

Abstract
Low-voltage-activated (LVA) Ca2+ channels are widely distributed throughout the CNS and are important determinants of neuronal excitability, initiating dendritic and somatic Ca2+ spikes that trigger and shape the pattern of action potential firing. Here, we define a molecular mechanism underlying the dynamic regulation of α1H channels (Cav3.2), by Ca2+/CaM-dependent protein kinase II (CaMKII). We show that channel regulation is selective for the LVA α1H Ca2+ channel subtype, depends on determinants in the α1H II-III intracellular loop, and requires the phosphorylation of a serine residue absent from unregulated α1G (Cav3.1) channels. These studies identify the α1H channel as a new substrate for CaMKII and provide the first molecular mechanism for the direct regulation of T-type Ca2+ channels by a protein kinase. Our data suggest a novel mechanism for modulating the integrative properties of neurons.
Keywords: calcium [Ca], calmodulin, channel, dendrite, hippocampus, protein kinase, T-type
Introduction
Low-voltage-activated (LVA), T-type, Ca2+ channels are important determinants of neuronal excitability (McCormick, 1992; Huguenard, 1996; Eilers and Konnerth, 1997; Hausser et al., 2000). They support the forward propagation of distal dendritic inputs in neocortical (Markram and Sakmann, 1994) and CA1 hippocampal pyramidal neurons (Magee and Johnston, 1995a,b) and mediate low-threshold spikes that support somatic burst firing in thalamocortical (Huguenard and McCormick, 1992; Destexhe et al., 1996) and CA3 hippocampal pyramidal neurons (Fisher et al., 1990; Migliore and Shepherd, 2002). Both dendritic boosting and neuronal bursting supported by LVA channels amplify subthreshold stimuli (Hausser et al., 2000). LVA Ca2+ channels can also facilitate long-term changes in neuronal plasticity as demonstrated in the dorsal horn (Ikeda et al., 2003) and CA1 pyramidal neurons (Su et al., 2002). Thus, LVA Ca2+ channel activity can contribute to the complex and diverse firing behavior of neurons.
LVA Ca2+ channels belong to the Cav3 family, of which there are three family members (Cav3.1, Cav3.2, and Cav3.3: α1G, α1H, and α1I) (Cribbs et al., 1998; Perez-Reyes et al., 1998; Lee et al., 1999). Little is known about the regulation of this Ca2+ channel class, in contrast to high voltage-activated Ca2+ channels that are well-established targets of G-proteins and kinases (Catterall, 2000). One defined mode of LVA channel regulation depends on the activity of Ca2+/CaM-dependent protein kinase II (CaMKII), an important regulator of ion channel activity (Soderling and Derkach, 2000; Hudmon and Schulman, 2002). Our previous work established that CaMKII modulates α1H but not α1G channel gating, preferentially increasing current at subthreshold potentials (Wolfe et al., 2002). Because the activity of multimeric CaMKII depends on Ca2+/CaM binding and intersubunit autophosphorylation, CaMKII activity can extend beyond the lifetime of the Ca2+ signal and enable the kinase to sum and thereby respond to small repeated Ca2+ transients (Soderling and Derkach, 2000; Hudmon and Schulman, 2002). Thus, CaMKII-dependent regulation of LVA channel activity could persist during weak synaptic stimulation.
CaMKII is ubiquitously distributed throughout the CNS and PNS and is a major constituent of postsynaptic densities of excitatory synapses (Kennedy et al., 1983; Kelly et al., 1984; Braun and Schulman, 1995; Liu and Jones, 1996; Hudmon and Schulman, 2002). The widespread localization of this kinase (Braun and Schulman, 1995) and LVA channels (Perez-Reyes, 2003) in both dendritic and somatic compartments (Liu and Jones, 1996; Shen et al., 1998; Thiagarajan et al., 2002) and their independent roles in modulating synaptic transmission suggested a direct mechanism for the regulation of α1H channels by CaMKII that would be of general importance to neuronal function.
Here, we show that the regulation of α1H channels by CaMKII depends on structural determinants contained within the intracellular linker connecting channel domains II and III. A serine residue (Ser1198) in the HII-III linker is a preferred CaMKII phosphorylation site and is critical to this modulation. These data identify the α1H Ca2+ channel as a direct effector for CaMKII and provide a new CaMKII-dependent mechanism for enhancing neuronal excitability.
Materials and Methods
Preparation of human α1Gand α1Hchimeric and mutant constructs. All constructs were prepared using a PCR-subcloning strategy in which PCR products were verified by DNA sequencing. A truncated α1H sequence (AF051946 version 1.0) was extended by PCR to add a sequence encoding for 50 amino acids using 5′-GACAGCCCTAGGGACAC-3′ [5′-CGAGAATGCACTCGAGCTACACGGGGTCATC-3′ (AF051946 modified)]. The II-III linker regions were amplified from α1H (AF051946 modified) and α1G (AF190860) (Cribbs et al., 2000) were generated by PCR (α1H, 5′-TCCTGTACAACGGCATGG-3′ and 5′-CTCTCTAGATATCAGCGTCTCCACC-3′; α1G, 5′-CCTGTACAATGGTATGGCCTCC-3′ and 5′-CTCTCTAGACACGTGGTCGAACATCTTG-3′) and subcloned into pUC18 at the SmaI-XbaI site, resulting in pUC18-HII-III and pUC18-GII-III. The PCR reaction introduced an upstream BsrGI site in the HII-III. The II-III linker of α1G was isolated from pUC18-GII-III by BsrGI-PmlI digestion and inserted into pUC18-HII-III at the corresponding site to replace HII-III, resulting in pUC18-GII-III-1. Finally, the BsrGI-EcoRV fragment containing the II-III linker of α1G was isolated from the last construct and subcloned back to pcDNA3-α1H, resulting in construct H-GII-III. Similarly, preparation of the construct G-HII-III involved an intermediate construct pUC18-HII-III-1 that was generated by subcloning a PCR (5′-CCAATGCATCTTTTCGGCTGCAAGTTC-3′ and 5′-CCATCTAGACACGTGATCAAACATCTTG-3′) product of HII-III into pUC18 at the SmaI-XbaI site, in which an NsiI site was introduced upstream of HII-III. The HII-III linker was isolated from an NsiI-PmlI digestion of pUC18-HII-III-1 and subcloned into pUC18-α1G replacing the original II-III linker, resulting in pUC18-G-HII-III. The chimearic channel was subcloned into pcDNA3.1(-) at the EcoRI-KpnI site, resulting in the construct G-HII-III. All mutations were introduced into the intermediate constructs pUC18-HII-III or pUC18-HII-III-1 using QuikChange XL Site-Directed Mutagenesis Kit (catalog #200517; Stratagene, La Jolla, CA). Last, the construct G-HCterminus was prepared by fusing the 5′ section of α1H and the 3′ section of α1G at the PmlI site in pcDNA3.
