

Abstract
Severe hypoglycemia causes neuronal death and cognitive impairment. Evidence suggests that hypoglycemic neuronal death involves excitotoxicity and DNA damage. Poly(ADP-ribose) polymerase-1 (PARP-1) normally functions in DNA repair, but promotes cell death when extensively activated by DNA damage. Cortical neuron cultures were subjected to glucose deprivation to assess the role of PARP-1 in hypoglycemic neuronal death. PARP-1-/- neurons and wild-type, PARP-1+/+ neurons treated with the PARP inhibitor 3,4-dihydro-5-[4-(1-piperidinyl)butoxy]-1(2H)-isoquinolinone both showed increased resistance to glucose deprivation. A rat model of insulin-induced hypoglycemia was used to assess the therapeutic potential of PARP inhibitors after hypoglycemia. Rats subjected to severe hypoglycemia (30 min EEG isoelectricity) accumulated both nitrotyrosine and the PARP-1 product, poly(ADP-ribose), in vulnerable neurons. Treatment with PARP inhibitors immediately after hypoglycemia blocked production of poly(ADP-ribose) and reduced neuronal death by >80% in most brain regions examined. Increased neuronal survival was also achieved when PARP inhibitors were administered up to 2 hr after blood glucose correction. Behavioral and histological assessments performed 6 weeks after hypoglycemia confirmed a sustained salutary effect of PARP inhibition. These results suggest that PARP-1 activation is a major factor mediating hypoglycemic neuronal death and that PARP-1 inhibitors can rescue neurons that would otherwise die after severe hypoglycemia.
Keywords: diabetes, glucose, glutamate, neuron, nitrotyrosine, water maze, 7-nitroindazole, 3-aminobenzamide
Introduction
Hypoglycemic brain injury is a common and serious complication of insulin therapy. Not surprisingly, hypoglycemic brain injury occurs most frequently in patients attempting tight glucose control (Davis et al., 1998). The only treatment for hypoglycemia is blood glucose repletion, and there is no currently available intervention for preventing the neuronal death that develops after hypoglycemia is corrected.
Certain neuronal populations are especially vulnerable to hypoglycemia, specifically neurons in the CA1, subiculum, and dentate gyrus of the hippocampus, and neurons in the outer layers of the cortex (Auer and Siesjo, 1993). These neurons receive a rich glutamatergic innervation, and evidence suggests that hypoglycemic injury in these neurons is precipitated almost entirely by sustained glutamate receptor activation (excitotoxicity) (Auer et al., 1985). Hypoglycemia causes several-fold elevations in brain extracellular glutamate and aspartate concentrations, and ablation of presynaptic glutamatergic terminals can prevent hypoglycemic neuronal death (Wieloch et al., 1985; Butcher et al., 1987). Pretreatment with glutamate receptor antagonists can also reduce hypoglycemic neuronal death (Wieloch, 1985), but these agents are less effective when administered after hypoglycemia has occurred (Nellgard and Wieloch, 1992). An additional limitation to the use of glutamate receptor antagonists in clinical settings is that these agents are themselves neurotoxic (Olney et al., 1989).
Poly(ADP-ribose) polymerase-1 (PARP-1) is a key downstream mediator of glutamate excitotoxicity (Cosi et al., 1994; Zhang et al., 1994; Mandir et al., 2000). The poly(ADP-ribose) polymerases (PARPs) are a family of enzymes that use the ADP-ribose groups in NAD+ to form branched ADP-ribose polymers on specific acceptor proteins in the vicinity of DNA strand breaks or kinks (Burzio et al., 1979; D'Amours et al., 1999). PARP-1, the most abundant of these family members, normally functions in DNA repair, but extensive PARP-1 activation can promote cell death through processes involving energy depletion and release of apoptosis-inducing factor (Ha and Snyder, 1999; Ying et al., 2002; Yu et al., 2002). Sustained action of glutamate at neuronal NMDA receptors leads to the production of peroxynitrite and other reactive oxygen species that damage DNA and activate PARP-1 (Dugan et al., 1995; Beckman and Koppenol, 1996; Bindokas et al., 1996; Mandir et al., 2000). Accordingly, PARP inhibitors have been shown to markedly reduce neuronal sensitivity to glutamate in cell culture preparations (Cosi et al., 1994; Mandir et al., 2000; Ying et al., 2001). Studies using animal models have similarly shown that PARP-1 inhibitors or gene disruption can reduce injury resulting from brain ischemia and trauma, which leads to glutamate neurotoxicity and oxidative stress (Eliasson et al., 1997; Endres et al., 1997; LaPlaca et al., 2001; Virag and Szabo, 2002).
In this study we investigated the possibility that hypoglycemic brain injury may be uniquely amenable to treatment with PARP inhibitors because, unlike ischemia and trauma, hypoglycemia produces an injury that is precipitated almost entirely by glutamate receptor stimulation. The findings suggest that PARP-1 activation is an important mediator of hypoglycemic neuronal death and that PARP inhibitors can be highly effective in preventing neuronal death when administered after hypoglycemia.
Materials and Methods
All studies were performed in accordance with protocols approved by the San Francisco Veterans Affairs Medical Center animal studies subcommittee. Rats were housed two per cage under conditions of constant temperature (18°C), light from 6:00 A.M. to 6:00 P.M., and ad libitum access to food and water except where noted otherwise. Reagents were obtained from Sigma-Aldrich (St. Louis, MO) except where noted otherwise.
Cell cultures. Cortical cultures were prepared as a mixture of neurons and astrocytes. Astrocyte cultures were prepared from cortices of 1-d-old wild-type (WT) Swiss-Webster mice (Simonsen, Gilroy, CA), as described previously (Ying et al., 2000), and plated on glass coverslips or 24-well Falcon culture plates. At confluence (day 12-15), cultures were used as plating surface for embryonic neurons. Embryonic (day 16) mouse forebrain cortices from either WT or PARP-1-/- mice (Wang et al., 1995) were dissociated as described previously (Ying et al., 2000) and plated on the confluent astrocyte cultures at a density of 1 × 106 cells/cm2. The cocultures were maintained in astrocyte-conditioned medium prepared by incubating Eagles Minimal Essential Medium (Invitrogen, Gaithersburg, MD) containing 5% fetal bovine serum (Hyclone, Logan, UT) with astrocyte cultures for 48 hr. Final glucose concentration was ∼5 mm. Partial medium exchanges were made twice weekly. The cultures were used at neuronal day 15-17 in vitro.
Glucose deprivation in vitro. Experiments were initiated by replacing the culture medium with a balanced salt solution (BSS). The BSS contained (in mm): 3.1 KCl, 134 NaCl, 1.2 CaCl2, 1.2 MgSO4, 0.25 KH2PO4, 15.7 NaHCO3, with or without 2 glucose. The pH was adjusted to 7.2 while the solution was equilibrated with 5% CO2 at 37°C. Osmolarity was verified at 290-310 mOsm with a Wescor vapor pressure osmometer (Logan, UT). Drugs were added from concentrated stocks prepared in BSS immediately before use and adjusted to pH 7.2 when necessary. Glucose deprivation was initiated by two washes in glucose-free BSS. The cultures were then incubated at 37°C in a 5% CO2 atmosphere until termination of glucose deprivation with 2 mm glucose. (Normal brain extracellular glucose concentration is 1-2 mm.) Drugs remained in the BSS until the cells were harvested for immunostaining or cell death determinations.
Cell death determinations. Neuron death in the astrocyte-neuron cultures was assessed by propidium iodide fluorescence at 18-24 hr after glucose restoration. Neurons were distinguished from the underlying astrocyte layer by their phase-bright, process-bearing morphology (Ying et al., 1999). Both the fluorescing dead neurons and nonfluorescing live neurons were counted, in a blinded manner, in four randomly picked optical fields. At least 400 neurons were counted in each well, and results from each well were expressed as percentage of neuronal survival.
