

Abstract
Presynaptic synthesis of acetylcholine (ACh) requires a steady supply of choline, acquired by a plasma membrane, hemicholinium-3-sensitive (HC-3) choline transporter (CHT). A significant fraction of synaptic choline is recovered from ACh hydrolyzed by acetylcholinesterase (AChE) after vesicular release. Although antecedent neuronal activity is known to dictate presynaptic CHT activity, the mechanisms supporting this regulation are unknown. We observe an exclusive localization of CHT to cholinergic neurons and demonstrate that the majority of CHTs reside on small vesicles within cholinergic presynaptic terminals in the rat and mouse brain. Furthermore, immunoisolation of presynaptic vesicles with multiple antibodies reveals that CHT-positive vesicles carry the vesicular acetylcholine transporter (VAChT) and synaptic vesicle markers such as synaptophysin and Rab3A and also contain acetylcholine. Depolarization of synaptosomes evokes a Ca2+-dependent botulinum neurotoxin C-sensitive increase in the Vmax for HC-3-sensitive choline uptake that is accompanied by an increase in the density of CHTs in the synaptic plasma membrane. Our study leads to the novel hypothesis that CHTs reside on a subpopulation of synaptic vesicles in cholinergic terminals that can transit to the plasma membrane in response to neuronal activity to couple levels of choline re-uptake to the rate of ACh release.
Keywords: choline, transport, hemicholinium-3, synaptic vesicle, acetylcholine, VAChT
Introduction
Acetylcholine (ACh) functions centrally in vertebrates as a modulator of cognitive processes, signals within both the sympathetic and parasympathetic branches of the autonomic nervous system, and is the principal neurotransmitter at the neuromuscular junction (Dani and De Biasi, 2001; Bymaster et al., 2003). ACh is synthesized from choline and acetyl-coenzyme A by choline acetyltransferase (ChAT; EC 2.3.1.7) (Wu and Hersh, 1994) and is packaged into synaptic vesicles by the vesicular acetylcholine transporter (VAChT) (Eiden, 1998). So that ACh synthesis can be sustained, cholinergic neurons efficiently recover choline from the synapse after ACh hydrolysis by acetylcholinesterase (AChE; EC 3.1.1.7) (Birks and MacIntosh, 1961). To this end, cholinergic neurons possess a high-affinity (Km = 1–2 μm) choline transport activity that is distinguished from a more widely expressed low-affinity choline transport activity by its sensitivity to hemicholinium-3 (HC-3; Ki = 5 nm) and dependence on extracellular Na+ and Cl– (Yamamura and Snyder, 1972; Guyenet et al., 1973; Haga and Noda, 1973; Simon and Kuhar, 1976). Blockade of choline transporter (CHT) with HC-3 abolishes ACh production and release, suggesting a presynaptic localization of the transporter. Whereas pharmacological studies reveal that in vivo CHT blockade is lethal (Quastel and Curtis, 1965), physiological studies indicate that HC-3-sensitive choline uptake represents a regulated step in sustaining ACh synthesis and release across a wide range of cholinergic firing rates (Guyenet et al., 1973; Maire and Wurtman, 1985). Indeed, choline availability, rather than intrinsic ChAT activity, is thought to be the rate-limiting determinant of ACh synthesis. Cholinergic neurons increase their rate of ACh synthesis to meet the demands of increased release mainly via an increase in HC-3-sensitive choline transport (Birks and MacIntosh, 1961; Simon and Kuhar, 1975; Collier et al., 1983; Lowenstein and Coyle, 1986). The cellular mechanisms supporting this link between neuronal activity and choline transport have not been defined.
In this study we demonstrate that CHT is expressed exclusively by central and peripheral cholinergic neurons, with a marked enrichment at presynaptic nerve terminals. Remarkably, we find CHT to be localized mainly on presynaptic vesicles that contain VAChT, synaptophysin, and Rab3A and store ACh. The predominant localization of CHT to presynaptic vesicles predicts an important role for vesicle trafficking and fusion in the regulation of presynaptic choline uptake. This hypothesis is supported by our findings that depolarization triggers an increase in synaptosomal CHT activity that is Ca2+-dependent, accompanied by an increase in CHT plasma membrane density, and is blocked by botulinum neurotoxin pretreatment. We propose that a subset of presynaptic vesicles in cholinergic terminals delivers CHTs to the plasma membrane, thereby coupling the retrieval of extracellular choline to the secretion of ACh. Our findings provide a mechanistic basis for the activity-dependent modulation of choline transport capacity (Kuhar and Murrin, 1978; Lowenstein and Coyle, 1986) and reveal a novel role for presynaptic vesicles in delivering proteins responsible for neurotransmitter homeostasis.
Materials and Methods
Generation of CHT antibodies. Rabbit polyclonal antisera (Ab20 and Ab21) were raised against the C-terminal 15 amino acids (VDSSPEGSGTEDNLQ, residues 566–580) fused to keyhole limpet hemocyanin (Research Genetics, Huntsville, AL). This sequence is common to the CHT proteins from human, mouse, and rat (Apparsundaram et al., 2000, 2001; Okuda et al., 2000). Antisera were affinity purified over a peptide-coupled Affigel column (Bio-Rad, Hercules, CA). Mouse monoclonal antibodies (hybridoma 62-2E8) were raised against a recombinant fusion protein consisting of the C-terminal 80 amino acids of human CHT (hCHT) fused to glutathione S-transferase (GST) (Smith and Johnson, 1988). Rat anti-m2 receptor monoclonal antibody and rabbit polyclonal anti-VAChT serum have been described previously (Levey et al., 1995; Gilmor et al., 1996).
Other antibodies and reagents. All chemicals were purchased from Sigma (St. Louis, MO) unless otherwise noted. Mouse anti-synaptophysin, goat anti-ChAT, goat anti-VAChT rabbit anti-VMAT2, and rabbit anti-NMDAR 2A/B were obtained from Chemicon (Temecula, CA). Mouse monoclonal anti-glutamic acid decarboxylase (GAD65) was purchased from the Developmental Studies Hybridoma Bank (University of Iowa, Iowa City, IA) (Chang and Gottlieb, 1988). Mouse anti-Na/K-ATPase α3 subunit monoclonal antibody was obtained from Affinity BioReagents (Golden, CO). Mouse anti-dopamine transporter (DAT) antibody was a gift from R. Vaughn (University of North Dakota School of Medicine and Health Sciences, Grand Forks, ND). Rabbit anti-GABA transporter 1 antiserum was a gift from N. Brecha (University of California at Los Angeles) and also was purchased from Chemicon. Mouse anti-synaptophysin (clone 7.2), anti-VAMP2 (Cl 69.1), anti-synaptotagmin I (Cl 41.1), and rabbit anti-vesicular GABA transporter (VGAT) were purchased from Synaptic Systems (Göttingen, Germany). Rabbit anti-Rab3A antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). CY2- and CY3-conjugated secondary antibodies were obtained from Jackson ImmunoResearch Laboratories (West Grove, PA); Alexa 488-conjugated goat anti-mouse antibody, Alexa 488-conjugated goat anti-rabbit antibody, and Alexa 488-conjugated α-bungarotoxin were purchased from Molecular Probes (Eugene, OR).
Immunofluorescence experiments. All experiments involving animals were approved by our respective institutional animal care and use committees. Immunofluorescent labeling of floating frozen brain sections was based on a previously described protocol (Schroeter et al., 2000). Briefly, adult C57Bl/6 mice (Harlan, Indianapolis, IN) were anesthetized deeply with Nembutal (80 mg/kg), heparinized (1000 U/kg), and perfused transcardially with 50 ml of saline containing heparin, followed by 4% paraformaldehyde or 4% paraformaldehyde plus 0.5% picric acid. After perfusion for 20 min the tissues were removed and placed in fixative overnight at 4°C. After a rinsing with Tris-buffered saline (TBS) the tissue (20 μm sections) was blocked and permeabilized with 2% normal donkey serum (NDS; Jackson ImmunoResearch) and 0.2% Triton X-100. Both primary and secondary antibody incubations also were performed in this buffer. After a washing with TBS the sections were mounted on slides and coverslipped with Aqua Polymount (Polysciences, Warrington, PA). A previously described protocol was adapted for studies of CHT localization at the neuromuscular junction (Keller-Peck et al., 2001). After perfusion and postfixation whole diaphragms were incubated overnight in 0.1 m glycine at 4°C. The tissues were blocked and permeabilized in TBS, 4% NDS, and 0.5% Triton X-100. Primary and secondary antibody incubations were performed in TBS, 2% NDS, and 0.5% Triton X-100. Nicotinic ACh receptor labeling was achieved with Alexa 488-conjugated α-bungarotoxin (0.05 μg/ml) in TBS plus 1% NDS (30 min at room temperature).
Immunohistochemical experiments. Floating brain sections (20 μm) were used for immunohistochemical staining with affinity-purified rabbit polyclonal CHT antibody (1:1000). Hydrogen peroxide (3%) was used to quench endogenous peroxidase activity, and nonspecific antibody binding was blocked by incubation in 4% normal goat serum (NGS) and 0.2% Triton X-100 for 1 hr, followed by incubation of the sections in solutions of primary antibodies containing 2% NGS and 0.2% Triton X-100 at room temperature overnight. Primary antibody was detected with the ABC method by using diaminobenzidine as a substrate (Elite ABC kit, Vector Laboratories, Burlingame, CA).
Image analysis. Bright-field images were captured on an Olympus BX50WI microscope with Kodak Elite 160 film (Kodak, Rochester, NY). Confocal analysis was performed with a Zeiss LSM 410 confocal imaging system equipped with internal He/Ne and external Ar/Kr lasers with output at 488 and 568 nm [Vanderbilt University Medical Center (VUMC) Cell Imaging Core Resource]. Z-series were collected by optical sectioning at an interval of 1 μm. Image processing and montage assembly were performed with Adobe Photoshop software (Mountain View, CA).
Electron microscopy analysis of CHT distribution in the rat brain. Adult rats were perfused with 3% paraformaldehyde/0.15% glutaraldehyde for 10 min, and brains were postfixed by immersion in 3% paraformaldehyde overnight. Vibratome sections were cut at 50 μm and treated with 0.1% sodium borohydride to inactivate unreacted aldehyde. After permeabilization with 0.05% Triton X-100 for 10 min the sections were blocked with 5% BSA, 0.1% coldwater fish skin gelatin, and 5% NGS in PBS for 1 hr. The sections were incubated in primary antibody for 24 hr and in ultra-small gold-conjugated secondary antibody for 16 hr. The buffer for primary and secondary antibody incubations and rinses contained acetylated BSA (BSA-c; Aurion, Wageningen, The Netherlands). After postfixation with 2.5% glutaraldehyde the immunogold particles were silver-enhanced with R-Gent SE-EM reagents (Aurion). Tissue sections were fixed with 0.5% osmium tetroxide for 15 min, dehydrated, and embedded for electron microscopy.
