

Abstract
Many studies have confirmed the time-limited involvement of the hippocampus in mnemonic processes and suggested that there is reorganization of the responsible brain circuitry during memory consolidation. To clarify such reorganization, we chose trace classical eyeblink conditioning, in which hippocampal ablation produces temporally graded retrograde amnesia. Here, we extended the temporal characterization of retrograde amnesia to other regions that are involved in acquisition during this task: the medial prefrontal cortex (mPFC) and the cerebellum. At a various time interval after establishing the trace conditioned response (CR), rats received an aspiration of one of the three regions. After recovery, the animals were tested for their CR retention. When ablated 1 d after the learning, both the hippocampal lesion and the cerebellar lesion group of rats exhibited a severe impairment in retention of the CR, whereas the mPFC lesion group showed only a slight decline. With an increase in interval between the lesion and the learning, the effect of the hippocampal lesion diminished and that of the mPFC lesion increased. When ablated 4 weeks after the learning, the hippocampal lesion group exhibited as robust CRs as its corresponding control group. In contrast, the mPFC lesion and the cerebellar lesion groups failed to retain the CRs. These results indicate that the hippocampus and the cerebellum, but only marginally the mPFC, constitute a brain circuitry that mediates recently acquired memory. As time elapses, the circuitry is reorganized to use mainly the mPFC and the cerebellum, but not the hippocampus, for remotely acquired memory.
Keywords: memory consolidation, classical conditioning, eyeblink, hippocampus, prefrontal cortex, cerebellum
Introduction
Memory is presumed to depend on synaptic changes in a distributed ensemble of neurons that belong to many different processing systems (Squire, 1987). Among these systems, the hippocampus and its related medial temporal lobe structure have been the target of the vigorous investigation since the finding of the dramatic retrograde amnesia produced by bilateral lesions to the medial temporal lobe in the patient H.M. (Scoville and Milner, 1957). Research in both humans and animals confirmed that damage restricted to the hippocampus also selectively impairs memory of recently acquired information but leaves more remote memories intact in numerous tasks (for review, see Squire, 1992; Squire et al., 2001). These results suggest two points: (1) the hippocampus may be important for a consolidation process that stabilizes memory from a temporary labile form to a long-lasting stable form (Squire, 1992; Knowlton and Fanselow, 1998) (but see Nadel and Moscovitch, 1997), and (2) the brain circuitry underlying memory retention is reorganized from a hippocampus-dependent form into a hippocampus-independent one during the post-learning period. As an important component for the latter, the neocortex is most commonly mentioned (McClelland et al., 1995; Squire and Alvarez, 1995); however, how the entire memory circuitry varies during the post-learning period has not been fully understood. To clarify such reorganization, we chose trace classical eyeblink conditioning as a model system. Because the sensory inputs (two kinds of stimuli) and the output (observed response) are simple and discrete in this system, it is especially suitable for identifying the essential neural circuitry for memory processing (Thompson and Kim, 1996).
Classical conditioning of the eyeblink response is a type of motor learning that depends critically on the cerebellum and brainstem (for review, see Thompson et al., 1997). The essential brain circuitry for eyeblink conditioning in these structures has been investigated intensively and clarified using the delay paradigm, in which the conditioned stimulus (CS) precedes and coterminates with the unconditioned stimulus (US). On the other hand, the trace paradigm, in which the CS and the US are separated by a long stimulus-free trace interval, requires the hippocampus (Solomon et al., 1986; Moyer et al., 1990; McGlinchey-Berroth et al., 1997; Weiss et al., 1999) and the medial prefrontal cortex (mPFC) for successful learning (Kronforst-Collins and Disterhoft, 1998; Weible et al., 2000; McLaughlin et al., 2002), in addition to the cerebellum (Woodruff-Pak et al., 1985). As in other hippocampus-dependent learning tasks, the hippocampal contribution to memory retention is time limited: the hippocampus is necessary for the recently acquired conditioned responses (CRs) to be retained but not for the remotely acquired responses in rabbits (Kim et al., 1995) and mice (Takehara et al., 2002). Here we have investigated the effects of aspiration of the hippocampus, the mPFC, or the cerebellum on memory retention at various time intervals after learning to address one of the unsolved questions regarding memory consolidation: how do brain regions other than the hippocampus participate in long-lasting memory retention according to the time elapsed after learning?
Materials and Methods
Subjects and surgery. The subjects were 208 male Wistar rats (Japan SLC, Inc., Hamamatsu, Shizuoka, Japan) weighing 180–250 gm (8 weeks old) at the time of the first surgery. The animals were housed in standard plastic cages in a colony room with a 12 hr light/dark cycle. Water and food were available ad libitum. All experiments were performed in accordance with the guidelines established by the Institutional Animal Investigation Committee at the University of Tokyo and the NIH Guide for the Care and Use of Laboratory Animals. All efforts were made to optimize comfort and minimize the use of animals.
Rats received two kinds of surgery in series: one for implanting electrodes before the acquisition sessions and one for lesioning of the targeted region after the acquisition sessions. In the first surgery, four Teflon-coated stainless steel wires (No. 7910; A-M systems, Carlsborg, WA) were implanted subcutaneously in the left upper eyelid under anesthesia with ketamine (87 mg/kg, i.p.; Sankyo, Tokyo, Japan) and xylazine (13 mg/kg, i.p.; Bayer, Tokyo, Japan). These wires were soldered to the pins of a connector that was secured to the skull anterior to bregma with dental acrylic resin and stainless steel screws. In the second surgery, the learned animals were anesthetized in the same way as in the first surgery and placed in a stereotaxic apparatus. In some experiments, the animals were pretreated with atropine (0.1 mg/kg, i.p.; Tanabe Pharmaceutical, Osaka, Japan). The skull was exposed by incision along the midline, and holes were made above the targeted regions. After the targeted regions were aspirated, the wounds were packed with surgicel (Johnson and Johnson, Tokyo, Japan), and the incised skins were sutured. The animals were injected with gentamycin sulfate (8 mg/kg, i.p.; Schering-Plough, Osaka, Japan) and warmed until they moved spontaneously. We chose aspiration rather than excitotoxic lesions because the latter might damage the memory stored in the structures (which receive afferents from the injection site) via the massive and sustained excitatory discharge transmitted from the injection site (Anagnostaras et al., 2001).
Detailed lesion methods. The animals received one of the lesions described below 1 d, 1 week, 2 weeks, or 4 weeks after their learning reached criterion (as described in Behavioral procedures).
Hippocampal and cortical lesions. On the skull posterior to the connector pins, holes were made bilaterally ∼2 mm from the midline to 1 mm inside the lateral ridge and from 1.5 mm posterior to bregma to 2 mm anterior to lambda. The hippocampal lesion groups received bilateral aspiration of the dorsal hippocampus and the overlying cortex. The cortical lesion groups received bilateral aspiration of the overlying cortex.
mPFC lesion. After removal of the connector pins and wires, a rectangular opening was made 2 mm from the midline on both sides and from 4 mm anterior to bregma to 1 mm posterior to bregma. The mPFC lesion groups received targeted cortical aspiration, with maximum care not to damage the septal area. For the sham operation groups, only a rectangular opening was made. At least 2 d before retraining, a new set of connector pins and wires was implanted in both groups of rats.
