

Abstract
Long-term treatment with nonsteroidal anti-inflammatory drugs (NSAIDs) reduces the risk for Alzheimer's disease (AD). To determine the mechanisms by which inflammation affects AD and how NSAIDs protect against it, we stimulated neuroblastoma cells stably transfected with amyloid precursor protein (APP) with proinflammatory cytokines, which increased the secretion of amyloid-β and APP ectodomain. Addition of ibuprofen, indomethacin, peroxisome proliferator-activated receptor-γ (PPARγ) agonists, or cotransfection with PPARγ cDNA reversed this effect. The inhibitory action of ibuprofen and indomethacin was suppressed by PPARγ antagonists. Finally, we observed that the mRNA levels, expression, and enzymatic activity of β-secretase were increased by immunostimulation and normalized by NSAIDs. In conclusion, proinflammatory cytokines activate β-secretase, and NSAIDs inhibit this effect through PPARγ.
Keywords: cytokine, amyloid precursor protein (APP), amyloid-β, NSAIDs, ibuprofen, PPAR-γ, BACE1
Introduction
The inflammatory response to neuritic plaques has been proposed to contribute to the pathogenesis of Alzheimer's disease (AD) and to increase neuronal damage. Activated microglia are invariably associated with amyloid deposits in AD brain (Perlmutter et al., 1990; McGeer et al., 1993), and amyloid peptides are potent glial activators (Dickson et al., 1993; Barger and Harmon, 1997). Several studies have implied an active role for microglia in the mediation of amyloid toxicity and subsequent secondary tissue damage via release of cytokines and cytotoxic molecules (Tan et al., 1999). Additional evidence supporting the involvement of inflammation in AD has emerged, such as the upregulation of cytokines and chemokines including interleukin (IL) 1β (Griffin et al., 1989), IL-6, tumor necrosis factor-α (TNFα), IL-8, TGF-β, and macrophage inflammatory protein-1α in the brains of AD patients compared with control individuals (Akiyama et al., 2000). Furthermore, recent confirmation of polymorphisms in a number of proinflammatory genes, including IL-1 (Nicoll et al., 2000), IL-6 (Papassotiropoulos et al., 1999), TNFα (McCusker et al., 2001; Perry et al., 2001), and α1-antichymotrypsin, an acute phase protein (Kamboh et al., 1995), has strengthened the hypothesis of a close involvement of inflammation with AD.
A number of epidemiological studies have provided evidence of a protective effect of long-term medication with nonsteroidal anti-inflammatory drugs (NSAIDs) against AD (Rogers et al., 1993; Rich et al., 1995, McGeer et al., 1996; Stewart et al., 1997; Akiyama et al., 2000; in t' Veld et al., 2001). These epidemiological findings are now well supported by recent data showing that long-term in vivo treatment of amyloid precursor protein (APP) transgenic mice with NSAIDs significantly diminished amyloid deposition (Lim et al., 2000) and improved behavior (Lim et al., 2001).
Recently, NSAIDs were shown to directly affect the generation of amyloid-β (Aβ)1–42 independently of cyclooxygenase (COX) activity (Weggen et al., 2001), substantiating the recent failure of a clinical trial with a selective COX-2 inhibitor and suggesting another mechanism behind the protective effect of NSAIDs. A potential target of NSAIDs is the ligand-activated nuclear receptor peroxisome proliferator-activated receptor-γ (PPARγ) (Lehmann et al., 1997; Willson et al., 2000). NSAIDs directly bind to PPARγ and activate its transcriptional regulatory activities. PPARγ has been shown to inhibit the expression of a wide range of proinflammatory genes (Jiang et al., 1998; Ricote et al., 1999; Murphy and Holder, 2000), thereby protecting neurons from cell death in vitro (Heneka et al., 1999; Combs et al., 2000) and in vivo (Heneka et al., 2000).
In this study, we tested the hypothesis that the protective action of NSAIDs is mediated by PPARγ. To this end, we evaluated the action of NSAIDs and PPARγ agonists in neuroblastoma cells exposed to cytokines. We demonstrate that inflammatory cytokines and NSAIDs have an important effect on a key enzyme responsible for the generation of Aβ. Our data also suggest that PPARγ could be a candidate to mediate the actions of NSAIDs in AD.
Materials and Methods
Materials and antibodies. The following immunostimulants and inhibitors were tested: IL-1β (Alexis, Grünberg, Germany), IL-6 (R & D Systems, Wiesbaden, Germany), interferon-γ (IFNγ) (Sigma, St. Louis, MO), TNFα (Roche Diagnostics GmbH, Mannheim, Germany), ibuprofen (Sigma), pioglitazone (Takeda Chemicals, Osaka, Japan), the COX-2 inhibitor NS-398 (Calbiochem, Bad Soden, Germany), the PPARγ agonist GW7845, and the antagonists GW0072 and GW5393 (Glaxo Wellcome, Research Triangle Park, NC). The monoclonal antibodies 6E10 (recognizes Aβ1–17) and 4G8 (recognizes Aβ17–24) were obtained from Signet Labs (Dedham, MA), and W0–2 was a generous gift from K. Beyreuther (University of Heidelberg, Heidelberg, Germany). The monoclonal antibody raised against PPARγ was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The polyclonal antibody 369 against the C-terminal (CT) of APP was a generous gift from Sam Gandy (Farber Institute for Neuroscience, Thomas Jefferson University, Philadelphia, PA). Polyclonal antibodies 5313 against N-terminal of APP, antibody against CT-β-secretase (BACE1) 7520 were generous gifts from C. Haass (Adolf Butenandt Institute, Ludwig-Maximilians-University, München, Germany). Tissue culture reagents were obtained from Invitrogen (Karlsruhe, Germany). All other chemicals were purchased from Sigma.
Cell lines, cell culture, and transient transfection. The human neuroblastoma cell line SK-N-SH was cultured in DMEM supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin sulfate. Mouse neuroblastoma N2a cells stably transfected with APP695 containing the Swedish mutation (APPsw, K595N/M596L) were obtained from G. Thinakaran (University of Chicago, Chicago, IL) and were cultured in 60% DMEM with 40% Opti-MEM and 5% fetal bovine serum. All cell lines were incubated at 37°C in a 5% CO2 atmosphere. Transfections were performed using the calcium phosphate method as described previously (Sastre et al., 1998) with APPsw and recombinant APP 99 C-terminal amino acids (CT-99) cDNAs (obtained from Dr. Efrat Levy, Nathan S. Kline Institute) and mouse PPARγ cDNA (from Dr. Ron Evans, The Salk Institute for Biological Studies, San Diego, CA). Cells were incubated for 20 hr with Opti-MEM alone or with the following cytokines: IL-1β (10 ng/ml), IL-6 (10 ng/ml), TNFα (30 ng/ml), IFNγ (1 ng/ml), IFNγ (1 ng/ml) plus IL-1β (10 ng/ml), or IFNγ (1 ng/ml) plus TNFα (30 ng/ml).
