

Abstract
Programmed cell death plays an important role both during the development of the CNS and in its homeostasis throughout adulthood. A complex balance between cell death- and survival-inducing signals determines the fate of individual neurons. Intracellular cAMP is thought to regulate neuronal survival, and previous studies have shown that the survival of retinal ganglion cells by brain-derived neurotrophic factor (BDNF) is dependent on cAMP. Here we report the surprising observation that cAMP attenuates the ability of BDNF to rescue cortical neurons from apoptosis after serum deprivation, a process mediated via the phosphatidylinositol 3 (PI3)-kinase signal transduction cascade. Depolarization by KCl, which increases cAMP in cortical neurons, also attenuates BDNF protection against serum withdrawal. Our data indicate that cAMP antagonizes neurotrophin protection from serum withdrawal by inhibiting the PI3-kinase signal transduction cascade. This study indicates that cAMP may inhibit some forms of neurotrophin-mediated neuronal survival and suggests that a number of PI3-kinase-regulated processes in neurons may be inhibited by cAMP.
Keywords: PI3-kinase, cAMP, neurotrophins, survival, apoptosis, coincident signaling
Introduction
The formation of the complex neural network that comprises the CNS is thought to result from a balance between the survival of neurons that make appropriate connections and the elimination of those that do not (Shatz, 1990; Oppenheim, 1991). Target-derived neurotrophic factors, including brain-derived neurotrophic factor (BDNF), ciliary neurotrophic factor (CNTF), as well as neurotrophins 3, 4, and 5, can provide signals that determine whether a neuron is maintained or lost (Levi-Montalcini and Booker, 1960; Barde, 1989). Additional factors, e.g., insulin-like growth factor-1 (IGF-1), produced by supporting non-neuronal cells, as well as neuronal activity itself contribute to neuronal survival (Ghosh et al., 1994; Miller et al., 1997). Those cells that lose trophic factor support undergo programmed cell death, an active process characterized by increased expression of specific genes, chromatin condensation, and DNA fragmentation (Clarke, 1988; Raff et al., 1993; Stefanis et al., 1997).
Neurotrophins, via binding to their cognate Trk receptors, activate several signal transduction cascades that regulate the cell death machinery (Segal and Greenberg, 1996; Kaplan, 1998; Friedman and Greene, 1999). The extracellular signal-regulated kinase/mitogen-activated protein kinase (Erk/MAP) cascade mediates trophic factor-supported survival in differentiated PC12 cells, retinal ganglion cells, cerebellar granule neurons, and cortical neurons (Meyer-Franke et al., 1995; Xia et al., 1995; Parrizas et al., 1997; Skaper et al., 1998; Hetman et al., 1999). The phosphatidylinositol 3 (PI3)-kinase pathway was implicated originally in nerve growth factor-dependent (NGF) and serum-dependent survival of PC12 cells (Yao and Cooper, 1995). Subsequent experiments showed that PI3-kinase mediates the NGF-dependent survival of dorsal root and superior cervical ganglion neurons (Crowder and Freeman, 1998; Klesse and Parada, 1998; Mazzoni et al., 1999; Kuruvilla et al., 2000) as well as BDNF protection of spinal cord motor neurons, neuroblastoma cells, and cortical neurons after serum deprivation (Dolcet et al., 1999; Encinas et al., 1999; Hetman et al., 1999). Activation of Akt/PKB, serum and glucocorticoid-inducible kinase (SGK), and p90Rsk by PI3-kinase-dependent mechanisms is proposed to promote survival via a number of contributing downstream effectors, including phosphorylation and subsequent inhibition of proapoptotic BAD-mediated signaling (Datta et al., 1997) as well as the inhibition of glycogen synthase kinase-β (GSK-β) (Hetman et al., 2000).
Given that neurons are exposed simultaneously to numerous stimuli during development, the ultimate fate of any given neuron in the CNS is dependent on the integration of coincident signal transduction cascades. The effect of cAMP on cell survival depends on cell type and the specific type of cellular stress and may be influenced by cross talk with other signaling pathways. cAMP is neuroprotective in cerebellar granule cells and can potentiate neuroprotection of dorsal root and retinal ganglion cells by neurotrophins (Rydel and Greene, 1988; D'Mello et al., 1993; Campard et al., 1997; Villalba et al., 1997). BDNF rescue of retinal ganglion cells from apoptosis caused by serum deprivation is also dependent on cAMP. This is attributable to cAMP-stimulated increases in TrkB receptor expression (Meyer-Franke et al., 1998).
Here we report the surprising finding that increased cAMP may attenuate BDNF protection of cortical neurons from apoptosis induced by serum withdrawal. Membrane depolarization, which produces a significant increase in cAMP, also blocks BDNF protection against serum withdrawal. These effects of cAMP on BDNF neuroprotection apparently are attributable to inhibition of the PI3-kinase pathway by cAMP. This novel form of signal transduction cross talk indicates that cAMP can promote either cell survival or death, depending on the specific type of cellular stress. Thus cAMP may play an important role in regulating the survival of cortical neurons during the development of the CNS.
Materials and Methods
Reagents. Forskolin, isoproterenol, and Hoechst 33258 (bis-benzimide) were purchased from Sigma (St. Louis, MO). BDNF was purchased from Invitrogen (San Diego, CA). RpcAMPS and SpcAMPS were purchased from Biolog (Hayward, CA). LY294002 and camptothecin were purchased from Calbiochem (La Jolla, CA).
Plasmids. SGK S422D and mycPDK-1 (Kobayashi and Cohen, 1999), p110* (Klippel et al., 1996), and R(AB) expression plasmids (Schecterson and McKnight, 1991) are described where indicated. The serum response element (SRE)–luciferase reporter is described in Johansen and Prywes (1994).
Cell culture. Cortical neurons were prepared as described (Chan et al., 1998). Briefly, cortical neurons were cultured from postnatal day 0 (P0) rats and plated at a density of 4 × 105 cells/well onto 24-well plates containing glass coverslips (Fisher Scientific, Pittsburgh, PA) coated with 50 μg/ml poly-d-lysine (Becton Dickinson, Mountain View, CA) and 1.0 μg/ml laminin (Invitrogen). Cells were maintained in Neurobasal medium (Invitrogen) supplemented with 3% fetal bovine serum (FBS), 35 mm glucose, 1 mm l-glutamine, 100 U/ml penicillin, 0.1 mg/ml streptomycin, and 2.5 μm cytosine arabinoside for 3 d before experimentation. Neurons used for reporter gene assays were cultured as above but without coverslips.