Preparation of glutathione S-transferase fusion proteins. The wild-type DNA construct of the glutathione S-transferase (GST)-HII-III fusion protein was generated by subcloning a PCR (5′-CATGGATCCCAGGCGGAGGGCGATG-3′ and 5′-CAAGATATCCGTGGGTCCAGGGACTC-3′) product of α1H (α1H3117-3696) into pGEX-2T at the BamHI-SmaI site. The mutations S1198A and/or S1153A were introduced by PCR. All PCR products were verified by DNA sequencing.
In vitro CaMKIIγc phosphorylation. GST-HII-III fusion proteins were purified from transformed Escherichia coli BL21 cell lysates by binding to 200 μl of 50% (w/v) slurry of glutathione-Sepharose 4B (Amersham Biosciences, Arlington Heights, IL). After repeated washes with PBS, the GST-HII-III fusion proteins were eluted from the beads with 20 mm reduced glutathione in a Tris-HCl buffer, pH 7.4. Purity of the isolated proteins was assessed by SDS-PAGE, and concentration was determined using a Coomassie Protein Assay (catalog #1856209; Pierce, Rockford, IL). It was estimated from Coomassie staining of SDS-PAGE samples that 50% of the final protein was full length. Phosphorylation of the GST-HII-III fusion proteins was performed in a final volume of 50 μl containing 1 μm fusion protein, 1 mm DTT, 10 mm HEPES, 0.5 mm CaCl2, 2 μm calmodulin, 10 mm MgCl2, 0.1% Triton X-100, 1 mg/ml BSA, and 100 μm [32P]ATP (10 mCi/ml). Reaction was initiated by the addition of 30 nm purified recombinant porcine CaMKIIγc, and samples were incubated at 37°C for up to 15 min. Reactions were terminated by addition of 20 μl of 4× SDS sample buffer containing 500 mm EGTA, followed by incubation at 95°C for 5 min before separation by SDS-PAGE. Gels were Coomassie stained, and, after autoradiography, bands corresponding to phosphorylated GST-HII-III fusion proteins were cut from the gel, and radioactivity was assessed using liquid scintillation counting.
Electrophysiology. Whole-cell currents were recorded from adherent HEK293 cells using a standard patch electrode voltage-clamp method (Lu et al., 1994). To eliminate K+ currents and fix free [calcium], we used internal solutions containing the following (in mm): 115 CsCl, 1 tetrabutylammonium chloride, 1 MgCl2, 5 Mg-ATP, 1 Li-GTP, 20 HEPES, pH.7.2 (adjusted with CsOH), and 11 BAPTA; added CaCl2 fixed the free Ca2+ at 27 nm (0.9 mm) or 1 μm (8.8 mm) with 2 μm CaM (Lu et al., 1994). HEK293 cells were superfused with a solution containing the following (in mm): 127 TEA-Cl, 10 CaCl2, 0.5 MgCl2, 10 HEPES, 5 dextrose, and 32 sucrose, pH 7.4 (adjusted with CsOH). Currents were filtered at 2.5 kHz and sampled at 12.5 kHz; leak subtraction was performed on line using P/4 protocol. Activation gating was determined using tail currents in response to 15 test depolarizations in 5 mV increments (-60 to +10 mV; 10.4 msec) from a holding potential of -90 mV during repolarization to -60 mV (45 msec). Interpulsing time was 6 sec to allow for recovery from inactivation. Tail currents were fitted to a single exponential plus a constant using the Chebyshev algorithim in pClamp 6.0 software (Axon Instruments, Foster City, CA). As described previously (Wolfe et al., 2002), activation gating fitted significantly better to a Boltzmann distribution raised to the second power: [I/Imax = 1/(1 + exp[(V0.25 - Vt)/k])2, where k is the slope factor (mV/e-fold change), Vt is the test potential, and Imax is the maxium current. Half-activation potential was calculated as follows: V0.5 = 0.882(k) + V0.25. Data obtained with the CaMKII-activating internal solution were fitted to the sum of two Boltzmanns. Averaged data sets obtained with activating or non-activating solutions were analyzed concurrently to obtain a single set of parameters for the non-modulated component that optimally describes both sets of data and another set of parameters for the second modulated component. Parameter estimations were performed by a nonlinear least-squares procedure (Johnson and Frasier, 1985) using boot strapping to determine confidence limits (Efron and Tibshirani, 1993). To evaluate the goodness-of-fit to a single versus a double Boltzmann distribution, the sum of the squared residuals was calculated and mapped into a probability by calculating a Z score for the runs test (Straume and Johnson, 1992).