Animal surgery and hypoglycemia. Procedures were adapted from those of Wieloch et al. (1984). Male Sprague Dawley rats (250-350 gm) were fasted overnight. Anesthesia was induced with 3% isoflurane in a 75:25 mixture of nitrous oxide and oxygen (Air Liquide America, Houston, TX). After intubation, rats were ventilated with a small animal respirator (Harvard Apparatus, South Natick, MA), and isoflurane content was reduced to 1%. The rats were kept under anesthesia during induction of hypoglycemia, during EEG isoelectricity, and for 3 hr after onset of glucose infusion to permit monitoring and control of temperature, blood pressure, and blood glucose concentration. A femoral artery line was placed for blood sampling and for blood pressure monitoring. Blood gases were measured at 1 hr intervals using an I-STAT machine (I-STAT, Princeton, NJ). Tidal volume and oxygen flow were adjusted to keep PaCO2 between 35 and 45 mmHg and PaO2 above 100 mmHg. Blood pressure and EEG were monitored continuously (BIOPAC System Inc., Santa Barbara, CA). For EEG monitoring, burr holes were made in the skull bilaterally over parietal cortex, and monopolar electrodes were placed beneath the dura. A reference needle was placed in neck muscle. A heating blanket/rectal probe servo-loop was used to maintain core temperature at 36.5-37.5°C until the rats had recovered from anesthesia, at which time they were euthermic. Hypoglycemia was induced by intraperitoneal injection of 15 U/kg of regular insulin (Novolin-R; Novo Nordisk, Princeton, NJ). Blood glucose was measured with a YSI 2700 glucose analyzer (YSI, Yellow Springs, OH) at 30 min intervals during induction of hypoglycemia and at 60 min intervals during recovery from hypoglycemia. Mean arterial blood pressure was maintained between 160 and 200 mmHg during the entire isoelectric period by adjusting the isoflurane concentration, and bradycardia during hypoglycemia was prevented with atropine (1 mg/kg; American Pharmaceutical Partners, Los Angeles, CA). Hypoglycemia was terminated by injection of 0.2 ml of 50% glucose via the femoral vein, followed by continuous infusion of 1:1 solution of 50% glucose and Krebs'-Henseleit buffer (1.5 ml/hr for 3 hr). This was supplemented by 1 ml of 25% glucose by intraperitoneal injection at 1 and 3 hr after beginning the intravenous glucose infusions. Rats were monitored under anesthesia for 3 hr after onset of glucose infusion. Anesthesia was discontinued after removal of the arterial and venous catheters, and the rats were returned to their cages when fully awake and ambulatory. Seizure activity was observed in 2 of 26 rats subjected to 45 min of isoelectric hypoglycemia and in 0 of 180 rats subjected to 30 min of isoelectric hypoglycemia. Rats with seizures were excluded from data analysis. The PARP inhibitors 3-aminobenzamide (3-AB) (Sigma-Aldrich) or 1,11b-dihydro-[2H] benzopyrano [4,3,2-de] isoquinolin-3-one (GPI 6150) (Guilford Pharmaceuticals, Baltimore, MD) were administered to the rats by intravenous or intraperitoneal injection, respectively, at the time of blood glucose correction or at designated later time points. 3-AB was dissolved 30 mg/ml in 10% ethanol, and GPI 6150 was dissolved 40 mg/ml in dimethyl sulfoxide (DMSO). The neuronal nitric oxide synthase (nNOS) inhibitor, 7-nitroindazole (7NI) (Sigma-Aldrich), was injected 50 mg/kg intraperitoneally 15 min after onset of EEG isoelectricity. 7NI was dissolved in peanut oil by sonication, 50 mg/ml (Yoshida et al., 1994). Control animals for these studies received the peanut oil vehicle only.
Histology. Rats were killed 7 d or 6 weeks after surgery. For hematoxylin-eosin staining, coronal 20 μm frozen sections were prepared, fixed in 70% ethanol, and conventionally stained (Suh et al., 2000). To quantify neuronal death, we collected five coronal sections from each animal 7 d after hypoglycemia, beginning 4.0 mm posterior to bregma and spaced 240 μm apart. A blinded observer counted the number of eosinophilic cells in the hippocampal CA1 cell field, subiculum, dentate gyrus, and perirhinal cortex in both hemispheres of each rat. A 10× microscopic field was centered on the structure of interest, and the number of eosinophilic neurons in the field was recorded for each section. Data from each animal were expressed as the mean number of degenerating (eosinophilic) cells per section. For Fluoro-Jade B staining, rats were transcardially perfused with a 0.9% saline flush followed by 4% paraformaldehyde (PFA) for 10 min, postfixation in 4% PFA for 1 hr, and then immersion in 30% sucrose for 24 hr. Coronal 20 μm sections were prepared on a cryostat and stained with Fluoro-Jade B (Histo-Chem Inc., Jefferson, AR) as described previously (Schmued and Hopkins, 2000). Fluoro-Jade B identifies degenerating cells in a manner similar to silver staining, probably by binding to multiple positive charge groups (Schmued and Hopkins, 2000). Fluorescent (degenerating) cells were detected with blue light (450-490 nm) excitation and a 515 nm barrier filter.
Immunostaining. Rats were killed at designated time points after hypoglycemia and transcardially perfused with a 0.9% saline flush followed by 4% PFA for 10 min. Brains were postfixed in 4% PFA for 1 hr, followed by 30% sucrose for 24 hr. Coronal 60 μm sections were prepared and immunostained as described previously (Ying et al., 2001), with modifications. Sections were blocked and permeablized with 10% goat serum/0.1% Triton X-100 in pH 7.4 PBS. Mouse anti-poly(ADP-ribose) monoclonal antibody (Trevigen, Gaitherburg, MD) and rabbit anti-nitrotyrosine polyclonal antibody (Chemicon, Temecula, CA) were added at 1:2000 and 1:5000 dilutions, respectively, and incubated at 4°C overnight. After washing, sections were incubated with Alexa Fluor 488-conjugated goat anti-mouse or Alexa Fluor 594-conjugated goat anti-rabbit IgG (Molecular Probes, Eugene, OR) at 1:500 dilution for 1 hr at room temperature. The sections were mounted and photographed with a Leica confocal laser-scanning microscope. Negative controls were prepared by omitting the primary antibodies. Immunostaining for poly(ADP)ribose in cell cultures was performed identically except that cultures were fixed in ice-cold 10% trichloroacetic acid, dehydrated in ethanol, and air dried. Propidium iodide was applied to the cultures after fixation to obtain nuclear staining.
Behavioral assessments. Rats were treated with hypoglycemia, hypoglycemia followed by 3-AB, or sham surgery. Six weeks later the rats were assessed by the Morris water-maze task to evaluate spatial learning and memory (Morris, 1984). A total of 24 rats were studied, prepared as three groups of 8 rats. To minimize the effects of social influences on behavior, rats were housed individually in the testing room beginning 3 d before testing. As a first step, spontaneous open field activity was assessed to establish any locomotor differences. Rats were placed individually in a Plexiglas enclosure (40 × 40 inches) equipped with two rows of infrared photocell panels interfaced with a computer (Hamilton Kinder, San Diego, CA). The bottom row detected horizontal exploratory activity, and the top row detected vertical exploratory activity or rearing. Light path breaks were recorded and assessed for measurements of the number of movements, active times, path length, and rearing events. On each of 3 consecutive days, open field activity was recorded for 10 min after an initial 1 min adaptation period. No significant differences were observed between the three treatment groups with respect to spontaneous open field activity.