Transient transfections and reporter assays. Cortical neurons were transfected with Lipofectamine 2000 (Invitrogen) for reporter gene assays. For each well of a 24-well plate, 800 ng of plasmid DNA was diluted into 50 μl of Neurobasal medium supplemented with 300 μm glutamine and B27. Lipofectamine (2 μl) was diluted into 50 μl and combined with the diluted DNA. The conditioned media from each well of the neurons were pooled and saved for later use. The DNA/Lipofectamine mixture was added to 500 μl of Neurobasal/B27 and placed onto the cells for 2–4 hr. The conditioned media were used to replace the Lipofectamine/DNA mixture after transfection. Reporter assays were conducted 36 hr after transfection. Cells were pretreated with forskolin for 1 hr and then treated with or without agonists for 5 hr. Luciferase and secreted alkaline phosphatase (SEAP) were measured with the Luciferase Assay kit and Phospha-Light assay as described by the manufacturer (Tropix, Bedford, MA).
cAMP assays. cAMP accumulation was measured by determining the ratio of [3H]cAMP to the total of [3H]cAMP, [3H]ATP, [3H]ADP, [3H]AMP pool. Media were supplemented with 5 μCi/ml [3H]adenine for 12 hr and then removed. Cells were washed with Krebs–Ringer–HEPES buffer [containing (in mm): 128 NaCl, 5 KCl, 1 NaHPO4, 10 glucose, 20 HEPES, pH 7.4, 1.2 MgSO4, 2.7 CaCl2], preincubated with Krebs–Ringer HEPES buffer containing 1 mm IBMX for 30 min, and then treated in buffer containing varying amounts of KCl for an additional 30 min. After treatment the reaction was terminated by the addition of 5% trichloroacetic acid containing 1 μm cAMP. After 30 min at room temperature the acid-soluble nucleotides were separated by ion exchange chromatography (Salomon et al., 1974; Salomon, 1991).
SGK kinase assays. Cortical neurons were pretreated with either vehicle or forskolin for 15 min and then stimulated for 5 min with 50 ng/ml BDNF. Cells were lysed in 100 μl of lysis buffer [containing (in mm): 20 Tris, pH 7.5, 150 NaCl, 1 EDTA, 1 EGTA, 2.5 sodium pyrophosphate, 1 β-glycerophosphate, 1 Na3VO4, 1 PMSF plus 1% Triton X-100]. Lysate (30 μg) was diluted to 500 μl and incubated with 2 μg of sheep anti-SGK antibody (Upstate Biotechnology, Lake Placid, NY) for 4 hr at 4°C, followed by incubation with 40 μl of protein G-agarose (Roche Bioscience, Palo Alto, CA) overnight at 4°C. Immunoprecipitated SGK was incubated with 1 μg of GSK3 peptide (Cell Signaling, Beverly, MA) in the presence of 200 μm ATP at 30°C for 30 min. The reaction was terminated by the addition of 3× boiling sample buffer, and samples were analyzed by SDS-PAGE and subsequent Western blotting for phosphorylation of the GSK3 peptide at Serine 21/9.
Western analysis. Agonist- and inhibitor-treated cortical neurons were lysed in 3× boiling SDS-PAGE sample buffer and boiled for 10 min. The samples were subjected to SDS-PAGE electrophoresis and blotted as described (Impey et al., 1998). Antibodies were used at the following dilutions: 1:1000 [rabbit anti-phospho PTEN, rabbit anti-phospho Erk, rabbit anti-phospho Akt, rabbit anti-phospho GSK3 (Cell Signaling), and goat anti-Erk (Santa Cruz Biotechnology, Santa Cruz, CA)] and 1:2000 [AP-conjugated anti-IgG (Cappel, West Chester, PA) and HRP-conjugated anti-IgG (Cappel)]. Western blots were developed by using alkaline phosphatase (Tropix) or horseradish peroxidase (Amersham Biosciences, Arlington Heights, IL) chemiluminescence as described by the manufacturers. Films were digitized by using Photoshop version 5.0.2 for the Macintosh; band intensities were quantitated by using NIH Image.
Apoptosis assays. Serum deprivation experiments were performed with neurons cultured on glass coverslips for 4 d in vitro. The conditioned medium from the culture was removed and saved (“serum-containing conditioned medium”). For serum deprivation the neurons were washed twice with serum-free Neurobasal medium and then incubated in serum-free Neurobasal medium supplemented with 35 mm glucose, 1 mm l-glutamine, 100 U/ml penicillin, 0.1 mg/ml streptomycin, 10 μm MK801, and 2.5 μm cytosine arabinoside. Control neurons were washed the same way and incubated in serum-containing conditioned medium. To visualize nuclear morphology, we fixed neurons in 4% formaldehyde and stained them with 2.5 μg/ml Hoechst 33258. Uniformly stained nuclei were scored as healthy, whereas condensed or fragmented nuclei were scored as apoptotic. To obtain unbiased counting, we coded the slides, and cells were scored blind. Statistical analysis of the data was performed with the two-tailed Student's t test, assuming equal variance. For p110*-dependent survival, cortical neurons were transfected on day 3 in vitro in 24-well plates containing glass coverslips coated with 50 μg/ml poly-d-lysine and 1 μg/ml laminin. Each well was transfected with 250 ng of UB6-lacZ expression vector and 750 ng of either p110* or empty control vector. After 48 hr the cells were serum-starved and treated as above. Transfected cells were identified by a β-galactosidase antibody (5 Prime→3 Prime, Boulder, CO). Cortical neurons were treated similarly for R(AB) expression experiments with the exception that a UB6-GFP expression vector was used to visualize transfected cells. For camptothecin-induced apoptosis the neurons were plated as above and pretreated with BDNF and either 0.01% DMSO or forskolin for 1 hr before treatment with camptothecin. Apoptosis then was scored as described above.
PI3-kinase assay. Cortical neurons were plated on 10 cm plates and treated as indicated on day 6 in vitro. After treatment PI3-kinase activity was assessed as described in (Myers et al., 1993). Briefly, cells were washed once with ice-cold PBS, twice with ice-cold Buffer A [containing (in mm): 20 Tris, pH 7.5, 137 NaCl, 1 MgCl2, 1 CaCl2 plus 100 μm Na3VO4], and then lysed in Buffer A supplemented with 1% NP-40 and 10% glycerol. Cell lysate (1 mg) was immunoprecipitated with anti-phosphotyrosine antibodies (PY-20, Transduction Laboratories, Lexington, KY). The immunopellet was resuspended in 50 μl of a (in mm) 10 Tris, pH 7.5, 100 NaCl, 1 EDTA, 100 MgCl2 solution. Then 10 μl of 2 mg/ml l-α-phosphatidylinositol (sonicated in 10 mm Tris, pH 7.5, 1 mm EDTA; Avanti Polar Lipids, Alabaster, AL) was added, and the reaction was started by adding 44 μm ATP and 30 μCi of γ32P-ATP. After 10 min of constant agitation at room temperature the reaction was terminated by the addition of 20 μl of 8N HCl. Then the phospholipids were extracted with 160 μl of 1:1 CHCl3/methanol and resolved by thin-layer chromatography (TLC). Phosphatidylinositol 4-monophosphate was run as a standard and visualized by exposing the TLC plate to I2.
Results
BDNF rescue of cortical neurons from serum deprivation is attenuated by cAMP
We examined the effect of cAMP on BDNF protection of cortical neurons because previous data indicated that neurotrophin protection of these neurons is mediated by either the PI3-kinase or Erk/MAP kinase pathway, depending on the type of cellular stress (Hetman et al., 1999). Consequently, cAMP may have different effects on BDNF neuroprotection, depending on the cell death paradigm. Withdrawal of serum from cortical neurons causes apoptosis and constitutes a useful model to study mechanisms underlying the neuroprotection afforded by target-derived neurotrophic support (Hetman et al., 1999; Yamada et al., 2001).