Determination of secreted APP ectodomain and Aβ. Secreted APP (sAPP) in the medium was measured by Western blotting after separation by SDS-PAGE (8% gels) followed by transfer to nitrocellulose membranes and immunodetection with antibodies 5313 or 6E10 and the ECL system (Amersham Biosciences, Freiburg, Germany). The protein bands were scanned and quantified (NIH Image J analysis program). Aβ was immunoprecipitated from conditioned media with protein A beads (Amersham Biosciences) and antibodies 4G8 or W0–2, separated on 10–20% Tris–tricine gels (Invitrogen), detected by immunoblotting with antibody 6E10 and quantified by densitometric scanning.
ELISA of amyloid peptides. ELISA for human Aβ40 and Aβ42 peptides was performed using the capture antiserum joint-reaction forces (JRF)/cAβ40/14 and 21F12, respectively, and was developed with monoclonal antibodies 3D6 and JRF/cAβ42/14, respectively, as described previously (Dewachter et al., 2002).
Analysis of APP C-terminal domains. C-terminal domains (CTFs) were analyzed by immunoblotting with antibody 369 of membrane extracts from APPsw stably transfected N2a cells or transiently transfected SK-N-SH cells, as described previously (Sastre et al., 2001). CTFγ was generated in vitro from membrane preparations of the indicated cell lines according to procedures described previously (Sastre et al., 2001). Membranes were pelleted by centrifugation for 20 min at 16,000 × g at 4°C, washed, and resuspended in assay buffer (150 mm sodium citrate, pH 6.4). To allow generation of CTFγ, samples were incubated at 37°C for 1 hr. After termination of the assay, samples were separated by centrifugation at 16,000 × g at 4°C. Supernatants were separated by electrophoresis using 10–20% Tris–tricine gels, and CTFγ was analyzed by immunoblotting with antibody 369 using ECL and quantified by densitometric analysis.
Determination of BACE1 expression and activity. Cells were harvested in PBS and lysed in radioimmunoprecipitation assay buffer (1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 150 mm NaCl, and 50 mm Tris-HCl, pH 7.2), and 100 μg protein samples were separated in 8% SDS gels, followed by immunoblotting with antibody 7520. Analysis of relative intensity was performed by a computer-assisted densitometry system. The enzymatic activity of BACE1 was measured by a fluorimetric reaction (R & D Systems).
Immunocytochemistry. The subcellular localization of PPARγ and APP was performed by immunocytochemistry in N2a cells transiently transfected with APPsw and PPARγ using fluorescent secondary antibodies. Transfected cells grown on coverslips were permeabilized and fixed in methanol at –20°C for 10 min. Cells were washed in PBS, pH 7.4, blocked with 1% bovine serum albumin in PBS for 10 min, and incubated with primary antibody in blocking buffer for 1 hr at 37°C. Cells were incubated with secondary antibodies for 1 hr at 37°C. The coverslips were mounted on glass slides using Vectashield mounting medium (Vector Laboratories, Burlingame, CA). For the detection of APP, we used polyclonal antibody 5313 (1:50) and for staining of PPARγ, we used the anti-PPARγ E-8 monoclonal antibody from Santa Cruz Biotechnology (1:100).
RNA preparation and reverse transcription-PCR. Total RNA was extracted using Trizol reagent according to the manufacturer's instructions (Sigma). Reverse transcription (RT)-PCR was performed as described previously (Heneka et al., 2000). The following primers were used: for BACE1, 5′-GGCGGGAGTGGTATTATGAGGTGA-3′ and 5′-TATTGCTGCGGAAGGATGGTGA-3′; for glyceraldehyde 3-phosphate dehydrogenase (GAPDH), 5′-ACGACAGTCCATGCCATCAC-3′ and 5′-TCCACCACCCTGTTGCTGTA-3′. PCR conditions were 35 cycles at 95°C for 30 sec, annealing at 63°C for 45 sec, and extension at 42°C for 45 sec. PCR products were separated by electrophoresis through 2% agarose-containing ethidium bromide, and the fluorescence was recorded and band densities were quantified using the NIH Image J program.
Statistical evaluation. Data were statistically analyzed by Systat (Evanston, IL) using ANOVA followed by Tukey's test.
Results
Effect of inflammatory cytokines on APP processing
Previous reports have indicated that some inflammatory cytokines can influence Aβ formation in cultured human neuroblastoma cells and astrocytes; however, they did not define the mechanisms involved (Blasko et al., 1999, 2000). Because this is fundamental to the proposed study, we reevaluated these experiments. Cytokines that have been linked to AD were analyzed for their effect on APP processing in mouse neuroblastoma N2a cells permanently transfected with APPsw. After incubation for 20 hr with IL-1β, IL-6, TNFα, IFNγ, and the combinations IFNγ plus IL-1β and IFNγ plus TNFα, the media were analyzed by Western blotting with antibody 5313 to determine the total soluble fraction of sAPP or 6E10 for α-sAPP. Total secreted APP as well as α-sAPP were increased by addition of the cytokines, most prominently with combinations of IFNγ and TNFα (Fig. 1A,B). Increased APP secretion was not attributable to increased APP expression, as observed in neural cells (Blasko et al., 1999) (Fig. 1C), but rather to a direct effect on APP processing causing increased Aβ generation and APP secretion.
Figure 1.

Proinflammatory cytokines affect sAPP secretion. A, Proinflammatory cytokines increase total sAPP (APPs) and α-sAPP (α-APPs) levels in N2a cells stably transfected with APPsw and immunostimulated with proinflammatory cytokines. B, Quantification of total sAPP andα-sAPP in N2a cells permanently transfected with APPsw by Western blotting experiments shows an increase after cytokine stimulation in six different experiments. C, Quantification of intracellular full-length APP in N2a cells permanently transfected with APPsw by Western blotting in nine experiments shows no effect after cytokine stimulation. Columns represent average ± SEM. *p ≤ 0.05; ANOVA, Tukey's post hoc test. c, Control.