Spatial learning and memory were tested in a circular pool (180 cm in diameter, 50 cm deep) filled with opaque water of constant temperature (24°C). The pool was divided into four quadrants, and the platform was always placed in the center of the designated quadrant. The rats were trained first to locate a visible platform (days 1-2) and then to locate a hidden platform (days 3-5). In a subsequent trial, the hidden platform was removed (probe trial, day 5). During the visible training, the platform was rotated clockwise to a new quadrant in each session. The platform remained in the same quadrant throughout all the sessions during hidden platform training. The rats received two training sessions per day for 5 consecutive days. Each session consists of three 1 min trials with a 10 min intertrial interval. The interval between the two daily sessions was 3 hr. Once the rats located the platform they were allowed to remain on it for 10 sec. Rats that failed to find the platform within 1 min were manually placed on the platform for 15 sec. Time to reach the platform (latency), distance traveled, and swim speed were recorded with an EthoVision video tracking system (Noldus Information Technology, Leesburg, VA) set to analyze two samples per second. Two rats were excluded from the analysis because of slow swim speed (<10 cm/sec) and significant periods of passive floating. Both of these rats had been randomized to the sham surgery group.
Statistical analyses. Data are expressed as means ± SEM. The water-maze data (swim speed and time to platform) were analyzed by mixed-model regression using SAS version 9 Proc MIXED (SAS Institute, Cary, NC), followed by post hoc tests adjusted for multiple comparisons when appropriate. All other data were assessed by one-way ANOVA followed by either the Tukey-Kramer test for multiple comparisons between groups or the Dunnett's test for comparisons of multiple groups against a control group.
Results
PARP inhibition attenuates neuronal cell death caused by glucose deprivation in vitro
Glucose deprivation caused neuronal death in a time-dependent manner, with >90% of neurons in the WT cultures killed by glucose-free intervals of 5 hr or longer (Fig. 1A). Astrocytes in these cultures showed no cell death, as judged by propidium iodide exclusion. The sensitivity to glucose deprivation was reduced in neurons prepared from PARP-1-/- mice. To exclude the possibility that this difference was caused by some other property of the PARP-1-/- neurons, we tested the effect of the PARP inhibitor 3,4-dihydro-5-[4-(1-piperidinyl)butoxy]-1(2H)-isoquinolinone (DPQ) in WT neurons subjected to glucose deprivation. As shown in Figure 1B, 25 μm DPQ reduced neuronal death to an extent similar to that observed with PARP-1-/- neurons. To directly confirm that glucose deprivation causes PARP activation, cell cultures were immunostained for the presence of poly(ADP-ribose), the enzymatic product of PARPs. Poly(ADP-ribose) was undetectable under control conditions but prominent in neuronal nuclei after 5 hr of glucose deprivation (Fig. 1C). Poly(ADP-ribose) accumulation was suppressed almost completely in cultures treated with DPQ.
Figure 1.
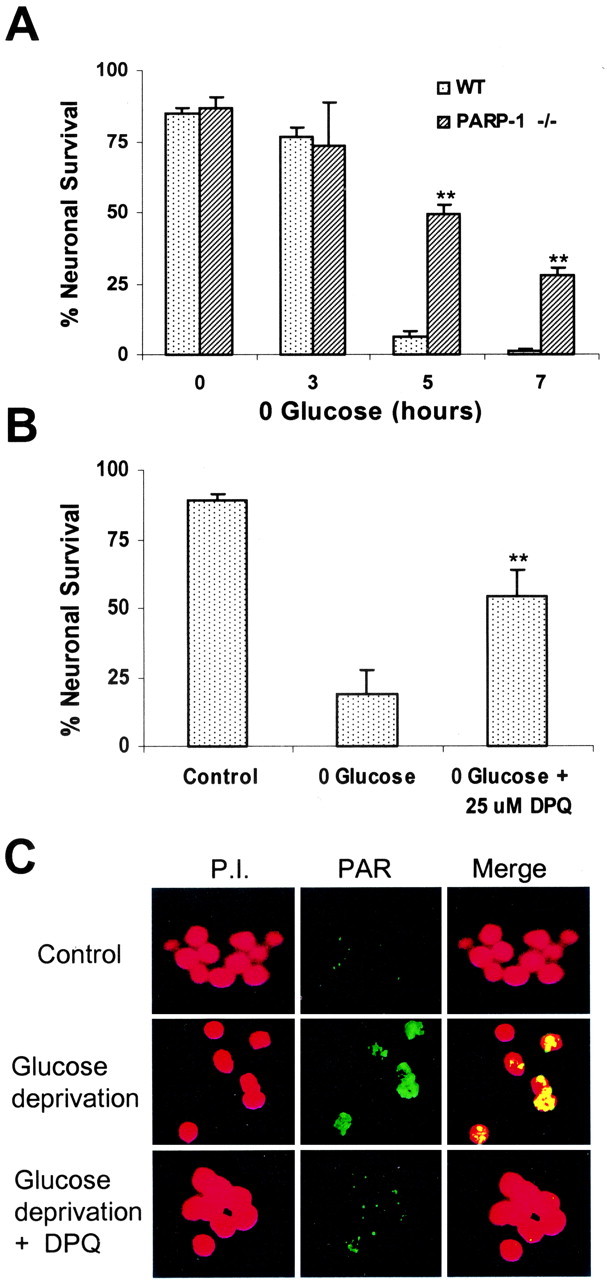
PARP inhibition reduces neuronal death after glucose deprivation. A, Survival of cortical neurons prepared from PARP1-1-/- and WT mice, assessed 18 hr after the designated intervals of glucose deprivation. B, Effect of the PARP inhibitor DPQ on survival of WT neurons exposed to 5 hr of glucose deprivation. DPQ remained in the culture medium until cell death was assessed (24 hr). C, Confocal images showing poly(ADP-ribose) formation in WT neurons after sham washes (control) and after 5 hr glucose deprivation. Poly(ADP-ribose), detected by immunostaining (green), was increased by glucose deprivation; this increase was blocked by DPQ. Neuronal nuclei are counterstained with propidium iodide (red). Yellow color in the overlaid images indicates poly(ADP-ribose) in the nuclei. n = 3-4; **p < 0.01. The photomicrographs are representative of three independent experiments.
Hypoglycemia-induced neuronal death in vivo
We used a rat model of insulin-induced hypoglycemia because reproducible hypoglycemic brain injury is difficult to achieve in the mouse. Because plasma glucose concentrations provide only a rough correlation with brain glucose concentrations and ultimate brain injury, fixed intervals of EEG isoelectricity were used to generate a reproducible injury pattern (Auer et al., 1984a; Friberg et al., 1998). As shown in Figure 2, the EEG patterns passed through progressive stages characteristic of severe hypoglycemia. The isoelectric EEG pattern developed after blood glucose concentrations had fallen below 0.5 mm for several minutes. EEG recovery began 12-15 min after onset of glucose infusion.
Figure 2.

EEG changes during hypoglycemia. A, Normal EEG pattern. B, With onset of hypoglycemia, EEG shows slowing and increased amplitude. C, Further slowing and increased amplitude. D, Paroxysmal activity is evident. E, Burst-suppression pattern. F, Isoelectricity. G, Thirty minutes after glucose infusion; EEG is beginning to normalize.
Hematoxylin-eosin staining of brain sections prepared 7 d after hypoglycemia showed that 30 min of hypoglycemia-induced EEG isoelectricity caused substantial neuronal death in the CA1 and subiculum regions of the hippocampus (Fig. 3), the brain regions most vulnerable to hypoglycemic injury (Auer et al., 1984a,b). A second marker, Fluoro-Jade B (Schmued and Hopkins, 2000), was used to confirm the pattern of cell death observed with standard hematoxylin-eosin histology (Fig. 3A).
Figure 3.