Cortical neurons maintained under serum-free conditions for 24 hr showed a significant increase in apoptosis as compared with neurons in serum-containing media (Fig. 1A,B). Consistent with previous studies, BDNF protected cortical neurons from serum withdrawal. However, the addition of forskolin, a general activator of adenylyl cyclases, significantly attenuated neuroprotection caused by BDNF. This inhibition was comparable to that seen with LY294002, an inhibitor of PI3-kinase activity. It is notable that forskolin alone caused a small but reproducible increase in neuronal survival (Fig. 1B). Likewise, treatment with the cAMP analog SpcAMPS also reduced BDNF neuroprotection under serum-free conditions (Fig. 1C).
Figure 1.

cAMP attenuates neurotrophin-mediated protection against serum deprivation. A, Representative fields of cortical neuron nuclear morphology visualized by Hoechst staining. Cortical neurons were washed twice with serum-free Neurobasal medium and then incubated in serum-containing conditioned medium (+ Serum) or serum-free Neurobasal medium (- Serum). Cells then were treated with 0.01% DMSO (vehicle), 10 μm forskolin, or 25 μmLY294002 in the presence or absence of 10 ng/ml BDNF. Apoptosis was scored 24 hr later. B, Bar graph representing the mean percentage of apoptosis per coverslip ± SEM from a count of at least 500 cells per coverslip of triplicate wells. C, Cortical neurons were washed twice with serum-free Neurobasal medium and then incubated in serum-containing conditioned Neurobasal medium (+ Serum) or serum-free Neurobasal medium (- Serum). Cells then were treated with 10 μm forskolin or 200 μm SpcAMPS in the presence or absence of 10 ng/ml BDNF. Apoptosis was scored 24 hr later by visualization of nuclear morphology. D, Same as above except that cells were treated with 10 μm isoproterenol. The data are derived by counting at least 500 cells per coverslip from three coverslips and are presented as the mean percentage of apoptosis per coverslip ± SEM. Statistics were determined via two-tailed Student's t test, assuming equal variance. *p < 0.01; ns, not significant.
Because β-adrenergic receptors couple to the activation of adenylyl cyclase in cortical neurons (Sibley and Lefkowitz, 1987; Ma et al., 1991), we also examined the effect of isoproterenol on BDNF protection after serum deprivation (Fig. 1D). Isoproterenol, a β-adrenergic receptor agonist, almost completely blocked BDNF neuroprotection, providing further evidence that cAMP antagonizes BDNF neuroprotection.
Membrane depolarization caused by KCl often is used as a model for the enhancement of neuronal survival because of activity-dependent Ca 2+ influx. This has been attributed to Ca 2+ activation of calmodulin-stimulated adenylyl cyclases (Meyer-Franke et al., 1995). In cortical neurons 40 mm KCl increased intracellular cAMP 2.4-fold (Fig. 2A). Under depolarizing conditions BDNF was unable to protect cortical neurons from serum deprivation (Fig. 2B). Collectively, these data suggest that cAMP increases, which can be neuroprotective on their own, antagonize the anti-apoptotic activity of BDNF.
Figure 2.

Depolarization-induced increases in cAMP inhibit BDNF neuroprotection after serum deprivation. A, Cortical neurons were treated with varying concentrations of KCl for 30 min and then assayed for cAMP accumulation. B, Cortical neurons were washed twice with serum-free Neurobasal medium and then incubated in serum-containing conditioned Neurobasal medium (+ Serum) or serum-free Neurobasal medium (- Serum). Cells then were treated with 40 mm KCl in the presence or absence of 10 ng/ml BDNF. Apoptosis was scored 24 hr later by visualization of nuclear morphology. The data are derived by counting at least 500 cells per coverslip and are presented as the mean percentage of apoptosis per coverslip ± SEM from three coverslips.
PKA activity is required for cAMP inhibition of BDNF neuroprotection
Because cAMP-dependent protein kinase (PKA) is one of the major downstream effectors of cAMP, the importance of PKA activity for cAMP attenuation of neurotrophin-dependent survival was investigated. RpcAMPS, a selective inhibitor of PKA (Botelho et al., 1988), prevented forskolin from interfering with BDNF rescue from apoptosis after serum withdrawal (Fig. 3A). KCl also had no effect on neuroprotection in the presence of RpcAMPS, suggesting that KCl antagonism of BDNF neuroprotection is attributable primarily to Ca 2+-stimulated cAMP increases and PKA activation. In addition, cells transfected with a dominant-negative regulatory subunit of PKA were resistant to forskolin inhibition of BDNF-dependent neuroprotection (Fig. 3B).
Figure 3.

PKA activity is required for cAMP inhibition of BDNF neuroprotection. A, Cortical neurons were placed in serum-free Neurobasal medium including BDNF with 0.01% DMSO, 10 μm forskolin, or 40 mm KCl. Then the cells were treated with saline (Vehicle) or 200 μm RpcAMPS and scored for apoptosis 24 hr later. The data are derived from counting at least 500 cells per coverslip and are presented as the mean percentage of apoptosis per coverslip ± SEM from three coverslips. B, Cortical neurons were transfected with a GFP expression vector and a three-fold excess of either empty vector or dominant-negative PKA expression vector [R(AB)]. At 48 hr after transfection the cells were washed twice with serum-free Neurobasal medium and then incubated in serum-containing conditioned medium (+ Serum) or serum-free Neurobasal medium (- Serum). Then the cells were pretreated with vehicle, 10 μm forskolin (Forsk), or 25 μm LY294002 (LY), followed by treatment with 10 ng/ml BDNF. Apoptosis was scored 24 hr later by visualization of nuclear morphology. The data are derived by counting at least 150 cells per coverslip of duplicate coverslips and are presented as the average percentage of apoptosis per coverslip.
cAMP inhibits PI3-kinase-mediated, but not MAP kinase-dependent, neuroprotection
Both MAP kinase and PI3-kinase are key survival-promoting pathways activated by neurotrophins. Because PI3-kinase activity is required for BDNF protection of cortical neurons after serum withdrawal (Hetman et al., 1999), we examined the effect of cAMP on neuroprotection caused by direct increases in PI3-kinase activity. This was accomplished by expressing a constitutively active PI3-kinase catalytic subunit, p110* (Klippel et al., 1996), in cortical neurons. Neurons expressing p110* showed increased resistance to apoptosis caused by serum deprivation (Fig. 4A). Forskolin treatment significantly reduced the neuroprotective effects of p110*, consistent with the idea that cAMP is inhibitory to PI3-kinase signaling in cortical neurons. In contrast, forskolin did not inhibit BDNF protection against camptothecin, a DNA-damaging agent (Fig. 4B). BDNF protection from camptothecin-induced apoptosis is mediated by the Erk/MAP kinase pathway with little contribution from PI3-kinase (Hetman et al., 1999). The slight enhancement in neuroprotection caused by forskolin alone is probably attributable to cAMP stimulation of Rap1, leading to increased Erk/MAP kinase activity (Vossler et al., 1997). These data suggest that cAMP specifically antagonizes neurotrophic protection that relies on signaling via the PI3-kinase pathway.
Figure 4.