Confirming results of Blasko et al. (1999, 2000), IFNγ plus IL-1β and IFNγ plus TNFα increased secretion of Aβ into the medium of N2a cells stably transfected with APPsw (Fig. 2A,B). A specific ELISA for Aβ1–40 and Aβ1–42 demonstrated both to be significantly increased in media from cells stimulated with TNFα, IFNγ plus IL-1β, or IFNγ plus TNFα (Fig. 2C). Incubation of N2a cells for different times demonstrated that this effect was already evident at 4 hr, was maximal at 20 hr, and persisted for up to 48 hr (data not shown). The same results were obtained in SK-N-SH cells transiently transfected with APPsw (data not shown).
Figure 2.

Proinflammatory cytokines affect Aβ secretion. A, Increased Aβ generation in media obtained from N2a cells overexpressing APPsw by proinflammatory cytokines, shown by immunoprecipitation and Western blotting. B, Quantification of total Aβ by immunoprecipitation–Western blotting in five similar experiments as above. C, Analysis of Aβ1–40 and Aβ1–42 secretion in N2a APPsw cells by ELISA (n = 6). Columns represent average ± SEM. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ANOVA, Tukey's post hoc test. c, Control.
Effect of NSAIDs and PPARγ-agonists on APP processing
N2a cells stably transfected with APPsw were immunostimulated with IFNγ plus TNFα for 20 hr followed by incubation with either ibuprofen or the specific PPARγ agonist pioglitazone for 4 hr. Ibuprofen (at 1–200 μm) and pioglitazone (10–100 μm) reversed the effects of inflammatory cytokines on the secretion of total sAPP and α-sAPP (Fig. 3A,B) without affecting total APP expression (Fig. 3C). In unstimulated cells, neither ibuprofen nor pioglitazone affected the expression or secretion of APP (Fig. 3A–C).
Figure 3.

Effect of Ibuprofen (IBU) and pioglitazone (PIO) on APP secretion. A, IBU and PIO decrease total sAPP (APPs) and α-sAPP (α-APPs) levels in N2a cells immunostimulated with proinflammatory cytokines but not under noninflammatory conditions. B, Quantification of sAPP by Western blot in six different experiments. C, Quantification of intracellular full-length APP by Western blotting shows no difference among treatments. Columns represent average ± SEM. Asterisks indicate significant differences between control and treatment. Number signs indicate significant differences between treatment with cytokines alone and with NSAIDs. **p ≤ 0.01; ***p ≤ 0.001; #p ≤ 0.05; ###p ≤ 0.001; ANOVA followed by a Tukey's post hoc test. C, Control.
Ibuprofen and pioglitazone also decreased the levels of secreted Aβ from stimulated cells, thereby reversing the effect of the proinflammatory cytokines IFNγ plus IL-1β (Fig. 4A,B) or IFNγ plus TNFα (Fig. 4C). Secretion of Aβ from unstimulated cells was not affected by ibuprofen or pioglitazone treatments (Fig. 4C). The same results were obtained in SK-N-SH cells transiently transfected with APPsw. In N2a cells, a nonthiazolidinedione PPARγ agonist (Willson et al., 2000), GW7845, caused the same effects as pioglitazone (data not shown). Ibuprofen and pioglitazone decreased the generation of both Aβ1–40 and Aβ1–42 levels (Fig. 4D). To determine whether NSAIDs other than ibuprofen could reduce Aβ generation, we examined the role of indomethacin, which acts as a nonselective COX inhibitor as well as a PPARγ agonist (Lehmann et al., 1997). Under inflammatory conditions, 4 hr incubation with 1–10 μm indomethacin decreased total Aβ levels (Fig. 4E), as demonstrated for ibuprofen.
Figure 4.

Effect of ibuprofen (IBU) and pioglitazone (PIO) on Aβ secretion. A, Ibuprofen and pioglitazone decrease Aβ secretion in immunoprecipitation–Western blotting experiments in N2a cells for which APPsw was stimulated with IFNγ (1ng/ml)plus IL-1β (10 ng/ml) overnight and then incubated with IBU (10μm) or PIO (10μm) for 4 hr. B, Quantification of total Aβ in six to nine experiments of cells stimulated with the same conditions as above. C, Quantification of total Aβ in six to nine experiments of cells stimulated with IFNγ (1 ng/ml) plus TNFα (30 ng/ml). D, Analysis of Aβ1–40 and Aβ1–42 secretion in APPsw-transfected N2A cells by ELISA. E, Indomethacine (INDO) exerts the same effect as ibuprofen on Aβ generation. Columns represent average ± SEM. Asterisks indicate significant differences between control and treatment. Number signs indicate significant differences between treatment with cytokines alone and with NSAIDs. *p ≤ 0.05; ***p ≤ 0.001; #p ≤ 0.05; ##p ≤ 0.01; ###p ≤ 0.001; ANOVA followed by a Tukey's post hoc test. C, Control.
To rule out the possibility that the effect of ibuprofen was mediated by COX-2 inhibition, we performed experiments using the COX-2 inhibitor NS-398 under the same conditions as above. The levels of secreted Aβ remained unchanged after addition of 10 or 50 μm NS-398 under inflammatory and noninflammatory conditions (data not shown).
Modulation of Aβ secretion by PPARγ
To study the effect of PPARγ on cytokine-induced APP processing, we transiently transfected PPARγ cDNA into the N2a cells that stably expressed APPsw. The efficiency of transfection and the subcellular localization of both proteins were monitored by immunofluorescence (Fig. 5A). The cellular distribution of APP was demonstrated with antibody 5313. Expression of PPARγ was observed in the cell nucleus as anticipated (Fig. 5A). Experiments were done with cultures that contained >80% fluorescent cells.
Figure 5.