Results
Activation of CaMKII induces an 11 mV hyperpolarizing shift in the half-activation potential (V1/2) of native and recombinant α1H channels (Lu et al., 1994; Schrier et al., 2001; Wolfe et al., 2002). This regulation depends on active kinase (Lu et al., 1994; Barrett et al., 2000) and is not observed with human recombinant α1G channels (Cav3.1) (Monteil et al., 2000) stably expressed in HEK293 cells (Wolfe et al., 2002). To test for the differential regulation of these two Cav3.0 family members in a transient transfection paradigm, we cotransfected CaMKII with either the α1H or the α1G channel construct. Channel activity was measured using internal solutions that either promote (1 μm free Ca2+; 2 μm CaM) or impair (27 nm free Ca2+) CaMKII activation (Wolfe et al., 2002). Ca2+/CaM potentiates α1H currents at potentials at which channel activation is incomplete (-60 to 0 mV) (Fig. 1b). Neither currents recorded at maximally activating voltages (Vtest =+10 mV; Vr = -60 mV) nor currents recorded from α1G channels (Fig. 1b,e) are increased with CaMKII-activating internal solutions. The gating behavior of α1H channels recorded with CaMKII-activating solutions is poorly fitted by a single squared Boltzmann function but is fitted significantly better (p < 0.05) by a double Boltzmann function that defines two channel populations. One population displays properties indistinguishable from channels recorded with impaired CaMKII activity [termed non-modulated (N-M)], whereas the other population [termed modulated (M)] displays a more hyperpolarized half-activation potential (V1/2) and an increased voltage sensitivity (k) (Fig. 1c, inset). Concurrent analysis of the averaged data sets reveals that CaMKII activation modulates α1H but not α1G channels, inducing a -14.3 ± 1.6 mV (p < 0.05) shift in the half-activation potential and an Δ -4.5 ± 1.4 mV/e-fold (p < 0.05) increase in the voltage sensitivity of gating, as indicated by the reduction in slope factor (k). A concomitant change in the midpoint and the apparent steepness of gating suggests that CaMKII may change the work required to open the channel, possibly by reducing the electrostatic surface potential sensed by the voltage-sensing module (Hille, 2001). On average, CaMKII modulates 45 ± 9% of the α1H channel population; however, in any given cell, modulation can be near complete or remain submaximal. An additional analysis of the regulation of α1H channels shows that the CaMKII-modulated channel population in individual cells may be as large as 99% or as small as 21% of the total channel population (46 ± 5%). We interpret this variability as a byproduct of transient transfection because the expression of GFP, one cotransfectant, is also variable. Nonetheless, the regulation of α1H but not α1G channels in transient transfection suggests that structural determinants in the channel protein may underlie CaMKII-induced regulation.
Figure 1.
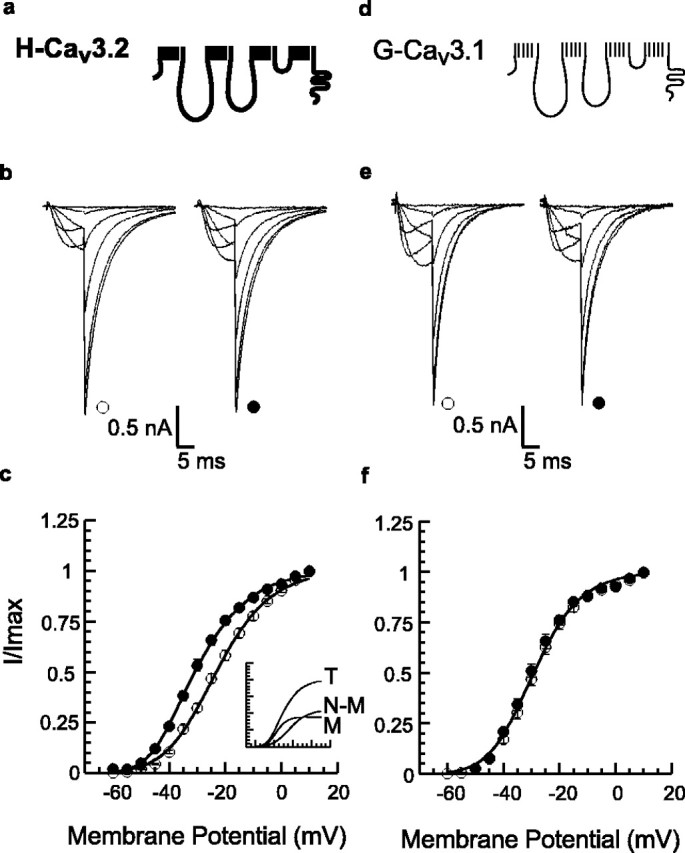
Effect of CaMKII activity on the voltage dependence of activation of T-type Ca2+ channels. a, d, Schematic representation of α1H (Cav3.2) and α1G (Cav3.1) channels. b, e, Whole-cell current traces recorded from α1H (b) or α1G (e) channels transiently expressed with CaMKII in HEK293 cells. Sample currents at Vtest = -55, -45, -35, -25, -10, and +10 mV recorded with intracellular free Ca2+ fixed at 27 nm Ca2+ (open symbols) to prevent CaMKII activation or 1 μm Ca2+ plus 2 μm CaM (filled symbols). Currents were normalized (Imax = 2.5 nA) to illustrate differences at hyperpolarized Vm between channel subtypes recorded with CaMKII activating solutions. c, f, Relative conductance (normalized tail current amplitudes, I/Imax) plotted (means ± SEM) versus Vt. Data sets were fitted to squared Boltzmann functions; single functions provided good fits to low Ca2+ data (open symbols) for α1H (c) [V1/2 = -23.2 ± 0.3mV; k = 10.9 ± 0.4 mV/e-fold change; n = 17 (27 nm)] and to both low- and high-Ca2+ data for α1G channels (f) [V1/2 = -29.2 ± 0.6 mV; k = 9.4 ± 0.4 mV/e-fold change; n = 16 (27 nm) and 11 (1 μm Ca2+)]. Fitting to the sum of two functions was significantly better (p < 0.05) for the high-Ca2+ data for α1H. Inset shows the contributions of the two components fitted to the high-Ca2+ α1H data set (T); one component is identical to the low-Ca2+ fit and is designated non-modulated (N-M), whereas the second modulated component (M) activates at more hyperpolarized voltages [M: V1/2 = -37.5 ± 0.3mV; k = 6.4 ± 1.5 mV/e-fold change; n = 15 (1 μm Ca2+)].