Hypoglycemic damage to hippocampal CA1 and subiculum is reduced by 3-AB. A, Photomicrographs of hippocampal cell fields from rats subjected to 30 min of isoelectric hypoglycemia show numerous degenerating (dead) neurons, identified as pyknotic, pink-red cells with hematoxylin-eosin staining, and as fluorescent cells with Fluoro-Jade B staining. Rats treated with 30 mg/kg of the PARP inhibitor 3-AB at the time of glucose correction sustained less neuronal death in both the CA1 (top panels) and subiculum (bottom panels). Scale bar, 100 μm. B, The effect of 3-AB was dose dependent, with maximal efficacy at 30 mg/kg. C, Increasing the isoelectric period from 30 to 45 min produced more extensive neuronal death in both the CA1 and subiculum but did not diminish the effect of 3-AB. D, 3-AB also reduced neuronal death when administered hours after correction of hypoglycemia. n = 3-8 for each condition. *p < 0.05 versus no 3-AB.
PARP inhibitors reduce neuronal death after hypoglycemia in vivo
3-AB inhibits PARPs by competing with NAD+ at the catalytic site (Purnell and Whish, 1980; Virag and Szabo, 2002). 3-AB was administered to rats at a dose of 30 mg/kg by intravenous injection after 30 min EEG isoelectricity, concurrent with intravenous infusion of glucose. Both the hematoxylin-eosin and the Fluoro-Jade B staining methods showed a large reduction in hippocampal neuronal death in rats treated with the 30 mg/kg 3-AB (Fig. 3A). Treatment groups did not differ with respect to blood glucose concentrations before, during, or after hypoglycemia (Table 1), and sham-treated rats given glucose along with insulin to prevent hypoglycemia showed no neuronal death (data not shown). The effect of 3-AB on neuronal survival was maximal at 30 mg/kg (Fig. 3B). At this dose, neuronal death was reduced by 90 ± 9% in the CA1 region and 85 ± 9% in the subiculum compared with rats receiving glucose alone.
Table 1.
Arterial blood glucose concentrations (micromolar)
|
Treatment group |
|
Before insulin |
During isoelectricity |
30 min after glucose infusion |
|---|---|---|---|---|
| No drug | (n=8) | 3.98 ± 0.35 | 0.45 ± 0.06 | 8.01 ± 0.43 |
| 3-AB, 10 mg/kg | (n=3) | 3.88 ± 0.63 | 0.53 ± 0.37 | 8.52 ± 1.09 |
| 3-AB, 30 mg/kg | (n=8) | 4.09 ± 0.28 | 0.53 ± 0.08 | 7.57 ± 0.96 |
| 3-AB, 90 mg/kg | (n=6) | 3.97 ± 0.14 | 0.49 ± 0.08 | 7.88 ± 0.50 |
| DMSO | (n=9) | 3.92 ± 1.58 | 0.46 ± 0.07 | 7.96 ± 4.74 |
| GPI 6150, 10 mg/kg, in DMSO
|
(n=7)
|
3.87 ± 1.11
|
0.45 ± 0.10
|
8.50 ± 2.66
|
Values are means ± SEM.
To determine whether this neuroprotective effect would also be obtained with more severe hypoglycemic injury, we extended the isoelectric period from 30 to 45 min in a second group of rats. Hypoglycemia of this duration killed >70% of the neurons in both the CA1 and subiculum regions, but the neuroprotective effect of 3-AB was not diminished (Fig. 3C). We also tested the effect of 3-AB when administered at time points after glucose correction because, in the emergency care setting, drug treatment might often be delayed until after glucose infusion has begun. These studies showed significant reductions in CA1 hippocampal neuron death with 3-AB treatment delayed for up to 2 hr after blood glucose correction (Fig. 3D).
Neurons in the outer layers of cerebral cortex and hippocampal granule cells are also highly vulnerable to hypoglycemic injury (Auer and Siesjo, 1993). 3-AB (30 mg/kg) also reduced death in these cell populations (Fig. 4A). Survival of cortical neurons, like hippocampal CA1 and subiculum neurons, was improved by 3-AB administration up to 2 hr after blood glucose correction (Fig. 4B); however, this was not true of the dentate granule cells, in which 3-AB added at time points of 1 hr or longer after glucose correction had no significant effect on cell survival.
Figure 4.
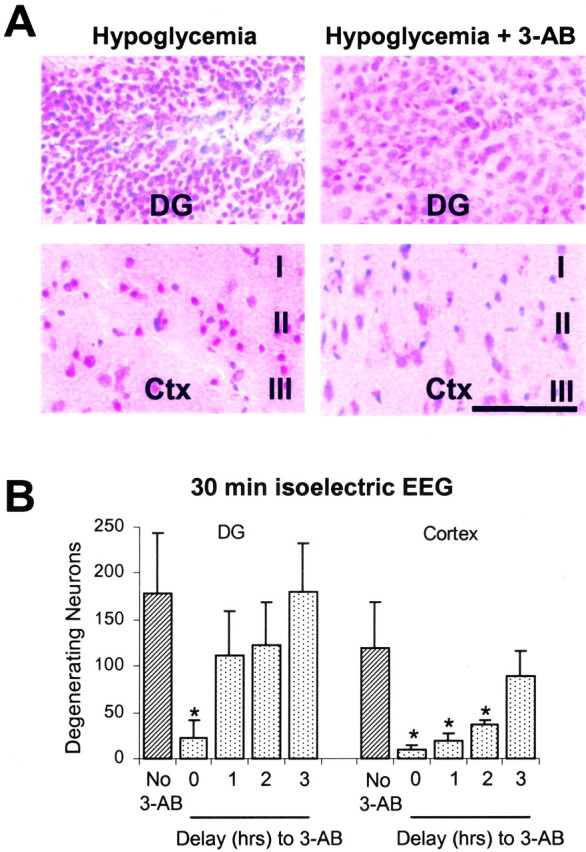
Hypoglycemic damage to dentate granule and cortical neurons is reduced by 3-AB. A, Photomicrographs of dentate granule (DG) cell and perirhinal cortex (Ctx) fields from rats subjected to 30 min of isoelectric hypoglycemia show numerous degenerating (dead) neurons, as evidenced by shrunken cell bodies stained dark red. Rats treated with 30 mg/kg of the PARP inhibitor 3-AB at the time of glucose correction sustained less neuronal death in both the dentate granule cell area (top panels) and cortex (bottom panels). Scale bar, 100 μm. B, 3-AB also reduced the death of cortical neurons but not dentate granule cell neurons when administered at time points after blood glucose correction. n = 3-8 for each condition. *p < 0.05 versus no 3-AB.
Hypoglycemia studies were also performed in a separate group of rats using GPI 6150 to confirm that the neuroprotective effects of 3-AB were caused by PARP inhibition and not some other action that could influence neuronal survival. GPI 6150 is a more potent PARP inhibitor that is chemically unrelated to 3-AB (Zhang et al., 2000; LaPlaca et al., 2001; Southan and Szabo, 2003). GPI 6150 produced large reductions in hypoglycemic neuronal death in both the CA1 and subiculum regions (Fig. 5A,B), similar to that observed with 3-AB. To assess the long-term prevention effects of 3-AB on hypoglycemia-induced neuronal cell death, brains from a separate group of rats (the rats used for behavioral studies) were harvested 7 weeks after the hypoglycemia. Hematoxylin-eosin staining of sections from these brains showed atrophy and loss of neurons in the CA1, dentate gyrus, and subiculum of the hippocampus and in layer II of the perirhinal cortex (Fig. 6). These changes were substantially attenuated in the rats treated with 3-AB after hypoglycemia, suggesting that neuronal death is prevented rather than simply delayed by this intervention.
Figure 5.