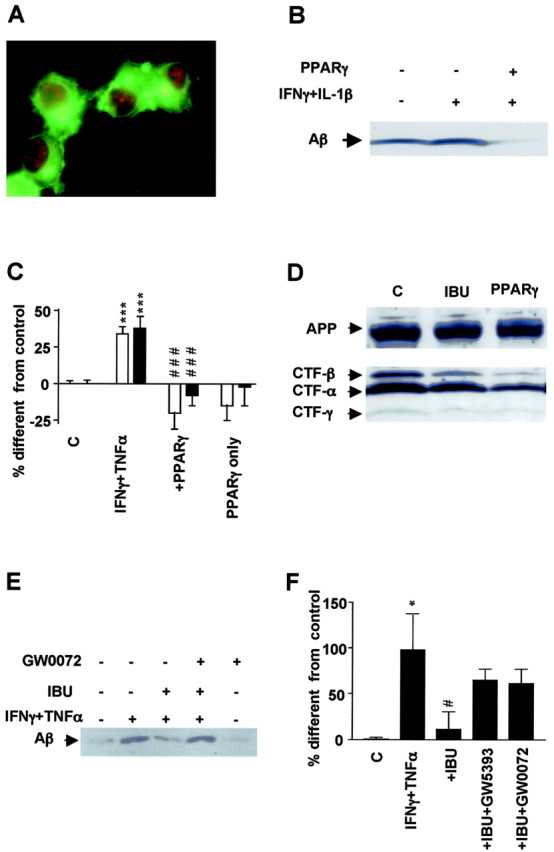
Effect of PPARγ on APP processing and Aβ secretion. A, Immunofluorescence detection of APP with 5313 antibody (green) and PPARγ with E-8 (red) in N2a cells overexpressing both proteins. B, Aβ detection by immunoprecipitation and Western blotting from media obtained from N2a cells overexpressing APPsw with or without transient transfection with PPARγ cDNA. PPARγ expression decreases generation of Aβ in cells stimulated overnight with the proinflammatory cytokines IFNγ (1 ng/ml) plus IL-1β (10 ng/ml). C, Analysis of Aβ1–40 and Aβ1–42 secretion by ELISA under the same conditions as above but in cells immunostimulated with IFNγ (1 ng/ml) plus TNFα (30 ng/ml). D, Detection of different CTFs of APP in N2a cells overexpressing APPsw and cotransfected with vector or PPARγ cDNA or incubated with ibuprofen (IBU). E, Total Aβ detection in N2A cells stimulated overnight with IFNγ plus TNFα and afterward with IBU (1μm) with or without PPARγ antagonist GW420072X (1μm) for 4 hr. F, Quantification of Aβ levels in eight experiments in which incubation with PPARγ antagonists GW0072X (1 μm) and GW5393X (1 μm) reversed the suppressive effect of ibuprofen (IBU) on APP processing. Columns represent average ± SEM. Asterisks indicate significant differences between control and treatment. Number signs represent differences between cytokines and NSAIDs. *p ≤ 0.05; ***p ≤ 0.001; #p ≤ 0.05; ###p ≤ 0.001; ANOVA followed by a Tukey's post hoc test. C, Control.
The amount of Aβ secreted into the medium by N2a cells stably transfected with APPsw, and that transiently expressed PPARγ, was decreased compared with cells untreated or treated with IFNγ plus IL-1β (Fig. 5B). This was confirmed by ELISA and found to be statistically significant (Fig. 5C).
To understand the action of PPARγ on the processing of APP, we analyzed the levels of the different APP C-terminal stubs by Western blotting of membrane preparations from the N2a cells permanently transfected with APPsw with antibody 369. The CTF-β fragment resulting from cleavage by BACE1 and the CTF-α fragments were decreased in cells treated with ibuprofen, as well as in cells overexpressing PPARγ (Fig. 5D). This effect did not result from altered expression of total APP (Fig. 5D) and was consistent with the decreased levels of sAPP and Aβ after incubation with NSAIDs and PPARγ agonists. The levels of CTF-γ fragments were similarly affected by the treatment (Fig. 5D), which is likely secondary to the decrease in β-stubs and α-stubs, their obligate precursors.
An additional argument proving the role of PPARγ in mediating the action of ibuprofen was found in the action of the PPARγ antagonists (Willson et al., 2000; Willson, 2003) GW0072 and GW5393 on N2a cells. Both drugs reversed the suppressive effect of ibuprofen on Aβ generation (Fig. 5E,F).
Effect of NSAIDs and PPARγ agonists on the secretases of APP
To confirm previous reports, which claimed that γ-secretase cleavage site is influenced by NSAID treatment (Takahashi et al., 2003), γ-secretase activity was measured by in vitro assays of membranes prepared from N2a cells stably transfected with APPsw. Incubation of ibuprofen and pioglitazone did not decrease the formation of the CTF-γ fragment (Fig. 6A,B), as observed by Weggen et al. (2003). Therefore, a direct inhibition of γ-secretase activity by these drugs at the ϵ-cleavage site seems to be unlikely.
Figure 6.

Effect of proinflammatory cytokines and NSAIDs on γ-secretase activity. A, No direct effect of NSAIDs onγ-secretase activity demonstrated by in vitro detection of CTF-γ from membrane preparations incubated for 1 hr with different concentrations of ibuprofen (IBU) or pioglitazone (PIO) was observed. B, Quantification of CTF-γ detected in vitro from three to five experiments. C, Aβ detection in N2A cells transfected with CT-99 mutant of APP shows no change with different treatments. D, Quantification of Aβ from four to five experiments from N2A cells transiently transfected with CT-99. E, Quantification of the ratio Aβ1–40/Aβ1–42from ELISA experiments performed in N2A transfected with APPsw cells shows an increase in this ratio after IBU treatment. Columns represent average ± SEM. Asterisks indicate significant differences. *p ≤ 0.05; ANOVA followed by a Tukey's post hoc test. C, Control.
Another measurement of γ-secretase activity was performed by measuring Aβ in cells transfected with a construct coding for a truncated APP consisting of the CT-99 amino acids, which can be cleaved only by γ-secretase to produce Aβ. The levels of total Aβ secreted by these cells were not affected by any of the cytokines or drug combinations applied (Fig. 6C,D). These experiments verify that neither the inflammatory cytokines nor ibuprofen or PPARγ activation affected the γ-secretase cleavage of APP under these conditions.
We evaluated the ratio Aβ1–40/Aβ1–42 from the ELISA experiments shown above. As stated above, inflammatory stimulation had little or no effect on the ratio of Aβ1–40/Aβ1–42 (Fig. 6E). However, ibuprofen increased somewhat the Aβ1–40/Aβ1–42 ratio (Fig. 6E), indicating a tendency toward less Aβ1–42 production, as observed by others (Weggen et al., 2001).
Having excluded γ-secretase as the target modulated by cytokines, ibuprofen, or pioglitazone under immunostimulated conditions, we evaluated their effect on BACE1. Incubation of N2a or SK-N-SH cells overexpressing APPsw with cytokines increased BACE1 protein levels significantly, as demonstrated by Western blotting using antibody 7520 (Fig. 7A,B). The increase in BACE1 protein levels was attributable to increased steady-state levels of RNA, as demonstrated by semiquantitative RT-PCR (Fig. 7C,D).
Figure 7.

