliminated by Nct RNAi treatment because the FITC lifetime becomes the same as in the PS1–FITC alone control.
Discussion
FRET (or FRET-based FLIM) provides information about the proximity between donor- and acceptor-labeled molecules. FRET arises when donor and acceptor molecules are brought into close (<10 nm) proximity after the formation of a protein complex.
Using this proximity approach, we studied how different pharmacological agents or genetic manipulations affect APP–PS1 interactions in intact cells. We found the following: (1) APP–PS1 interactions are tighter near the cell membrane rather than in the Golgi–ER (despite greater expression and apparent colocalization in the latter compartments), (2) neither aspartate mutations nor γ-secretase inhibitors prevent the association of APP with PS1, suggesting the presence of both a docking site and an active site on the PS1–γ-secretase complex, and (3) nicastrin is important for the docking of APP to the PS1–γ-secretase.
These results confirm and extend the cell-free assay findings that APP CTFs and PS1 “pull down” in coimmunoprecipitation experiments (Xia et al., 2000), PS1 and APP CTFs coisolate when bound to a column containing a transition-state analog (Esler et al., 2002) and that isolated γ-secretase shows nonlinear kinetics with various inhibitors (Tian et al., 2002). However, in these reconstitution experiments, varying detergent conditions could have altered the extent to which various constituents were present.
We show that, in intact cells, inhibition of γ-secretase activity, either by the introduction of aspartate mutations in PS1 or by treatment with some γ-secretase inhibitors, did not affect association of APP with the PS1–γ-secretase complex. In both cases, APP and PS1 still interact at least as strongly as with untreated wild-type PS1 (Table 1). Moreover, occupation of the γ-secretase active site by the inhibitor WPE-III-31C, a well characterized transition state analog that can be used to isolate a functional γ-secretase complex from cell homogenates (Esler et al., 2002), did not prevent association of APP with PS1 in intact cells. These data show clearly that a very tight association between APP and PS1 remains, despite active site occupation, which is consistent with the presence of a docking site. In addition, we show that WPE-III-31C, which binds to the active site of γ-secretase, is closely associated with the PS1 loop domain but not with the N-terminal epitope. These results are consistent with copurification data (Esler et al., 2002) and strongly support the notion that the γ-secretase active site is located between TM6 and TM7, where the two critical aspartates lie near the loop region of PS1.
Our data also address the subcellular localization of APP–PS1 interactions. The spatial paradox reflects the observation that APP and PS1 overlap most strongly in the Golgi and ER but that γ-secretase function and APP cleavage are not prominent in the site of the greatest overlap, the ER (Cupers et al., 2001). This discrepancy has been used to support the argument that PS1 is not γ-secretase. Our imaging technique confirms that, by conventional immunostaining, APP and PS1 overlap to the greatest extent in a perinuclear location (Fig. 2). However, they come into closest proximity, as detected by the FLIM assay, in the distal compartments near the cell surface. We hypothesize that PS1 adopts an active conformation only in distal compartments. If so, these data provide an alternative explanation for the spatial paradox in which PS1 in distal cellular compartments is an important component of a γ-secretase complex.
We also tested the role of nicastrin in the formation of an APP–PS1 complex. Nicastrin knock-down using RNAi has been reported to disrupt PS1 processing to heterodimeric forms and γ-secretase function (Edbauer et al., 2002). We asked whether RNAi-mediated knock-down of nicastrin would alter APP–PS1 interactions, because other manipulations that inhibit γ-secretase, including the D257A PS1 mutation that also precludes heterodimer formation, still allow for APP–PS1 interactions that are detectable by coimmunoprecipitation or FRET. Interestingly, we could not detect any FRET between APP and PS1 in cells treated with nicastrin RNAi. We interpret this result to mean that the absence of nicastrin diminishes APP–PS1 interactions. Our current data do not differentiate between the possibilities that nicastrin contributes directly to the docking site or that it acts upstream of docking in the formation of the γ-secretase complex.
Our model suggests that APP interacts with γ-secretase at a docking site, as well as at a proteolytic cleavage site. We propose that the docking site may confer specificity or play a modulatory role in γ-secretase–substrate interactions. If APP and Notch (or other substrates) are docked at different sites or if docking of each substrate is subserved by separate docking proteins, it may be possible to selectively inhibit APP–γ-secretase interactions. There have been a number of proteins reported to undergo PS1-dependent γ-secretase cleavage (Baki et al., 2001; Ni et al., 2001; Lee et al., 2002; Marambaud et al., 2002; May et al., 2002). Therefore, because direct inhibition of γ-secretase cleavage could disrupt many substrates, a better understanding of specific APP–γ-secretase interactions at a noncatalytic site may ultimately have therapeutic implications, because drugs targeted to a docking site might better discriminate among substrates than active site γ-secretase inhibitors.
Footnotes
This work was supported by National Institutes of Health Grants AG15379, AG 08487, EB00768, and NS 41355 (M.S.W.) and a Pioneer Award from the Alzheimer Association.
Correspondence should be addressed to Dr. Oksana Berezovska, 114 16th Street, Room 2750, Massachusetts General Hospital, Charlestown, MA 02129. E-mail: oberezovska@partners.org.