To determine the molecular basis for this regulation, we generated chimeric channel constructs from wild-type α1H (Cav3.2) and α1G (Cav3.1) channels. Replacement of the HII-III linker with the GII-III linker abolishes CaMKII-dependent regulation (Fig. 2a-c) [H/GII-IIIlinker: V1/2 = -22.4 ± 0.3 mV, k = 11.4 ± 0.5 mV; n = 15 (27 nm) and 11 (1 μm Ca2+)], suggesting that the determinants for the regulation of activation gating by CaMKII are located in the HII-III linker. Despite the absence of regulation, other channel properties remain unchanged. Neither the voltage dependency of inactivation (-65 ± 0.2 mV) nor the τ of deactivation (3.9 ± 0.3 msec at -60 mV) of the H/GII-III linker chimera differs from that of α1H wild-type channels (-64.5 ± 0.2 mV, 4.2 ± 0.5 msec; NS).
Figure 2.

The II-III cytoplasmic linker of α1H channels confers modulation in cells coexpressing CaMKII. a, d, Schematic representation of channel chimeras in which the II-III linker plus a single transmembrane helix is swapped. b, e, Sample currents at Vt = -55, -45, -35, -25, -10, and +10 mV recorded with solutions that impair (open symbols) or promote (filled symbols) CaMKII activation as presented in Figure 1, b and e. c, f, Normalized tail current amplitudes (I/Imax) plotted (means ± SEM) versus Vt. Single squared Boltzman functions provided good fits for both low- and high-Ca2+ data for H/GII-III linker (c) [V1/2 = -22.4 ± 0.3 mV; k = 11.4 ± 0.5 mV; n = 15 (27 nm) and 11 (1 μm Ca2+)] and low-Ca2+ data (open symbols) for G/HII-III linker. High Ca2+ data for G/H II-III linker fitted significantly better (p < 0.05) to the sum of two functions (filled symbols) (f) [G/HII-III linker, M: V1/2 = -37.8 ± 0.6 mV; k = 5.3 ± 0.6 mV; n = 17 (1 μm Ca2+)]. Inset shows the fitted contribution of modulated (M) and non-modulated (N-M) channels to the voltage dependence of activation during CaMKII simulation (T). T, Sum of both fitted components for high Ca2+ data.
On the basis of the previous studies, we predicted that the HII-III linker, when transferred to the α1G backbone, would result in a gain of regulation and tested for CaMKII-dependent regulation of this G/HII-IIIlinker chimera. CaMKII activity shifts the half-activation potential of this chimera by -9.4 ± 0.4 mV (p < 0.05) and also increases the voltage sensitivity by Δ -3.3 ± 0.6 mV/e-fold change (p < 0.05). On average, 75 ± 11% of the G chimeric channel population is modulated, displaying a half-activation potential that is nearly identical to that recorded for CaMKII-modulated α1H wild-type channels [G/HII-III linker: M, V1/2 = -37.8 ± 0.6 mV, k = 5.3 ± 0.6 mV, n = 17 (1 μm Ca2+)]. An additional analysis of the regulation of G-chimeric channels shows that the CaMKII-modulated channel population in individual cells may vary from 89 to 46% of the total channel population (74 ± 4%). This broad distribution differs but overlaps that observed for wild-type α1H channels. The singular importance of the HII-III linker to the CaMKII-dependent change in channel gating was corroborated by the construction of a second G/H chimera that replaces a large segment of the α1G channel from IIIS1 to the C terminus (residues 1294-2353), with the corresponding regions from α1H channels. CaMKII-dependent regulation is not conferred to this G/HCterminus chimera (Fig. 3c). On the basis of these findings, we conclude that residue(s) critical for CaMKII-dependent regulation are located in the HII-III linker.
Figure 3.
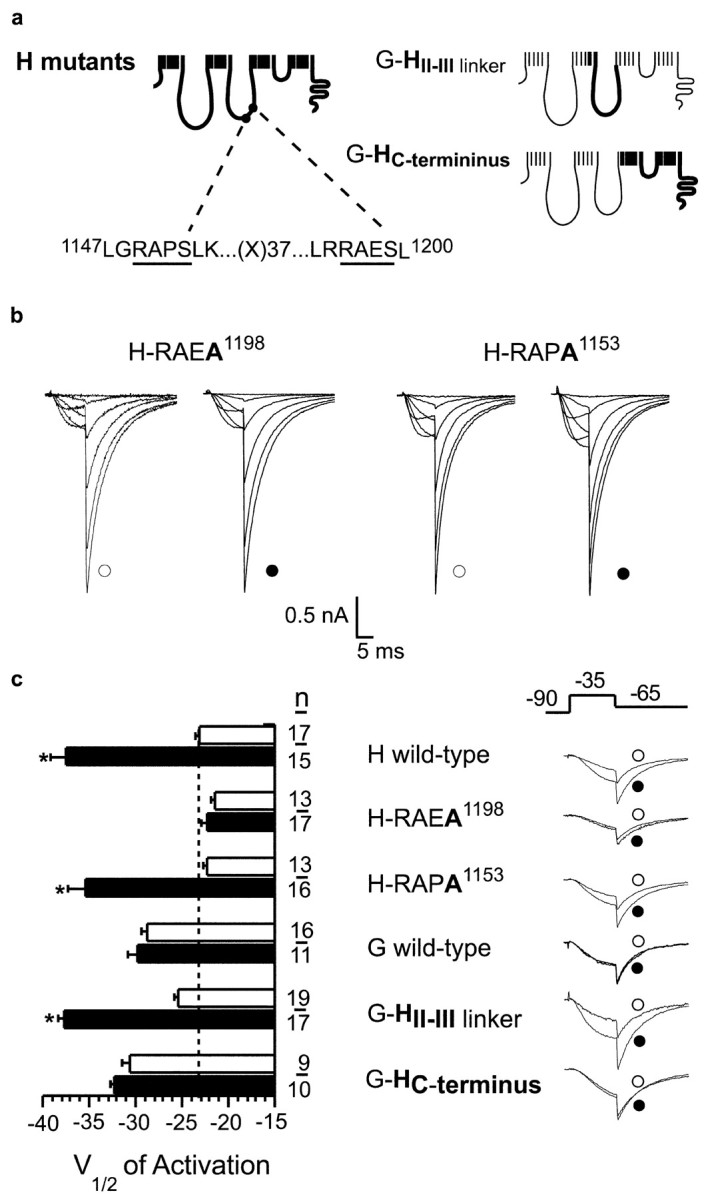
Effect of single point mutations on CaMKII-induced changes in channel gating. a, Schematic representation of α1H chimeras in which CaMKII-consensus motifs on the HII-III domain linker are mutated (RAPA1153 and RAEA1198), alone and in combination, and α1G chimeras with transferred α1H domains. b, Sample currents at Vt = -55, -45, -35, -25, -10, and +10 mV recorded with solutions that impair (open symbols) or promote (filled symbols) CaMKII activation as presented in Figure 1, b and e. c, Fitted values for half-activation potential; open bars indicate low-Ca2+ fitted parameters (open bars), and filled bars indicate high-Ca2+ fitted parameters. Values are mean ± SE; *p < 0.05; n indicates number of cells. Normalized current waveforms recorded at Vt = -35 mV (10 msec, Vrepolarization of -60 mV) with free Ca2+ fixed at 27 nm Ca2+ (open symbols) or 1 μm Ca2+ plus 2 μm CaM (filled symbols).