Effect of proinflammatory cytokines on BACE1 expression, mRNA levels, and activity. A, Proinflammatory cytokines increase BACE1 expression in SK-N-SH cells by Western blotting experiments.B, Quantification of BACE1 expression in eight to nine experiments performed in N2A cells. C, Proinflammatory cytokines increase BACE1 steady-state mRNA levels shown in this semiquantitative RT-PCR representation from N2a cells treated with different proinflammatory cytokines. As a control, PCR was performed for GAPDH with the same samples. D, Quantification of BACE1 steady-state mRNA levels in six to nine experiments performed in N2A cells. E, Proinflammatory cytokines increase BACE1 activity in N2A cells. Columns represent average ± SEM. *p ≤ 0.05; **p < 0.01. c, Control.
To determine whether the increase in protein levels led to an increase in enzyme activity, we also measured BACE1 enzymatic activity in the cellular extracts in cells immunostimulated previously with proinflammatory cytokines. The results demonstrated that cytokine treatment increased BACE1 activity in N2a cells, which was statistically significant for the combination of IFNγ plus IL-1β and IFNγ plus TNFα (Fig. 7E). The same combinations of cytokines that led to increased levels of Aβ and sAPP also increased expression and mRNA levels of BACE1.
We also evaluated whether ibuprofen and pioglitazone affected BACE1 transcription, expression, and activity. In N2a cells immunostimulated with IFNγ plus TNFα, BACE1 expression was modulated negatively by ibuprofen and pioglitazone (Fig. 8A,B). This decrease was likely the consequence of decreased steady-state levels of mRNA, because BACE1 mRNA was similarly decreased after treatment with ibuprofen and pioglitazone (Fig. 8C). Other NSAIDs such as indomethacin also reduced BACE1 mRNA under inflammatory conditions (data not shown). Addition of PPARγ antagonists reversed the inhibitory action of ibuprofen and indomethacin on BACE1 expression and transcription (Fig. 8D,E). Finally, the suppressive effect of ibuprofen and pioglitazone was paralleled by a fall of BACE1 enzymatic activity (Fig. 8F).
Figure 8.

Effect of ibuprofen (IBU) and pioglitazone (PIO) on BACE1 expression, mRNA levels, and activity. A, Increased BACE1 expression in SK-N-SH cells incubated with IFNγ (1 ng/ml) plus TNFα (30ng/ml) is reversed with IBU (10μm) in Western blotting experiments. B, Quantification of BACE1 expression in four to five experiments performed in N2A cells. Cells were incubated with IFNγ (1 ng/ml) plus TNFα (30 ng/ml) with or without IBU (10μm) or PIO(10 μm). C, Modulation of BACE1 steady-state mRNA levels by the same treatments is shown in semiquantitative RT-PCR representation of BACE1 mRNA from SK-N-SH cells. As a negative control, PCR was performed for β-actin with the same samples. D, PPARγ antagonists GW0072X (1 μm) reversed the effect of IBU and indomethacin (INDO) on BACE1 mRNA levels. E, Quantification of BACE1 steady-state mRNA levels in four to five experiments performed in N2A cells. F, BACE1 activity from N2A cells is affected by treatment with similar cytokines and NSAIDs. Asterisks indicate significant differences between control and treatment. Number signs represent differences between cytokines and NSAIDs. *p < 0.05. Columns represent average ± SEM; #p ≤ 0.05; ##p ≤ 0.01; ###p ≤ 0.001; ANOVA followed by a Tukey's post hoc test. C, Control.
Discussion
The present study seeks to determine the molecular mechanisms by which inflammatory insults interfere with the generation of amyloid peptides and how NSAIDs exert their beneficial effects on AD progression. Combinations of inflammatory cytokines have been shown to increase secretion of Aβ in cultured human neuroblastoma cells and astrocytes (Blasko et al., 1999, 2000). We demonstrate here that this is not attributable to increased APP expression, as observed previously (Amara et al., 1999; Blasko et al., 1999; Rogers et al., 1999), but to a direct effect on APP processing causing increased amyloid peptide generation and APP secretion. This work also demonstrates that mRNA levels, expression, and activity of BACE1 in neural cells is modulated by proinflammatory cytokine stimulation. Moreover, we show that anti-inflammatory drug treatment with NSAIDs and PPARγ agonists downregulates immunostimulated Aβ generation by reducing BACE1 expression and activity.
Our results strongly suggest that expression of BACE1 can be regulated by inflammatory stimuli and that ibuprofen and a PPARγ agonist such as pioglitazone decreased its steady-state mRNA levels, expression, and, subsequently, enzymatic activity. This evidently would explain how NSAIDs affect the amyloidogenic processing of APP to decrease Aβ formation under inflammatory conditions. BACE1 and presenilin-1 are key enzymes in neuronal Aβ formation, because in their absence, Aβ synthesis is either abolished or considerably reduced (Walter et al., 2001). We did not find direct or indirect evidence for any effect of the proinflammatory cytokines on the activity of γ-secretase on APP processing. Recently, several groups reported an increased BACE1 expression and activity as well as CTF-β in brains of sporadic AD patients (Fukumoto et al., 2002; Holsinger et al., 2002; Sun et al., 2002; Yang et al., 2003). The results are in line with data concerning increased expression and activity of BACE1 in NT2 neurons exposed to oxidative stress (Tamagno et al., 2003) and increased BACE1 immunoreactivity in reactive astrocytes in chronic models of gliosis (Hartlage-Rubsamen et al., 2003).
There is compelling epidemiological evidence that inflammation significantly contributes to the pathogenesis of AD. Indeed, sustained anti-inflammatory drug therapy dramatically reduces the disease risk and delays the onset and progression of symptoms. The common anti-inflammatory drug ibuprofen crosses the blood–brain barrier (Kunsman and Rohrig, 1993), but the mechanism by which NSAIDs reduce AD brain pathology is unknown (Alafuzoff et al., 2000). The suggestion that ibuprofen would prevent amyloid load and plaque formation is based on experiments in cell culture in which ibuprofen could decrease secretion of amyloid peptides, although these findings are contradictory and heavily debated (Blasko et al., 2001, Weggen et al., 2001; Avramovich et al., 2002; Takasashi et al., 2003). The problem can partially be explained by the use of different approaches and methods and by the use of different concentrations of ibuprofen in conjunction with different cell culture conditions. The use of inflammatory stimuli in the present study was chosen because an increasing body of evidence suggests that AD brains contain a wide range of proinflammatory mediators including various cytokines (Akiyama et al., 2000). Therefore, we believe that cells and enzymatic systems involved in APP processing are under constant influence of inflammatory molecules; thus, APP processing needs to be studied under conditions reflecting the presence of inflammation in AD brains. Any or all of these could be critical factors in demonstrating the effects and in analyzing the underlying molecular mechanisms.