Cerebellar lesion. A fan-shaped hole was made by removing the ipsilateral side of the skull above the cerebellum. The dura was cut, and the ipsilateral cerebellar hemisphere, including the deep nuclei, was aspirated. The sham operation groups received only the removal of the skull.
Conditioning apparatus. Four cylindrical Plexiglas containers (15 cm in diameter and 25 cm high) were placed in a sound- and light-attenuated chamber. One rat was placed in each container. A lightweight cable was connected to the connector pins secured on the animal's head. The CS was a 350 msec tone (5 kHz, 85 dB) with a rise–fall time of 10 msec; it was delivered from a speaker (16.5 cm in diameter) placed above the containers. The US was a 100 msec peri-orbital shock (1.5 mA, 100 Hz square pulses) that was delivered through a pair of electrodes implanted in the left upper eyelid. A stimulus-free trace interval of 500 msec was interposed between the end of the CS and the US onset. The CR was monitored through electromyographic (EMG) activity recorded with another pair of implanted electrodes.
Behavioral procedures. Two to 4 d after the first surgery, spontaneous eyeblink was recorded for 2 session days in the same way as during the conditioning session, except that no stimuli were presented. We started the conditioning beginning 4–6 d after the first surgery. A daily session consisted of 100 trials grouped into 10 blocks, which included 9 CS–US paired trials followed by one CS-alone trial. Trials were separated by a variable intertrial interval pseudorandomized between 20 and 40 sec, with a mean of 30 sec. The acquisition session was continued until the average adaptive CR% over the consecutive three sessions reached >60%, or for a maximum of 10 d. One hundred eleven rats exhibited an average frequency of adaptive CRs over 3 d of >60% within 9 d of acquisition (66.3 ± 1.2%). The other 97 rats did not reach the criterion and received 10 d of the acquisition session. Among these, 59 rats exhibited CRs of >30% (48.7 ± 2.5%) and were used for additional experiments, but the 38 rats with frequency of CR that was <30% were excluded from further study. One hundred seventy rats in total were divided into the 20 groups: the cortical lesion group (1 d, n = 14; 1 week, n = 7; 2 week, n = 7; 4 week, n = 7), the hippocampal lesion group (1 d, n = 22; 1 week, n = 8; 2 week, n = 7; 4 week, n = 10), the sham operation (mPFC) group (1 d, n = 7; 2 week, n = 6; 4 week, n = 7), the mPFC lesion group (1 d, n = 11; 2 week, n = 9; 4 week, n = 8), the sham operation (cerebellum) group (1 d, n = 7; 2 week, n = 6; 4 week, n = 6), and the cerebellar lesion group (1 d, n = 6; 2 week, n = 9; 4 week, n = 6). Then each group received the corresponding second surgery. After ∼2 weeks, the rats were conditioned again in the trace paradigm for3dto test their memory retention. Some rats of the 1 d cortical and hippocampal lesion groups received 10 d of the post-lesion conditioning sessions [1 d cortical lesion group (post-10), n = 6; 1 d hippocampal lesion group (post-10), n = 10] to test their ability to relearn.
There was no obvious difference in the lesion effects between rats that underwent 10 d of the pre-lesion sessions and those that achieved the criterion within 9 d. Therefore, the data from the two groups were combined for subsequent analysis. All experiments were performed during the light phase of the light/dark cycle.
Histology. At the end of the experiment, each animal was injected intraperitoneally with an excess amount of sodium pentobarbital (>80 mg/kg; Dainippon Pharmaceutical, Osaka, Japan) and perfused intracardially with 0.9% saline, followed by phosphate-buffered 10% formalin. The brain was removed from the skull and stored in 10% formalin for a few days. After infiltration with 30% sucrose, the brain was frozen, sectioned at 60 μm, and stained with cresyl violet. The largest and smallest lesioned areas were reconstructed from the sections according to the stereotaxic atlas of the rat brain (Paxinos and Watson, 1986), and we adopted the terminology of that atlas.
EMG analysis. The EMG activity was bandpass filtered between 0.15 and 1.0 kHz and fed into a computer with a sampling rate of 10 kHz. The maximum amplitude of EMG signals during a time period of t ± 1 msec was calculated and designated the EMG amplitude at t. Mean + SD of the EMG amplitudes during the pre-CS period (0–300 msec before CS onset) of 100 trials was defined as the threshold. If the average EMG amplitudes above the threshold during the pre-CS period exceeded 10% of this threshold, the trial was regarded as a “hyperactivity trial” and excluded from further analysis. A trial was assumed to contain an adaptive CR if the average EMG amplitude above the threshold during the 200 msec period before US onset exceeded 10% of the threshold and exceeded 10 times that of the pre-CS period. In the CS-alone trials, the period for averaging EMG amplitude was extended by 100 msec to the end of the expected US. The ratio of the number of trials containing the adaptive CR to that of total trials excluding the hyperactivity trials was calculated and denoted as the adaptive CR%. The frequency of the adaptive CRs (adaptive CR%) was expressed as a percentage for a session.
Further analyses described below were made for all groups except the 1 d cortical and hippocampal lesion groups (post-10). We analyzed the EMG activity using another definition of the CR%. All sets of the 100 msec periods of EMG data between 50 msec after CS onset and the onset of US were used for detection of the CR with the same criterion as described for the adaptive CR. In CS-alone trials, the 100 msec EMG data sets that included the data during the expected US were also used.
To show the temporal pattern of the CR, the EMG amplitude data for each rat were averaged over total trials (excluding the hyperactivity trials) for each day. These trial-averaged EMG amplitude data were normalized by the time-averaged values for 300 msec before the CS onset. Because the data were calculated from total trials excluding the hyperactivity trials, the temporal pattern of the normalized EMG amplitude data does not depend on the criterion used for detecting the CR.
To characterize changes in the temporal pattern of the CR, the latency of the CR peak was measured. The CR peak latency was defined as the time interval from the CS onset to the peak of the EMG amplitude. These values were averaged over the trials, which were judged to contain a CR, in a session.
We also analyzed the frequency of hyperactivity trials, which reflects spontaneous blinking during the pre-CS period. The ratio of the number of hyperactivity trials to that of total trials was calculated.
Statistical analysis. All data were expressed as the mean ± SEM. Statistical significance was determined by a two-way ANOVA with repeated measures or by a t test using SPSS statistical software. Some data were analyzed by a two-way ANOVA and then by a linear trend analysis. Differences with a p value of <0.05 were considered significant.
Results
Histology
The extent of the lesion in each rat was examined after completion of all the conditioning. Thirty-five rats in the cortical lesion groups had bilateral lesions of the neocortex overlying the hippocampus (Fig. 1A). One rat suffered severe damage to the dorsal hippocampus and was excluded from the analysis. Therefore, seven rats were analyzed in each of the 1 d, 1 week, 2 weeks, and 4 weeks cortical lesion groups, and six rats were analyzed in the 1 d cortical lesion group (post-10). Forty-seven rats in the hippocampal lesion groups had a bilateral lesion of most of the dorsal hippocampus, including the fimbria (Fig. 1B). The lateral part of the dorsal hippocampus was spared in 12 rats, and in 1 rat the damage extended to the septal area. After these 13 rats were excluded, 7 rats were analyzed in each of the 1 d, 1 week, 2 weeks, and 4 weeks hippocampal lesion groups, and 6 rats were analyzed in the 1 d hippocampal lesion group (post-10). Figure 1C represents the extent of the lesion in the mPFC groups. In most cases, the damaged area covered the prelimbic cortex, the anterior cingulate cortex, and part of the secondary motor cortex. There were some animals with damage that also reached the infralimbic cortex. After excluding seven animals with a disrupted septal area and two animals with unilateral damage, seven rats were analyzed in the 1 d mPFC lesion group and six rats were analyzed in the 2 and 4 weeks mPFC lesion groups. Twentyone rats in the cerebellar lesion group had an ipsilateral lesion of most of the hemicerebellum, including both the cerebellar cortex and the deep cerebellar nuclei (Fig. 1D). After excluding three rats with incomplete removal of the deep cerebellar nuclei, six rats were analyzed in each of the 1 d, 2 weeks, and 4 weeks cerebellar lesion groups.