The α1H channel is a large 259 kDa membrane protein containing 22 sites that conform to the minimal consensus sequence for CaMKII phosphorylation, RXXS/T (Soderling, 1996). We considered each site as a putative substrate for CaMKII using criteria established on the basis of the rank order of phosphorylation efficiency (Vmax/Km) of a combinatorial peptide library (White et al., 1998). The 1192LRRAESL1200 recognition motif, located in the 275 residue HII-III linker, contains hydrophobic residues at -5 and +1, a non-basic amino acid residue at -3, and an alanine (Ala) at the -2 position, determinants that are known to enhance substrate recognition (White et al., 1998). Notably, no other motif in the channel protein has as many advantageous residues for substrate recognition, and this motif is not found in the α1G sequence. To determine whether the LRRAES1198L recognition motif on the HII-III linker is a site of CaMKII phosphorylation, we expressed GST fusion proteins containing ∼70% of the HII-III linker that includes Ser1198 as well as Ser1153, a strong but less optimal substrate site (1147LGRAPSLK1156). Recombinant CaMKII (at 30 nm) phosphorylates the full-length wild-type linker protein (47 kDa) as well as a proteolytic fragment (44 kDa) lacking the RAES1198 motif that resolves as a lower band of a doublet on SDS-PAGE (Fig. 4a,b). The full-length wild-type linker protein is phosphorylated to a stoichiometry of 1.7 mol of PO4 incorporated per moles of protein at 1 min, indicating that more than a single residue is phosphorylated, and reaches saturation at 15 min, to attain a final stoichiometry of 2.3. Despite the presence of five CaMKII phosphorylation motifs on the HII-III linker protein, the majority of incorporated phosphate at 1 and 15 min is distributed between Ser1198 and Ser1153. The combined mutation of these residues to Ala reduces phosphorylation by ∼80%. At 1 min, Ser1198 is preferentially phosphorylated to full stoichiometry as determined by mutational analysis. In contrast, mutation of Ser1153, which is embedded in a less optimal substrate recognition motif, does not result in significant loss of PO4 incorporation at 1 min. Longer incubation allows for the slower phosphorylation of the RAPS1153 motif and the recognition of other poorer substrates. Because rapid stoichiometric phosphorylation of a motif is a signature feature of a preferred kinase substrate, our findings suggested that the phosphorylation of Ser1198 on the HII-III linker could be a critical event that mediates CaMKII-dependent changes in channel gating.
Figure 4.

Phosphorylation of GST-domain II-III linker by CaMKII. A, GST fusion proteins containing residues 1039-1232 of domain linker II-III (wild-type or mutated at RAPA1153 and/or RAEA1198) were phosphorylated with purified recombinant CaMKIIγC in the presence of [γ-32P]ATP. Reactions were subjected to SDS-PAGE analysis. Representative autoradiograph showing phosphorylation state at 15 min (n = 2). Autophosphorylated CaMKIIγC resolves above the linker proteins. b, Analysis of phosphorylation stoichiometry of full-length linker protein (47 kDa) at 1 and 15 min of incubation (mean ± SE). The apparent phosphorylation stoichiometry of the RAES1198 or RAPS1153 motifs is indicated by the difference in phosphorylation state between the single-mutated (hatched or stripped bars) and double-mutated (open bars) fusion proteins. Phosphate incorporation into mutant GST-linker fusion proteins were compared with wild-type using ANOVA with post hoc Dunnett's testing, in which significance was p < 0.05.
Finally, to test the functionality of the RAES1198 motif, we mutated Ser1198 in the full-length channel protein and assayed for CaMKII-dependent regulation of channel activity. Mutation of Ser1198 to Ala abrogates CaMKII-dependent modulation of α1H channels (Fig. 3c). Neither the half-activation potential nor the voltage sensitivity differs from non-modulated wild-type α1H channels [V1/2 = -21.8 ± 0.4 mV, k = 11.5 ± 0.5 mV; n = 17 (1 μm Ca2+); NS]. In contrast, mutant α1H channels harboring a Ser1153 to Ala mutation retain CaMKII-dependent modulation (M: V1/2 = -35.4 ± 1.9 mV; k = 5.3 ± 2.0 mV/e-fold change; 34 ± 11%; p < 0.05).
Modification of protein function by phosphorylation can be the result of the accommodation of the bulk of the phosphate moiety (Busch et al., 2002) or the introduction of negative charge (Buchbinder et al., 1997). We mutated Ser1198 to glutamine (Gln) to test first for the possible importance of the loss of serine hydroxyl interactions during phosphate incorporation. Gln1198 failed to change either the half-activation potential or the voltage sensitivity of this mutant α1H channel [V1/2 = -23.5 ± 1.2 mV; k = 9.4 ± 0.3 mV; n = 10 (27 nm)]. Moreover, as observed previously with mutation of the RAES1198 motif, the loss of Ser1198 prevents regulation by CaMKII [V1/2 = -25.2 ± 0.7 mV; k = 9.4 ± 0.2 mV; n = 11 (1 μm)]. In a similar manner, mutation of Ser1198 to glutamic acid (Glu) creates a channel with gating properties that mimic those of the Gln1198 mutant. Both the V1/2 of activation and the voltage sensitivity of this mutant channel also remain unaltered in the absence and presence of CaMKII activation [V1/2 = -24.8 ± 0.8 mV, k = 9.8 ± 0.4 mV, n = 12 (27 nm); V1/2 = -23.3 ± 0.5 mV, k = 9.7 ± 0.4 mV, n = 11., (1 μm)], suggesting that the monoanionic carboxylate group of Glu cannot replace the dianionic phosphate of phosphoserine in reproducing either the exact charge balance or the phosphopeptide positioning that is required to change channel gating (Buchbinder et al., 1997). Collectively, our mutagenesis studies provide support for our in vitro phosphorylation data and identify the key role played by Ser1198 in the observed changes in α1H channel gating induced by CaMKII.