Ibuprofen has no effect on the cleavage of APP that generates CTF-γ (present work; Weggen et al., 2003) or on the corresponding cleavage of Notch (Weggen et al., 2001). The action of ibuprofen is demonstrated in the present work to reside in the inhibition of the inflammatory response that increased BACE1 activity. It should be noted that both of our reported findings were totally unexpected and appeared to be unrelated until the combination of immunological and pharmacological effects led us to probe and demonstrate a role of PPARγ in transcriptional control of BACE1 and its activity on APP. We hypothesize, therefore, that the beneficial effect of ibuprofen is evident only under inflammatory conditions, at least as far as the generation of Aβ is concerned.
Other observations suggested that NSAIDs decreased Aβ secretion by mediation of COX, but specific COX-2 inhibitors are ineffective in AD and do not affect Aβ generation (Skovronsky et al., 2001; Aisen et al., 2003). Furthermore, clinical studies suggest that NSAID doses showing optimal efficiency seem to correlate with the range of occupancy of these drugs for PPARγ, which are much higher than the concentrations required to inhibit COX (Jiang et al., 1998). In addition, ibuprofen inhibits COX activity at much lower concentrations than those needed to affect APP processing and must therefore operate via different molecular mechanisms (Heneka et al., 2001; Landreth and Heneka, 2001). Ibuprofen acting as a PPARγ agonist inhibits inflammatory cytokine synthesis and blocks Aβ-stimulated expression of IL-1β, IL-6, TNFα, COX-2 (Combs et al., 2000, 2001), and the chemokine monocyte chemoattractant protein-1 gene (Murao et al., 2000). Two other PPARγ agonists, pioglitazone and GW3478, mimicked the effects of ibuprofen in this study. Importantly, overexpression of PPARγ itself led to the identical effect. Furthermore, the PPARγ antagonists GW0072X and GW5393X reversed the suppressive effect of ibuprofen, completing this combination of four independent arguments to firmly establish the direct action of PPARγ on BACE1 expression and thereby on amyloidogenic APP processing.
In conclusion, the current work supports the hypothesis that inflammation affects AD pathology by increasing the generation of amyloid peptides attributable to increased steady-state mRNA levels, expression, and activity of BACE1. An important fundamental question for future investigation concerns the downstream signal cascades that control the BACE1 gene promoter in response to immunostimulation. Our work also indicates the existence of a vicious cycle that accelerates the development of AD, because amyloid peptides cause microglial activation and astrocytosis and, therefore, increased secretion of a number of cytokines. These cytokines may subsequently upregulate BACE1 expression and increase Aβ generation, which serves to drive a fulminating, feedforward mechanism. Second, we observed an effect on the generation of Aβ1–42 by ibuprofen, reported previously by other groups. The third important point of this study is that ibuprofen is able to decrease total Aβ levels only under inflammatory conditions, and this effect seems to be mediated by PPARγ. Therefore, the development of new drugs targeting PPARγ would be of great interest for future treatments of AD.
Footnotes
This investigation was supported by the Sonderforschungsbereich 400 (SFB 400, Teilprojekt A8) to M.T.H. and T.K., by Fonds voor Wetenschappelijk Onderzoek (FWO)-Vlaanderen, by (KULeuven) Research Fund, and by KULeuven R&D. I.D.W. is a postdoctoral fellow at FWO-Vlaanderen. We thank Drs. Sam Gandy (Farber Institute for Neuroscience, Thomas Jefferson University, Philadelphia, PA), K. Beyreuther (University of Heidelberg, Heidelberg, Germany), and Christian Haass (University of Munich, Munich, Germany) for their generous gift of antibodies; Dr. Gopal Thinakaran (University of Chicago, Chicago, IL) for providing N2a Swedish-transfected cells; Dr. Efrat Levy (Nathan Kline Institute) for APPsw and C-99 construct cDNA; Dr. Ron Evans (The Salk Institute for Biological Studies, San Diego, CA) for PPAR-γ cDNA; Dr. Vitalyi Gavrilyuk (University of Illinois at Chicago, Chicago, IL), Drs. Jochen Walter and Thomas A. Bayer (University of Bonn, Bonn, Germany) for stimulating discussions; and René Mérou for technical assistance. We also thank the Alexander von Humboldt Foundation for stipendium to M.S.
Correspondence should be addressed to Magdalena Sastre or Michael T. Heneka, Department of Neurology, University of Bonn, Sigmund-Freud-Strasse 25,53127 Bonn, Germany. E-mail: Magdalena.Sastre@ukb.uni-bonn.de or m.heneka@uni-bonn.de.
Copyright © 2003 Society for Neuroscience 0270-6474/03/239796-09$15.00/0
References
- Aisen PS, Schafer KA, Grundman M, Pfeiffer E, Sano M, Davis KL, Farlow MR, Jin S, Thomas RG, Thal LJ ( 2003) Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. JAMA 289: 2819–2826. [DOI] [PubMed] [Google Scholar]
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, et al. ( 2000) Inflammation and Alzheimer's disease. Neurobiol Aging 21: 383–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alafuzoff I, Overmyer M, Helisalmi S, Soininen H ( 2000) Lower counts of astroglia and activated microglia in patients with Alzheimer's disease with regular use of non-steroidal anti-inflammatory drugs. J Alzheimers Dis 2: 37–46. [DOI] [PubMed] [Google Scholar]
- Amara FM, Junaid A, Clough RR, Liang B ( 1999) TGF-beta(1), regulation of alzheimer amyloid precursor protein mRNA expression in a normal human astrocyte cell line: mRNA stabilization. Brain Res Mol Brain Res 71: 42–49. [DOI] [PubMed] [Google Scholar]
- Avramovich Y, Amit T, Youdim MB ( 2002) Non-steroidal anti-inflammatory drugs stimulate secretion of non-amyloidogenic precursor protein. J Biol Chem 277: 31466–31473. [DOI] [PubMed] [Google Scholar]