Figure 1.
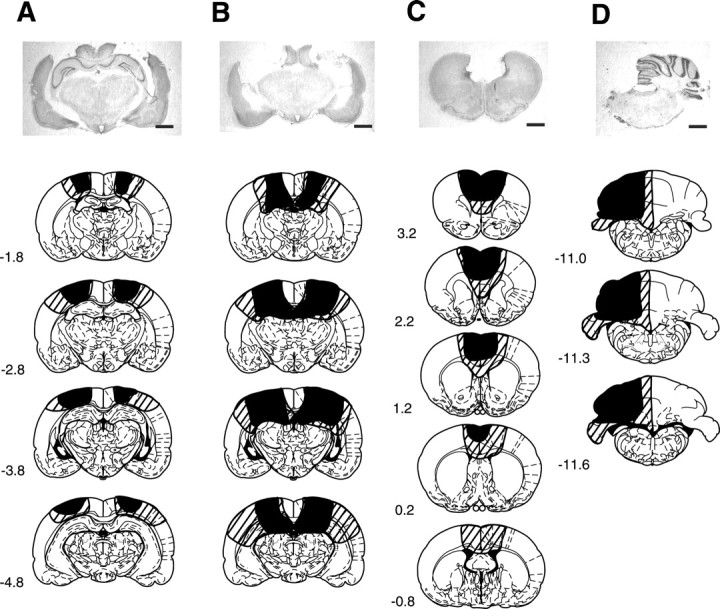
Representative photomicrographs of coronal brain sections stained with cresyl violet and extent of the lesion reconstructed from brain sections. A, The cortical lesion group. B, The hippocampal lesion group. C, The medial prefrontal cortical lesion group. D, The cerebellar lesion group. Scale bars, 2 mm. The black and striped areas indicate the smallest and largest lesions, respectively. Numbers to the left indicate stereotaxic coordinates relative to bregma (Paxinos and Watson, 1986).
Hippocampal lesion disrupts a recently acquired CR but not a remotely acquired one
Figure 2 shows the percentage of the adaptive CR during the pre-lesion and post-lesion sessions of the rats that received a hippocampal or cortical lesion. Before aspiration, all of the hippocampal lesion groups exhibited sufficient adaptive CRs identical to the corresponding cortical lesion groups during the last 3 d of pre-lesion sessions (Figs. 2A–D) (all F <1.7; NS). During the post-lesion sessions, as shown in Figure 2A, the 1 d hippocampal lesion group exhibited significantly lower adaptive CR% than the 1 d cortical lesion group (groups by sessions, F(2,24) = 0.000, NS; groups, F(1,12) = 17.0, p < 0.01; sessions, F(2,24) = 3.93, p < 0.05). On the other hand, the 1 week and 2 weeks hippocampal lesion groups did not differ in the adaptive CR percentages from the corresponding cortical lesion group through the post-lesion sessions, although they tended to show lower adaptive CR% in the first session (groups by sessions, F(2,24) = 0.578, NS; groups, F(1,12) = 4.12, NS; sessions, F(2,24) = 1.87, NS in 1 week group; groups by sessions, F(2,24) = 3.09, NS; groups, F(1,12) = 0.734, NS; sessions, F(2,24) = 0.459, NS in 2 weeks group) (Figs. 2B,C). The 4 weeks hippocampal lesion group exhibited as robust an adaptive CR as the 4 weeks cortical lesion group through the postlesion sessions (groups by sessions, F(2,24) = 0.809, NS; groups, F(1,12) = 0.173, NS; sessions, F(2,24) = 3.38, NS) (Fig. 2D). These results indicate that the 1 week, 2 weeks, and 4 weeks hippocampal lesion groups could retain the adaptive CR in a manner comparable with the corresponding cortical lesion group, whereas the 1 d hippocampal lesion group was severely impaired in its retention of the adaptive CR.
Figure 2.
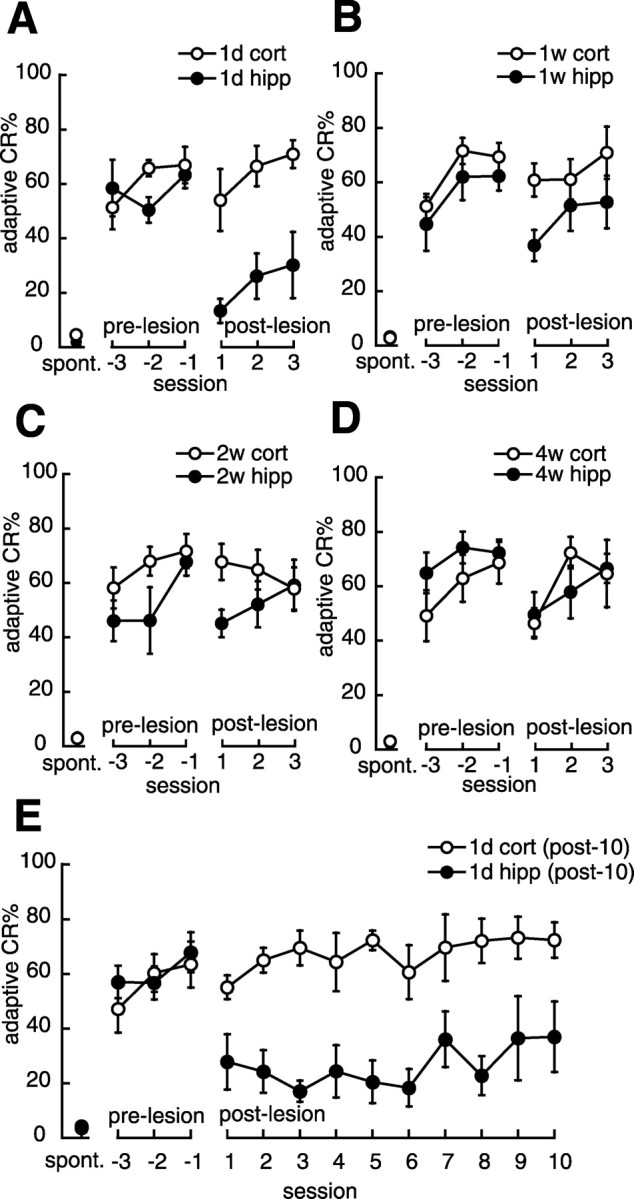
Effect of a hippocampal lesion on memory retention. A, Mean percentage of adaptive CRs in the 1 d group. The abscissa shows the session number after the lesion was made; the session at which the rat reached the criterion is denoted –1. Negative and positive values indicate the pre-lesion and post-lesion sessions. spont., Spontaneous eyeblink frequency. The hippocampal lesion group (•, 1d hipp; n = 7) showed a significantly lower adaptive CR% through the post-lesion sessions compared with the cortical lesion group (○, 1d cort; n = 7). B, Mean percentage of adaptive CRs in the 1 week group. The hippocampal lesion group (•, 1w hipp; n = 7) tended to exhibit a lower adaptive CR% than the cortical lesion group (○, 1w cort; n = 7) through the post-lesion sessions, although this difference was not statistically significant. C, Mean percentage of adaptive CRs in the 2 weeks group. The hippocampal lesion group (•, 2w hipp; n = 7) exhibited a slightly lower adaptive CR% than the cortical lesion group (○, 2w cort; n = 7) throughout the post-lesion sessions, although this difference was not statistically significant. D, Mean percentage of adaptive CRs in the 4 weeks group. The hippocampal lesion group (•, 4w hipp; n = 7) exhibited as robust an adaptive CR as the cortical lesion group (○, 4w cort; n = 7) through the post-lesion sessions. E, Mean percentage of adaptive CRs in the 1 d group (post-10). The hippocampal lesion group [•, 1d hipp (post-10); n = 6] did not recover to the level of the control group [○, 1d cort (post-10); n = 6] during 10 d of the post-lesion sessions. All data points represent group mean ± SEM.