Discussion
The present data provide a molecular explanation for how CaMKII differentially regulates LVA Ca2+ channels. We described a remarkably simple way by which CaMKII changes α1H activation gating. Here, we identify a single serine residue (Ser1198) within the II-III cytoplasmic linker that is unique to α1H channels, phosphorylated by CaMKII, and critical for channel regulation. This mechanism for Ca2+ channel regulation differs from that reported for α1C (Cav1.2) channels. Feedforward regulation (facilitation) of α1C channels can be supported by a CaMKII-independent mechanism that requires CaM binding to an IQ motif (Zuhlke et al., 2000), a domain absent from the T-type Ca2+ channel family, and/or by a CaMKII-dependent mechanism that may require cytoskeletal intermediates (Yuan and Bers, 1994; Dzhura et al., 2000, 2002; Wu et al., 2001). However, the critical substrate(s) targeted by CaMKII remains undetermined. Our data support previous observations showing that the opening frequency of single α1H channels in excised patches can be increased by membrane-associated CaMKII (Barrett et al., 2000). These studies showed that the participation of a cytosolic kinase cascade is not required for CaMKII-dependent regulation and implied the possibility of a direct modulation of α1H Ca2+ channels by CaMKII. Our data provide the first evidence for direct regulation of the pore-forming subunit of a voltage-dependent Ca2+ channel by CaMKII.
The II-III cytoplasmic linker of the pore-forming channel subunit of other voltage-gated Ca2+ channels forms important structural associations with signaling effectors. In the Cav1 family, the II-III linker of α1S couples the voltage sensor to the ryanodine-sensitive Ca2+ release channel (Tanabe et al., 1988, 1990), whereas in the Cav2 family, the II-III linker facilitates efficient delivery of Ca2+ to sites of neurotransmitter release by binding SNARE (soluble N-ethylmaleimide-sensitive factor attached protein receptor) proteins that concomitantly modify inactivation gating (Sheng et al., 1994; Catterall, 2000; Jarvis and Zamponi, 2001). Our data highlight a role for the II-III linker of T-type Ca2+ channels and show that, in the Cav3 family, the II-III linker is an important site for the control of activation gating. Recent studies from our laboratory indicate that the HII-III linker also transduces current inhibition by directly binding G-protein β2-containing βγ-dimers (Wolfe et al., 2003). Thus, in the Cav3 family, the HII-III linker may serve as a center for integrating effects of multiple stimuli.
Although mRNA transcripts encoding each of the T-type Ca2+ channel subtypes are widely expressed throughout the CNS and PNS, the pattern of expression of each channel gene is unique, albeit overlapping (Talley et al., 1999). α1H mRNA is expressed at high levels in restricted regions of the hippocampus (pyramidal cell layers and granule layer of the dentate gyrus), cerebral cortex (a subset of layer V neocortical cells), and thalamus (reticular cells), as well as in sensory ganglia, dorsal horn neurons (external lamina), and the olfactory bulb. In contrast to other members of the Cav3 Ca2+ channel family whose currents are blocked by Ni2+ with an IC50 that overlaps that for block of HVA Ca2+ channels, α1H channels are inhibited by a Ni2+ concentration that is 20- to 50-fold lower (IC50 of 10 μm) (Perez-Reyes, 2003).
The Ni2+ sensitivity of low-threshold, slowly deactivating Ca2+ channel currents has revealed the specific importance of the α1H channel isoform in the following: hippocampal-neocortical dendritic low-threshold Ca2+ spike initiation (Markram and Sakmann, 1994; Magee and Johnston, 1995b), regenerative potential conduction, and somatic burst firing (Gillessen and Alzheimer, 1997; Larkum et al., 2001), as well as in both synaptic (lamina I projection neurons of the dorsal horn) and nonsynaptic (CA1 pyramidal neurons) forms of activity-dependent potentiation (Su et al., 2002; Ikeda et al., 2003). Given the broad distribution of α1H channels and CaMKII in nervous tissue, the operation of the direct phosphorylation-dependent regulatory mechanism described here may be widespread, supporting the enhanced participation of α1H channels in these diverse neuronal settings.
Footnotes
This study was supported by National Institutes of Health-National Heart, Lung, and Blood Institute Grant R01 36977 (P.Q.B.). J.T.W. was supported by predoctoral fellowships from the University of Virginia Cardiovascular Research Center and the American Heart Association (Mid-Atlantic Affiliate). We thank E. Perez-Reyes for α1H and α1G clones and D. Bayliss, J. J. Zhu, and E. M. Talley for their careful reading of this manuscript.
P.J.W. and H.W. contributed equally to this work.