Cell culture. PC12 cells were grown in DMEM plus 10% horse serum, 5% fetal bovine serum, 2 mm l-glutamine, penicillin, and streptomycin at 37°C in a humidified incubator containing 5% CO2 (Greene and Tischler, 1976). For the retroviral transduction of PC12 cells the hCHT was subcloned into the vector pLZRS-MS-Neo (provided by Al Reynolds, VUMC Department of Cancer Biology), and this construct or the empty pLZRS-MS-Neo vector was transfected into the amphotropic Phoenix packaging cell line (American Type Culture Collection, Manassas, VA), using Fugene 6 transfection reagent (Roche Molecular Biochemicals, Indianapolis, IN) (Kinsella and Nolan, 1996). PC12 cells were transduced with the supernatant from the packaging cells plus 5 μg/ml hexadimethrine bromide (Sigma), and polyclonal populations of hCHT-expressing cells were selected in PC12 media supplemented with 0.5 mg/ml G418 (Cellgro, Herndon, VA). For immunoblot analyses a postnuclear supernatant (PNS) was prepared from PC12 cells by rinsing with PBS and scraping the cells in PBS plus 1 mm EDTA and a protease inhibitor mixture [PIC; 4-(2-aminoethyl)benzenesulfonyl fluoride (AEBSF), pepstatin A, E-64, bestatin, leupeptin, and aprotinin]. The cells were lysed by Dounce homogenization, intact cells and nuclei were removed by centrifugation at 1000 × g for 10 min, and the supernatant was saved for immunoblotting.
Immunoblot analyses. Samples were denatured for 15 min at 37°C in Laemmli sample buffer [1% SDS, 31.25 mm Tris, pH 6.8, 5% glycerol, 200 mm 2-mercaptoethanol (Laemmli, 1970)], resolved by standard SDS-PAGE, and transferred electrophoretically to polyvinylidene difluoride (PVDF) membranes (Amersham Biosciences, Arlington Heights, IL). The membranes were blocked with PBS containing 0.5% Tween-20 (PBS-T) and 5% fat-free skim milk. Antibody incubations also were performed in this buffer. Primary antibodies were detected with the appropriate horseradish peroxidase-conjugated secondary antibody. After extensive washing in PBS-T the signals were detected with Western Lightning Chemiluminescence Reagent Plus (PerkinElmer Life Sciences, Boston, MA) or Amersham ECL Plus and Hyperfilm ECL (Amersham Biosciences). To analyze single PVDF membranes for multiple proteins, we stripped blots with 2% SDS, 62.5 mm Tris-HCl, pH 6.8, and 100 mm 2-mercaptoethanol at 55°C for 20 min. After a washing with PBS-T the blots were blocked in 5% milk PBS-T before analysis with the next antibody. Where noted, before SDS-PAGE the PC12 postnuclear supernatant samples were digested with peptide N-glycosidase F (PNGase F) per the supplier's directions (New England Biolabs, Beverly, MA).
Subcellular fractionation. Our subcellular fractionation strategy was adapted from previously described methods for the study of presynaptic vesicular proteins (Huttner et al., 1983). Briefly, mouse brain tissue was homogenized in 0.32 m sucrose and 5 mm HEPES-NaOH, pH 7.3, in a Wheaton Instruments (Millville, NJ) Potter Elvejhem homogenizer (10 strokes at 500 rpm) and centrifuged at 1000 × g for 10 min. The supernatant was centrifuged at 13,000 × g to yield a crude synaptosomal pellet (P2). Synaptosomes in this P2 fraction were lysed by homogenization (5 strokes, 500 rpm) in either ice-cold 5 mm Tris-acetate, pH 8, or 5 mm HEPES-NaOH, pH 7.4, plus PIC. Synaptic plasma membranes (LP1) and other large membranes were collected at 15,000 × g for 20 min. The LP2 pellet was obtained after centrifugation of the resulting supernatant (LS1 fraction) for 30 min at 200,000 × g in a S100AT4 rotor (Kendro-Sorvall, Newtown, CT). Proteins were extracted from each fraction with 1% SDS plus (in mm) 5 HEPES-KOH, pH 7.3, 1 EDTA, 1 EGTA, and protease inhibitor mixture. Protein concentrations were determined by the bicinchoninic acid method (Pierce, Rockford, IL), and equal quantities of protein from each fraction were subjected to immunoblot analysis. Further fractionation of the vesicles in the LS1 and LP2 fractions was performed by sedimentation on 10–35% glycerol and 50–800 mm sucrose gradients, based on previously described protocols (Lee et al., 2001). Briefly, the LP2 fraction was resuspended in gradient buffer (GB) [containing (in mm): 5 HEPES-KOH, pH 7.3, 1 EDTA, 1 EGTA, 1 dithiothreitol, and protease inhibitor mixture (Sigma)] with 25 strokes of a tight pestle in a Dounce homogenizer (Wheaton Instruments). Then 500 μl (∼1 mg of protein/ml) of this vesicle-enriched suspension was layered onto either an 11 ml 10–35% glycerol gradient and centrifuged for 1 hr at 173,000 × g in an SW40Ti rotor (Beckman Instruments) or onto a 9 ml 50–800 mm sucrose gradient and centrifuged at 65,000 × g for 3 hr. After the centrifugation 12 equal fractions were collected from the top of each gradient. Before immunoblot analysis, protein was precipitated from each fraction with 6% trichloroacetic acid (TCA) plus 0.02% deoxycholate. For the analysis of striatal LS1 fraction vesicles, the LS1 fraction was mixed with 1/10 vol of 10× PBS and 10 mm EDTA with a Potter Elvejhem homogenizer (10 strokes at 1000 rpm), followed by repeated extrusion through 23 gauge and then 27 gauge needles. These steps were essential to observe the nearly complete separation of synaptic vesicle markers from larger membranes that sedimented toward the bottom of the gradient.
To separate the CHT-containing membranes further, we used a self-generating iodixanol gradient protocol previously described for the characterization of GLUT4 glucose transporter-containing vesicles (Hashiramoto and James, 2000). The synaptic vesicle-containing fractions (fractions 5–8) from the sucrose velocity gradient were pooled and adjusted to 250 mm sucrose, 14% iodixanol (Optiprep gradient media, Greiner Bio-One, Longwood, FL), and protease inhibitor mixture in GB. Samples were centrifuged for 4 hr at 265,000 × g in a Sorvall TV-865 rotor, and 12 equal fractions were collected from the top of the gradient. Then 300 μl aliquots of these fractions were TCA precipitated and subjected to Western analysis as described above.
Vesicle immunoisolation. For immunoisolation of CHT-containing vesicles rabbit antisera against CHT (Ab21), VAChT or Rab3A was coupled to protein A-Sepharose beads (Amersham Biosciences) or to either sheep anti-rabbit IgG-coated or protein A-coupled paramagnetic Dynabeads (Dynal, Lake Success, NY) in PBS and 1 mm EDTA, pH 7.4, for at least 4 hr at 4°C. The mouse monoclonal anti-CHT and anti-synaptophysin were bound to protein G-Sepharose beads (5 μg of IgG per 2.5 mg of dry beads; Amersham Biosciences). After being rinsed three times, the coated beads were added to mouse striatal LS1 fractions (125 μg of protein) and allowed to rotate overnight at 4°C in PBS, 1 mm EDTA, 1 mm EGTA, and protease inhibitor mixture. Sepharose beads and bound vesicles were pelleted by centrifugation for 1 min at 10,000 × g. Magnetic beads and bound vesicles were separated magnetically (Magnesphere, Promega, Madison, WI) from the supernatant, and the supernatant was collected. Membranes in the supernatant were pelleted by centrifugation for 30 min at 200,000 × g and resuspended in PBS, followed by the addition of SDS sample buffer. The beads were washed five times with PBS and 1 mm EDTA; immunoisolated proteins were eluted during a 15 min incubation at 37°C in SDS sample buffer before SDS-PAGE and immunoblot analysis. To quantify the extent of CHT and VAChT colocalization, we scanned the films and measured the band densities with NIH Image 1.6 software.
For electron microscopy (EM) analysis of immunoisolated vesicles, bead-bound vesicles were fixed with 2% glutaraldehyde, washed twice with 0.1 m cacodylate buffer, postfixed with 1.0% osmium tetroxide, dehydrated via ascending ethanol series, infiltrated with propylene oxide, and embedded in Spurr's resin. Ultrathin sections were cut at 60 nm, stained with lead citrate and uranyl acetate, and examined on a Hitachi H-7500 transmission electron microscope (Tokyo, Japan).
Immunoisolation of radiolabeled synaptic vesicles. Mouse striatal synaptosomes (P2 fraction) were prepared, resuspended in Krebs–Ringer HEPES (KRH) buffer [containing (in mm): 130 NaCl, 3 KCl, 2.2 CaCl2, 1.2 MgSO4, 1.2 KH2PO4, 10 glucose, 10 HEPES, pH 7.4], and were preincubated with or without 40 μm ± vesamicol (Alexis, San Diego, CA) for 10 min at 37°C before the addition of [3H]-choline (83 Ci/mmol; Amersham Biosciences) to a final concentration of 100 nm. After 30 min the synaptosomes were collected by centrifugation for 20 min at 20,000 × g and lysed in 5 mm HEPES-NaOH, pH 7.4. Heavy membranes were cleared by spinning the samples for 15 min at 20,000 × g; labeled synaptic vesicles remaining in the supernatant (LS1) were saved for the immunoisolation assay. Each [3H]-ACh-labeled vesicle sample, corresponding to 1/12 of the yield from one mouse striatum P2 fraction, was subjected to immunoisolation for 90 min with sheep anti-rabbit IgG M280 Dynabeads (Dynal) coated with either CHT-specific serum or preimmune serum in a buffer of PBS, 1 mm EDTA, 1% BSA. After being washed six times with PBS-EDTA, the immunoisolation of vesicular [3H]-ACh was quantified by liquid scintillation spectrometry. Total vesicle labeling was determined by filtration over 0.3% polyethylenimine-coated glass fiber filters (Whatman, Maidstone, UK) on a Brandel cell harvester (Gaithersburg, MD) and was quantified by liquid scintillation spectrometry. Specific synaptic vesicle [3H]-ACh radioactivity was defined as the difference between assays performed on synaptosomes incubated with and without vesamicol. Synaptosomes loaded in parallel with 100 nm [3H]-dopamine (31.8 Ci/mmol; PerkinElmer Life Sciences) ± 5 μm reserpine (Sigma) served as a negative control for specific immunoisolation of cholinergic vesicles (Eiden, 2000).
Synaptosomal choline transport assays. For the kinetic analysis of HC-3-sensitive choline uptake after depolarization-evoked synaptic vesicle turnover, rat hippocampal synaptosomes were resuspended in KRH at a concentration of 3 mg of protein/ml and incubated for 15 min at 37°C, after which time 1 vol of KRH or 1 vol of high K + KRH (40 mm KCl, 96 mm NaCl; equimolar reductions in Na + were required to maintain osmolarity in the high K + incubation medium) was added, and the synaptosomes were incubated for a further 15 min at 37°C. The incubation was terminated by centrifugation for 2 min at 4°C. For the measurement of choline uptake the synaptosomes were resuspended to 2 mg of protein/ml in uptake assay buffer [containing (in mm): 130 NaCl, 3 KCl, 1 EGTA, 1.2 MgSO4, 1.2 KH2PO4, 10 glucose, 10 HEPES, pH 7.4]. The synaptosome suspension was added to [3H]-choline containing uptake assay buffer to give a final protein concentration of 1 mg/ml in a volume of 100 μl. Stock solutions of [3H]-choline (83 Ci/mmol) were mixed with cold choline to yield a specific activity of 2.5 Ci/mmol, and [3H]-choline concentrations from 0.25 to 6 μm were used to examine the saturation kinetics for choline uptake. After 5 min at 37°C the uptake assay was terminated, and the samples were collected onto 0.3% polyethylenimine-coated glass fiber filters and washed with a Brandel cell harvester. All uptake assays were performed in triplicate, and CHT-specific uptake was determined by subtracting uptake that was measured in the presence of 1 μm HC-3. Km and Vmax values were calculated by nonlinear regression analysis with GraphPad Prism software (San Diego, CA).