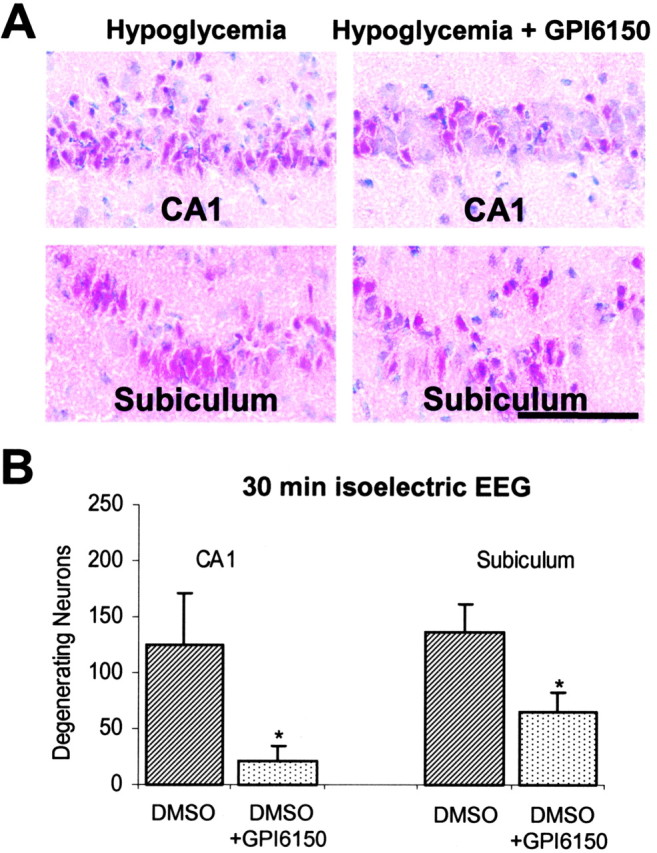
The PARP inhibitor GPI 6150 reduces neuronal death after hypoglycemia. A, Photomicrographs show degenerating hippocampal neurons in rats treated with glucose or with glucose plus GPI 6150 after 30 min of hypoglycemic isoelectricity. Both groups also received the DMSO vehicle used to deliver GPI 6150. Scale bar, 100 μm. B, Quantification of neuronal death showed the effect of GPI 6150 to be significant in both CA1 and subiculum cell fields. n = 7-9 for each condition. *p < 0.05 versus DMSO alone.
Figure 6.
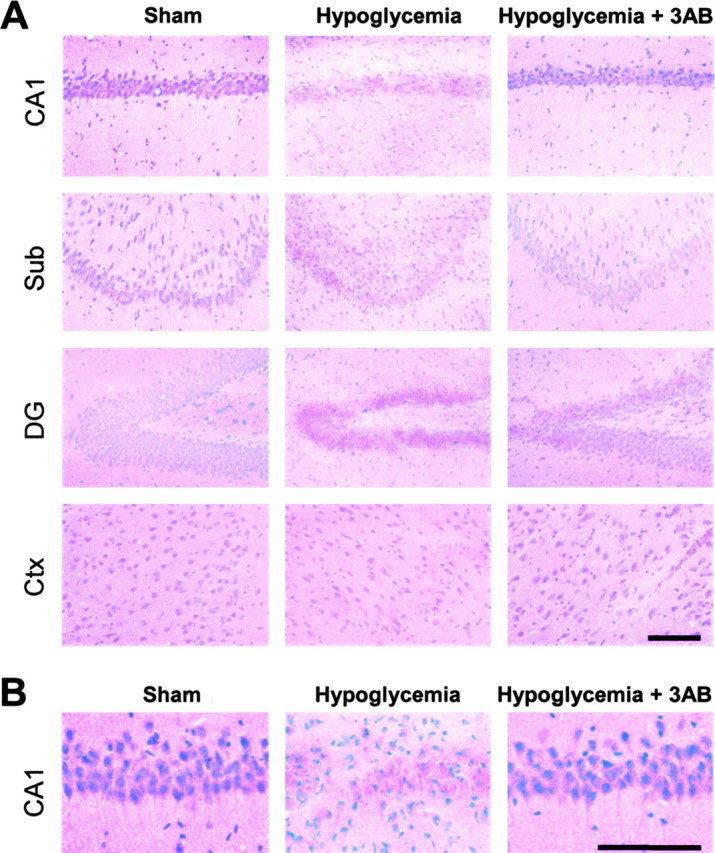
Long-term neuroprotective effects of 3-AB after hypoglycemia. A, Hematoxylin-eosin staining of brain sections harvested 7 weeks after sham hypoglycemia, 30 min of hypoglycemic EEG isoelectricity, or 30 min of hypoglycemic EEG isoelectricity followed by 30 mg/kg 3-AB. Hypoglycemia caused a loss of neurons in the CA1, subiculum, and dentate gyrus regions of the hippocampus and in layer II of perirhinal cortex. Sections from 3-AB-treated rats showed improved neuronal survival in each of these regions. B, Higher magnification of hippocampal CA1 pyramidal area shows 3-AB-treated rats to have nearly normal cytoarchitecture 7 weeks after hypoglycemia. Sections are representative of four rats. Scale bars, 100 μm.
Hypoglycemia induces poly(ADP-ribose) and nitrotyrosine formation in vulnerable neurons
The effect of the two dissimilar PARP inhibitors on neuron survival after hypoglycemia suggests that PARP activation is an important downstream mechanism of cell death after hypoglycemia. To directly confirm that hypoglycemia causes PARP activation, brains taken at serial time points after hypoglycemia were immunostained for the presence of poly(ADP-ribose) (Endres et al., 1997; Ying et al., 2001), the enzymatic product of PARP, in the same manner as done with the cortical cell cultures. These studies demonstrated PARP activation in the neuron populations that are vulnerable to hypoglycemic injury. As shown in Figure 7A, poly(ADP-ribose) accumulation was increased only modestly in brains harvested immediately after termination of isoelectricity (0 hr) but became markedly increased by 3 hr after isoelectricity and declined slowly after that point. Poly(ADP-ribose) accumulation was reduced in rats treated with 30 mg/kg 3-AB at the time of glucose correction (Fig. 7A). Poly(ADP-ribose) accumulation was not observed in glia, in neuronal populations not highly vulnerable to hypoglycemia, or in animals subjected to sham hypoglycemia (data not shown).
Figure 7.
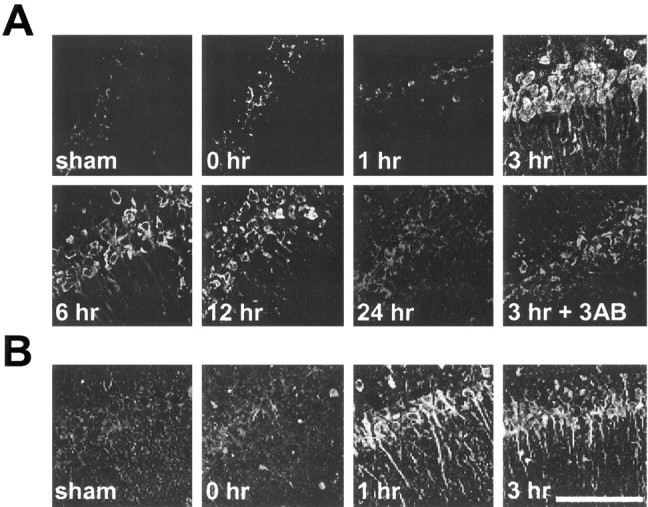
Hypoglycemia-induced poly(ADP-ribose) and nitrotyrosine formation in CA1 hippocampus. A, Poly(ADP-ribose) immunoreactivity was increased only modestly at the termination of 30 min of EEG isoelectricity (0 hr) but was increased markedly 3 hr after isoelectricity and declined slowly after that point. Poly(ADP-ribose) formation at 3 hr was reduced by administration of 3-AB at the time of glucose correction (3 hr + 3AB). B, Nitrotyrosine formation in CA1 neurons was also negligible immediately after 30 min EEG isoelectricity but was robust by 1 hr after glucose correction. Photomicrographs are representative of three rats under each condition. Scale bar, 100 μm.