Correspondence should be addressed to Paula Q. Barrett at the above address. E-mail: pqb4b@virginia.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/2310116-06$15.00/0
References
- Barrett PQ, Lu HK, Colbran R, Czernik A, Pancrazio JJ ( 2000) Stimulation of unitary T-type Ca2+ channel currents by calmodulin-dependent protein kinase II. Am J Physiol 279: C1694-C1703. [DOI] [PubMed] [Google Scholar]
- Braun AP, Schulman H ( 1995) The multifunctional calcium/calmodulin-dependent protein kinase: from form to function. Annu Rev Physiol 57: 417-445. [DOI] [PubMed] [Google Scholar]
- Buchbinder JL, Luong CB, Browner MF, Fletterick RJ ( 1997) Partial activation of muscle phosphorylase by replacement of serine 14 with acidic residues at the site of regulatory phosphorylation. Biochemistry 36: 8039-8044. [DOI] [PubMed] [Google Scholar]
- Busch JL, Bessay EP, Francis SH, Corbin JD ( 2002) A conserved serine juxtaposed to the pseudosubstrate site of Type 1 cGMP-dependent protein inase contributes stronly to autoinhibition and lower cGMP affinity. J Biol Chem 277: 34048-34054. [DOI] [PubMed] [Google Scholar]
- Catterall WA ( 2000) Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol 16: 521-555. [DOI] [PubMed] [Google Scholar]
- Cribbs LL, Lee JH, Yang J, Satin J, Zhang Y, Daud A, Barclay J, Williamson MP, Fox M, Rees M, Perez-Reyes E ( 1998) Cloning and characterization of α1H from human heart, a member of the T-type Ca2+ channel gene family. Circ Res 83: 103-109. [DOI] [PubMed] [Google Scholar]
- Cribbs LL, Gomora JC, Daud AN, Lee JH, Perez-Reyes E ( 2000) Molecular cloning and functional expression of Ca(v)3.1c, a T-type calcium channel from human brain. FEBS Lett [Erratum (2000) 470:378] 466: 54-58. [DOI] [PubMed] [Google Scholar]
- Destexhe A, Contreras D, Steriade M, Sejnowski TJ, Huguenard JR ( 1996) In vivo, in vitro, and computational analysis of dendritic calcium currents in thalamic reticular neurons. J Neurosci 16: 169-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzhura I, Wu Y, Colbran RJ, Balser JR, Anderson ME ( 2000) Calmodulin kinase determines calcium-dependent facilitation of L-type calcium channels. Nat Cell Biol 2: 173-177. [DOI] [PubMed] [Google Scholar]
- Dzhura I, Wu Y, Colbran RJ, Corbin JD, Balser JR, Anderson ME ( 2002) Cytoskeletal disrupting agents prevent calmodulin kinase, IQ domain, and voltage-dependent facilitation of L-type Ca2+ channels. J Physiol (Lond) 545 2: 399-406. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Efron B, Tibshirani RJ ( 1993) An introduction to the bootstrap. New York: Chapman and Hall.
- Eilers J, Konnerth A ( 1997) Dendritic signal integration. Curr Opin Neurobiol 7: 385-390. [DOI] [PubMed] [Google Scholar]
- Fisher RE, Gray R, Johnston D ( 1990) Properties and distribution of single voltage-gated calcium channels in adult hippocampal neurons. J Neurophysiol 64: 91-104. [DOI] [PubMed] [Google Scholar]
- Gillessen T, Alzheimer C ( 1997) Amplification of EPSPs by low Ni2+- and amiloride-sensitive Ca2+ channels in apical dendrites of rat CA1 pyramidal neurons. J Neurophysiol 77: 1639-1643. [DOI] [PubMed] [Google Scholar]
- Hausser M, Spruston N, Stuart GJ ( 2000) Diversity and dynamics of dendritic signaling. Science 290: 739-744. [DOI] [PubMed] [Google Scholar]
- Hille B ( 2001) Gating: voltage sensing and inactivation. In: Ion channels of excitable membranes, Chap 19, Ed 3. Sunderland, MA: Sinauer.
- Hudmon A, Schulman H ( 2002) Neuronal Ca2+/calmodulin-dependent protein kinase II: the role of structure and autoregulation in cellular function. Annu Rev Biochem 71: 473-510. [DOI] [PubMed] [Google Scholar]
- Huguenard JR ( 1996) Low-threshold calcium currents in central nervous system neurons. Annu Rev Physiol 58: 329-348. [DOI] [PubMed] [Google Scholar]
- Huguenard JR, McCormick DA ( 1992) Simulation of the currents involved in rhythmic oscillations in thalamic relay neurons. J Neurophysiol 68: 1373-1383. [DOI] [PubMed] [Google Scholar]
- Ikeda H, Heinke B, Ruscheweyh R, Sandkuhler J ( 2003) Synaptic plasticity in spinal lamina I projection neurons that mediate hyperalgesia. Science 299: 1237-1240. [DOI] [PubMed] [Google Scholar]
- Jarvis SE, Zamponi GW ( 2001) Interactions between presynaptic Ca2+ channels, cytoplasmic messengers and proteins of the synaptic vesicle release complex. Trends Pharmacol Sci 22: 519-525. [DOI] [PubMed] [Google Scholar]
- Johnson ML, Frasier SG ( 1985) Nonlinear least-squares analysis. In: Enzyme structure, Pt J (Hirs CHW, Timasheff SN, eds). London: Academic.