To investigate whether animals with hippocampal lesions given 1 d after training can eventually learn the association if they are provided additional training trials, another group of 1 d hippocampal and cortical lesioned rats [the 1 d cortical and hippocampal lesion group (post-10)] underwent 10 d of the post-lesion sessions. As shown in Figure 2E, the 1 d hippocampal lesion group (post-10) did not recover to the level of the corresponding control group even after 10 d of retraining, although it exhibited some tendency toward increasing adaptive CR% (groups by sessions, F(9,90) = 1.06, NS; groups, F(1,10) = 17.1, p < 0.01; sessions, F(9,90) = 1.86, NS). This result suggests that the 1 d hippocampal lesion group could not fully relearn the adaptive CRs during the 10 d of retraining.
mPFC lesion affects a remotely acquired CR more severely than a recently acquired one
Figure 3 shows the frequency of the adaptive CR during the pre-lesion acquisition sessions and the post-lesion sessions of the rats that received an mPFC lesion or sham operation. Before the lesions, there were no significant differences between the mPFC lesion group and its corresponding sham operation group at any of the learning–lesion intervals (Figs. 3A–C) (all F < 1.3; NS). During the post-lesion sessions, the 1 d mPFC lesion group exhibited slightly fewer adaptive CRs than the 1 d sham operation group (groups by sessions, F(2,24) = 2.28, NS; groups, F(1,12) = 5.98, p < 0.05; sessions, F(2,24) = 4.25, p < 0.05) (Fig. 3A). The 2 weeks mPFC lesion group was mildly impaired in its adaptive CR% compared with the corresponding sham group during the post-lesion sessions (groups by sessions, F(2,20) = 0.093, NS; groups, F(1,10) = 5.28, p < 0.05; sessions, F(2,20) = 8.33, p < 0.01) (Fig. 3B). In contrast, the 4 weeks mPFC lesion group exhibited great impairment in its retention of the adaptive CR compared with the 4 weeks sham operation group (groups by sessions, F(2,22) = 1.45, NS; groups, F(1,11) = 65.1, p < 0.001; sessions, F(2,22) = 3.87, p < 0.05) (Fig. 3C). These results suggest that mPFC ablation impaired retention of the acquired adaptive CR at every learning–lesion interval, and the extent of this impairment increased with lengthening of the interval between learning and the lesion.
Figure 3.

Effect of a mPFC lesion on memory retention. A, Mean percentage of adaptive CRs in the 1 d group. The abscissa shows the session number after the lesion was made; the session at which the rat reached the criterion is denoted –1. Negative and positive values indicate the pre-lesion and post-lesion sessions. spont., Spontaneous eyeblink frequency. The mPFC lesion group (•, 1d mPFC; n = 7) exhibited a slightly lower adaptive CR% than the sham operation group (○, 1d sham; n = 7) through the post-lesion sessions. B, Mean percentage of adaptive CRs in the 2 weeks group. The mPFC lesion group (•, 2w mPFC; n = 6) was mildly impaired in its adaptive CR% compared with the sham operation group (○, 2w sham; n = 6) through the post-lesion sessions. C, Mean percentage of the adaptive CRs in the 4 weeks group. The mPFC lesion group (•, 4w mPFC; n = 6) exhibited a great impairment in retention of the adaptive CR% compared with its corresponding sham operation group (○, 4w sham; n = 7). All data points represent group mean ± SEM.
Cerebellar lesion disrupts both recently and remotely acquired CRs
Figure 4 shows the frequency of the adaptive CR during the pre-lesion acquisition sessions and the post-lesion sessions of the rats that received a cerebellar lesion or sham. Before the lesions, all of the cerebellar lesion groups exhibited an adaptive CR% that was comparable with the corresponding sham operation group at any of the learning–lesion intervals (Figs. 4A–C) (all F < 0.5; NS). As shown in Figure 5A–C, however, during the post-lesion sessions the adaptive CR% of the cerebellar lesion group were significantly lower than the percentages of the corresponding sham operation group at all learning–lesion intervals (groups by sessions, F(2,22) = 1.57, NS; groups, F(1,11) = 19.0, p < 0.01; sessions, F(2,22) = 1.18, NS in 1 d group; groups by sessions, F(2,20) = 0.093, NS; groups, F(1,10) = 18.2, p < 0.01; sessions, F(2,20) = 1.83, NS in 2 weeks group; groups by sessions, F(2,20) = 0.628, NS; groups, F(1,10) = 26.8, p < 0.001; sessions, F(2,20) = 5.36, p < 0.05 in 4 weeks group). These results indicate that a cerebellar lesion severely impaired retention of the acquired memory regardless of the interval between learning and the lesion.
Figure 4.

Effect of a cerebellar lesion on memory retention. A, Mean percentage of adaptive CRs in the 1 d group. The abscissa shows the session number after the lesion was made; the session at which the rat reached the criterion is denoted –1. Negative and positive values indicate the pre-lesion and post-lesion sessions. spont., Spontaneous eyeblink frequency. The cerebellar lesion group (•, 1d cere; n = 6) showed a significantly lower CR% compared with its sham operation group (○, 1d sham; n = 7) during the post-lesion sessions. B, Mean percentage of adaptive CRs in the 2 weeks group. The cerebellar lesion group (•, 2w cere; n = 6) was greatly impaired in its retention of the adaptive CR% compared with the cerebellar sham operation group (○, 2w sham; n = 6). C, Mean percentage of the adaptive CRs in the 4 weeks group. The cerebellar lesion group (•, 4w cere; n = 6) exhibited significantly fewer adaptive CRs compared with the sham operation group (○, 4w sham; n = 6) through the post-lesion sessions. All data points represent group mean ± SEM.
Figure 5.