Copyright © 2003 Society for Neuroscience 0270-6474/03/234560-07$15.00/0
References
- Bacskai BJ, Skoch J, Kickey GA, Allen R, Hyman BT ( 2003) FRET determinations using multiphoton fluorescence lifetime imaging microscopy (FLIM) to characterize amyloid-beta plaques. J Biomed Optics, in press. [DOI] [PubMed]
- Baki L, Marambaud P, Efthimiopoulos S, Georgakopoulos A, Wen P, Cui W, Shioi J, Koo E, Ozawa M, Friedrich Jr VL, Robakis NK ( 2001) Presenilin-1 binds cytoplasmic epithelial cadherin, inhibits cadherin/p120 association, and regulates stability and function of the cadherin/catenin adhesion complex. Proc Natl Acad Sci USA 98: 2381–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berezovska O, Jack C, McLean P, Aster JC, Hicks C, Xia W, Wolfe MS, Kimberly WT, Weinmaster G, Selkoe DJ, Hyman BT ( 2000) Aspartate mutations in presenilin and γ-secretase inhibitors both impair Notch1 proteolysis and nuclear translocation with relative preservation of Notch signaling. J Neurochem 75: 583–593. [DOI] [PubMed] [Google Scholar]
- Cupers P, Bentahir M, Craessaerts K, Orlans I, Vanderstichele H, Saftig P, De Strooper B, Annaert W ( 2001) The discrepancy between presenilin subcellular localization and gamma-secretase processing of amyloid precursor protein. J Cell Biol 154: 731–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F ( 1998) Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 391: 387–390. [DOI] [PubMed] [Google Scholar]
- Dovey HF, John V, Anderson JP, Chen LZ, de Saint Andrieu P, Fang LY, Freedman SB, Folmer B, Goldbach E, Holsztynska EJ, Hu KL, Johnson-Wood KL, Kennedy SL, Kholodenko D, Knops JE, Latimer LH, Lee M, Liao Z, Lieberburg IM, Motter RN, et al. ( 2001) Functional gamma-secretase inhibitors reduce beta-amyloid peptide levels in brain. J Neurochem 76: 173–181. [DOI] [PubMed] [Google Scholar]
- Edbauer D, Winkler E, Haass C, Steiner H ( 2002) Presenilin and nicastrin regulate each other and determine amyloid beta-peptide production via complex formation. Proc Natl Acad Sci USA 99: 8666–8671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esler WP, Kimberly WT, Ostaszewski BL, Diehl TS, Moore CL, Tsai JY, Rahmati T, Xia W, Selkoe DJ, Wolfe MS ( 2000) Transition-state analogue inhibitors of gamma-secretase bind directly to presenilin-1. Nat Cell Biol 2: 428–434. [DOI] [PubMed] [Google Scholar]
- Esler WP, Kimberly WT, Ostaszewski BL, Ye W, Diehl TS, Selkoe DJ, Wolfe MS ( 2002) Activity-dependent isolation of the presenilin–gamma-secretase complex reveals nicastrin and a gamma substrate. Proc Natl Acad Sci USA 99: 2720–2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis R, McGrath G, Zhang J, Ruddy DA, Sym M, Apfeld J, Nicoll M, Maxwell M, Hai B, Ellis MC, Parks AL, Xu W, Li J, Gurney M, Myers RL, Himes CS, Hiebsch R, Ruble C, Nye JS, Curtis D ( 2002) aph-1 and pen-2 are required for Notch pathway signaling, gamma-secretase cleavage of betaAPP, and presenilin protein accumulation. Dev Cell 3: 85–97. [DOI] [PubMed] [Google Scholar]
- Goutte C, Tsunozaki M, Hale VA, Priess JR ( 2002) APH-1 is a multipass membrane protein essential for the Notch signaling pathway in Caenorhabditis elegans embryos. Proc Natl Acad Sci USA 99: 775–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C, DeStrooper B ( 1999) The presenilins in Alzheimer's disease–proteolysis holds the key. Science 286: 916–919. [DOI] [PubMed] [Google Scholar]
- Jack C, Berezovska O, Wolfe MS, Hyman BT ( 2001) Effect of PS1 deficiency and an APP gamma-secretase inhibitor on Notch1 signaling in primary mammalian neurons. Brain Res Mol Brain Res 87: 166–174. [DOI] [PubMed] [Google Scholar]
- Kinoshita A, Whelan CM, Smith CJ, Mikhailenko I, Rebeck GW, Strickland DK, Hyman BT ( 2001) Demonstration by fluorescence resonance energy transfer of two sites of interaction between the low-density lipoprotein receptor-related protein and the amyloid precursor protein: role of the intracellular adapter protein Fe65. J Neurosci 21: 8354–8361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Jung KM, Huang YZ, Bennett LB, Lee JS, Mei L, Kim TW ( 2002) Presenilin-dependent gamma-secretase-like intramembrane cleavage of ErbB4. J Biol Chem 277: 6318–6323. [DOI] [PubMed] [Google Scholar]