To analyze the Ca 2+ dependence of CHT regulation, we tested the ability of Cd 2+ to block voltage-gated Ca 2+ channel-mediated Ca 2+ influx during the high K + stimulation of choline uptake (Lonart et al., 1998). In these experiments 100 μm CdCl2 was added along with the high K + buffer or the control KRH buffer during the second 15 min incubation.
Botulinum neurotoxin C treatment. Rat hippocampal synaptosomes were prepared as described above and resuspended in KRH buffer (3 mg/ml). Botulinum neurotoxin C (BoNT/C; Calbiochem, La Jolla, CA) was added to a final concentration of 66 nm, and the synaptosomes were incubated for 45 min at 37°C. Control synaptosomes were processed in parallel and were exposed to a mock incubation in BoNT/C-free KRH. After this treatment the samples were centrifuged at 13,000 × g to pellet the synaptosomes, and the toxin remaining in the supernatant was aspirated. The synaptosomes were washed once with 1 ml of KRH buffer, resuspended in KRH buffer to a final incubation of 2.0 mg/ml, and processed as described above for KCl treatment and uptake assays
CHT trafficking in synaptosomes. Synaptosomes were prepared and treated as for the uptake assays. However, after stimulation and centrifugation they were resuspended in (in mm) 5 HEPES-NaOH, 1 EDTA, 1 EGTA, pH 7.4, and homogenized with a Dounce homogenizer. Heavy membranes were pelleted at 15,000 × g, and the supernatant was discarded. After a second resuspension, homogenization, and centrifugation the synaptosomal plasma membrane-enriched pellets were processed for immunoblot analysis.
Results
Development and validation of CHT antibodies
To isolate and localize CHT proteins, we generated polyclonal and monoclonal antibodies against CHT peptides and fusion proteins (Fig. 1A). Rabbit polyclonal antisera were raised against the C-terminal 15 amino acids of the transporter (VDSSPEGSGTEDNLQ), a sequence that is conserved completely among human, mouse, and rat CHT proteins. We also raised a mouse monoclonal antibody (62-2E8) against an hCHT C-terminal GST fusion protein for which the amino acid sequence is 94 and 92% conserved in mouse and rat CHT, respectively. Both the polyclonal and monoclonal antibodies specifically recognize a 65 kDa band in hCHT-transduced PC12 cells for which the size shifts to 45 kDa after PNGase F-mediated deglycosylation (Fig. 1B,C).
Figure 1.
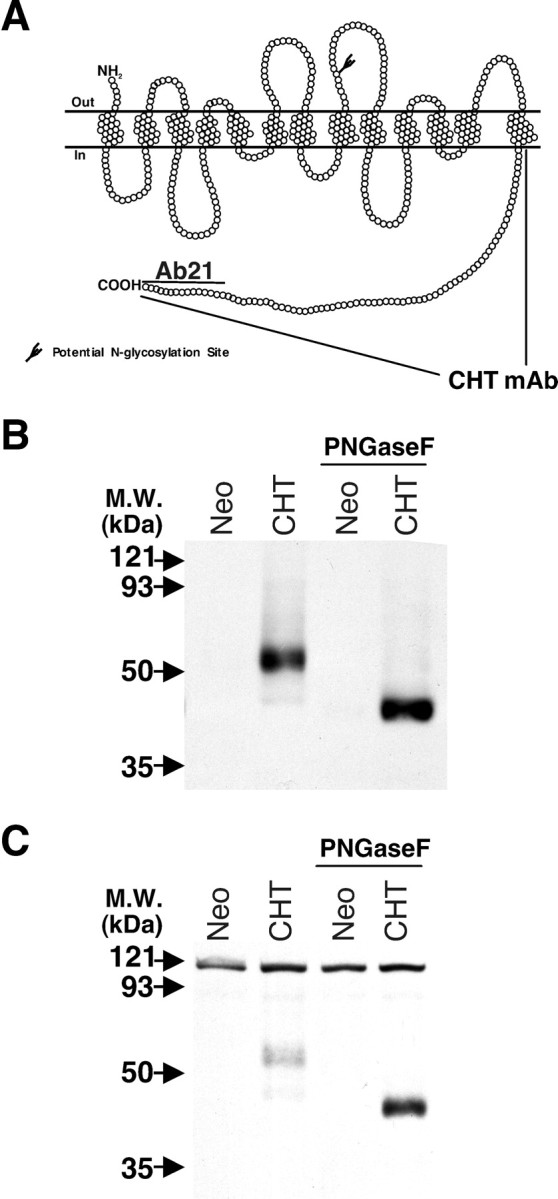
Generation of CHT-specific antibodies. A, The 580 amino acids of mammalian CHTs are predicted to yield proteins with 13 transmembrane-spanning domains, an extracellular N terminus, and an intracellular C terminus. Rabbit polyclonal antibodies (Ab21) and a mouse monoclonal antibody were raised against a C-terminal 15 amino acid peptide and the C-terminal 80 amino acids of hCHT fused to GST, respectively. B, The affinity-purified anti-CHT antibody (Ab21; 1:1000) recognizes a 65 kDa glycoprotein in the postnuclear supernatant (PNS) fraction (10 μg of protein per lane) from hCHT-transduced PC12 cells, but not in negative controls that were transduced with an empty vector containing the neomycin (Neo) resistance gene. Deglycosylation of PNS extracts results in a shift in the migration of the CHT band to ∼45 kDa. C, The CHT-specific monoclonal antibody recognizes the same 65 kDa glycoprotein as well as the 45 kDa form after deglycosylation with PNGase F.
CHT expression is limited to cholinergic neurons, where it is localized to presynaptic nerve terminals
To support cholinergic neurotransmission, we held an expectation that CHT should be found in cholinergic neurons with enrichment at presynaptic terminals. The affinity-purified CHT polyclonal antibody detected CHT immunoreactivity in all major cholinergic nuclei and their projections (Fig. 2). Moreover, we observed extensive overlap between CHT and the cholinergic neuron-specific marker ChAT (Levey et al., 1983) throughout our preparations, as evident in the cell bodies of large aspiny striatal interneurons (Fig. 3A–C). In contrast, no colocalization was evident between CHT and the presynaptic GABA-synthesizing enzyme GAD65 (Fig. 3D–F). In addition to ChAT, the vesamicol-sensitive VAChT protein is an established marker of cholinergic neurons (Gilmor et al., 1996). A confocal reconstruction of CHT and VAChT dual-labeled neurons in the ventral spinal cord demonstrates the colocalization of these two transporters in both the motor neurons as well as in the large cholinergic C-terminals that contact them (Fig. 3G–I). In addition to labeling motor neurons in the spinal cord, we also detected CHT in sympathetic preganglionic neurons in the intermediolateral nucleus (data not shown). The reliable and exclusive localization of CHT to ChAT- and VAChT-positive cells confirms that CHT is a specific marker of cholinergic neurons. A presynaptic enrichment of CHT was further evident in our labeling of the neuromuscular junction of the mouse diaphragm with antibodies to CHT and fluorescently labeled α-bungarotoxin. Intense presynaptic CHT labeling was evident on muscle fibers in a pattern overlying α-bungarotoxin labeling of the muscle (Fig. 3J–L). A small degree of CHT immunoreactivity is evident along the preterminal segment of motor axons, although the intensity of staining in axons is much reduced as compared with the signal detected in the presynaptic arborization. The CHT monoclonal antibody (62-2E8) used in our studies also has been demonstrated recently to label cholinergic neurons selectively in the primate CNS (Kus et al., 2003). As a final note pertaining to the specificity of our polyclonal and monoclonal anti-CHT antibodies, in our recently generated CHT knock-out mice these antibodies no longer immunohistochemically label cholinergic neurons, and the 65 kDa CHT-specific band observed in immunoblot analysis is absent (S. Ferguson, V. Savchenko, J. Wright, and R. D. Blakely, unpublished observations).
Figure 2.

Characterization of CHT-immunoreactive neurons in the mouse CNS. A, The striatum exhibits intense CHT immunoreactivity in coronal sections of the forebrain. B, Immunoabsorption of the CHT antibody Ab21 with the immunizing peptide abolishes the detection of CHT immunoreactivity in the striatum on a serial section to that presented in A. C, A confocal image from the striatum shows CHT expression in large aspiny interneurons. D, An example of CHT-immunoreactive neurons in the vertical limb of the diagonal band. E, This confocal reconstruction illustrates the labeling for CHT in varicosities (arrowheads) along nerve fibers in the cerebral cortex. F, Fiber labeling for CHT in the dentate gyrus is most intense in the molecular layer. G, Confocal reconstruction of CHT-IR demonstrates the extremely dense innervation of the amygdala by CHT-positive fibers. H, In the thalamus small CHT-positive cell bodies were found in the medial habenula nucleus. I, Detection of CHT immunoreactivity in neuronal cell bodies in the pendunculopontine tegmental nucleus. J, CHT immunoreactivity is intense in the facial nucleus. K, L, Higher-power magnification indicates that the CHT is present in motor neuron cell bodies as well in the large C boutons (arrows) that contact these motor neurons in the facial nucleus and the motor trigeminal nucleus, respectively. Scale bars: A, B, 400μm; C–G, I, 50 μm; H, 20μm; J, 250 μm; K, L, 30 μm.
Figure 3.

CHT colocalizes with markers of cholinergic neurons. A, The somata of large striatal interneurons are CHT-positive in this confocal section. B, The same cell bodies exhibit ChAT immunoreactivity. C, Merging the images for CHT and ChAT in this section emphasizes the dual labeling of these cholinergic neurons. D, CHT immunoreactivity is detected in the somata of striatal cholinergic interneurons as well as in fibers and terminals distributed throughout the neuropil in this confocal reconstruction. E, Double labeling of this section with a GAD65-specific antibody also shows labeling of GABAergic terminals (arrowheads). F, When the images from D and E are merged, it is clear that GAD and CHT do not colocalize in the same terminals. G, Confocal reconstruction of CHT labeling in the somata of motor neurons in the ventral horn of the spinal cord as well as in the large presynaptic C-terminals that innervate these cell bodies (arrowheads). H, Double labeling also shows VAChT to be localized to both motor neuron somata as well as the C boutons in this section. I, Overlaying the images from G and H demonstrates an extensive overlap between the immunoreactivity for CHT and VAChT, especially in the presynaptic C-terminals. J–L, Sections of mouse diaphragm were stained for CHT and Alexa 488-conjugated α-bungarotoxin (BgTX) to establish the presynaptic localization of CHT in motor neurons. The concentrated localization of CHT in presynaptic arborizations overlaps with the postsynaptic binding of BgTX to nicotinic receptors in the motor endplate (L). CHT labeling in this figure was performed with the polyclonal CHT antibody. Scale bars: A–C, G–L, 20 μm; D–F, 5 μm.