Percentage of adaptive CRs in the first post-lesion session as a function of the time interval between learning and the lesion. A, Mean percentage of the adaptive CRs of the hippocampal lesion group (•, hipp) and the cortical lesion group (○, cort). The abscissa indicates the interval between the last acquisition session and the second surgery (lesion). The hippocampal lesion produced temporally graded retrograde amnesia. B, Mean percentage of the adaptive CRs of the mPFC lesion group (•, mPFC) and its corresponding sham operation group (○, sham). Ablation of the mPFC produced retrograde amnesia with temporal characteristics that were the reverse of those produced by a hippocampal lesion. C, Mean percentage of the adaptive CRs of the cerebellar lesion group (•, cere) and the sham operation group (○, sham). A cerebellar lesion produced retrograde amnesia without a temporal gradient. All data points represent group mean ± SEM.
Ablation of one of the three structures produces retrograde amnesia with a different temporal gradient
To clarify the temporal gradient of retrograde amnesia produced by ablation, we summarize the adaptive CR% of the first post-lesion session. The dependency of the adaptive CR% on the learning–lesion interval in the hippocampal lesion group was quite different from what we observed in the cortical lesion group (groups by learning–lesion intervals, F(3,48) = 3.44, p < 0.05) (Fig. 5A). The hippocampal lesion group retained the adaptive CR more successfully with a longer learning–lesion interval (a linear trend analysis 1 d, 1 week, 2 weeks, and 4 weeks; F(3,24) = 15.6, p < 0.001) than with a shorter learning–lesion interval; this is evidence that a hippocampal lesion produces temporally graded retrograde amnesia. The learning–lesion interval dependency of the adaptive CR% of the mPFC lesion group was markedly different from that of the sham operation group. (groups by learning–lesion intervals, F(2,33) = 3.26, p = 0.051; groups, F(1 33) = 25.8, p < 0.001; learning–lesion intervals, F(2,33) = 3.12, NS) (Fig. 5B). The mPFC lesion group retained the acquired adaptive CR less well, with a lengthening interval between the lesion and the learning (a linear trend analysis 1 d, 2 weeks, and 4 weeks; F(2,16) = 11.6, p < 0.001). Therefore, mPFC ablation produced temporal characteristics of retrograde amnesia that were the reverse of that produced by a hippocampal lesion. As can be seen, a cerebellar lesion produced severe impairment in retention of the adaptive CR at every learning–lesion interval (groups by learning–lesion intervals, F(2,31) = 0.108, NS; groups, F(1,31) = 38.5, p < 0.001; learning–lesion intervals, F(2,31) = 2.90, NS) (Fig. 5C). The impairment of the cerebellar lesion group did not vary with the length of the learning–lesion interval (a linear trend analysis 1 d, 2 weeks, and 4 weeks; F(2,15) = 0.039, NS), clearly suggesting that the cerebellar lesion produced retrograde amnesia without a temporal gradient.
Ablation does not affect the CR timing
In the present study, an adaptive CR was defined as a significant change in EMG amplitude during the 200 msec period just before the US onset. It is possible, therefore, that the observed deficit was caused by a failure to express a CR at an adaptive timing, rather than a failure to retain the association of the CS and US. Thus, we also analyzed the change in EMG amplitude from the CS onset to just before the US onset to detect any short-latency CR as well as any adaptive one (for details, see Materials and Methods). The dependency of the CR% on the learning–lesion interval was similar to that of the adaptive CR% in all groups (data not shown). Consistent with this, there was no obvious difference in the temporal pattern of CRs among the last pre-lesion session, the first post-lesion session, and the third post-lesion session in most groups, except the third post-lesion session of the 1 d hippocampal lesion group and the 4 weeks cerebellar lesion group (Fig. 6). The temporal pattern of the CRs tended to recover to that of the last pre-lesion session in most groups. To quantify this observation, we calculated the peak latency of these CRs to assess their CR timing. Tables 1 and 2 show the CR peak latency of each of the lesion groups in the first and third post-lesion sessions. At no time point did the CR peak latency of any lesion group differ from its corresponding control group in the first post-lesion session (t test; all p > 0.05). Thus all lesion groups show CRs with timing comparable with their corresponding control groups in this session. On the other hand, in the third post-lesion session, the 1 d hippocampal lesion group and the 4 weeks cerebellar lesion group exhibited significantly shorter CR peak latency than their corresponding control groups (t test; each p < 0.05), whereas the CR peak latency of the other groups did not differ from their corresponding control groups (t test; all p > 0.05). The significant difference was attributable to the recovery of CR peak latency in the corresponding control groups, which did not occur in the 1 d hippocampal lesion group and the 4 weeks cerebellar lesion group. This difference, however, was small and did not affect the detection of CRs. We conclude that the impairment described above was caused by a failure to retain a memory of the trace CR rather than by a failure to express a CR with adaptive timing.
Figure 6.

The temporal pattern of the CR of the last pre-lesion session (pre –1), the first post-lesion session (post 1), and the third post-lesion session (post 3). A, Averaged EMG amplitude of the cortical lesion group (thin line, cort) and hippocampal lesion group (thick line, hipp). There was no obvious difference of the temporal pattern of CRs between groups, except the third post-lesion session of the 1 d hippocampal lesion group. B, Averaged EMG amplitude of the sham operation group (thin line, sham) and the mPFC lesion group (thick line, mPFC). There was no obvious difference of the temporal pattern of CRs between groups. C, Averaged EMG amplitude of the sham operation group (thin line, sham) and the cerebellar lesion group (thick line, cere). There was no obvious difference of the temporal pattern of CRs between groups, except the third post-lesion session of the 4 weeks cerebellar lesion group. The solid line under each trace indicates the timing of the 350 msec CS. The vertical scale indicates the average EMG amplitude before the CS presentation; the horizontal scale indicates 100 msec.
Table 1.
Summary of CR peak latency in the first post-lesion session
|
Group |
1 d (msec) |
1 week (msec) |
2 weeks (msec) |
4 weeks (msec) |
|---|---|---|---|---|
| Cortical lesion | 516 ± 23.2 | 514 ± 16.9 | 575 ± 26.3 | 546 ± 34.8 |
| Hippocampal lesion | 500 ± 28.9 | 546 ± 27.1 | 582 ± 46.0 | 569 ± 18.6 |
| Sham operation (mPFC) | 574 ± 30.4 | 566 ± 35.1 | 581 ± 13.1 | |
| mPFC lesion | 616 ± 32.8 | 582 ± 24.7 | 608 ± 23.2 | |
| Sham operation (cerebellum) | 571 ± 23.1 | 625 ± 36.3 | 583 ± 29.9 | |
| Cerebellar lesion
|
574 ± 30.9
|
|
621 ± 12.0
|
547 ± 29.9
|
No lesion group differed from its corresponding control group in CR peak latency. All data points represent group mean ± SEM.
Table 2.
Summary of CR peak latency in the third post-lesion session
|
Group |
1 d (msec) |
1 week (msec) |
2 weeks (msec) |
4 weeks (msec) |
|---|---|---|---|---|
| Cortical lesion | 563 ± 20.2 | 497 ± 18.2 | 578 ± 30.5 | 555 ± 27.3 |
| Hippocampal lesion | 468 ± 24.4* | 598 ± 42.9 | 591 ± 21.0 | 577 ± 27.4 |
| Sham operation (mPFC) | 617 ± 13.1 | 591 ± 24.9 | 586 ± 9.20 | |
| mPFC lesion | 629 ± 12.6 | 592 ± 36.0 | 620 ± 26.4 | |
| Sham operation (cerebellum) | 619 ± 19.8 | 644 ± 27.9 | 644 ± 23.6 | |
| Cerebellar lesion
|
581 ± 29.7
|
|
627 ± 51.3
|
555 ± 27.6*
|
Most groups did not differ from their corresponding control groups in CR peak latency except the 1 d hippocampal and the 4 weeks cerebellar lesion group. All data points represent group mean ± SEM. *p<0.05.