cAMP inhibits PI3-kinase, but not MAP kinase-dependent, neuronal survival. A, Cortical neurons were transfected with a β-galactosidase expression vector and a threefold excess of either control vector or p110* expression vector. Cells were starved and treated as described in Fig 1 A, 48 hr after transfection. Data are derived by counting at least 50 transfected cells per condition and are presented as the mean percentage of apoptosis per coverslip ± SEM for three coverslips. B, Forskolin does not block BDNF rescue of camptothecin-induced apoptosis. Cortical neurons were treated with BDNF and 0.01% DMSO (Vehicle) or BDNF and 10 μm forskolin in the presence or absence of 5 μm camptothecin (CPT) as indicated. Apoptosis was scored 24 hr later. Data are derived by counting at least 500 cells per coverslip and are presented as the mean percentage of apoptosis per coverslip ± SEM from three coverslips. Where applicable, the statistics were determined by two-tailed Student's t test, assuming equal variance. *p < 0.01.
cAMP negatively regulates PI3-kinase signal transduction cascades
Downstream effectors of PI3-kinase signaling are critical components of the cell survival machinery, the most prominent example being Akt (Yao and Cooper, 1995; Dudek et al., 1997). To determine the effects of cAMP on Akt activation, we examined the phosphorylation of Akt at Ser473 and Thr308, because phosphorylation of both residues is required for the activation of Akt (Alessi et al., 1996). BDNF caused a rapid increase in the phosphorylation of Akt at Ser473 and Thr308 in cortical neurons (Fig. 5A,B). Pretreatment with forskolin inhibited BDNF stimulation of both phosphorylation events, indicating that cAMP inhibits the activation of Akt by PI3-kinase. This was not the case with MAP kinase, which was activated by forskolin (Fig. 5A), as previously described (Vossler et al., 1997).
Figure 5.
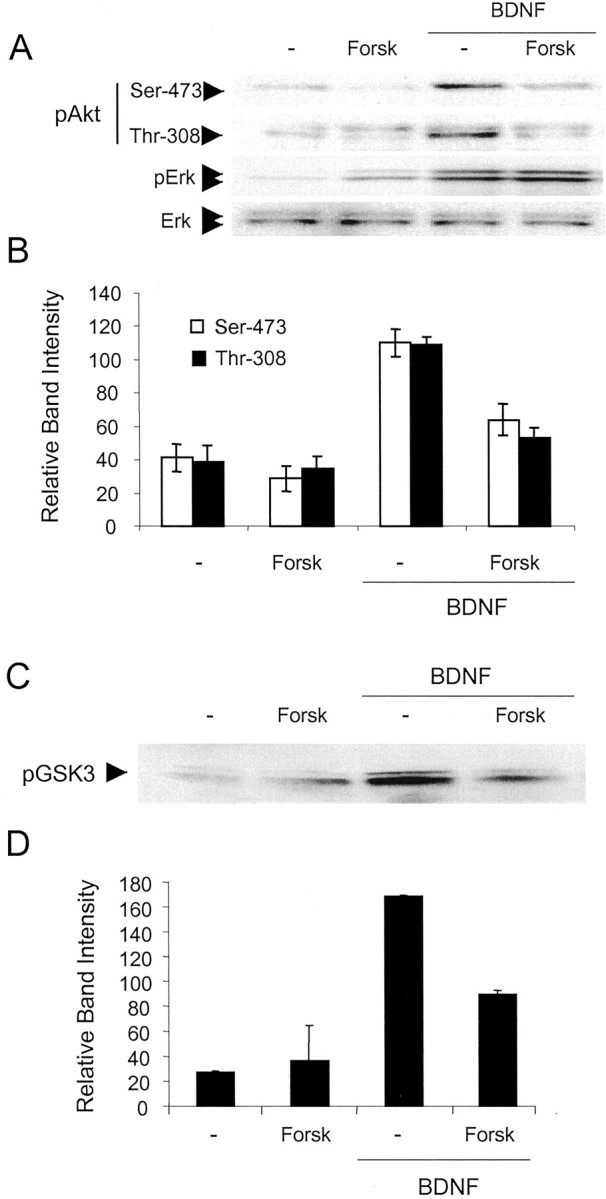
BDNF stimulation of Akt and SGK activity in cortical neurons is inhibited by cAMP. A, Cortical neurons were pretreated with 10 μm forskolin (Forsk) and stimulated with 20 ng/ml BDNF for 5 min. Cell lysates were submitted to Western analysis to quantitate the phosphorylation of Akt at Ser 473 and Thr 308 and Erk phosphorylation at Thr 202/Tyr 204. Western analysis for Erk protein was performed to show equal protein loading. B, Quantitation of pAkt band intensity normalized to Erk ± SD (n = 5). C, SGK activity was attenuated by increased cAMP. Cell lysates were collected from cortical neurons pretreated with vehicle or 10 μm forskolin and stimulated with 20 ng/ml BDNF. SGK was immunoprecipitated, and kinase activity was assessed by measuring the phosphorylation of GSK3 on Western blots. D, Average (with variation) of the GSK3 phosphorylation band intensities from two separate experiments.
Another effector of PI3-kinase is SGK, which is thought to be important for transducing survival signals within cells (Brunet et al., 2001). SGK activity, monitored by the phosphorylation of GSK3, was increased in cortical neurons treated with BDNF (Fig. 5C,D). Forskolin reduced this activity, providing further evidence for the cAMP inhibition of PI3-kinase signal transduction pathways.
To determine whether cAMP inhibits PI3-kinase signaling upstream from Akt or SGK, we coexpressed expression vectors for constitutively active members of the PI3-kinase signal transduction cascade with a SRE reporter construct in cortical neurons. SRE-mediated transcription was used as a downstream reporter for the activity of the PI3-kinase pathway, because previous work showed that neurotrophin stimulation of SRE-dependent gene expression is dependent on PI3-kinase activity in PC12 cells (Poser et al., 2000). Stimulation of SRE-dependent gene expression by BDNF was inhibited by forskolin (Fig. 6A). Likewise, p110* stimulation of SRE-mediated gene expression was inhibited by forskolin, suggesting that cAMP/PKA acts downstream of PI3-kinase activity. In contrast, stimulation of SRE-mediated transcription by the expression of constitutively active SGK (SGK S422D) or wild-type phospholipid-dependent protein kinase-1, PDK-1, was not inhibited by forskolin. PDK-1 directly binds the phospholipid products of PI3-kinase and activates SGK (Alessi et al., 1997). These data suggest that PI3-kinase coupling to SRE-mediated gene expression is inhibited by cAMP at a point upstream from PDK or SGK but subsequent to PI3-kinase activation. Supporting this notion, BDNF-stimulated PI3-kinase activity was not inhibited by forskolin (Fig. 6B,C).
Figure 6.