- Kelly PT, McGuinness TL, Greengard P ( 1984) Evidence that the major postsynaptic density protein is a component of a Ca2+/calmodulin-dependent protein kinase. Proc Natl Acad Sci USA 81: 945-949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy MB, Bennett MK, Erondu NE ( 1983) Biochemical and immunochemical evidence that the “major postsynaptic density protein” is a subunit of a calmodulin-dependent protein kinase. Proc Natl Acad Sci USA 80: 7357-7361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkum ME, Zhu JJ, Sakmann B ( 2001) Dendritic mechanisms underlying the coupling of the dendritic with the axonal action potential initiation zone of adult rat layer 5 pyramidal neurons. J Physiol (Lond) 533: 447-466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Daud AN, Cribbs LL, Lacerda AE, Pereverzev A, Klockner U, Schneider T, Perez-Reyes E ( 1999) Cloning and expression of a novel member of the low voltage-activated T-type calcium channel family. J Neurosci 19: 1912-1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XB, Jones EG ( 1996) Localization of alpha type II calcium calmodulin-dependent protein kinase at glutamatergic but not gamma-aminobutyric acid (GABAergic) synapses in thalamus and cerebral cortex. Proc Natl Acad Sci USA 93: 7332-7336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu HK, Fern RJ, Nee JJ, Barrett PQ ( 1994) Ca2+-dependent activation of T-type Ca2+ channels by calmodulin-dependent protein kinase II. Am J Physiol 267: F183-F189. [DOI] [PubMed] [Google Scholar]
- Magee JC, Johnston D ( 1995a) Characterization of single voltage-gated Na+ and Ca2+ channels in apical dendrites of rat CA1 pyramidal neurons. J Physiol (Lond) 487: 67-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC, Johnston D ( 1995b) Synaptic activation of voltage-gated channels in the dendrites of hippocampal pyramidal neurons. Science 268: 301-304. [DOI] [PubMed] [Google Scholar]
- Markram H, Sakmann B ( 1994) Calcium transients in dendrites of neocortical neurons evoked by single subthreshold excitatory postsynaptic potentials via low-voltage-activated calcium channels. Proc Natl Acad Sci USA 91: 5207-5211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA ( 1992) Neurotransmitter actions in the thalamus and cerebral cortex and their role in neuromodulation of thalamocortical activity. Prog Neurobiol 39: 337-388. [DOI] [PubMed] [Google Scholar]
- Migliore M, Shepherd GM ( 2002) Emerging rules for the distributions of active dendritic conductances. Nat Rev Neurosci 3: 362-370. [DOI] [PubMed] [Google Scholar]
- Monteil A, Chemin J, Bourinet E, Mennessier G, Lory P, Nargeot J ( 2000) Molecular and functional properties of the human alpha(1G) subunit that forms T-type calcium channels. J Biol Chem 275: 6090-6100. [DOI] [PubMed] [Google Scholar]
- Perez-Reyes E ( 2003) Molecular physiology of low voltage-activated T-type calcium channels. Physiol Rev 83: 117-161. [DOI] [PubMed] [Google Scholar]
- Perez-Reyes E, Cribbs LL, Daud A, Lacerda AE, Barclay J, Williamson MP, Fox M, Rees M, Lee JH ( 1998) Molecular characterization of a neuronal low-voltage-activated T-type calcium channel. Nature 391: 896-900. [DOI] [PubMed] [Google Scholar]
- Schrier AD, Wang H, Talley EM, Perez-Reyes E, Barrett PQ ( 2001) α1H T-type Ca2+ channel is the predominant subtype expressed in bovine and rat zona glomerulosa. Am J Physiol 280: C265-C272. [DOI] [PubMed] [Google Scholar]
- Shen K, Teruel MN, Subramanian K, Meyer T ( 1998) CaMKIIbeta functions as an F-actin targeting module that localizes CaMKIIalpha/beta heterooligomers to dendritic spines. Neuron 21: 593-606. [DOI] [PubMed] [Google Scholar]
- Sheng ZH, Rettig J, Takahashi M, Catterall WA ( 1994) Identification of a syntaxin-binding site on N-type calcium channels. Neuron 13: 1303-1313. [DOI] [PubMed] [Google Scholar]
- Soderling TR ( 1996) Structure and regulation of calcium/calmodulin-dependent protein kinases II and IV. Biochim Biophys Acta 1297: 131-138. [DOI] [PubMed] [Google Scholar]
- Soderling TR, Derkach VA ( 2000) Postsynaptic protein phosphorylation and LTP. Trends Neurosci 23: 75-80. [DOI] [PubMed] [Google Scholar]
- Straume M, Johnson M, eds ( 1992) Numerical computer methods. San Diego: Academic.
- Su H, Sochivko D, Becker A, Chen J, Jiang Y, Yaari Y, Beck H ( 2002) Upregulation of a T-type Ca2+ channel causes a long-lasting modification of neuronal firing mode after status epilepticus. J Neurosci 22: 3645-3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talley EM, Cribbs LL, Lee JH, Daud A, Perez-Reyes E, Bayliss DA ( 1999) Differential distribution of three members of a gene family encoding low voltage-activated (T-type) calcium channels. J Neurosci 19: 1895-1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe T, Beam KG, Powell JA, Numa S ( 1988) Restoration of excitation-contraction coupling and slow calcium current in dysgenic muscle by dihydropyridine receptor complementary DNA. Nature 336: 134-139. [DOI] [PubMed] [Google Scholar]
- Tanabe T, Beam KG, Adams BA, Niidome T, Numa S ( 1990) Regions of the skeletal muscle dihydropyridine receptor critical for excitation-contraction coupling. Nature 346: 567-569. [DOI] [PubMed] [Google Scholar]
- Thiagarajan TC, Piedras-Renteria ES, Tsien RW ( 2002) alpha- and beta-CaMKII. Inverse regulation by neuronal activity and opposing effects on synaptic strength. Neuron 36: 1103-1114. [DOI] [PubMed] [Google Scholar]
- White RR, Kwon YG, Taing M, Lawrence DS, Edelman AM ( 1998) Definition of optimal substrate recognition motifs of Ca2+-calmodulin-dependent protein kinases IV and II reveals shared and distinctive features. J Biol Chem 273: 3166-3172. [DOI] [PubMed] [Google Scholar]
- Wolfe JT, Wang H, Perez-Reyes E, Barrett PQ ( 2002) Stimulation of recombinant Ca(v)3.2, T-type, Ca2+ channel currents by CaMKIIγC. J Physiol (Lond) 538: 343-355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe JT, Wang H, Howard J, Garrison JC, Barrett PQ ( 2003) T-type calcium channel regulation by specific G-protein βγ subunits. Nature 424: 209-213. [DOI] [PubMed] [Google Scholar]
- Wu Y, Dzhura I, Colbran RJ, Anderson ME ( 2001) Calmodulin kinase and a calmodulin-binding “IQ” domain facilitate L-type Ca2+ current in rabbit ventricular myocytes by a common mechanism. J Physiol (Lond) 535: 679-687. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Yuan W, Bers DM ( 1994) Ca-dependent facilitation of cardiac Ca current is due to Ca-calmodulin-dependent protein kinase. Am J Physiol 267: H982-H993. [DOI] [PubMed] [Google Scholar]
- Zuhlke RD, Pitt GS, Tsien RW, Reuter H ( 2000) Ca2+-sensitive inactivation and facilitation of L-type Ca2+ channels both depend on specific amino acid residues in a consensus calmodulin-binding motif in the α1C subunit. J Biol Chem 275: 21121-21129. [DOI] [PubMed] [Google Scholar]