Previous studies suggest that peroxynitrite formation is an important link between glutamate receptor stimulation and PARP activation (Zhang et al., 1994; Szabo and Dawson, 1998; Mandir et al., 2000). Nitrotyrosine is a useful marker of peroxynitrite formation (Beckman and Koppenol, 1996). Evidence supporting a role for peroxynitrite as a link between glutamate excitotoxicity and PARP activation in hypoglycemia was provided here by nitrotyrosine immunostaining in sections from the same rat brains used for poly(ADP-ribose) immunostaining. Nitrotyrosine immunoreactivity was prominent in CA1 hippocampal neurons at 1 hr after glucose correction and became less apparent at later time points (Fig. 7B). Double-label immunostaining for nitrotyrosine and poly(ADP-ribose) in CA1 neurons (Fig. 8A) showed that almost all CA1 neurons immunoreactive for nitrotyrosine were also immunoreactive for poly(ADP-ribose). These markers were also examined in piriform cortex, which is less vulnerable to hypoglycemic injury. In contrast to CA1, neurons in layers 3 and 4 of piriform cortex showed only modest nitrotyrosine and barely detectable poly(ADP-ribose) immunoreactivity (Fig. 8B), consistent with the relative vulnerability of these structures to hypoglycemic injury. Intraperitoneal injection of the neuronal nNOS inhibitor 7NI (50 mg/kg) during the isoelectric EEG period blocked both nitrotyrosine formation and poly(ADP-ribose) accumulation in the CA1 hippocampal area when evaluated at 2 hr after hypoglycemia (Fig. 8C). 7NI also showed significant neuroprotective effects in the hippocampus and cerebral cortex when evaluated at 7 d after hypoglycemia (Fig. 8D), suggesting that nNOS activation is a key intermediary step between hypoglycemia and PARP activation.
Figure 8.
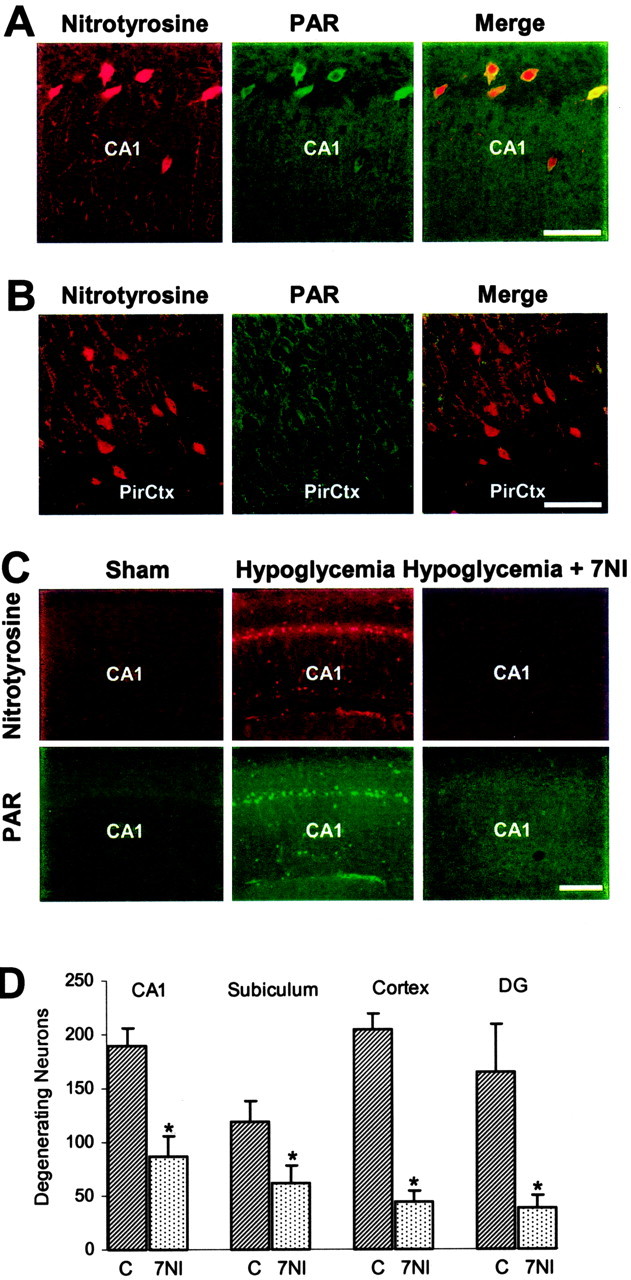
Effects of 7NI on nitrotyrosine and poly(ADP-ribose) accumulation in hippocampal CA1 neurons after hypoglycemia. A, Double-label immunostaining for nitrotyrosine (red) and PAR (green) indicated that almost all CA1 neurons immunoreactive for nitrotyrosine were also immunoreactive for poly(ADP-ribose). Panels are representative of three rats in each group. Scale bar, 100 μm. B, Neurons in layers III and IV of piriform cortex (PirCtx), which are relatively resistant to hypoglycemia, show less intense nitrotyrosine staining and barely detectable PAR staining after hypoglycemia. Sections are from the same brain as shown in A. Scale bar, 100 μm. C, Hypoglycemia increased both nitrotyrosine and PAR immunoreactivity in CA1 neurons 2 hr after glucose correction. This was prevented in rats treated with the nNOS inhibitor 7NI (50 mg/kg) administered during the EEG isoelectric period. Scale bar, 200 μm. D, Histological assessment 7 d after hypoglycemia showed substantially less neuronal death in vulnerable brain regions of the 7NI-treated rat brains. Cortex, Layer II of perirhinal cortex; DG, hippocampal dentate granule cells. n = 6-7; *p < 0.05.
PARP inhibition prevents spatial learning and memory deficits after hypoglycemia
Rats that had been subjected to hypoglycemia were not distinguishable from sham-operated rats with respect to spontaneous activity or locomotor speed, but significant learning and memory deficits were identified with behavioral testing 6 weeks after the hypoglycemia. These rats were slower to find the platform during the visible platform trial of the water maze and slower to learn the position of the platform during the hidden platform trial (Fig. 9A). Rats treated with 3-AB after hypoglycemia did not show any significant deficit in their ability to locate the platform compared with sham-operated animals during either the visible or hidden platform trials (p = 0.80 and 0.44, respectively). There was no significant difference in swim speed among the treatment groups during visible or hidden platform sessions (p = 0.11 and 0.96, respectively). After the hidden platform training, rats were subjected to a probe trial, a putative measure of memory retention. In the probe trial, both the sham-operated and 3-AB-treated animals showed memory retention and spent significantly more time searching in the target quadrant than in any other quadrant (p < 0.005 and < 0.05, respectively), whereas the hypoglycemic rats not treated with 3-AB failed to spend significantly more time searching in the target quadrant (Fig. 9B).
Figure 9.