- Barger SW, Harmon AD ( 1997) Microglial activation by Alzheimer amyloid precursor protein and modulation by apolipoprotein E. Nature 388: 878–881. [DOI] [PubMed] [Google Scholar]
- Blasko I, Marx F, Steiner E, Hartmann T, Grubeck-Loebenstein B ( 1999) TNFα plus IFNγ induce the production of Alzheimer β-amyloid peptides and decrease the secretion of APPs. FASEB J 13: 63–68. [DOI] [PubMed] [Google Scholar]
- Blasko I, Veerhuis R, Stampfer-Kountchev M, Saurwein-Teissl M, Eikelenboom P, Grubeck-Loebenstein B ( 2000) Costimulatory effects of interferongamma and interleukin-1β or tumor necrosis factor alpha on the synthesis of Aβ1–40 and Aβ1–42 by human astrocytes. Neurobiol Dis 7: 682–689. [DOI] [PubMed] [Google Scholar]
- Blasko I, Apochal A, Boeck G, Hartmann T, Grubeck-Loebenstein B, Ransmayr G ( 2001) Ibuprofen decreases cytokine-induced amyloid beta production in neuronal cells. Neurobiol Dis 8: 1094–1101. [DOI] [PubMed] [Google Scholar]
- Combs CK, Johnson DE, Karlo JC, Cannady SB, Landreth GE ( 2000) Inflammatory mechanisms in Alzheimer's disease: inhibition of β-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARγ agonists. J Neurosci 20: 558–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combs CK, Bates P, Karlo JC, Landreth GE ( 2001) Regulation of beta-amyloid stimulated pro-inflammatory responses by peroxisome proliferator-activated receptor alpha. Neurochem Int 39: 449–457. [DOI] [PubMed] [Google Scholar]
- Dewachter I, Reverse D, Caluwaerts N, Ris L, Kuiperi C, Van den Haute C, Spittaels K, Umans L, Serneels L, Thiry E, Moechars D, Mercken M, Godaux E, Van Leuven F ( 2002) Neuronal deficiency of presenilin 1 inhibits amyloid plaque formation and corrects hippocampal long-term potentiation but not a cognitive defect of amyloid precursor protein [V717I] transgenic mice. J Neurosci 22: 3445–3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson DW, Lee SC, Mattiace LA, Yen SH, Brosnan C ( 1993) Microglia and cytokines in neurological disease, with special reference to AIDs and Alzheimer's disease. Glia 7: 75–83. [DOI] [PubMed] [Google Scholar]
- Fukumoto H, Cheung BS, Hyman BT, Irizarry MC ( 2002) Beta-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch Neurol 59: 1381–1389. [DOI] [PubMed] [Google Scholar]
- Griffin WS, Stanley LC, Ling C, White L, MacLeod V, Perrot LJ, White III CL, Araoz C ( 1989) Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc Natl Acad Sci USA 86: 7611–7615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartlage-Rubsamen M, Zeitschel U, Apelt J, Gartner U, Franke H, Stahl T, Gunther A, Schliebs R, Penkowa M, Bigl V, Rossner S ( 2003) Astrocytic expression of the Alzheimer's disease beta-secretase (BACE1) is stimulus-dependent. Glia 4: 169–179. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Feinstein DL, Galea E, Gleichmann M, Wüllner U, Klockgether T ( 1999) Peroxisome proliferator-activated receptor gamma agonists protect cerebellar granule cells from cytokine-induced apoptotic cell death by inhibition of inducible nitric oxide synthase. J Neuroimmunol 100: 156–168. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Klockgether T, Feinstein DL ( 2000) Peroxisome proliferator-activated receptor γ agonists reduce inducible nitric oxide synthase expression in cerebellar granule neurons in vivo J Neurosci 20: 6862–6867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Landreth GE, Feinstein DL ( 2001) Role for peroxisome-activated receptor-γ in Alzheimer's disease. Ann Neurol 49: 276. [DOI] [PubMed] [Google Scholar]
- Holsinger RM, McLean CA, Beyreuther K, Masters CL, Evin G ( 2002) Increased expression of the amyloid precursor beta-secretase in Alzheimer's disease. Ann Neurol 51: 783–786. [DOI] [PubMed] [Google Scholar]
- in t' Veld BA, Ruitenberg A, Hofman A, Launer LJ, van Duijn CM, Stijnen T, Breteler MM, Stricker BH ( 2001) Nonsteroidal anti-inflammatory drugs and the risk of Alzheimer's disease. N Engl J Med 345: 1515–1521. [DOI] [PubMed] [Google Scholar]
- Jiang C, Ting AT, Seed B ( 1998) PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature 391: 82–86. [DOI] [PubMed] [Google Scholar]
- Kamboh MI, Sanghera DK, Ferrell RE, DeKosky ST ( 1995) APOE*4-associated Alzheimer's disease risk is modified by alpha 1-antichymotrypsin polymorphism. Nat Genet 10: 486–488. [DOI] [PubMed] [Google Scholar]
- Kunsman GW, Rohrig TP ( 1993) Tissue distribution of ibuprofen in a fatal overdose. Am J Forensic Med Pathol 14: 48–50. [DOI] [PubMed] [Google Scholar]
- Landreth GE, Heneka MT ( 2001) Anti-inflammatory actions of peroxisome proliferator-activated receptor gamma agonists in Alzheimer's disease. Neurobiol Aging 22: 937–944. [DOI] [PubMed] [Google Scholar]
- Lehmann JM, Lenhard JM, Oliver BB, Ringold GM, Kliewer SA ( 1997) Peroxisome proliferator-activated receptors alpha and gamma are activated by indomethacin and other non-steroidal anti-inflammatory drugs. J Biol Chem 272: 3406–3410. [DOI] [PubMed] [Google Scholar]
- Lim GP, Yang F, Chu T, Chen P, Beech W, Teter B, Tran T, Ubeda O, Hsiao Ashe K, Frautschy SA, Cole GM ( 2000) Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer's disease. J Neurosci 20: 5709–5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim GP, Yang F, Chu T, Gahtan E, Ubeda O, Beech W, Overmier JB, Hsiao Ashe K, Frautschy SA, Cole GM ( 2001) Ibuprofen effects on Alzheimer pathology and open field activity in APPsw transgenic mice. Neurobiol Aging 22: 983–991. [DOI] [PubMed] [Google Scholar]
- McCusker SM, Curran MD, Dynan KB, McCullagh CD, Urquhart DD, Middleton D, Patterson CC, Mcllroy SP, Passmore AP ( 2001) Association between polymorphism in regulatory region of gene encoding TNFα and risk of Alzheimer disease and vascular dementia: a case-control study. Lancet 357: 436–439. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Kawamata T, Walker DG, Akiyama H, Tooyama I, McGeer EG ( 1993) Microglia in degenerative neurological disease. Glia 7: 84–92. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Schulzer M, McGeer EG ( 1996) Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer's disease: a review of 17 epidemiologic studies. Neurology 47: 425–432. [DOI] [PubMed] [Google Scholar]