CHT predominantly resides on presynaptic vesicles
Our immunofluorescence experiments indicate a significant localization of CHT to cholinergic presynaptic terminals. However, the dense labeling and small size of mammalian cholinergic terminals preclude an investigation of CHT subcellular localization with light microscopy. To visualize CHT subcellular distribution, we used gold particle-conjugated secondary antibodies and silver enhancement to localize CHT immunoreactivity by EM. By this technique we detected CHT immunoreactivity in cholinergic nerve terminals, consistent with the light microscopic studies. Our labeling of CHT-positive terminals was consistent with the expected abundance and morphology of cholinergic terminals (Gilmor et al., 1996). For example, in accordance with the localization of CHT at the light level (Fig. 2J,K), the large and distinct cholinergic C-terminals contacting motor neurons in the facial nucleus exhibited significant CHT labeling (Fig. 4A–C; polyclonal CHT antibody). Consistent with the known function for CHT, a fraction of immunogold particles was found in association with the plasma membrane (arrows in Fig. 4A–C). To our surprise, the majority of CHT immunoreactivity is located intracellularly and is associated predominantly with small, clear vesicles. A similar pattern of CHT localization to cytoplasmic vesicles was observed in cholinergic terminals in the striatum (Fig. 4D,E; CHT monoclonal antibody). In an analysis of 69 CHT-positive striatal terminals we observed an average terminal area of 0.23 μm2 and, on average, 10.8 silver-enhanced gold particles per terminal (total, 745). With respect to subcellular localization, the majority of these particles (71%) was intracellular. Although we acknowledge that occlusion of the epitopes recognized by our CHT antibodies could result in an underestimation of the abundance of CHT at the plasma membrane, further biochemical fractionation studies (see below) support the conclusions from our labeling studies. Control experiments using preimmune serum or peptide preabsorption afforded little or no labeling. An essentially identical pattern was evident with either polyclonal or monoclonal CHT antibodies.
Figure 4.
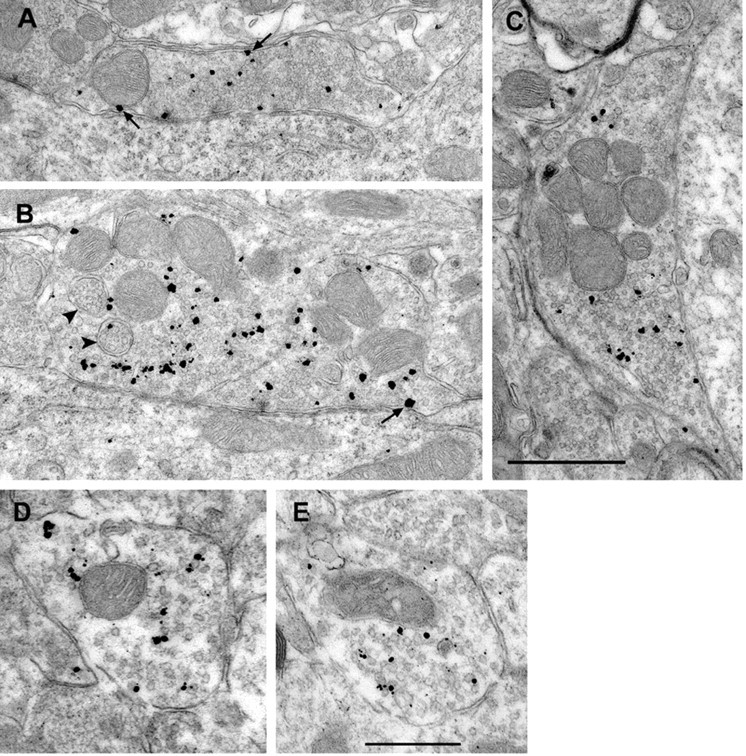
EM immunolocalization of CHT in the rat brain. A–C, Representative images demonstrating the distribution of CHT immunoreactivity in large morphologically distinct cholinergic presynaptic terminals in the facial nucleus. The silver-enhanced gold particles are predominantly intracellular and overlap with the distribution of numerous synaptic vesicles. Consistent with the function of CHT in Na+-dependent choline transport, CHT immunoreactivity also is detected at the plasma membrane (arrows in A–C). We also observed some rare CHT immunoreactivity associated with multivesicular bodies indicated by arrowheads in B. In cell bodies the CHT labeling was associated with the cytoplasmic faces of endoplasmic reticulum and Golgi membranes, consistent with the proposed cytoplasmic localization of the CHT C terminus epitope used for antibody generation (data not shown). D and E show a vesicular pattern of CHT distribution in striatal presynaptic terminals, with only occasional particles in close proximity to the plasma membrane. Scale bars: A–C, 1 μm; D, E, 500 nm.
Biochemical support for an enrichment of CHT on synaptic vesicles
The surprising vesicular localization of CHT in our EM studies suggested that a significant reserve pool of CHT vesicles exists in cholinergic terminals and offered an explanation to the longstanding, but unaccounted for, observation that the CHT Vmax is tightly coupled to levels of ACh release (Kuhar and Murrin, 1978). To validate these findings, we proceeded with a biochemical characterization of presynaptic CHT vesicles. In a survey of crude synaptosome extracts (P2 fraction) from various mouse brain regions (Fig. 5A), immunoblotting with polyclonal CHT antibody detected a 65 kDa band for which the regional distribution matched expectations for the distribution of cholinergic terminals (Gilmor et al., 1996). Thus CHT immunoreactivity was most abundant in the striatum, with intermediate expression evident in the spinal cord, brainstem, midbrain, hippocampus, and cortex and a low level of expression in the cerebellum. Renal tissue lacked CHT-immunoreactive protein. The same 65 kDa band was detected by the mouse anti-CHT monoclonal antibody (see below, Fig. 7).
Figure 5.

Subcellular fractionation indicates a vesicular enrichment for CHT in presynaptic terminals. A, Immunoblot analysis with CHT polyclonal antibody detects CHT as a 65 kDa band for which the intensity is most robust in cholinergic-rich brain regions (25 μg of P2 fraction detergent extract per lane). Whole mouse brain (B) or striatal tissue (C) was fractionated by differential centrifugation as described in Materials and Methods to yield a total homogenate fraction (Tot.), a large membrane and synaptosome-enriched P2 fraction, a large membrane and synaptic plasma membrane fraction (LP1), and a presynaptic intracellular vesicle-enriched fraction (LP2). The strong enrichment for CHT in the LP2 fraction versus the LP1 fraction most closely resembles that of the synaptic vesicle markers VAChT, synaptophysin (Syp), VMAT2, and VGAT as compared with the m2 muscarinic receptor, dopamine transporter (DAT), GABA transporter (GAT1), the Na+/K+-ATPase α3 subunit (Na/K-ATPase), and the NR 2A/B subunits of the NMDA receptor.
Figure 7.

Immunoisolation of CHT-containing vesicles. A, Compared with non-antibody-coupled protein A Dynabeads, VAChT polyclonal antiserum-coated beads efficiently isolate vesicles containing CHT and VAChT from the presynaptic vesicle-enriched striatal LS1 preparation. In the reciprocal experiment the antiserum directed against the cytoplasmic C terminus of CHT also effectively captures both CHT and a large fraction of the VAChT immunoreactivity. None of these assays captures appreciable quantities of the general synaptic vesicle marker synaptotagmin I or the dopaminergic neuron-specific dopamine transporter (DAT). B, Immunodepletion analysis of the synaptic vesicle-containing fractions from glycerol velocity gradients demonstrates that, when the CHT polyclonal antiserum clears the majority of CHT-containing vesicles (91±2%) from this fraction, VAChT immunoreactivity representing cholinergic synaptic vesicles also is depleted by one-half (45±5%). The depletion is specific for cholinergic synaptic vesicles because VMAT2 levels are unaltered (4± 6% depletion) between the control beads and the CHT antibody-coated beads (n = 4). C, In reciprocal experiments using the VAChT antiserum for the immunodepletion of cholinergic synaptic vesicles, depletion of VAChT immunoreactivity by 88 ± 2% is accompanied by a proportional and near-complete (89 ± 2%) depletion of CHT-containing vesicles. Immunoblotting for VMAT2 demonstrates the selective immunodepletion of cholinergic synaptic vesicles (n = 3). D, The monoclonal antibody raised against the C terminus of CHT also isolates CHT-containing vesicles that are positive for VAChT. Immunoisolation of synaptic vesicles with a synaptophysin antibody (clone 7.2) also results in the purification of CHT- and VAChT-containing vesicles. Notably, VMAT2 immunoreactivity is absent from the nonspecific mouse IgG-coated control protein G Sepharose beads and the CHT antibody beads but is present on the synaptophysin antibody beads. E, Closer examination of CHT monoclonal antibody beads with longer film exposures reveals the enrichment for CHT, VAChT, synaptophysin, and Rab3A as compared with nonspecific mouse IgG beads. Equivalent amounts of IgG heavy chain are present in each lane. F, Immunoisolation of Rab3A-positive vesicles depletes striatal LS1 fractions of CHT, Rab3A, and synaptophysin, but not DAT or GAT1 (results representative of 3 independent experiments). G, CHT-containing vesicles exhibit vesamicol-sensitive ACh uptake. After the loading of striatal synaptosomes with [3H]-choline ± vesamicol, CHT-specific antiserum is effective at immunoisolating vesamicol-sensitive [3H]-ACh-containing vesicles (2.8 ± 0.4 vs 1.1 ± 0.2 fmol of ACh; n = 3) but is no better than the preimmune serum with respect to the isolation of vesicles containing reserpine-sensitive dopamine (DA) counts (14.1 ± 3.7 vs 14.2 ± 2.6 fmol DA for anti-CHT serum and preimmune serum, respectively). Averaged data are presented from three separate experiments performed in duplicate ± SEM; *p < 0.05 as determined by Student's t test. The mean of the total vesamicol-sensitive [3H]-ACh and reserpine-sensitive [3H]-DA input for each immunoisolation assay is 42.3 ± 3.1 and 162.3 ± 11.8 fmol, respectively.
To elucidate the subcellular localization of CHT further, we fractionated mouse brain tissue (whole brain and striatum) by homogenization and differential centrifugation (Huttner et al., 1983) (Fig. 5B,C). As expected, we found enrichment for CHT immunoreactivity in the synaptosome containing P2 fraction relative to the total homogenate. Consistent with the EM studies, CHT was enriched in the LP2 fractions that also are enriched for well characterized synaptic vesicle proteins, including synaptophysin (Navone et al., 1986), VAChT, the vesicular monoamine transporter (VMAT2) (Eiden, 2000), and the vesicular GABA transporter (VGAT) (Takamori et al., 2000). In contrast, the presynaptic plasma membrane transporters for dopamine and GABA (DAT and GAT1, respectively) as well as the m2 muscarinic receptor and Na/K-ATPase were relatively more abundant in the plasma membrane-enriched LP1 fraction. The differing striatal pattern of fractionation for CHT compared with the m2 muscarinic receptor is notable because both proteins are expressed by cholinergic interneurons; however, the m2 receptor is not present on small presynaptic vesicles (Rouse et al., 1997). In addition to our whole brain and striatal fractionations, we achieved similar enrichments of CHT in the LP2 fractions from hippocampus and cerebral cortex (data not shown), suggesting that this vesicular localization is a common property of CHTs throughout the CNS.