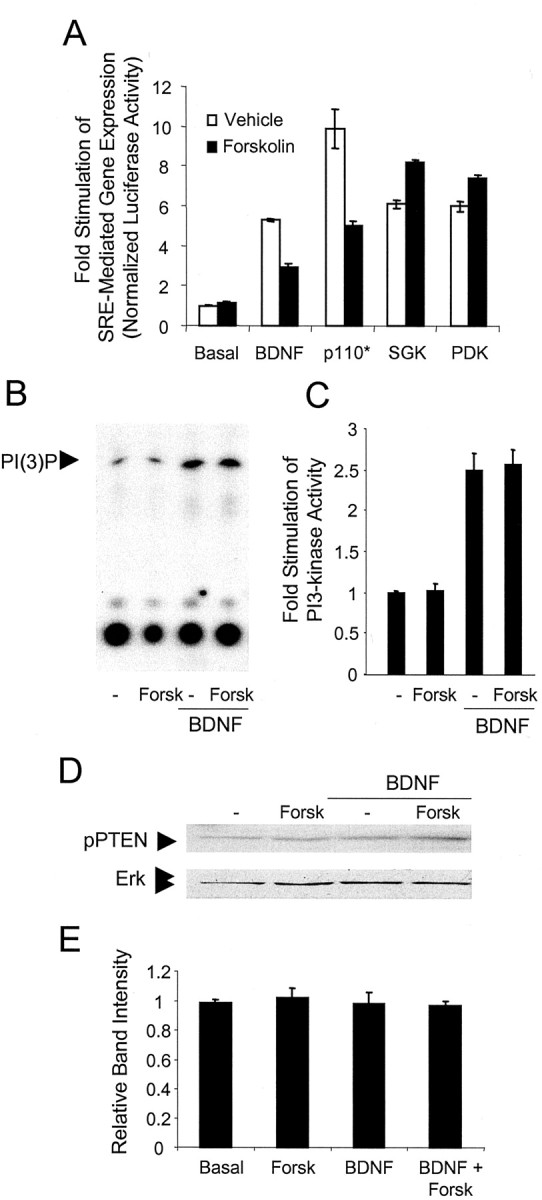
cAMP inhibits the PI3-kinase signal transduction cascade downstream of PI3-kinase via a PTEN-independent mechanism. A, Cortical neurons were transfected with an SRE–luciferase reporter construct and a threefold excess of control, wild-type PDK-1, SGK S422D, or p110* expression vectors. After 36 hr the neurons were treated with 10 μm forskolin. When present, BDNF was at 20 ng/ml. Neurons were lysed and assayed for luciferase activity. Data are the mean of luciferase activity ± SD of triplicate assays normalized to EF1 α-SEAP expression. B, Cortical neurons were pretreated with 0.01% DMSO or 10 μm forskolin (Forsk) and then stimulated with 10 ng/ml BDNF. PI3-kinase was immunoprecipitated from cell lysates by an anti-phosphotyrosine antibody. Phospholipids were resolved by TLC. C, PI3-kinase activity was assessed by quantitation of radiolabeled spots corresponding to phosphatidylinositol 3-monophosphate from triplicate experiments. D, Cell lysates were collected from cortical neurons pretreated with 0.01% DMSO or 10 μm forskolin (Forsk) and stimulated with 10 ng/ml BDNF. PTEN activity was analyzed by Western analysis for phosphorylation of PTEN at Ser 308 and Thr 382/383. Lysates also were analyzed for Erk to measure protein loading. E, Quantitation of pPTEN band intensity normalized to Erk ± SD (n = 3).
Inhibition of the PI3-kinase pathway by cAMP could be attributable to PKA activation of the phospholipid phosphatase and tensin homolog (PTEN), which is responsible for terminating PI(3,4,5)P3 phosphoinositol lipid-dependent signaling (Wu et al., 1998). To test this possibility, we measured the phosphorylation of PTEN in cortical neurons after mitogen and forskolin exposure. Phosphorylation at Ser380 and Thr382/383 within PTEN, a measure of PTEN activity (Vazquez et al., 2000), was unchanged by treatment with BDNF or forskolin (Fig. 6D,E), indicating that neither cAMP nor BDNF affects PTEN activity in cortical neurons.
Discussion
The development of the CNS depends on a complex program of cellular events including cell proliferation, differentiation, and apoptosis that are regulated by signal transduction cross talk. It has become increasingly evident that one of the major roles of cAMP is to modulate other regulatory pathways and that cAMP often serves as a critical checkpoint for coincidence detection. For example, Ca 2+ stimulation of the CREB/CRE transcriptional pathway in neurons, a process that contributes to late-phase long-term potentiation and long-term memory, depends on the activation of MAP kinase (for review, see Impey et al., 1999). The activation and nuclear translocation of MAP kinase is regulated by cAMP (Vossler et al., 1997; Impey et al., 1998). In this study we discovered a novel mechanism by which cAMP negatively regulates the survival of neuronal cells. We show that cAMP antagonizes BDNF protection of cortical neuron apoptosis induced by serum deprivation. Our data indicate that this is attributable to the inhibition of neurotrophin-stimulated PI3-kinase signaling.
Although previous studies have established that cAMP and KCl-induced membrane depolarization often promote the survival of different neuronal populations (Shatz, 1990; Oppenheim, 1991; Ghosh et al., 1994; Ruchaud et al., 1997; Villalba et al., 1997; Hanson et al., 1998; Meyer-Franke et al., 1998; Shen et al., 1999; Vaillant et al., 1999; Li et al., 2000), none of these studies examined the effect of cAMP on neurotrophin protection of cortical neurons against serum withdrawal. cAMP is required for retinal ganglion cells to respond to BDNF after serum deprivation because of cAMP-induced increases in TrkB receptors that mediate the effects of BDNF (Meyer-Franke et al., 1998). This is likely not the case in cortical neurons. The signal transduction machinery of cortical neurons differs from other neuron populations in a number of significant ways. For example, cortical neurons are fully competent to respond to BDNF and, in contrast to retinal ganglion cells, do not require additional signals to support BDNF actions. Furthermore, BDNF neuroprotection against serum withdrawal in cortical neurons is mediated primarily by the PI3-kinase pathway. In contrast, BDNF stimulation of the MAP kinase pathway plays a major role in protection of retinal ganglion cells from serum deprivation (Meyer-Franke et al., 1998). This explains why cAMP antagonizes BDNF neuroprotection against serum withdrawal in cortical neurons, but not in retinal ganglion cells. Ghosh and colleagues showed that depolarization enhances the survival of cortical neurons after the dissociation and plating of the cells (Ghosh et al., 1994). Protection against cellular stress after dissociation and plating of neurons may be mediated by multiple survival pathways and is clearly a different paradigm than serum deprivation. In addition, cerebellar granule cells, often used as models for central neurons, require general depolarization for survival (Gallo et al., 1987), whereas cortical neurons do not. Our data, coupled with other studies examining the effects of cAMP or depolarization on neurotrophin-induced survival, indicate that survival mechanisms vary among different populations of neurons.
Because PI3-kinase does not contribute to neurotrophin protection against all forms of apoptosis, cAMP is not a general inhibitor of the neuroprotective activity of neurotrophins in cortical neurons. For example, BDNF protection of cortical neurons against apoptosis induced by camptothecin is mediated by MAP kinase, whereas the PI3-kinase pathway is the dominant survival mechanism against serum withdrawal (Hetman et al., 1999). cAMP does not decrease neurotrophin attenuation of apoptosis caused by camptothecin-induced DNA damage. In fact, cAMP enhances BDNF neuroprotection against camptothecin, presumably because cAMP activates the MAP kinase pathway and supports the nuclear translocation of MAP kinase (Vossler et al., 1997; Impey et al., 1998). This suggests that cAMP can be either pro-apoptotic or anti-apoptotic, depending on which survival pathway is dominant.
How does cAMP inhibit PI3-kinase signaling in neurons? Our data are consistent with previous reports demonstrating inhibitory cross talk between the cAMP and PI3-kinase signal transduction cascades in non-neuronal cell lines (Ahmed et al., 1995; Kim et al., 2001; Wang et al., 2001; Mei et al., 2002). However, the mechanism for cAMP inhibition of PI3-kinase signaling in neurons appears to be distinct from that described in other studies. Unlike in COS cells in which cAMP directly inhibits PI3-kinase activity (Kim et al., 2001), our data indicate that PKA acts downstream of PI3-kinase in cortical neurons after the generation of 3′-phosphorylated inositol membrane lipids. Regardless of the exact locus for cAMP inhibition of the PI3-kinase signaling pathway, the fact that cAMP inhibits neurotrophin stimulation of both of the downstream survival pathways, SGK and Akt, explains why cAMP antagonizes BDNF neuroprotection.