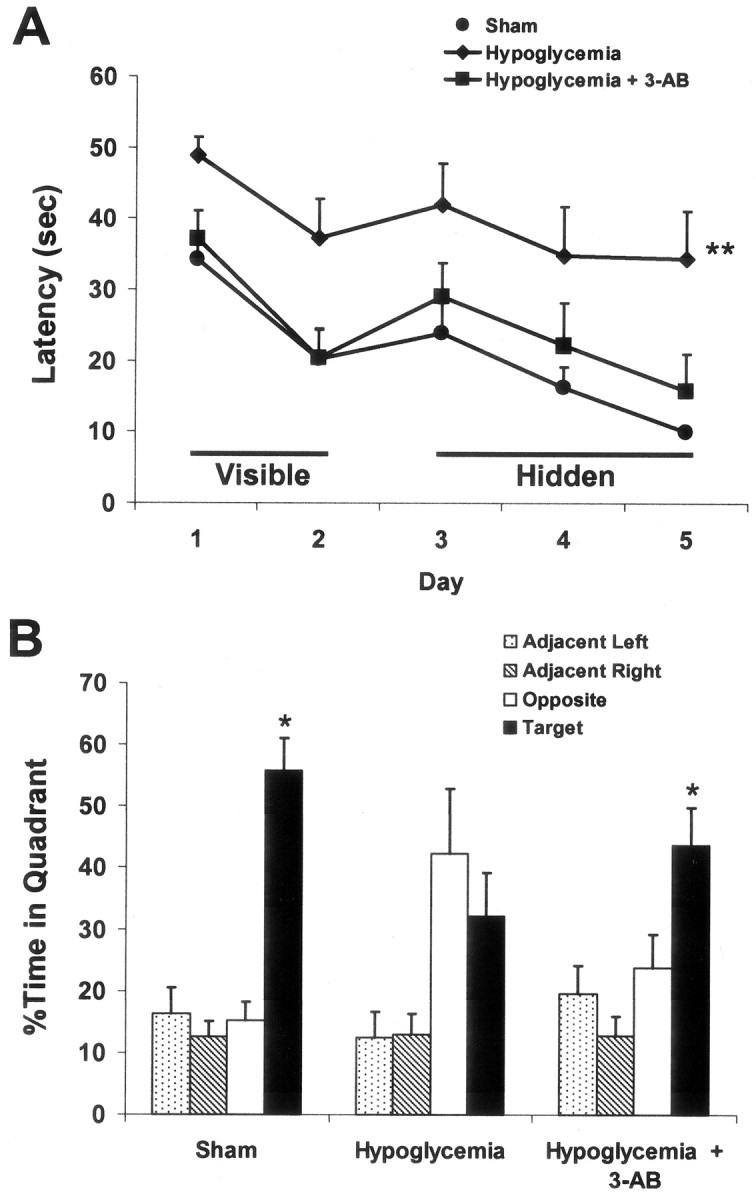
3-AB prevented hypoglycemia-induced spatial learning and memory impairments assessed 6 weeks after hypoglycemia. Rats were treated with 30 min EEG isoelectricity with (n = 8) or without (n = 8) 30 mg/kg 3-AB administered at the time of glucose correction, or with sham surgery only (n = 6). A, During both the visible and hidden platform sessions, rats subjected to hypoglycemia showed a significant impairment in ability to locate the platform when compared with either the sham-treated or 3-AB-treated animals (**p < 0.01). B, In the probe trial, in which the platform was removed from the tank, both sham-operated and 3-AB-treated groups spent significantly more time searching in the target quadrant than in any of the other quadrants (*p < 0.05), whereas the hypoglycemia only group did not.
Discussion
Hypoglycemic neuronal death is not a direct and immediate consequence of low-energy substrate but results instead from a cascade of events precipitated by the lack of substrate. Sustained activation of glutamate receptors has been established as a necessary upstream event in this cascade (Auer and Siesjo, 1993). The results of the present study suggest that PARP activation is an important downstream mediator of hypoglycemic neuronal death and a target for therapeutic intervention.
PARP activation was demonstrated by the accumulation of poly(ADP-ribose) in both cell culture and in vivo models of hypoglycemia and by the suppression of poly(ADP-ribose) accumulation by PARP inhibitors. Poly(ADP-ribose) accumulation has similarly been used as a marker of PARP activation after brain ischemia (Tokime et al., 1998; Love et al., 1999). Poly(ADP) ribose immunoreactivity was seen in the cytosol as well as in the nucleus at later time points after hypoglycemia (Fig. 7). This pattern has been observed in other settings (Tokime et al., 1998; Love et al., 1999; Ducrocq et al., 2000; Weise et al., 2001; Ying et al., 2001) and may reflect movement of cleaved poly(ADP-ribose) fragments from the nucleus into the cytosol. Specificity of the anti-polyADP-ribose antibody is confirmed by the absence of poly(ADP)-ribose immunoreactivity under sham conditions and its suppression by PARP-1 gene disruption or pharmacological inhibition.
The immunostaining approach can be used to identify cells in which significant PARP activation has occurred, but it cannot be used to quantify PARP activation because of the rapid and possibly variable rate of poly(ADP-ribose) turnover (Alvarez-Gonzalez and Althaus, 1989; Jacobson and Jacobson, 1999; Ying et al., 2001). The effects of PARP inhibitors provide a more quantitative measure of the importance of PARP activation on hypoglycemic neuronal death. In cell culture, both PARP-1-/- neurons and WT neurons treated with the PARP inhibitor DPQ showed increased resistance to glucose deprivation. The roughly equivalent effects of PARP-1 gene knock-out and the general PARP inhibitor DPQ suggest that PARP-1 is the major PARP family member mediating hypoglycemic neuronal death.
Comparable results were obtained in the rat model of hypoglycemic brain injury. The pattern and extent of neuronal injury induced by hypoglycemia were similar to that reported previously (Auer et al., 1984a; Friberg et al., 1998). The PARP inhibitor 3-AB reduced neuronal death by >85% in the cortex, CA1, and dentate granule cell fields and by >50% in the subiculum. The structurally unrelated PARP inhibitor, GPI 6150, showed similar efficacy in regions where the two agents were compared. Moreover, the effect of 3-AB on neuronal survival is not unique to the injury pattern produced by the 30 min isoelectric interval because a comparable effect was observed when the isoelectric interval was extended to 45 min (Fig. 3C). It is likely that these results may underestimate the role of PARP activation in hypoglycemic neuronal death because the PARP inhibitors were administered only after the hypoglycemic intervals and consequently were not active in the rat brains during the period of EEG isoelectricity.
Histological assessments were also performed at 7 weeks after hypoglycemia in the rats used for behavioral and cognitive studies. The assessments at this later time point, like those at the 7 d time point, showed neuronal loss to be substantially attenuated in rats treated with 3-AB, suggesting that neuronal death was prevented rather than simply delayed by this intervention. Behavioral and cognitive assessments are useful adjuncts to histological end points because interventions that preserve neuronal survival may not necessarily preserve normal neuronal function (Corbett and Nurse, 1998). The brain regions most vulnerable to hypoglycemia are important for learning and memory. Accordingly, patients who recover from severe hypoglycemia may be left with difficulties in cognition, particularly short-term memory, out of proportion to their gross motor disability (Langan et al., 1991). The behavioral studies presented here showed this pattern to hold for the rat model also. The rats showed no impairment in locomotion but did exhibit deficits in a standard measure of learning and spatial memory. These findings validated the utility of the rat model and permitted assessment of the effects of PARP inhibitor on the cognitive deficits. Rats treated with 3-AB after hypoglycemia exhibited much better performance in the cognitive testing than did rats treated with glucose alone. Together, the behavioral and histological studies performed 6-7 weeks after hypoglycemia suggest that the neuroprotection provided by PARP inhibition leads to long-lasting preservation of neuronal survival and function.
Previous studies have identified peroxynitrite as an important intermediate between glutamate receptor activation and PARP activation (Zhang et al., 1994; Tokime et al., 1998; Mandir et al., 2000). Glutamate or aspartate activation of neuronal NMDA-type glutamate receptors leads to production of nitric oxide and superoxide, which combine to form peroxynitrite and related species with high reactivity toward DNA and other cell constituents (Zhang et al., 1994; Beckman and Koppenol, 1996; Bindokas et al., 1996; Szabo et al., 1996). Nitrotyrosine is produced as a result of peroxynitrite reaction with tyrosine residues and thus serves as a useful marker for peroxynitrite formation (Beckman and Koppenol, 1996). In the present study, nitrotyrosine immunoreactivity was induced by hypoglycemia in the same CA1 neuron population that accumulated poly(ADP-ribose) and was rescued by PARP inhibitors after hypoglycemia. In addition, administration of the nitric oxide synthase inhibitor 7NI (Yoshida et al., 1994; Tokime et al., 1998) reduced both nitrotyrosine and poly(ADP-ribose) accumulation and improved neuronal survival. Given the established role of glutamate excitotoxicity in hypoglycemic brain injury, the present results suggest that in hypoglycemia as in other settings, peroxynitrite formation is an important intermediate between glutamate receptor stimulation and PARP-mediated neuronal death.