Although the enrichment of CHT in the LP2 fraction is similar to synaptic vesicle markers, the presence of nonsynaptic vesicle proteins such as DAT, m2 receptor, GAT1, and Na/K-ATPase proteins in this fraction also suggests the presence of endosomal structures and/or plasma membrane fragments in the LP2 preparation. Previous studies have used velocity gradients to separate the small, homogeneous synaptic vesicles from larger membranes present in the LP2 fraction (Huttner et al., 1983). Thus we further resolved the LP2 fraction on a sucrose velocity gradient to distinguish between synaptic vesicles and presynaptic endosomes or plasma membrane fragments generated during tissue homogenization. These gradients generated two distinct pools of CHT immunoreactivity (Fig. 6A). We stripped these membranes and reprobed them for synaptic vesicle and plasma membrane markers and once again confirmed an overlap of CHT immunoreactivity with the signals for VAChT and synaptophysin (fractions 5–8). Notably, DAT and Na/K-ATPase fractionated predominantly with large membranes that accumulated at the bottom of the gradient. We further resolved the synaptic vesicle-enriched fractions 5–8 from the sucrose velocity gradient by pooling and centrifugation on a self-generating gradient of 14% iodixanol (Fig. 6B). Despite our ability to extend the distribution of synaptophysin immunoreactivity, we maintained a tight overlap between CHT- and VAChT-containing membranes. The separation of membranes on these gradients suggests that, in presynaptic cholinergic nerve terminals, CHT and VAChT are distributed to populations of vesicles with virtually identical physical characteristics. The broader distribution of synaptophysin on iodixanol gradients is consistent with its presence on a more heterogeneous population of synaptic vesicles, as expected, given that multiple neurotransmitter-specific vesicles exist in our starting material and that VAChT and CHT expression is restricted to cholinergic neurons.
Figure 6.
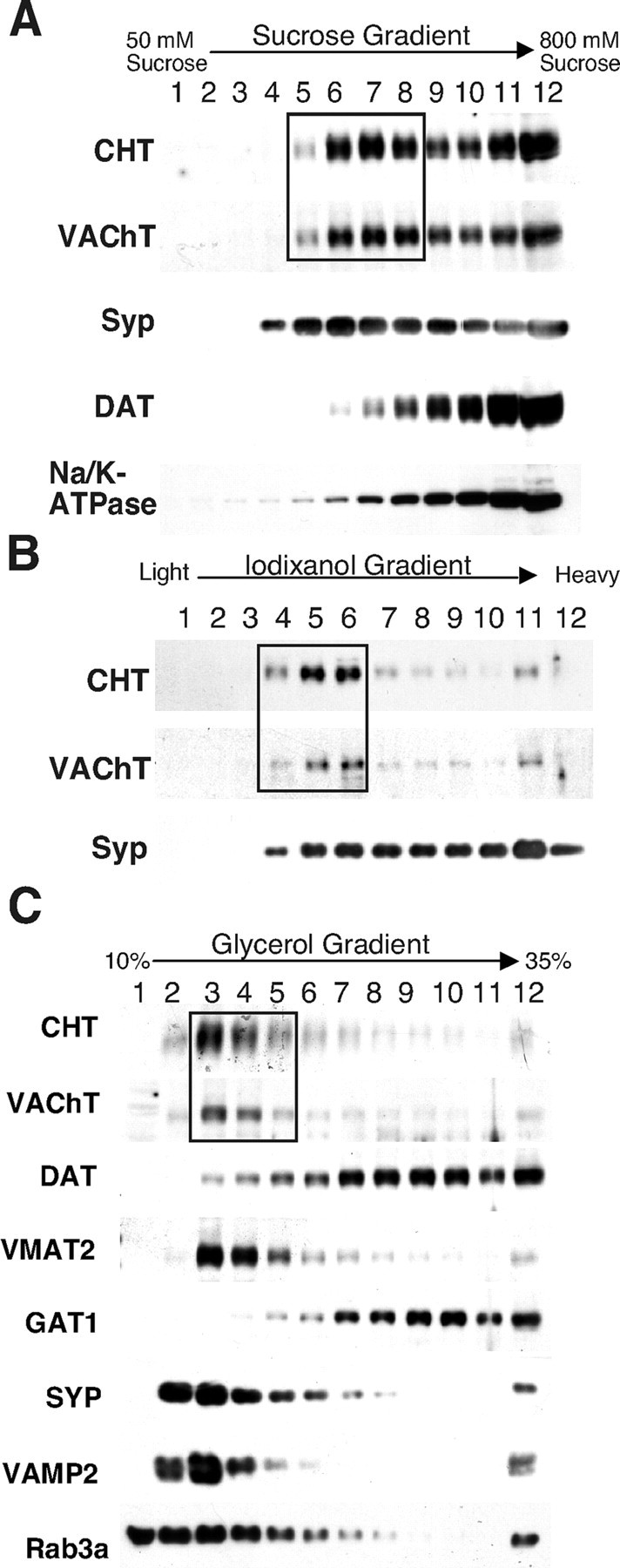
Gradient analysis of CHT-containing vesicles. A, Sucrose velocity gradient sedimentation of whole brain LP2 fraction membranes demonstrates that CHT exists on similar vesicle populations as VAChT (boxed region) and synaptophysin. B, The synaptic vesicle-enriched fractions 5–8 (CHT and VAChT in box) from the sucrose velocity gradient in A were pooled and resolved on a self-generating iodixanol gradient. Once again, CHT and VAChT are distributed in an identical pattern that overlaps with a subset of the synaptophysin immunoreactivity. C, Glycerol velocity gradient separation of the striatal LS1 fraction yields a pattern of distribution for CHT that is indistinguishable from that of VAChT. The box highlights the close match between CHT and VAChT in fractions 3–5 that represent the peak of synaptic vesicle proteins because synaptophysin, VMAT2, VAMP2, and Rab3A also are found in these fractions, but DAT and GAT1 are excluded. Equivalent results were obtained in four separate experiments.
Although we observed a tight overlap between CHT and VAChT on sucrose and iodixanol gradients, a considerable fraction of the CHT and VAChT immunoreactivity overlapped with nonsynaptic vesicle markers that pelleted at the bottom of the gradient. We were concerned that this could represent an artifact arising from the high-speed centrifugation required to generate the LP2 fraction. We thus repeated our fractionation studies with striatal preparations (to take advantage of the high density of cholinergic terminals in this brain region) and directly analyzed vesicles generated from the hypotonic lysis of synaptosomes (LS1 fraction). Here we also adopted glycerol velocity gradients for the separation of synaptic vesicles from larger membranes such as endosomes, plasma membrane fragments, and artifactual membrane aggregates (Clift-O'Grady et al., 1990; Lee et al., 2001). These experiments reveal a clear major peak of CHT immunoreactivity that overlaps completely with VAChT (Fig. 6C). The vesicles representing this peak of CHT and VAChT immunoreactivity are likely to exhibit synaptic vesicle morphology because we observe a similar distribution for known synaptic vesicle markers such as the VMAT2, synaptophysin, vesicle-associated membrane protein 2 (VAMP2) (Sudhof, 1995), and the synaptic vesicle-specific small GTPase Rab3A (Fischer von Mollard et al., 1991). Notably, other known presynaptic plasma membrane neurotransmitter transporters, such as DAT and GAT1, peak in fractions corresponding to larger vesicles (note particularly the readily discernible difference between DAT and the vesicular biogenic amine transporter VMAT2).
CHT immunoreactivity defines a subset of cholinergic synaptic vesicles
The cofractionation and cosedimentation of CHT and VAChT raise the possibility that VAChT and CHT might reside on the same vesicles whereby ACh release could be coupled to the delivery of CHT to the plasma membrane in response to neural activity. To explore this possibility, we first immunoprecipitated VAChT-positive vesicles from a presynaptic vesicle-enriched fraction derived from lysed striatal synaptosomes (LS1 fraction), using VAChT polyclonal antibody (Gilmor et al., 1996), and immunoblotted them for CHT (Fig. 7A). Notably, the majority of the CHT immunoreactivity was recovered specifically on VAChT antibody-coated beads. Furthermore, the reciprocal experiment whereby rabbit polyclonal anti-CHT antiserum was used for vesicle isolation effectively recovered both CHT and a significant fraction of VAChT. Negative controls in which the antiserum was omitted or in which the preimmune serum was used failed to immunoprecipitate CHT- or VAChT-containing vesicles. Moreover, the lack of DAT-containing vesicles recovered in our immunoadsorption experiments further attests to the specificity of our vesicle isolations. In these experiments, however, immunoblotting for synaptic vesicle markers failed to detected substantial amounts of general synaptic vesicle markers such as synaptotagmin I (Sudhof, 1995) or synaptophysin (data not shown), which we believe likely reflects the fact that only a small subset (1–2%) of striatal neurons is cholinergic (Zhou et al., 2002). Importantly, we were unable to coimmunoprecipitate CHT and VAChT in the presence of 1% Triton X-100, suggesting that, although they are present on the same vesicles, our co-isolation of CHT and VAChT does not arise because the two are associated physically but are rather colocalized on the same vesicles.
To quantify the extent of CHT/VAChT colocalization on synaptic vesicles, we immunodepleted striatal LS1 vesicle preparations with anti-CHT polyclonal antibody-coupled beads and analyzed the vesicles remaining in the supernatant on glycerol velocity gradients (Fig. 7B). We specifically examined the gradient fractions representing the peak of synaptic vesicle-specific enrichment (see fractions 3 and 4, Fig. 6A). The effectiveness of these CHT antibody-mediated immunodepletions was determined by comparison with the synaptic vesicle-enriched fractions from a control velocity gradient produced in parallel in which a mock immunoprecipitation was performed with beads lacking antibody. Under conditions that depleted 91 ± 2% of the CHT from the synaptic vesicle-specific gradient fractions, VAChT immunoreactivity was depleted by 45 ± 5% (n = 4; Fig. 7B). In the reciprocal experiment, immunodepletion with the VAChT antiserum removed equivalent proportions of both CHT and VAChT (89 ± 2 and 88 ± 2%, respectively; n = 3; Fig. 7C). In neither case did we observe significant VMAT2 immunodepletion (4 ± 6 and –4 ± 7% for CHT and VAChT antisera, respectively). Similar results were obtained by using whole brain LP2 rather than striatal vesicle fractions for immunodepletion studies (83 ± 4% CHT depleted with a corresponding 58 ± 5% immunodepletion of VAChT; n = 6). Because of the near-complete localization of CHT on VAChT-positive synaptic vesicles, it seems unlikely that CHT is associated with any other presynaptic vesicle population in these fractions. Rather, our results are most consistent with the presence of two, nearly equally abundant, subgroups of cholinergic synaptic vesicles that contain either VAChT alone or that contain both VAChT and CHT.