Multiple aspects of neurobiology regulated by PI3-kinase potentially could be modulated by coincident cAMP-elevating signals. Inhibitory cross talk between cAMP and PI3-kinase signal transduction cascades may play an important role in the regulation of neuronal survival after ischemic injury to the neocortex. There is elevated cAMP in the neocortex after the occlusion of cerebral blood flow (Domanska-Janik and Pylova, 1989, 1992; Prado et al., 1992; Cai et al., 2002). This cAMP increase may contribute to the magnitude of delayed apoptosis seen after the induction of cerebral infarction, leading to the elimination of severely damaged neurons that otherwise might be spared by BDNF secreted from neighboring cells (Narumiya et al., 1998; Ferrer et al., 2001). Consequently, the increase in cAMP would ensure the removal of all damaged neurons. In addition, cAMP inhibition of BDNF-stimulated PI3-kinase signaling may be critical during cortical development. Studies that used a mouse strain deficient in type 1 adenylyl cyclase indicate that cAMP may regulate pattern formation in the somatosensory cortex (Abdel-Majid et al., 1998). These mutant mice lack the specific cAMP signal necessary during a critical period of development that may be important for pruning of the neural network. Without this cAMP signal the pattern development in layer IV of the somatosensory cortex does not occur normally (Woolsey and Van der Loos, 1970). PI3-kinase also is implicated in regulating the proliferation of cortical neuronal precursors (Groszer et al., 2001; Li et al., 2001). cAMP is known to inhibit the proliferation of these cells (Lu and DiCicco-Bloom, 1997; Palmer et al., 1997). Therefore, coincident cAMP-generating signals may influence neurogenesis in the developing cortex by blocking proliferative signals that use PI3-kinase.
In summary, cAMP may inhibit a number of processes in CNS neurons that are regulated positively by the PI3-kinase pathway, including neurotrophin-stimulated survival and synaptic plasticity (Levi-Montalcini and Booker, 1960; Barde, 1989; Datta and Greenberg, 1998; Lin et al., 2001). Although previous studies suggest that a combination of neurotrophins and cAMP might be useful therapeutically to promote the survival of neurons, our data indicate that such combinations may not always provide synergistic neuroprotection.
Footnotes
This research was supported by National Institutes of Health Grants NS 20498 and DC 04158. We thank members of the Storm and Xia laboratories for constructive reading of this manuscript and editorial input.
Correspondence should be addressed to Daniel R. Storm at the above address. E-mail: dstorm@u.washington.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/234420-08$15.00/0
References
- Abdel-Majid RM, Leong WL, Schalkwyk LC, Smallman DS, Wong ST, Storm DR, Fine A, Dobson MJ, Guernsey DL, Neumann PE ( 1998) Loss of adenylyl cyclase I activity disrupts patterning of mouse somatosensory cortex. Nat Genet 19: 289–291. [DOI] [PubMed] [Google Scholar]
- Ahmed MU, Hazeki K, Hazeki O, Katada T, Ui M ( 1995) Cyclic AMP-increasing agents interfere with chemoattractant-induced respiratory burst in neutrophils as a result of the inhibition of phosphatidylinositol 3-kinase rather than receptor-operated Ca 2+ influx. J Biol Chem 270: 23816–23822. [DOI] [PubMed] [Google Scholar]
- Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA ( 1996) Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J 15: 6541–6551. [PMC free article] [PubMed] [Google Scholar]
- Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P ( 1997) Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα. Curr Biol 7: 261–269. [DOI] [PubMed] [Google Scholar]
- Barde YA ( 1989) Trophic factors and neuronal survival. Neuron 2: 1525–1534. [DOI] [PubMed] [Google Scholar]
- Botelho LH, Rothermel JD, Coombs RV, Jastorff B ( 1988) cAMP analog antagonists of cAMP action. Methods Enzymol 159: 159–172. [DOI] [PubMed] [Google Scholar]
- Brunet A, Park J, Tran H, Hu LS, Hemmings BA, Greenberg ME ( 2001) Protein kinase SGK mediates survival signals by phosphorylating the Forkhead transcription factor FKHRL1 (FOXO3a). Mol Cell Biol 21: 952–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z, Lin S, Rhodes PG ( 2002) Neuroprotective effects of N-acetylaspartyl-glutamate in a neonatal rat model of hypoxia-ischemia. Eur J Pharmacol 437: 139–145. [DOI] [PubMed] [Google Scholar]
- Campard PK, Crochemore C, Rene F, Monnier D, Koch B, Loeffler JP ( 1997) PACAP type I receptor activation promotes cerebellar neuron survival through the cAMP/PKA signaling pathway. DNA Cell Biol 16: 323–333. [DOI] [PubMed] [Google Scholar]
- Chan GC, Hinds TR, Impey S, Storm DR ( 1998) Hippocampal neurotoxicity of Δ9-tetrahydrocannabinol. J Neurosci 18: 5322–5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke PGH ( 1988) Apoptosis versus necrosis: how valid a dichotomy for neurons? In: Cell death and diseases of the nervous system (Koliatsos V, Ratan RR, eds), pp 3–28. Totowa, NJ: Humana.
- Crowder RJ, Freeman RS ( 1998) Phosphatidylinositol 3-kinase and Akt protein kinase are necessary and sufficient for the survival of nerve growth factor-dependent sympathetic neurons. J Neurosci 18: 2933–2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta SR, Greenberg ME ( 1998) Molecular mechanisms of neuronal survival and apoptosis. In: Hormones and signaling (O'Malley B, ed), pp 257–306. San Diego: Academic.