- Murao K, Ohyama T, Imachi H, Ishida T, Cao WM, Namihira H, Sato M, Wong NC, Takahara J ( 2000) TNF-alpha stimulation of MCP-1 expression is mediated by the Akt/PKB signal transduction pathway in vascular endothelial cells. Biochem Biophys Res Commun 276: 791–796. [DOI] [PubMed] [Google Scholar]
- Murphy GJ, Holder JC ( 2000) PPAR-gamma agonists: therapeutic role in diabetes, inflammation and cancer. Trends Pharmacol Sci 21: 469–474. [DOI] [PubMed] [Google Scholar]
- Nicoll JA, Mrak RE, Graham DI, Steward J, Wilcock G, MacGowan S, Esiri MM, Murray LS, Dewar D, Love S, Moss T, Griffin WS ( 2000) Association of interleukin-1 gene polymorphisms with Alzheimer's disease. Ann Neurol 47: 365–368. [PMC free article] [PubMed] [Google Scholar]
- Papassotiropoulos A, Bagli M, Jessen F, Bayer TA, Maier W, Rao ML, Heun R ( 1999) Genetic variation of the inflammatory cytokine IL-6 delays the initial onset, and reduces the risk for sporadic Alzheimer's disease. Ann Neurol 45: 666–668. [DOI] [PubMed] [Google Scholar]
- Perlmutter LS, Barron E, Chui HC ( 1990) Morphologic association between microglia and senile plaque amyloid in Alzheimer's disease. Neurosci Lett 119: 32–36. [DOI] [PubMed] [Google Scholar]
- Perry RT, Collins JS, Wiener H, Acton R, Go RCP ( 2001) The role of TNF and its receptors in Alzheimer's disease. Neurobiol Aging 22: 873–883. [DOI] [PubMed] [Google Scholar]
- Rich JB, Rasmusson DX, Folstein MF, Carson KA, Kawas C, Brandt J ( 1995) Nonsteroidal anti-inflammatory drugs in Alzheimer's disease. Neurology 45: 51–55. [DOI] [PubMed] [Google Scholar]
- Ricote M, Huang JT, Welch JS, Glass CK ( 1999) The peroxisome proliferator-activated receptor (PPARgamma) as a regulator of monocyte/macrophage function. J Leukoc Biol 66: 733–739. [DOI] [PubMed] [Google Scholar]
- Rogers J, Kirby LC, Hempelman SR, Berry DL, McGeer PL, Kaszniak AW, Zalinski J, Cofield M, Mansukhani L, Willson P, Kogan F ( 1993) Clinical trial of indomethacin in Alzheimer's disease. Neurology 43: 1609–1611. [DOI] [PubMed] [Google Scholar]
- Rogers JT, Leiter LM, McPhee J, Cahill CM, Zhan SS, Potter H, Nilsson LN ( 1999) Translation of the alzheimer amyloid precursor protein mRNA is upregulated by interleukin-1 through 5′-untranslated region sequences. J Biol Chem 274: 6421–6431. [DOI] [PubMed] [Google Scholar]
- Sastre M, Turner RS, Levy E ( 1998) X11 interaction with β-amyloid precursor protein modulates its cellular stabilization and reduces amyloid β-protein secretion. J Biol Chem 273: 22351–22357. [DOI] [PubMed] [Google Scholar]
- Sastre M, Steiner H, Fuchs K, Capell A, Multhaup G, Condron MM, Teplow DB, Haass C ( 2001) Presenilin-dependent γ-secretase processing of β-amyloid precursor protein at a site corresponding to the S3 cleavage of Notch. EMBO Rep 2: 835–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skovronsky DM, Lee VM, Pratico D ( 2001) Amyloid precursor protein and amyloid beta peptide in human platelets. Role of cyclooxygenase and protein kinase C. J Biol Chem 276: 17036–17043. [DOI] [PubMed] [Google Scholar]
- Stewart WF, Kawas C, Corrada M, Metter EJ ( 1997) Risk of Alzheimer's disease and duration of NSAID use. Neurology 48: 626–632. [DOI] [PubMed] [Google Scholar]
- Sun A, Koelsch G, Tang J, Bing G ( 2002) Localization of beta-secretase Memapsin 2 in the brain of Alzheimer's patients and normal aged controls. Exp Neurol 175: 10–22. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Hayashi I, Tominari Y, Rikimaru K, Morohashi Y, Kan T, Natsugari H, Fukuyama T, Tomita T, Iwatsubo T ( 2003) Sulindac sulfide is a non-competitive γ-secretase inhibitor that preferentially reduces Aβ42 generation. J Biol Chem 278: 18664–18670. [DOI] [PubMed] [Google Scholar]
- Tamagno E, Bardini P, Obbili A, Vitali A, Borghi R, Zaccheo D, Pronzato MA, Danni O, Smith MA, Perry G, Tabaton M ( 2003) Oxidative stress increases expression andactivity of BACE in NT2 neurons. Neurol Dis 10: 279–288. [DOI] [PubMed] [Google Scholar]
- Tan J, Town T, Paris D, Mori T, Suo Z, Crawford F, Mattson MP, Flavell RA, Mullan M ( 1999) Microglial activation resulting from CD40–CD40L interaction after beta-amyloid stimulation. Science 286: 2352–2355. [DOI] [PubMed] [Google Scholar]
- Walter J, Kaether C, Steiner H, Haass C ( 2001) The cell biology of Alzheimer's disease: uncovering the secret of secretases. Curr Opin Neurobiol 11: 585–590. [DOI] [PubMed] [Google Scholar]
- Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, Findlay KA, Smith TE, Murphy MP, Bulter T, Kang DE, Marquez-Sterling N, Golde TE, Koo EH ( 2001) A subset of NSAIDs lower amyloidogenic Aβ42 independently of cyclooxygenase activity. Nature 414: 212–216. [DOI] [PubMed] [Google Scholar]
- Weggen S, Eriksen JL, Sagi SA, Pietrzik CU, Golde TE, Koo EH ( 2003) Aβ42-lowering nonsteroidal anti-inflammatory drugs preserve intramembrane cleavage of the amyloid precursor protein (APP) and ErbB-4 receptor and signaling through the APP intracellular domain. J Biol Chem 278: 30748–30754. [DOI] [PubMed] [Google Scholar]
- Willson TM ( 2003) Chemical genomics of the orphan nuclear receptors. Ernst Schering Res Found Workshop 42: 29–40. [DOI] [PubMed] [Google Scholar]
- Willson TM, Brown PJ, Sternbach DD, Henke BR ( 2000) The PPARs: from orphan receptors to drug discovery. J Med Chem 43: 527–550. [DOI] [PubMed] [Google Scholar]
- Yang LB, Lindholm K, Yan R, Citron M, Xia W, Yang XL, Beach T, Sue L, Wong P, Price D, Li R, Shen Y ( 2003) Elevated β-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat Med 9: 3–4. [DOI] [PubMed] [Google Scholar]