Ablation does not affect spontaneous blinking
To assess the effects of lesions on spontaneous blinking during conditioning, we calculated the frequency of hyperactive trials, which reflects blinking during the pre-CS period (for details, see Materials and Methods), and compared the values of the last pre-lesion session and the first post-lesion session. The averaged value of each session of each lesion group ranged from 9 to 18%. In all groups, there were no statistically significant differences between spontaneous blinking during the pre-CS period before and after lesions (t test; all p > 0.05). We conclude that lesions did not affect spontaneous blinking during conditioning.
Discussion
We investigated the effects of post-learning lesion of the hippocampus, the mPFC, and the cerebellum on memory retention at several time intervals after trace eyeblink conditioning. We found that the mPFC plays an important role in CR retention, with temporal characteristics that are the reverse of those of the hippocampus. The mPFC is involved more in retention of a remotely acquired CR than in retention of a recently acquired one, whereas the hippocampus plays an important role in retention of a recently acquired CR but not in retention of a remotely acquired one. In contrast, the cerebellum was always necessary for CR retention, regardless of the time that had elapsed after the learning. These results suggest that the brain circuitry that mediates memory retention of the trace CR is dynamically reorganized during the post-learning period.
The time-limited nature of hippocampal participation in memory retention has been reported in various types of hippocampus-dependent learning tasks, including trace eyeblink conditioning (Kim et al., 1995; Takehara et al., 2002). If one begins with the idea that information contained within the hippocampus directs consolidation by gradually changing the organization of cortical representations (McClelland et al., 1995; Squire and Alvarez, 1995), ablation of a candidate area for permanent memory will result in retrograde amnesia in a temporal manner that is reverse that of the hippocampus. Our results, along with recent reports of neocortical participation in long-lasting memory retention (Bontempi et al., 1999; Frankland et al., 2001), clearly support this idea. We suggest that the mPFC is one such candidate in trace eyeblink conditioning. Several studies offer support for this view. (1) The mPFC receives a direct anatomical projection from the CA1/subicular region of the hippocampus (Jay and Witter, 1991), and this pathway exhibits NMDA receptor-mediated long-term potentiation (Laroche et al., 1990, Jay et al., 1995), which might enable the hippocampus to exert enduring effects on the mPFC. (2) This pathway is activated during a critical phase of consolidation, as suggested by a delayed increase in synaptic transmission and by learning-induced change in the expression of genes associated with plasticity in this pathway (for review, see Laroche et al., 2000). (3) Network oscillations in the mPFC (spindle waves) have a correlation with the high-frequency transient bursts of CA1 pyramidal cell activity (ripples) during slow-wave sleep (Siapas and Wilson, 1998), indicating a functional connection between the hippocampus and the mPFC. The importance of this type of interaction has been discussed in relation to memory consolidation (for review, see Sutherland and McNaughton, 2000).
This is not the only possible interpretation of our data, however. First, it is unlikely that the observed deficits reflect a motor deficit resulting from motor cortex damage, because motor cortex damage does not impair trace eyeblink conditioning (Ivkovich and Thompson, 1997). Therefore, the impairment observed in the mPFC lesion group was most likely caused by disruption of the prelimbic cortex or the anterior cingulate cortex, or both, which are important for acquisition during trace conditioning (Kronforst-Collins and Disterhoft, 1998; Weible et al., 2000; McLaughlin et al., 2002). Second, these areas of the prefrontal cortex in rodents have been thought to be important for working memory, attention, and decision making (Broersen, 2000; De Bruin et al., 2000), indicating functional similarity with the primate prefrontal cortex (Kolb, 1984; Goldman-Rakic, 1990; Fuster, 1997). Furthermore, several reports have argued that the mPFC is important for retrieval of the memory stored within other brain regions (Seamans et al., 1995; Schacter et al., 1996; Tomita et al., 1999; Powell et al., 2001). This presumptive requirement of the mPFC in the effortful recall of a remotely acquired trace CR could account for the severe impairment that we observed in the 4 weeks mPFC lesion group, because these subjects may have used greater retrieval effort to access the acquired CR than did the 1 d mPFC lesion group. Further studies will be necessary to clarify the exact role of the mPFC in remotely acquired memory.
The present study clarified that the hippocampus has already become unnecessary for memory retention 1 week after the learning. This result suggests that some parts of the circuitry that mediates the long-lasting memory are formed outside the hippocampus during this period. Consistent with this time course, the neuronal excitability in CA1 and CA3 increases during a limited period of 1 week after trace eyeblink conditioning (Moyer et al., 1996; Thompson et al., 1996). Therefore, it is possible that enhanced excitability is involved in a formation of extrahippocampal memory circuitry by facilitating propagation of selective afferent information through the hippocampus to extrahippocampal regions.
It is worth noting that3dofthe post-lesion sessions increased the adaptive CR% in the hippocampal and mPFC lesion groups, which may reflect the remaining ability of relearning caused by the paired presentation of the CS and US. Unlike other hippocampal lesion groups, the 1 d hippocampal lesion group showed incomplete relearning, and its CR peak latency did not improve compared with that of the 1 d cortical lesion group (Table 2). We also confirmed this incomplete relearning in another group of 1 d hippocampal lesioned rats with adaptive CR% and CR peak latency that never fully recovered to the level of the corresponding control group even after 10 d of retraining (Fig. 2E). This is consistent with the previous rabbit study (Kim et al., 1995). The relearning observed in the 1 week, 2 weeks, and 4 weeks hippocampal lesion groups might be mediated by the gradually organized extrahippocampal circuitry, probably including the mPFC and the cerebellum, which may not have been formed sufficiently in the 1 d hippocampal lesion group. Similarly, all of the mPFC lesion groups tended to relearn the CR; this is especially apparent in the 4 weeks mPFC lesion group attributable to its great impairment in the first post-lesion session. The reason for this relearning is not clear; however, one possibility is that the circuitry involving the hippocampus and the cerebellum (and mediating the recently acquired memory) is reactivated and exerts an effect on this relearning. One recent report supports this view: the hippocampus becomes essential again when the acquired memory is reactivated, although the memory has already become independent of the hippocampus (Debiec et al., 2002). Taken together, these results suggest the possibility that the hippocampus and the mPFC are interchangeable with each other for relearning once the learning-related changes are partially consolidated within the whole circuitry in the presence of both structures.
In contrast, the effect of the cerebellar lesion on memory retention was profound, regardless of the interval between learning and the lesion. No consistent relearning was observed during the post-lesion sessions, although the 4 weeks lesion group showed a trend toward relearning. These results clearly suggest that the ipsilateral cerebellum is indispensable for memory retention, which is consistent with the report in rabbits (Woodruff-Pak et al., 1985), and expands its importance to long-lasting memory retention in the hippocampus-dependent trace paradigm as well as the delay paradigm (McCormick et al., 1982; Steinmetz et al., 1992; Attwell et al., 2002). The cerebellar lesion group in the present study, however, exhibited a relatively higher CR% (17–30%) through the post-lesion sessions compared with that in the rabbit study (10%) (Woodruff-Pak et al., 1985). This may have been caused by the difference in the US (peri-orbital shock vs air puff), the CR monitoring (EMG activity vs nictitating membrane response), the conditioning chamber (free moving vs restrained), or the contribution of the contralateral cerebellum (for review, see Ivarsson et al., 2000).