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME ( 1997) Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91: 231–241. [DOI] [PubMed] [Google Scholar]
- D'Mello SR, Galli C, Ciotti T, Calissano P ( 1993) Induction of apoptosis in cerebellar granule neurons by low potassium: inhibition of death by insulin-like growth factor I and cAMP. Proc Natl Acad Sci USA 90: 10989–10993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolcet X, Egea J, Soler RM, Martin-Zanca D, Comella JX ( 1999) Activation of phosphatidylinositol 3-kinase, but not extracellular-regulated kinases, is necessary to mediate brain-derived neurotrophic factor-induced motoneuron survival. J Neurochem 73: 521–531. [DOI] [PubMed] [Google Scholar]
- Domanska-Janik K, Pylova S ( 1989) Rapid enhancement of cAMP accumulation in rat brain particulate fraction after ischaemia. Int J Tissue React 11: 73–79. [PubMed] [Google Scholar]
- Domanska-Janik K, Pylova S ( 1992) Postreceptor modulation of cAMP accumulation in rat brain particulate fraction after ischemia—involvement of protein kinase C. Mol Chem Neuropathol 17: 65–77. [DOI] [PubMed] [Google Scholar]
- Dudek H, Datte SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME ( 1997) Regulation of neuronal survival by the serine–threonine protein kinase Akt. Science 275: 628–630. [DOI] [PubMed] [Google Scholar]
- Encinas M, Iglesias M, Llecha N, Comella JX ( 1999) Extracellular-regulated kinases and phosphatidylinositol 3-kinase are involved in brain-derived neurotrophic factor-mediated survival and neuritogenesis of the neuroblastoma cell line SH-SY5Y. J Neurochem 73: 1409–1421. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Krupinski J, Goutan E, Marti E, Ambrosio S, Arenas E ( 2001) Brain-derived neurotrophic factor reduces cortical cell death by ischemia after middle cerebral artery occlusion in the rat. Acta Neuropathol (Berl) 101: 229–238. [DOI] [PubMed] [Google Scholar]
- Friedman WJ, Greene LA ( 1999) Neurotrophin signaling via Trks and p75. Exp Cell Res 253: 131–142. [DOI] [PubMed] [Google Scholar]
- Gallo V, Kingsbury A, Balazs R, Jorgensen OS ( 1987) The role of depolarization in the survival and differentiation of cerebellar granule cells in culture. J Neurosci 7: 2203–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Carnahan J, Greenberg ME ( 1994) Requirement for BDNF in activity-dependent survival of cortical neurons. Science 263: 1618–1623. [DOI] [PubMed] [Google Scholar]
- Groszer M, Erickson R, Scripture-Adams DD, Lesche R, Trumpp A, Zack JA, Kornblum HI, Liu X, Wu H ( 2001) Negative regulation of neural stem/progenitor cell proliferation by the PTEN tumor suppressor gene in vivo Science 294: 2186–2189. [DOI] [PubMed] [Google Scholar]
- Hanson Jr MG, Shen S, Wiemelt AP, McMorris FA, Barres BA ( 1998) Cyclic AMP elevation is sufficient to promote the survival of spinal motor neurons in vitro J Neurosci 18: 7361–7371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetman M, Kanning K, Cavanaugh JE, Xia Z ( 1999) Neuroprotection by brain-derived neurotrophic factor is mediated by extracellular signal-regulated kinase and phosphatidylinositol 3-kinase. J Biol Chem 274: 22569–22580. [DOI] [PubMed] [Google Scholar]
- Hetman M, Cavanaugh JE, Kimelman D, Xia Z ( 2000) Role of glycogen synthase kinase-3β in neuronal apoptosis induced by trophic withdrawal. J Neurosci 20: 2567–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Impey S, Obrietan K, Wong ST, Poser S, Yano S, Wayman G, Deloulme JC, Chan G, Storm DR ( 1998) Cross talk between ERK and PKA is required for Ca 2+ stimulation of CREB-dependent transcription and ERK nuclear translocation. Neuron 21: 869–883. [DOI] [PubMed] [Google Scholar]
- Impey S, Obrietan K, Storm DR ( 1999) Making new connections: role of ERK/MAP kinase signaling in neuronal plasticity. Neuron 23: 11–14. [DOI] [PubMed] [Google Scholar]
- Johansen FE, Prywes R ( 1994) Two pathways for serum regulation of the c-fos serum response element require specific sequence elements and a minimal domain of serum response factor. Mol Cell Biol 14: 5920–5928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan DR ( 1998) Studying signal transduction in neuronal cells: the Trk/NGF system. Prog Brain Res 117: 35–46. [DOI] [PubMed] [Google Scholar]
- Kim S, Jee K, Kim D, Koh H, Chung J ( 2001) Cyclic AMP inhibits Akt activity by blocking the membrane localization of PDK1. J Biol Chem 276: 12864–12870. [DOI] [PubMed] [Google Scholar]
- Klesse LJ, Parada LF ( 1998) p21 ras and phosphatidylinositol 3-kinase are required for survival of wild-type and NF1 mutant sensory neurons. J Neurosci 18: 10420–10428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klippel A, Reinhard C, Kavanaugh WM, Apell G, Escobedo MA, Williams LT ( 1996) Membrane localization of phosphatidylinositol 3-kinase is sufficient to activate multiple signal-transducing kinase pathways. Mol Cell Biol 16: 4117–4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Cohen P ( 1999) Activation of serum- and glucocorticoid-regulated protein kinase by agonists that activate phosphatidylinositide 3-kinase is mediated by 3-phosphoinositide-dependent protein kinase-1 (PDK1) and PDK2. Biochem J 339: 319–328. [PMC free article] [PubMed] [Google Scholar]
- Kuruvilla R, Ye H, Ginty DD ( 2000) Spatially and functionally distinct roles of the PI3-K effector pathway during NGF signaling in sympathetic neurons. Neuron 27: 499–512. [DOI] [PubMed] [Google Scholar]
- Levi-Montalcini R, Booker B ( 1960) Destruction of sympathetic ganglia in mammals by an antisera to nerve growth factor protein. Proc Natl Acad Sci USA 46: 384–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li BS, Ma W, Zhang L, Barker JL, Stenger DA, Pant HC ( 2001) Activation of phosphatidylinositol-3 kinase (PI-3K) and extracellular regulated kinases (Erk1/2) is involved in muscarinic receptor-mediated DNA synthesis in neural progenitor cells. J Neurosci 21: 1569–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Wang X, Meintzer MK, Laessig T, Birnbaum MJ, Heidenreich KA ( 2000) Cyclic AMP promotes neuronal survival by phosphorylation of glycogen synthase kinase-3β. Mol Cell Biol 20: 9356–9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CH, Yeh SH, Lu KT, Leu TH, Chang WC, Gean PW ( 2001) A role for the PI-3 kinase signaling pathway in fear conditioning and synaptic plasticity in the amygdala. Neuron 31: 841–851. [DOI] [PubMed] [Google Scholar]
- Lu N, DiCicco-Bloom E ( 1997) Pituitary adenylate cyclase-activating polypeptide is an autocrine inhibitor of mitosis in cultured cortical precursor cells. Proc Natl Acad Sci USA 94: 3357–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma FH, Ohkuma S, Kishi M, Kuriyama K ( 1991) Ontogeny of β-adrenergic receptor-mediated cyclic AMP-generating system in primary cultured neurons. Int J Dev Neurosci 9: 347–356. [DOI] [PubMed] [Google Scholar]
- Mazzoni IE, Said FA, Aloyz R, Miller FD, Kaplan D ( 1999) Ras regulates sympathetic neuron survival by suppressing the p53-mediated cell death pathway. J Neurosci 19: 9716–9727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei FC, Qiao J, Tsygankova OM, Meinkoth JL, Quilliam LA, Cheng X ( 2002) Differential signaling of cyclic AMP: opposing effects of exchange protein directly activated by cyclic AMP and cAMP-dependent protein kinase on protein kinase B activation. J Biol Chem 277: 11497–11504. [DOI] [PubMed] [Google Scholar]