To substantiate further the surprising colocalization between CHT and VAChT, we performed additional immunoisolation experiments by using the CHT monoclonal antibody and consistently found that it also pulled down VAChT protein on CHT-containing vesicles (Fig. 7D,E). This immunoisolation is selective for cholinergic synaptic vesicles because the CHT monoclonal antibody beads are devoid of detectable VMAT2 immunoreactivity. To visualize better the presence of synaptophysin and synaptic vesicle-specific small GTPase Rab3A on CHT monoclonal antibody-coated beads, we found it necessary to prepare immunoblots in which the small cholinergic neuron-specific signals would not be obscured by large signals in the neighboring lanes after long film exposures (Fig. 7D vs E). In doing so, we clearly detected the presence of synaptophysin (Fig. 7E) as well as Rab3A (Fischer von Mollard et al., 1991). Moreover, when vesicle immunoprecipitation experiments were performed by using a synaptophysin monoclonal antibody (clone 7.2) that has been characterized extensively for the immunoisolation of synaptic vesicles (Burger et al., 1989; Takamori et al., 2000), we recovered CHT and VAChT in equal proportions (Fig. 7D). Furthermore, CHT-positive vesicles also were recovered with VAMP2 and synaptotagmin I monoclonal antibodies (data not shown), providing additional evidence that CHT resides on synaptic vesicles. The presence of Rab3A on CHT-positive vesicles (Fig. 7E) serves as an indication that these are indeed mature cholinergic synaptic vesicles because Rab3A is thought to dissociate from synaptic vesicles during exocytosis (Fischer von Mollard et al., 1991) and is absent from endocytic synaptic vesicle precursors such as clathrin-coated vesicles (Maycox et al., 1992). To evaluate this localization of CHT to Rab3A-positive vesicles further, we performed vesicle immunoadsorption experiments with a Rab3A antibody and examined the clearance of CHT from the supernatants (Fig. 7F). Under conditions that effectively clear the supernatant of Rab3A and synaptophysin, there is also a profound depletion of CHT, whereas the vesicles that recycle other plasma membrane transporters such as DAT and GAT1 are not removed from the supernatant in these experiments. Finally, direct EM analysis of the vesicles immunoisolated from the striatal LS1 fraction that used CHT or synaptophysin antibodies revealed an indistinguishable vesicle morphology. Vesicle diameters (n = 42) measured from random sections from CHT or synaptophysin antibody-coated beads yielded dimensions of 28.7 ± 8 and 28.1 ± 8 nm, respectively (data not shown).
If the vesicles we isolate on CHT antibody beads represent a fraction of intact cholinergic synaptic vesicles, they should contain ACh. It is well described that, after transport into cholinergic terminals, [3H]-choline is converted efficiently to [3H]-ACh (Mulder et al., 1974). Thus we actively loaded synaptosomes with [3H]-choline to generate [3H]-ACh-labeled cholinergic synaptic vesicles, performing incubations ± the VAChT inhibitor vesamicol to define [3H]-ACh-labeled cholinergic synaptic vesicles (Prior et al., 1992). After hypotonic lysis of synaptosomes the radiolabeled synaptic vesicle fraction (LS1) was immunoisolated by using either preimmune serum or anti-CHT antibody-coupled magnetic beads. Unlike material captured on preimmune beads, vesicles isolated with anti-CHT serum were loaded with radiolabel in a vesamicol-sensitive manner (p < 0.05, Student's unpaired t test; Fig. 7G). Importantly, CHT antibodies failed to capture [3H]-dopamine-labeled vesicles (Fig. 7G).
Evidence of vesicular trafficking supporting CHT regulation
The presence of CHT on a portion of VAChT-positive cholinergic synaptic vesicles raises the expectation that HC-3-sensitive choline uptake should be responsive to treatments that support or elicit vesicular release of ACh. In support of a contribution of vesicular fusion to the maintenance of basal levels of choline transport, we observed diminished basal uptake when hippocampal synaptosomes were preincubated in the presence of Cd2+ to block voltage-dependent calcium channels (Fig. 8B). More conclusively, BoNT/C pretreatments, which cleave syntaxin 1A proteins critical to the fusion of synaptic vesicles (Blasi et al., 1993), significantly reduced basal levels of choline uptake (Fig. 8B; BoNT/C-treated, 55 ± 5% of control; n = 3). Furthermore, when synaptosomes were treated with 20 mm KCl to trigger ACh release and synaptic vesicle turnover, we measured a significant increase in the choline transport Vmax (2.1 ± 0.03 vs 4.7 ± 0.2 pmol/mg protein/min) with little or no change in Km (0.62 ± 0.03 vs 0.71 ± 0.09 μm; Fig. 8A). A similar elevation in CHT activity was observed by using 150 μm 4-aminopyridine (4-AP) to evoke synaptosome depolarization (Fig. 8B). As observed with basal levels of choline uptake, the depolarization-evoked increase in choline transport was attenuated significantly in the presence of Cd2+ or with BoNT/C pretreatment (Fig. 8B). Finally, we blotted synaptic plasma membrane-enriched fractions (washed LP1) after K+ depolarization and observed a consistent and significant increase in CHT immunoreactivity (40 ± 5%; n = 3) as compared with unstimulated controls (Fig. 8C). Interestingly, we detected no increase in VAChT abundance in this fraction, suggesting that the two transporters may exhibit different retention characteristics after vesicle fusion. Moreover, we found no change in GAT1 GABA transporter abundance, consistent both with an even loading of our samples and with previous reports of a lack of depolarization-evoked increases in GABA uptake (Simon et al., 1976).
Figure 8.
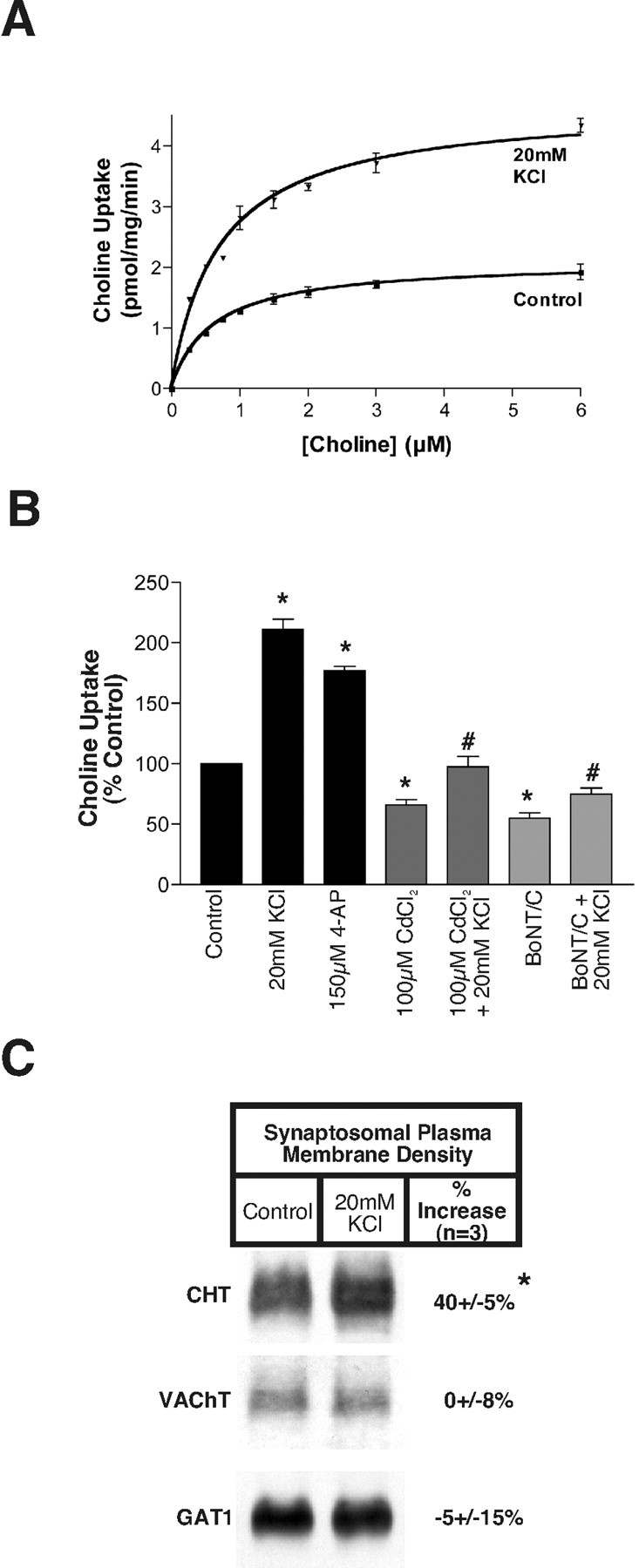
Synaptic vesicle exocytosis and selective plasma membrane retention of CHT support activity-dependent regulation of choline uptake. A, Saturation analysis indicates that the 20 mm KCl pretreatment-evoked increase in HC-3-sensitive choline uptake is associated with an increase in CHT Vmax. B, HC-3-sensitive choline uptake is enhanced after treatments that evoke neurotransmitter release from hippocampal synaptosomes (20 mm KCl and 150 μm 4-AP). The effect of 20 mm KCl is attenuated when CdCl2 is added to block voltage-gated Ca 2+ channels or when synaptosomes are preincubated with BoNT/C to block syntaxin 1A-dependent neurotransmitter release (*p < 0.05 compared with control treatment and #p < 0.001 compared with 20 mm KCl treatment by ANOVA with Bonferroni's post test; n = 3 for measurements performed in triplicate). C, The 20 mm KCl pretreatment evokes a selective increase in CHT density in the synaptosomal plasma membrane (*p < 0.05 compared with control treatment, Student's t test; n = 3), whereas VAChT and GAT1 levels are unchanged in response to this treatment.
Discussion
The recent cloning of CHT cDNAs has provided new opportunities with respect to the study of presynaptic choline transport regulation. The initial establishment of CHT as a presynaptic protein residing on cholinergic terminals was deduced from radiolabeled choline transport assays and [3H]-HC-3 autoradiography in conjunction with the lesion of the proposed pathways (Kuhar et al., 1975; Rainbow et al., 1984). The generation of antibodies that specifically recognize unique epitopes in the CHT C terminus has now allowed us to visualize CHT in all major mammalian cholinergic cell groups and their projections. The colocalization of CHT with ChAT and VAChT unambiguously establishes the cholinergic nature of these terminals (Fig. 3) and corroborates other recent reports of the immunohistochemical localization of CHT (Misawa et al., 2001; Kus et al., 2003).
Our EM analysis of the subcellular distribution of CHT demonstrated a surprising enrichment for CHT on small presynaptic vesicles rather than at the plasma membrane (Fig. 4). This observation is supported by the biochemical evidence showing a coenrichment of CHT with VAChT and other synaptic vesicle markers in subcellular fractions from the mouse brain (Fig. 5). The parallel distributions of CHT, VAChT, and other synaptic vesicle markers (e.g., synaptophysin, VGAT, VMAT2) between the synaptic plasma membrane (LP1) and vesicular fraction (LP2) provided initial evidence that, at steady state, CHT has a mainly intracellular localization (Fig. 5). Moreover, when the vesicle-enriched LS1 or LP2 fractions were defined further via velocity gradients, CHT fractionated in a pattern that was indistinguishable from VAChT, the only previously characterized specific marker of cholinergic synaptic vesicles (Fig. 6). The direct immunoisolation of vesicles that are positive for both CHT and VAChT with three distinct antibodies (CHT polyclonal, CHT monoclonal, and VAChT polyclonal) firmly establishes the colocalization of these two proteins on presynaptic vesicles (Fig. 7). CHT localization to synaptic vesicles is substantiated further by the immunoisolation of CHT- and VAChT-containing vesicles with the use of antibodies against the ubiquitous synaptic vesicle proteins synaptophysin and Rab3A. This synaptophysin antibody has been tested rigorously with respect to its ability to isolate very pure preparations of synaptic vesicles from crude brain homogenates and lysed synaptosome preparations achieving purifications equivalent if not superior to controlled pore glass resin chromatography (Burger et al., 1989; Takamori et al., 2000).