Although the lesion method, per se, cannot serve to localize a memory trace, we propose that there are two memory circuitries for trace eyeblink conditioning. One involves the hippocampus and the cerebellum and mediates recently acquired memory; the other involves the mPFC and the cerebellum and mediates remotely acquired memory. As suggested in other articles, the basic circuitry for acquisition, storage, and generation of the CR might exist within the cerebellum in the trace paradigm just as in the delay paradigm, and the forebrain may provide input to the cerebellum that is essential for acquisition and stable performance (Weiss and Disterhoft, 1996; Green and Woodruff-Pak, 2000; Clark et al., 2002; Kotani et al., 2003). According to this view, the present results suggest that this essential input to the cerebellum was memorized and provided first by the hippocampus and, as time elapsed, was gradually provided instead by the mPFC. At the same time, the hippocampus must receive some learning-related information from the cerebellum, because the cerebellar nuclei lesions abolished the learning-related hippocampal activity, which forms a temporal model of the CR (Clark et al., 1984; Sears and Steinmetz, 1990). The mPFC also may have some contact with the cerebellum through the mediodorsal thalamus (Powell and Churchwell, 2002). These bi-directional interactions between the cerebellum and the forebrain might mediate acquisition and storage of relatively complicated motor memory.
In conclusion, the present study suggests that the brain regions that participate in the memory for trace eyeblink conditioning are gradually reorganized. The hippocampus and cerebellum are important for recently acquired memory, and the mPFC and cerebellum are important for remotely acquired memory. Further studies using trace eyeblink conditioning should be useful for the investigation of the mechanism of memory consolidation.
Footnotes
This work was supported by the Ministry of Education, Culture, Sports, Science and Technology of Japan (Grants 13210036 and 13680734), the Core Research for Evolution Science and Technology of Japan Science and Technology Corporation, and the Kato Memorial Bioscience Foundation.
Correspondence should be addressed to Dr. Shigenori Kawahara, Laboratory of Neurobiophysics, School of Pharmaceutical Sciences, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: kawahara@mol.f.u-tokyo.ac.jp.
Copyright © 2003 Society for Neuroscience 0270-6474/03/239897-09$15.00/0
References
- Anagnostaras SG, Gale GD, Fanselow MS ( 2001) Hippocampus and contextual fear conditioning: recent controversies and advances. Hippocampus 11: 8–17. [DOI] [PubMed] [Google Scholar]
- Attwell PJ, Cooke SF, Yeo CH ( 2002) Cerebellar function in consolidation of a motor memory. Neuron 34: 1011–1020. [DOI] [PubMed] [Google Scholar]
- Bontempi B, Laurent-Demir C, Destrade C, Jaffard R ( 1999) Time-dependent reorganization of brain circuitry underlying long-term memory storage. Nature 400: 671–675. [DOI] [PubMed] [Google Scholar]
- Broersen LM ( 2000) Attentional processes and learning and memory in rats: the prefrontal cortex and hippocampus compared. Prog Brain Res 126: 79–94. [DOI] [PubMed] [Google Scholar]
- Clark GA, McCormick DA, Lavond DG, Thompson RF ( 1984) Effects of lesions of cerebellar nuclei on conditioned behavioral and hippocampal neuronal responses. Brain Res 291: 125–136. [DOI] [PubMed] [Google Scholar]
- Clark RE, Manns JR, Squire LR ( 2002) Classical conditioning, awareness, and brain systems. Trends Cogn Sci 6: 524–531. [DOI] [PubMed] [Google Scholar]
- Debiec J, LeDoux JE, Nader K ( 2002) Cellular and systems reconsolidation in the hippocampus. Neuron 36: 527–538. [DOI] [PubMed] [Google Scholar]
- De Bruin JP, Feenstra MG, Broersen LM, Van Leeuwen M, Arens C, De Vries S, Joosten RN ( 2000) Role of the prefrontal cortex of the rat in learning and decision making: effects of transient inactivation. Prog Brain Res 126: 103–113. [DOI] [PubMed] [Google Scholar]
- Fuster JM ( 1997) The prefrontal cortex: anatomy, physiology, and neuropsychology of the frontal lobe, Ed 3. Philadelphia: Lippincott-Raven.
- Frankland PW, O'Brien C, Ohno M, Kirkwood A, Silva AJ ( 2001) Alpha-CaMKII-dependent plasticity in the cortex is required for permanent memory. Nature 411: 309–313. [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS ( 1990) Cellular and circuit basis of working memory in prefrontal cortex of nonhuman primates. Prog Brain Res 85: 325–335. [DOI] [PubMed] [Google Scholar]
- Green JT, Woodruff-Pak DS ( 2000) Eyeblink classical conditioning: hippocampal formation is for neutral stimulus associations as cerebellum is for association-response. Psychol Bull 126: 138–158. [DOI] [PubMed] [Google Scholar]
- Ivarsson M, Svensson P, Hesslow G ( 2000) Contralateral cerebellar involvement in conditioned eyeblink responses. Prog Brain Res 124: 309–316. [DOI] [PubMed] [Google Scholar]
- Ivkovich D, Thompson RF ( 1997) Motor cortex lesions do not affect learning or performance of the eyeblink response in rabbits. Behav Neurosci 111: 727–738. [DOI] [PubMed] [Google Scholar]
- Jay TM, Witter MP ( 1991) Distribution of hippocampal CA1 and subicular efferents in the prefrontal cortex of the rat studied by means of anterograde transport of Phaseolus vulgaris-leucoagglutinin. J Comp Neurol 313: 574–586. [DOI] [PubMed] [Google Scholar]
- Jay TM, Burette F, Laroche S ( 1995) NMDA receptor-dependent long-term potentiation in the hippocampal afferent fibre system to the prefrontal cortex in the rat. Eur J Neurosci 7: 247–250. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Clark RE, Thompson RF ( 1995) Hippocampectomy impairs the memory of recently, but not remotely, acquired trace eyeblink conditioned responses. Behav Neurosci 109: 195–203. [DOI] [PubMed] [Google Scholar]
- Knowlton BJ, Fanselow MS ( 1998) The hippocampus, consolidation and on-line memory. Curr Opin Neurobiol 8: 293–296. [DOI] [PubMed] [Google Scholar]
- Kolb B ( 1984) Functions of the frontal cortex of the rat: a comparative review. Brain Res Brain Res Rev 8: 65–98. [DOI] [PubMed] [Google Scholar]
- Kotani S, Kawahara S, Kirino Y ( 2003) Trace eyeblink conditioning in decerebrate guinea pigs. Eur J Neurosci 17: 1445–1454. [DOI] [PubMed] [Google Scholar]
- Kronforst-Collins MA, Disterhoft JF ( 1998) Lesions of the caudal area of rabbit medial prefrontal cortex impair trace eyeblink conditioning. Neurobiol Learn Mem 69: 147–162. [DOI] [PubMed] [Google Scholar]
- Laroche S, Jay TM, Thierry AM ( 1990) Long-term potentiation in the prefrontal cortex following stimulation of the hippocampal CA1/subicular region. Neurosci Lett 114: 184–190. [DOI] [PubMed] [Google Scholar]
- Laroche S, Davis S, Jay TM ( 2000) Plasticity at hippocampal to prefrontal cortex synapses: dual roles in working memory and consolidation. Hippocampus 10: 438–446. [DOI] [PubMed] [Google Scholar]
- McClelland JL, McNaughton BL, O'Reilly RC ( 1995) Why there are complementary learning systems in the hippocampus and neocortex: insights from the successes and failures of connectionist models of learning and memory. Psychol Rev 102: 419–457. [DOI] [PubMed] [Google Scholar]
- McCormick DA, Clark GA, Lavond DG, Thompson RF ( 1982) Initial localization of the memory trace for a basic form of learning. Proc Natl Acad Sci USA 79: 2731–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlinchey-Berroth R, Carrillo MC, Gabrieli JD, Brawn CM, Disterhoft JF ( 1997) Impaired trace eyeblink conditioning in bilateral, medial-temporal lobe amnesia. Behav Neurosci 111: 873–882. [DOI] [PubMed] [Google Scholar]
- McLaughlin J, Skaggs H, Churchwell J, Powell DA ( 2002) Medial prefrontal cortex and Pavlovian conditioning: trace versus delay conditioning. Behav Neurosci 116: 37–47. [PubMed] [Google Scholar]
- Moyer JR, Deyo RA, Disterhoft JF ( 1990) Hippocampectomy disrupts trace eye-blink conditioning in rabbits. Behav Neurosci 104: 243–252. [DOI] [PubMed] [Google Scholar]
- Moyer JR, Thompson LT, Disterhoft JF ( 1996) Trace eyeblink conditioning increases CA1 excitability in a transient and learning-specific manner. J Neurosci 16: 5536–5546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadel L, Moscovitch M ( 1997) Memory consolidation, retrograde amnesia and the hippocampal complex. Curr Opin Neurobiol 7: 217–227. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C ( 1986) The rat brain in stereotaxic coordinates. San Diego: Academic.