- Li YM, Xu M, Lai MT, Huang Q, Castro JL, DiMuzio-Mower J, Harrison T, Lellis C, Nadin A, Neduvelil JG, Register RB, Sardana MK, Shearman MS, Smith AL, Shi XP, Yin KC, Shafer JA, Gardell SJ ( 2000) Photoactivated gamma-secretase inhibitors directed to the active site covalently label presenilin 1. Nature 405: 689–694. [DOI] [PubMed] [Google Scholar]
- Marambaud P, Shioi J, Serban G, Georgakopoulos A, Sarner S, Nagy V, Baki L, Wen P, Efthimiopoulos S, Shao Z, Wisniewski T, Robakis NK ( 2002) A presenilin-1/gamma-secretase cleavage releases the E-cadherin intracellular domain and regulates disassembly of adherens junctions. EMBO J 21: 1948–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May P, Reddy YK, Herz J ( 2002) Proteolytic processing of low density lipoprotein receptor-related protein mediates regulated release of its intracellular domain. J Biol Chem 277: 18736–18743. [DOI] [PubMed] [Google Scholar]
- McLean PJ, Kawamata H, Ribich S, Hyman BT ( 2000) Membrane association and protein conformation of alpha-synuclein in intact neurons: effect of Parkinson's disease-linked mutations. J Biol Chem 275: 8812–8816. [DOI] [PubMed] [Google Scholar]
- Micchelli CA, Esler WP, Kimberly WT, Jack C, Berezovska O, Kornilova A, Hyman BT, Perrimon N, Wolfe MS ( 2003) Gamma-secretase/presenilin inhibitors for Alzheimer's disease phenocopy Notch mutations in Drosophila FASEB J 17: 79–81. [DOI] [PubMed] [Google Scholar]
- Ni CY, Murphy MP, Golde TE, Carpenter G ( 2001) Gamma-secretase cleavage and nuclear localization of ErbB-4 receptor tyrosine kinase. Science 294: 2179–2181. [DOI] [PubMed] [Google Scholar]
- Seiffert D, Bradley JD, Rominger CM, Rominger DH, Yang F, Meredith Jr JE, Wang Q, Roach AH, Thompson LA, Spitz SM, Higaki JN, Prakash SR, Combs AP, Copeland RA, Arneric SP, Hartig PR, Robertson DW, Cordell B, Stern AM, Olson RE et al. ( 2000) Presenilin-1 and -2 are molecular targets for gamma-secretase inhibitors. J Biol Chem 275: 34086–34091. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ ( 2001) Presenilin, Notch, and the genesis and treatment of Alzheimer's disease. Proc Natl Acad Sci USA 98: 11039–11041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe D, Podlisny M, Joachim C, Vickers E, Lee G, Fritz L, Oltersdor T ( 1988) Beta-amyloid precursor protein of Alzheimer disease occurs as 110- to 135-kilodalton membrane-associated proteins in neural and nonneural tissues. Proc Natl Acad Sci USA 85: 7341–7345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian G, Sobotka-Briner CD, Zysk J, Liu X, Birr C, Sylvester MA, Edwards PD, Scott CD, Greenberg BD ( 2002) Linear non-competitive inhibition of solubilized human gamma-secretase by pepstatin A methylester, L685458, sulfonamides, and benzodiazepines. J Biol Chem 277: 31499–31505. [DOI] [PubMed] [Google Scholar]
- Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ ( 1999) Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature 398: 513–517. [DOI] [PubMed] [Google Scholar]
- Xia W, Zhang J, Perez R, Koo EH, Selkoe D ( 1997) Interaction between amyloid precursor protein and presenilins in mammalian cells: implications for the pathogenesis of Alzheimer disease. Proc Natl Acad Sci USA 94: 8208–8213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia W, Ray WJ, Ostaszewski BL, Rahmati T, Kimberly WT, Wolfe MS, Zhang J, Goate AM, Selkoe DJ ( 2000) Presenilin complexes with the C-terminal fragments of amyloid precursor protein at the sites of amyloid beta-protein generation. Proc Natl Acad Sci USA 97: 9299–9304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Chen F, Nishimura M, Steiner H, Tandon A, Kawarai T, Arawaka S, Supala A, Song YQ, Rogaeva E, Holmes E, Zhang DM, Milman P, Fraser PE, Haass C, George-Hyslop PS ( 2000a) Mutation of conserved aspartates affects maturation of both aspartate mutant and endogenous presenilin 1 and presenilin 2 complexes. J Biol Chem 275: 27348–27353. [DOI] [PubMed] [Google Scholar]
- Yu G, Nishimura M, Arawaka S, Levitan D, Zhang L, Tandon A, Song YQ, Rogaeva E, Chen F, Kawarai T, Supala A, Levesque L, Yu H, Yang DS, Holmes E, Milman P, Liang Y, Zhang DM, Xu DH, Sato C, Rogaev E, Smith M, Janus C, Zhang Y, Aebersold R, Farrer LS, Sorbi S, Bruni A, Fraser P, St. George-Hyslop P ( 2000b) Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and betaAPP processing. Nature 407: 48–54. [DOI] [PubMed] [Google Scholar]