Having established the specificity of the colocalization of CHT and VAChT, we turned our attention to quantifying this phenomenon and found that, although essentially all of the CHT-positive vesicles are VAChT-positive, only one-half of the VAChT-positive vesicles possess CHT. We speculate that two major subpopulations of cholinergic synaptic vesicles exist that are distinguished by their CHT content. We speculate that the “VAChT only” pool preferentially supports low rates of ACh release, whereas the “CHT/VAChT” pool might be recruited in response to higher firing rates. Multiple synaptic vesicle populations have been defined on the basis of vesicle size (Zimmermann and Whittaker, 1977), FM1-43 release, and loading kinetics (Richards et al., 2000) as well as their sensitivity to changes in cytoplasmic cAMP (Kuromi and Kidokoro, 2000) and calcium (Kuromi and Kidokoro, 2002), although molecular distinctions between pools and their differences for chemically distinct terminals are unknown. Targeting of CHT and VAChT to a reserve pool would allow for CHTs to be recruited after strong depolarization, conditions under which elevated choline transport activity might be essential to sustain further ACh synthesis and release. Additional studies are needed to investigate this possibility.
A mainly vesicular localization of CHT might seem contradictory to the evidence that HC-3-sensitive choline uptake is essential for the sustained synthesis and release of ACh (Guyenet et al., 1973). However, there exists a tight relationship linking cholinergic neuronal activity to the regulation of CHT-mediated choline uptake that could be explained by activity-dependent vesicular trafficking to the plasma membrane and subsequent CHT retention. For example, anesthetics such as pentobarbital decrease both ACh release and choline uptake in the hippocampus, whereas they are without effect on ACh release in the striatum, where they also do not affect choline uptake (Simon and Kuhar, 1975). Furthermore, direct electrical stimulation of the septal–hippocampal pathway reverses the effects of pentobarbital on hippocampal choline uptake. CHT function also is inhibited in response to in vivo muscarinic receptor activation with oxotremorine that inhibits ACh release (Atweh et al., 1975; Lowenstein and Coyle, 1986). In contrast, choline transport and HC-3 binding are increased in response to in vivo administration of muscarinic receptor antagonists such as atropine and scopolamine that increase ACh release (Atweh et al., 1975; Lowenstein and Coyle, 1986). As in our studies (Fig. 8), kinetic analyses of choline uptake by the aforementioned modulators are supported by changes in choline uptake Vmax and HC-3 binding Bmax, respectively. CHT function also can be modulated ex vivo in sympathetic ganglia, brain slice, and synaptosome preparations by treatments that are known to increase vesicular ACh release such as direct electrical stimulation and high K+ buffers (Murrin et al., 1977; Collier et al., 1983; Saltarelli et al., 1987). Our findings that activity-dependent CHT regulation is BoNT/C-sensitive (Fig. 8B) and our detection of a shift of CHT protein density in synaptic membranes (Fig. 8C) provide compelling evidence for vesicular trafficking supporting CHT regulation. In this model increasing the rate of ACh release will enhance delivery of CHTs to the plasma membrane. Conversely, a reduction in exocytosis would contribute to the decrease in choline transport when neuronal activity is suppressed and CHT endocytosis might exceed exocytosis. These results also suggest the presence of a mechanism to regulate the retention of CHT in the plasma membrane after periods of increased neuronal activity. The increase in synaptic plasma membrane CHT levels may underestimate the true magnitude of the CHT retention in the plasma membrane because our synaptosome preparation is likely to contain a substantial background level of CHT on broken membranes or nonfunctional synaptosomes that cannot support CHT trafficking and membrane insertion. Although the evident vesicular localization of CHT indicates an important role for trafficking in the acute regulation of CHT function, our findings do not rule out additional modes of CHT regulation after insertion into the plasma membrane, as suggested by the modulation of HC-3 binding with the use of synaptic plasma membrane preparations (Ferguson and Collier, 1994). Modulation of transporter activity after insertion into the plasma membrane is an emerging theme for the insulin-dependent glucose transporter (GLUT4) (Sweeney et al., 1999), the presynaptic GABA transporter (Deken et al., 2000) and the cocaine and antidepressant-sensitive norepinephrine transporter (Sung et al., 2003).
One question that arises from these findings asks why levels of CHT protein at the plasma membrane are not set constitutively at maximal levels to ensure efficient recycling of choline for ACh biosynthesis, regardless of ACh release rates. One possibility implicates a role for choline itself as a neurotransmitter for which the extracellular levels are modulated via the regulation of CHT activity. This idea is supported by the existence of choline-sensitive nicotinic ACh receptors that might respond to the choline produced after acetylcholinesterase-mediated hydrolysis of synaptically released ACh (Guo and Chiappinelli, 2002). Alternatively, it has been proposed that in cholinergic-rich brain regions there is sufficient choline uptake capacity to starve noncholinergic neurons of extracellular choline if CHT is not sequestered during periods of diminished ACh release (Farber et al., 1996). Thus tight neuronal activity-dependent regulation of CHT localization could act to maintain a balance between the high-affinity CHT-mediated uptake of choline for neurotransmitter synthesis and the dependence of surrounding cells on low-affinity uptake mechanisms to provide choline for membrane phospholipid synthesis. The efficient recycling of CHT into a synaptic vesicle pool that can be called on in an activity-dependent manner is reminiscent of the insulin-dependent regulation of GLUT4 in muscle and adipose tissue (Bryant et al., 2002). The insulin-dependent regulation of GLUT4 ensures that the large glucose uptake capacity of skeletal muscle and adipocytes does not out-compete other tissues for glucose during times of fasting. Although the presence of CHT on cholinergic synaptic vesicles is attractive from the point of view of a trafficking model for the activity-dependent regulation of cholinergic function, we acknowledge the possibility that CHT also could function as a vesicular transporter for choline or some as yet unidentified substrate.
Our studies provide a foundation for future experiments assessing the role of CHT trafficking in the dysregulation of presynaptic choline transport and ACh synthesis in disease states. In humans CHT is encoded by a single gene on chromosome 2 (Apparsundaram et al., 2000), and polymorphisms or mutations in this gene could impair cholinergic signaling. The large intracellular pool of CHT protein predicts that mutations that impair CHT vesicular localization, trafficking, or endocytic recycling might have important consequences resulting from diminished ACh synthesis and release. In vivo pharmacological blockade of CHT is lethal (Quastel and Curtis, 1965), and recently we have observed neonatal lethality in CHT knock-out mice (Ferguson, Savchenko, Wright, and Blakely, unpublished observations). Unlike l-DOPA therapy for the treatment of dopaminergic deficits in Parkinson's disease, choline supplementation for disorders with cholinergic deficits may be limited by insufficient surface density of transporters because of efficient endocytosis of CHT to synaptic vesicles. Agents that enhance CHT levels at the plasma membrane would be predicted to increase ACh production and release and thus could complement current approaches, for example in Alzheimer's disease, that focus on limiting ACh degradation.
Footnotes
This work was supported by a predoctoral fellowship from the Vanderbilt Brain Institute (to S.F.) and National Center for Research Resources Grant P20RR15592 (to S.A.) as well as by the Alzheimer Association and National Institutes of Health (NIH) Grants NS30454 (to A.L.) and MH58921 (to R.B.). Experiments were performed in part via the use of the Vanderbilt University Medical Center (VUMC) Cell Imaging Core Resource (supported by NIH Grants CA68485 and DK20593) and the Neural Histology and Imaging Core of the Center for Molecular Neuroscience (VUMC). We thank Lou DeFelice for his helpful comments on this manuscript. We also acknowledge the contributions of Howard Rees, Laura Volpicelli-Daley, and Dana Michelle Hines.
Correspondence should be addressed to Randy D. Blakely, Department of Pharmacology, Suite 4160, Medical Research Building III, Vanderbilt University School of Medicine, Nashville, TN 37232-8548. E-mail: randy.blakely@vanderbilt.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/239697-13$15.00/0
References
- Apparsundaram S, Ferguson SM, George Jr AL, Blakely RD ( 2000) Molecular cloning of a human hemicholinium-3-sensitive choline transporter. Biochem Biophys Res Commun 276: 862–867. [DOI] [PubMed] [Google Scholar]
- Apparsundaram S, Ferguson SM, Blakely RD ( 2001) Molecular cloning and characterization of a murine hemicholinium-3-sensitive choline transporter. Biochem Soc Trans 29: 711–716. [DOI] [PubMed] [Google Scholar]
- Atweh S, Simon JR, Kuhar MJ ( 1975) Utilization of sodium-dependent high affinity choline uptake in vitro as a measure of the activity of cholinergic neurons in vivo Life Sci 17: 1535–1544. [DOI] [PubMed] [Google Scholar]
- Birks RI, MacIntosh FC ( 1961) Acetylcholine metabolism of a sympathetic ganglion. Can J Biochem Physiol 39: 787–827. [Google Scholar]
- Blasi J, Chapman ER, Yamasaki S, Binz T, Niemann H, Jahn R ( 1993) Botulinum neurotoxin C1 blocks neurotransmitter release by means of cleaving HPC-1/syntaxin. EMBO J 12: 4821–4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant NJ, Govers R, James DE ( 2002) Regulated transport of the glucose transporter GLUT4. Nat Rev Mol Cell Biol 3: 267–277. [DOI] [PubMed] [Google Scholar]
- Burger PM, Mehl E, Cameron PL, Maycox PR, Baumert M, Lottspeich F, De Camilli P, Jahn R ( 1989) Synaptic vesicles immunoisolated from rat cerebral cortex contain high levels of glutamate. Neuron 3: 715–720. [DOI] [PubMed] [Google Scholar]
- Bymaster FP, McKinzie DL, Felder CC, Wess J ( 2003) Use of m1–m5 muscarinic receptor knockout mice as novel tools to delineate the physiological roles of the muscarinic cholinergic system. Neurochem Res 28: 437–442. [DOI] [PubMed] [Google Scholar]