- Powell DA, Churchwell J ( 2002) Mediodorsal thalamic lesions impair trace eyeblink conditioning in the rabbit. Learn Mem 9: 10–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell DA, Skaggs H, Churchwell J, McLaughlin J ( 2001) Posttraining lesions of the medial prefrontal cortex impair performance of Pavlovian eyeblink conditioning but have no effect on concomitant heart rate changes in rabbits (Oryctolagus cuniculus). Behav Neurosci 115: 1029–1038. [DOI] [PubMed] [Google Scholar]
- Schacter DL, Alpert NM, Savage CR, Rauch SL, Albert MS ( 1996) Conscious recollection and the human hippocampal formation: evidence from positron emission tomography. Proc Natl Acad Sci USA 93: 321–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scoville WB, Milner B ( 1957) Loss of recent memory after bilateral hippocampal lesions. J Neurol Neurosurg Psychiatry 20: 11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seamans JK, Floresco SB, Phillips AG ( 1995) Functional differences between the prelimbic and anterior cingulate regions of the rat prefrontal cortex. Behav Neurosci 109: 1063–1073. [DOI] [PubMed] [Google Scholar]
- Sears LL, Steinmetz JE ( 1990) Acquisition of classically conditioned-related activity in the hippocampus is affected by lesions of the cerebellar interpositus nucleus. Behav Neurosci 104: 681–692. [DOI] [PubMed] [Google Scholar]
- Siapas AG, Wilson MA ( 1998) Coordinated interactions between hippocampal ripples and cortical spindles during slow-wave sleep. Neuron 21: 1123–1128. [DOI] [PubMed] [Google Scholar]
- Solomon PR, Vander Schaaf ER, Thompson RF, Weisz DJ ( 1986) Hippocampus and trace conditioning of the rabbit's classically conditioned nictitating membrane response. Behav Neurosci 100: 729–744. [DOI] [PubMed] [Google Scholar]
- Squire LR ( 1987) Memory and brain. New York: Oxford UP.
- Squire LR ( 1992) Memory and the hippocampus: a synthesis from findings with rats, monkeys, and humans. Psychol Rev 99: 195–231. [DOI] [PubMed] [Google Scholar]
- Squire LR, Alvarez P ( 1995) Retrograde amnesia and memory consolidation: a neurobiological perspective. Curr Opin Neurobiol 5: 169–177. [DOI] [PubMed] [Google Scholar]
- Squire LR, Clark RE, Knowlton BJ ( 2001) Retrograde amnesia. Hippocampus 11: 50–55. [DOI] [PubMed] [Google Scholar]
- Steinmetz JE, Lavond DG, Ivkovich D, Logan CG, Thompson RF ( 1992) Disruption of classical eyelid conditioning after cerebellar lesions: damage to a memory trace system or a simple performance deficit? J Neurosci 12: 4403–4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland GR, McNaughton B ( 2000) Memory trace reactivation in hippocampal and neocortical neuronal ensembles. Curr Opin Neurobiol 10: 180–186. [DOI] [PubMed] [Google Scholar]
- Takehara K, Kawahara S, Takatsuki K, Kirino Y ( 2002) Time-limited role of the hippocampus in the memory for trace eyeblink conditioning in mice. Brain Res 951: 183–190. [DOI] [PubMed] [Google Scholar]
- Thompson LT, Moyer JR, Disterhoft JF ( 1996) Transient changes in excitability of rabbit CA3 neurons with a time course appropriate to support memory consolidation. J Neurophysiol 76: 1836–1849. [DOI] [PubMed] [Google Scholar]
- Thompson RF, Kim JJ ( 1996) Memory systems in the brain and localization of a memory. Proc Natl Acad Sci USA 93: 13438–13444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RF, Bao S, Chen L, Cipriano BD, Grethe JS, Kim JJ, Thompson JK, Tracy JA, Weninger MS, Krupa DJ ( 1997) Associative learning. Int Rev Neurobiol 41: 151–189. [DOI] [PubMed] [Google Scholar]
- Tomita H, Ohbayashi M, Nakahara K, Hasegawa I, Miyashita Y ( 1999) Topdown signal from prefrontal cortex in executive control of memory retrieval. Nature 401: 699–703. [DOI] [PubMed] [Google Scholar]
- Weible AP, McEchron MD, Disterhoft JF ( 2000) Cortical involvement in acquisition and extinction of trace eyeblink conditioning. Behav Neurosci 114: 1058–1067. [DOI] [PubMed] [Google Scholar]
- Weiss C, Disterhoft JF ( 1996) Eyeblink conditioning, motor control, and the analysis of limbic-cerebellar interactions. Behav Brain Sci 19: 479–481. [Google Scholar]
- Weiss C, Bouwmeestev H, Power JM, Disterhoft JF ( 1999) Hippocampal lesions prevent trace eyeblink conditioning in the freely moving rat. Behav Brain Res 99: 123–132. [DOI] [PubMed] [Google Scholar]
- Woodruff-Pak DS, Lavond DG, Thompson RF ( 1985) Trace conditioning: abolished by cerebellar nuclear lesions but not lateral cerebellar cortex aspirations. Brain Res 348: 249–260. [DOI] [PubMed] [Google Scholar]