- Meyer-Franke A, Kaplan MR, Pfrieger FW, Barres BA ( 1995) Characterization of the signaling interactions that promote the survival and growth of developing retinal ganglion cells in culture. Neuron 15: 805–819. [DOI] [PubMed] [Google Scholar]
- Meyer-Franke A, Wilkinson GA, Kruttgen A, Hu M, Munro E, Hanson Jr MG, Reichardt LF, Barres BA ( 1998) Depolarization and cAMP elevation rapidly recruit TrkB to the plasma membrane of CNS neurons. Neuron 21: 681–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller TM, Tansey MG, Johnson Jr EM, Creedon DJ ( 1997) Inhibition of phosphatidylinositol 3-kinase activity blocks depolarization- and insulin-like growth factor I-mediated survival of cerebellar granule cells. J Biol Chem 272: 9847–9853. [DOI] [PubMed] [Google Scholar]
- Myers Jr MG, Sun XJ, Cheatham B, Jachna BR, Glasheen EM, Backer JM, White MF ( 1993) IRS-1 is a common element in insulin and insulin-like growth factor-I signaling to the phosphatidylinositol 3′-kinase. Endocrinology 132: 1421–1430. [DOI] [PubMed] [Google Scholar]
- Narumiya S, Ohno M, Tanaka N, Yamano T, Shimada M ( 1998) Enhanced expression of full-length TrkB receptors in young rat brain with hypoxic/ischemic injury. Brain Res 797: 278–286. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW ( 1991) Cell death during development of the nervous system. Annu Rev Neurosci 14: 453–501. [DOI] [PubMed] [Google Scholar]
- Palmer TD, Takahashi J, Gage FH ( 1997) The adult rat hippocampus contains primordial neural stem cells. Mol Cell Neurosci 8: 389–404. [DOI] [PubMed] [Google Scholar]
- Parrizas M, Saltiel AR, LeRoith D ( 1997) Insulin-like growth factor 1 inhibits apoptosis using the phosphatidylinositol 3′-kinase and mitogen-activated protein kinase pathways. J Biol Chem 272: 154–161. [DOI] [PubMed] [Google Scholar]
- Poser S, Impey S, Trinh K, Xia Z, Storm DR ( 2000) SRF-dependent gene expression is required for PI3-kinase-regulated cell proliferation. EMBO J 19: 4955–4966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prado R, Busto R, Globus MY ( 1992) Ischemia-induced changes in extracellular levels of striatal cyclic AMP: role of dopamine neurotransmission. J Neurochem 59: 1581–1584. [DOI] [PubMed] [Google Scholar]
- Raff MC, Barres BA, Burne JF, Coles HS, Ishizaki Y, Jacobson MD ( 1993) Programmed cell death and the control of cell survival: lessons from the nervous system. Science 262: 695–700. [DOI] [PubMed] [Google Scholar]
- Ruchaud S, Seite P, Foulkes NS, Sassone-Corsi P, Lanotte M ( 1997) The transcriptional repressor ICER and cAMP-induced programmed cell death. Oncogene 15: 827–836. [DOI] [PubMed] [Google Scholar]
- Rydel RE, Greene LA ( 1988) cAMP analogs promote survival and neurite outgrowth in cultures of rat sympathetic and sensory neurons independently of nerve growth factor. Proc Natl Acad Sci USA 85: 1257–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon Y ( 1991) Cellular responsiveness to hormones and neurotransmitters: conversion of [3H]adenine to [3H]cAMP in cell monolayers, cell suspensions, and tissue slices. Methods Enzymol 195: 22–28. [DOI] [PubMed] [Google Scholar]
- Salomon Y, Londos C, Rodbell M ( 1974) A highly sensitive adenylate cyclase assay. Anal Biochem 58: 541–548. [DOI] [PubMed] [Google Scholar]
- Schecterson LC, McKnight GS ( 1991) Role of cyclic adenosine 3′, 5′-monophosphate-dependent protein kinase in hormone-stimulated β-endorphin secretion in AtT20 cells. Mol Endocrinol 5: 170–178. [DOI] [PubMed] [Google Scholar]
- Segal RA, Greenberg ME ( 1996) Intracellular signaling pathways activated by neurotrophic factors. Annu Rev Neurosci 19: 463–489. [DOI] [PubMed] [Google Scholar]
- Shatz CJ ( 1990) Impulse activity and the patterning of connections during CNS development. Neuron 5: 745–756. [DOI] [PubMed] [Google Scholar]
- Shen S, Wiemelt AP, McMorris FA, Barres BA ( 1999) Retinal ganglion cells lose trophic responsiveness after axotomy. Neuron 23: 285–295. [DOI] [PubMed] [Google Scholar]
- Sibley DR, Lefkowitz RJ ( 1987) β-Adrenergic receptor-coupled adenylate cyclase. Biochemical mechanisms of regulation. Mol Neurobiol 1: 121–154. [DOI] [PubMed] [Google Scholar]
- Skaper SD, Floreani M, Negro A, Facci L, Giusti P ( 1998) Neurotrophins rescue cerebellar granule neurons from oxidative stress-mediated apoptotic death: selective involvement of phosphatidylinositol 3-kinase and the mitogen-activated protein kinase pathway. J Neurochem 70: 1859–1868. [DOI] [PubMed] [Google Scholar]
- Stefanis L, Burke RE, Greene LA ( 1997) Apoptosis in neurodegenerative disorders. Curr Opin Neurol 10: 299–305. [DOI] [PubMed] [Google Scholar]
- Vaillant AR, Mazzoni I, Tudan C, Boudreau M, Kaplan DR, Miller FD ( 1999) Depolarization and neurotrophins converge on the phosphatidylinositol 3-kinase–Akt pathway to synergistically regulate neuronal survival. J Cell Biol 146: 955–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez F, Ramaswamy S, Nakamura N, Sellers WR ( 2000) Phosphorylation of the PTEN tail regulates protein stability and function. Mol Cell Biol 20: 5010–5018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalba M, Bockaert J, Journot L ( 1997) Pituitary adenylate cyclase-activating polypeptide (PACAP-38) protects cerebellar granule neurons from apoptosis by activating the mitogen-activated protein kinase (MAP kinase) pathway. J Neurosci Res 17: 83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossler MR, Yao H, York RD, Pan MG, Rim CS, Stork PJ ( 1997) cAMP activates MAP kinase and Elk-1 through a B-Raf- and Rap1-dependent pathway. Cell 89: 73–82. [DOI] [PubMed] [Google Scholar]
- Wang L, Liu F, Adamo ML ( 2001) Cyclic AMP inhibits extracellular signal-regulated kinase and phosphatidylinositol 3-kinase/Akt pathways by inhibiting Rap1. J Biol Chem 276: 37242–37249. [DOI] [PubMed] [Google Scholar]
- Woolsey TA, Van der Loos H ( 1970) The structural organization of layer IV in the somatosensory region (SI) of mouse cerebral cortex. The description of a cortical field composed of discrete cytoarchitectonic units. Brain Res 17: 205–242. [DOI] [PubMed] [Google Scholar]
- Wu X, Senechal K, Neshat MS, Whang YE, Sawyers CL ( 1998) The PTEN/MMAC1 tumor suppressor phosphatase functions as a negative regulator of the phosphoinositide 3-kinase/Akt pathway. Proc Natl Acad Sci USA 95: 15587–15591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME ( 1995) Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 270: 1326–1331. [DOI] [PubMed] [Google Scholar]
- Yamada M, Tanabe K, Wada K, Shimoke K, Ishikawa Y, Ikeuchi T, Koizumi S, Hatanaka H ( 2001) Differences in survival-promoting effects and intracellular signaling properties of BDNF and IGF-1 in cultured cerebral cortical neurons. J Neurochem 78: 940–951. [DOI] [PubMed] [Google Scholar]
- Yao R, Cooper GM ( 1995) Requirement for phosphatidylinositol 3-kinase in the prevention of apoptosis by nerve growth factor. Science 267: 2003–2006. [DOI] [PubMed] [Google Scholar]