- Chang YC, Gottlieb DI ( 1988) Characterization of the proteins purified with monoclonal antibodies to glutamic acid decarboxylase. J Neurosci 8: 2123–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clift-O'Grady L, Linstedt AD, Lowe AW, Grote E, Kelly RB ( 1990) Biogenesis of synaptic vesicle-like structures in a pheochromocytoma cell line PC-12. J Cell Biol 110: 1693–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier B, Kwok YN, Welner SA ( 1983) Increased acetylcholine synthesis and release following presynaptic activity in a sympathetic ganglion. J Neurochem 40: 91–98. [DOI] [PubMed] [Google Scholar]
- Dani JA, De Biasi M ( 2001) Cellular mechanisms of nicotine addiction. Pharmacol Biochem Behav 70: 439–446. [DOI] [PubMed] [Google Scholar]
- Deken SL, Beckman ML, Boos L, Quick MW ( 2000) Transport rates of GABA transporters: regulation by the N-terminal domain and syntaxin 1A. Nat Neurosci 3: 998–1003. [DOI] [PubMed] [Google Scholar]
- Eiden LE ( 1998) The cholinergic gene locus. J Neurochem 70: 2227–2240. [DOI] [PubMed] [Google Scholar]
- Eiden LE ( 2000) The vesicular neurotransmitter transporters: current perspectives and future prospects. FASEB J 14: 2396–2400. [DOI] [PubMed] [Google Scholar]
- Farber SA, Savci V, Wei A, Slack BE, Wurtman RJ ( 1996) Choline's phosphorylation in rat striatal slices is regulated by the activity of cholinergic neurons. Brain Res 723: 90–99. [DOI] [PubMed] [Google Scholar]
- Ferguson SS, Collier B ( 1994) Stereoselectivity of the inhibition of [3H]hemicholinium-3 binding to the sodium-dependent high-affinity choline transporter by the enantiomers of α- and β-methylcholine. J Neurochem 62: 1449–1457. [DOI] [PubMed] [Google Scholar]
- Fischer von Mollard G, Sudhof TC, Jahn R ( 1991) A small GTP-binding protein dissociates from synaptic vesicles during exocytosis. Nature 349: 79–81. [DOI] [PubMed] [Google Scholar]
- Gilmor ML, Nash NR, Roghani A, Edwards RH, Yi H, Hersch SM, Levey AI ( 1996) Expression of the putative vesicular acetylcholine transporter in rat brain and localization in cholinergic synaptic vesicles. J Neurosci 16: 2179–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene LA, Tischler AS ( 1976) Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci USA 73: 2424–2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JZ, Chiappinelli VA ( 2002) A novel choline-sensitive nicotinic receptor subtype that mediates enhanced GABA release in the chick ventral lateral geniculate nucleus. Neuroscience 110: 505–513. [DOI] [PubMed] [Google Scholar]
- Guyenet P, Lefresne P, Rossier J, Beaujouan JC, Glowinski J ( 1973) Inhibition by hemicholinium-3 of [14C]acetylcholine synthesis and [3H]choline high-affinity uptake in rat striatal synaptosomes. Mol Pharmacol 9: 630–639. [PubMed] [Google Scholar]
- Haga T, Noda H ( 1973) Choline uptake systems of rat brain synaptosomes. Biochim Biophys Acta 291: 564–575. [DOI] [PubMed] [Google Scholar]
- Hashiramoto M, James DE ( 2000) Characterization of insulin-responsive GLUT4 storage vesicles isolated from 3T3–L1 adipocytes. Mol Cell Biol 20: 416–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttner WB, Schiebler W, Greengard P, De Camilli P ( 1983) Synapsin I (protein I), a nerve terminal-specific phosphoprotein. III. Its association with synaptic vesicles studied in a highly purified synaptic vesicle preparation. J Cell Biol 96: 1374–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller-Peck CR, Feng G, Sanes JR, Yan Q, Lichtman JW, Snider WD ( 2001) Glial cell line-derived neurotrophic factor administration in postnatal life results in motor unit enlargement and continuous synaptic remodeling at the neuromuscular junction. J Neurosci 21: 6136–6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsella TM, Nolan GP ( 1996) Episomal vectors rapidly and stably produce high-titer recombinant retrovirus. Hum Gene Ther 7: 1405–1413. [DOI] [PubMed] [Google Scholar]
- Kuhar MJ, Murrin LC ( 1978) Sodium-dependent, high affinity choline uptake. J Neurochem 30: 15–21. [DOI] [PubMed] [Google Scholar]
- Kuhar MJ, DeHaven RN, Yamamura HI, Rommel-Spacher H, Simon JR ( 1975) Further evidence for cholinergic habenulo-interpeduncular neurons: pharmacologic and functional characteristics. Brain Res 97: 265–275. [DOI] [PubMed] [Google Scholar]
- Kuromi H, Kidokoro Y ( 2000) Tetanic stimulation recruits vesicles from reserve pool via a cAMP-mediated process in Drosophila synapses. Neuron 27: 133–143. [DOI] [PubMed] [Google Scholar]
- Kuromi H, Kidokoro Y ( 2002) Selective replenishment of two vesicle pools depends on the source of Ca 2+ at the Drosophila synapse. Neuron 35: 333–343. [DOI] [PubMed] [Google Scholar]
- Kus L, Borys E, Ping Chu Y, Ferguson SM, Blakely RD, Emborg ME, Kordower JH, Levey AI, Mufson EJ ( 2003) Distribution of high affinity choline transporter immunoreactivity in the primate central nervous system. J Comp Neurol 463: 341–357. [DOI] [PubMed] [Google Scholar]
- Laemmli UK ( 1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680–685. [DOI] [PubMed] [Google Scholar]
- Lee SH, Valtschanoff JG, Kharazia VN, Weinberg R, Sheng M ( 2001) Biochemical and morphological characterization of an intracellular membrane compartment containing AMPA receptors. Neuropharmacology 41: 680–692. [DOI] [PubMed] [Google Scholar]
- Levey AI, Armstrong DM, Atweh SF, Terry RD, Wainer BH ( 1983) Monoclonal antibodies to choline acetyltransferase: production, specificity, and immunohistochemistry. J Neurosci 3: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levey AI, Edmunds SM, Hersch SM, Wiley RG, Heilman CJ ( 1995) Light and electron microscopic study of m2 muscarinic acetylcholine receptor in the basal forebrain of the rat. J Comp Neurol 351: 339–356. [DOI] [PubMed] [Google Scholar]
- Lonart G, Janz R, Johnson KM, Sudhof TC ( 1998) Mechanism of action of Rab3A in mossy fiber LTP. Neuron 21: 1141–1150. [DOI] [PubMed] [Google Scholar]
- Lowenstein PR, Coyle JT ( 1986) Rapid regulation of [3H]hemicholinium-3 binding sites in the rat brain. Brain Res 381: 191–194. [DOI] [PubMed] [Google Scholar]
- Maire JC, Wurtman RJ ( 1985) Effects of electrical stimulation and choline availability on the release and contents of acetylcholine and choline in superfused slices from rat striatum. J Physiol (Lond) 80: 189–195. [PubMed] [Google Scholar]
- Maycox PR, Link E, Reetz A, Morris SA, Jahn R ( 1992) Clathrin-coated vesicles in nervous tissue are involved primarily in synaptic vesicle recycling. J Cell Biol 118: 1379–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misawa H, Nakata K, Matsuura J, Nagao M, Okuda T, Haga T ( 2001) Distribution of the high-affinity choline transporter in the central nervous system of the rat. Neuroscience 105: 87–98. [DOI] [PubMed] [Google Scholar]
- Mulder AH, Yamamura HI, Kuhar MJ, Snyder SH ( 1974) Release of acetylcholine from hippocampal slices by potassium depolarization: dependence on high affinity choline uptake. Brain Res 70: 372–376. [DOI] [PubMed] [Google Scholar]
- Murrin LC, DeHaven RN, Kuhar MJ ( 1977) On the relationship between [3H]choline uptake activation and [3H]acetylcholine release. J Neurochem 29: 681–687. [DOI] [PubMed] [Google Scholar]
- Navone F, Jahn R, Di Gioia G, Stukenbrok H, Greengard P, De Camilli P ( 1986) Protein p38: an integral membrane protein specific for small vesicles of neurons and neuroendocrine cells. J Cell Biol 103: 2511–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda T, Haga T, Kanai Y, Endou H, Ishihara T, Katsura I ( 2000) Identification and characterization of the high-affinity choline transporter. Nat Neurosci 3: 120–125. [DOI] [PubMed] [Google Scholar]
- Prior C, Marshall IG, Parsons SM ( 1992) The pharmacology of vesamicol: an inhibitor of the vesicular acetylcholine transporter. Gen Pharmacol 23: 1017–1022. [DOI] [PubMed] [Google Scholar]
- Quastel DM, Curtis DR ( 1965) A central action of hemicholinium. Nature 208: 192–194. [DOI] [PubMed] [Google Scholar]
- Rainbow TC, Parsons B, Wieczorek CM ( 1984) Quantitative autoradiography of [3H]hemicholinium-3 binding sites in rat brain. Eur J Pharmacol 102: 195–196. [DOI] [PubMed] [Google Scholar]
- Richards DA, Guatimosim C, Betz WJ ( 2000) Two endocytic recycling routes selectively fill two vesicle pools in frog motor nerve terminals. Neuron 27: 551–559. [DOI] [PubMed] [Google Scholar]
- Rouse ST, Thomas TM, Levey AI ( 1997) Muscarinic acetylcholine receptor subtype m2: diverse functional implications of differential synaptic localization. Life Sci 60: 1031–1038. [DOI] [PubMed] [Google Scholar]
- Saltarelli MD, Lowenstein PR, Coyle JT ( 1987) Rapid in vitro modulation of [3H]hemicholinium-3 binding sites in rat striatal slices. Eur J Pharmacol 135: 35–40. [DOI] [PubMed] [Google Scholar]
- Schroeter S, Apparsundaram S, Wiley RG, Miner LH, Sesack SR, Blakely RD ( 2000) Immunolocalization of the cocaine- and antidepressant-sensitive l-norepinephrine transporter. J Comp Neurol 420: 211–232. [PubMed] [Google Scholar]
- Simon JR, Kuhar MG ( 1975) Impulse-flow regulation of high affinity choline uptake in brain cholinergic nerve terminals. Nature 255: 162–163. [DOI] [PubMed] [Google Scholar]
- Simon JR, Kuhar MJ ( 1976) High affinity choline uptake: ionic and energy requirements. J Neurochem 27: 93–99. [DOI] [PubMed] [Google Scholar]
- Simon JR, Atweh S, Kuhar MJ ( 1976) Sodium-dependent high affinity choline uptake: a regulatory step in the synthesis of acetylcholine. J Neurochem 26: 909–922. [DOI] [PubMed] [Google Scholar]
- Smith DB, Johnson KS ( 1988) Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene 67: 31–40. [DOI] [PubMed] [Google Scholar]
- Sudhof TC ( 1995) The synaptic vesicle cycle: a cascade of protein–protein interactions. Nature 375: 645–653. [DOI] [PubMed] [Google Scholar]
- Sung U, Apparsundaram S, Galli A, Kahlig K, Savchenko V, Schroeter S, Quick MW, Blakely RD ( 2003) A regulated interaction of syntaxin 1A with the antidepressant-sensitive norepinephrine transporter establishes catecholamine clearance capacity. J Neurosci 23: 1697–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney G, Somwar R, Ramlal T, Volchuk A, Ueyama A, Klip A ( 1999) An inhibitor of p38 mitogen-activated protein kinase prevents insulin-stimulated glucose transport but not glucose transporter translocation in 3T3–L1 adipocytes and L6 myotubes. J Biol Chem 274: 10071–10078. [DOI] [PubMed] [Google Scholar]
- Takamori S, Riedel D, Jahn R ( 2000) Immunoisolation of GABA-specific synaptic vesicles defines a functionally distinct subset of synaptic vesicles. J Neurosci 20: 4904–4911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Hersh LB ( 1994) Choline acetyltransferase: celebrating its fiftieth year. J Neurochem 62: 1653–1663. [DOI] [PubMed] [Google Scholar]
- Yamamura HI, Snyder SH ( 1972) Choline: high-affinity uptake by rat brain synaptosomes. Science 178: 626–628. [DOI] [PubMed] [Google Scholar]
- Zhou FM, Wilson CJ, Dani JA ( 2002) Cholinergic interneuron characteristics and nicotinic properties in the striatum. J Neurobiol 53: 590–605. [DOI] [PubMed] [Google Scholar]
- Zimmermann H, Whittaker VP ( 1977) Morphological and biochemical heterogeneity of cholinergic synaptic vesicles. Nature 267: 633–635. [DOI] [PubMed] [Google Scholar]