Glutamate receptor antagonists effectively prevent neuronal death when administered before the onset of hypoglycemia but become substantially less effective when administered after hypoglycemia has already occurred (Wieloch, 1985; Nellgard and Wieloch, 1992). Presumably this is because the cell death cascade triggered by glutamate receptor stimulation has already been set in motion. By contrast, the PARP inhibitor 3-AB produced large reductions in cell death when administered immediately after hypoglycemia and also exhibited significant effects in most brain regions when administered as long as 2 hr after blood glucose correction. This suggests that PARP activation continues in most brain regions for extended periods of time after blood glucose has been normalized. Given this efficacy of PARP inhibitors at delayed time points, it may be significant that both poly(ADP-ribose) and nitrotyrosine accumulation did not increase until after blood glucose concentrations were corrected, and further that poly(ADP-ribose) accumulation was nearly abolished by 3-AB administered at the time of blood glucose correction (Fig. 7). The reason for the delayed appearance of these markers is not established by these studies but may be related to delayed calcium dysregulation after NMDA receptor stimulation (Castilho et al., 1998). Reduced delivery of substrates to neuronal mitochondria during hypoglycemia may limit mitochondrial electron flux and superoxide production (and hence peroxynitrite formation) until after the glucose supply is reestablished. Similarly, Wieloch and colleagues (Friberg et al., 1998; Ferrand-Drake et al., 1999) have shown that DNA fragmentation, which can trigger PARP activation, is a delayed consequence of hypoglycemia and that inhibition of mitochondrial permeability transition with cyclosporin A can reduce DNA fragmentation and reduce hypoglycemia-induced neuronal death.
The present results suggest a pivotal role for PARP-1 activation in the development of hypoglycemic neuronal death and provide proof of principle that PARP inhibition can rescue neurons that would otherwise go on to die. Hypoglycemic brain injury may be a clinical disorder especially amenable to treatment with PARP inhibitors, given the effectiveness of these agents at time points after the hypoglycemic insults.
Footnotes
This work was supported by the Juvenile Diabetes Foundation, the National Institutes of Health (RO1 NS41421), and the Department of Veterans Affairs. We thank Dr. Charles McCulloch for assistance with the statistical analyses and David Burns for expert technical assistance.
Correspondence should be addressed to Dr. Raymond A. Swanson, (127) Neurology, Veterans Affairs Medical Center, 4150 Clement Street, San Francisco, CA 94121. E-mail: ray@itsa.ucsf.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/2310681-10$15.00/0
References
- Alvarez-Gonzalez R, Althaus FR ( 1989) Poly(ADP-ribose) catabolism in mammalian cells exposed to DNA-damaging agents. Mutat Res 218: 67-74. [DOI] [PubMed] [Google Scholar]
- Auer R, Kalimo H, Olsson Y, Wieloch T ( 1985) The dentate gyrus in hypoglycemia: pathology implicating excitotoxin-mediated neuronal necrosis. Acta Neuropathol (Berl) 67: 279-288. [DOI] [PubMed] [Google Scholar]
- Auer RN, Siesjo BK ( 1993) Hypoglycaemia: brain neurochemistry and neuropathology. Baillieres Clin Endocrinol Metab 7: 611-625. [DOI] [PubMed] [Google Scholar]
- Auer RN, Olsson Y, Siesjo BK ( 1984a) Hypoglycemic brain injury in the rat. Correlation of density of brain damage with the EEG isoelectric time: a quantitative study. Diabetes 33: 1090-1098. [DOI] [PubMed] [Google Scholar]
- Auer RN, Wieloch T, Olsson Y, Siesjo BK ( 1984b) The distribution of hypoglycemic brain damage. Acta Neuropathol 64: 177-191. [DOI] [PubMed] [Google Scholar]
- Beckman JS, Koppenol WH ( 1996) Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol 271: C1424-1437. [DOI] [PubMed] [Google Scholar]
- Bindokas VP, Jordan J, Lee CC, Miller RJ ( 1996) Superoxide production in rat hippocampal neurons: selective imaging with hydroethidine. J Neurosci 16: 1324-1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burzio LO, Riquelme PT, Koide SS ( 1979) ADP ribosylation of rat liver nucleosomal core histones. J Biol Chem 254: 3029-3037. [PubMed] [Google Scholar]
- Butcher SP, Sandberg M, Hagberg H, Hamberger A ( 1987) Cellular origins of endogenous amino acids released into the extracellular fluid of the rat striatum during severe insulin-induced hypoglycemia. J Neurochem 48: 722-728. [DOI] [PubMed] [Google Scholar]
- Castilho RF, Hansson O, Ward MW, Budd SL, Nicholls DG ( 1998) Mitochondrial control of acute glutamate excitotoxicity in cultured cerebellar granule cells. J Neurosci 18: 10277-10286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett D, Nurse S ( 1998) The problem of assessing effective neuroprotection in experimental cerebral ischemia. Prog Neurobiol 54: 531-548. [DOI] [PubMed] [Google Scholar]
- Cosi C, Suzuki H, Milani D, Facci L, Menegazzi M, Vantini G, Kanai Y, Skaper SD ( 1994) Poly(ADP-ribose) polymerase: early involvement in glutamate-induced neurotoxicity in cultured cerebellar granule cells. J Neurosci Res 39: 38-46. [DOI] [PubMed] [Google Scholar]
- D'Amours D, Desnoyers S, D'Silva I, Poirier GG ( 1999) Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J 342: 249-268. [PMC free article] [PubMed] [Google Scholar]
- Davis EA, Keating B, Byrne GC, Russell M, Jones TW ( 1998) Impact of improved glycaemic control on rates of hypoglycaemia in insulin dependent diabetes mellitus. Arch Dis Child 78: 111-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducrocq S, Benjelloun N, Plotkine M, Ben-Ari Y, Charriaut-Marlangue C ( 2000) Poly(ADP-ribose) synthase inhibition reduces ischemic injury and inflammation in neonatal rat brain. J Neurochem 74: 2504-2511. [DOI] [PubMed] [Google Scholar]
- Dugan LL, Sensi SL, Canzoniero LM, Handran SD, Rothman SM, Lin TS, Goldberg MP, Choi DW ( 1995) Mitochondrial production of reactive oxygen species in cortical neurons following exposure to N-methyl-d-aspartate. J Neurosci 15: 6377-6388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliasson MJ, Sampei K, Mandir AS, Hurn PD, Traystman RJ, Bao J, Pieper A, Wang ZQ, Dawson TM, Snyder SH, Dawson VL ( 1997) Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat Med 3: 1089-1095. [DOI] [PubMed] [Google Scholar]
- Endres M, Wang ZQ, Namura S, Waeber C, Moskowitz MA ( 1997) Ischemic brain injury is mediated by the activation of poly(ADP-ribose)polymerase. J Cereb Blood Flow Metab 17: 1143-1151. [DOI] [PubMed] [Google Scholar]
- Ferrand-Drake M, Friberg H, Wieloch T ( 1999) Mitochondrial permeability transition induced DNA-fragmentation in the rat hippocampus following hypoglycemia. Neuroscience 90: 1325-1338. [DOI] [PubMed] [Google Scholar]
- Friberg H, Ferrand-Drake M, Bengtsson F, Halestrap AP, Wieloch T ( 1998) Cyclosporin A, but not FK 506, protects mitochondria and neurons against hypoglycemic damage and implicates the mitochondrial permeability transition in cell death. J Neurosci 18: 5151-5159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha HC, Snyder SH ( 1999) Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc Natl Acad Sci USA 96: 13978-13982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson MK, Jacobson EL ( 1999) Discovering new ADP-ribose polymer cycles: protecting the genome and more. Trends Biochem Sci 24: 415-417. [DOI] [PubMed] [Google Scholar]
- Langan SJ, Deary IJ, Hepburn DA, Frier BM ( 1991) Cumulative cognitive impairment following recurrent severe hypoglycaemia in adult patients with insulin-treated diabetes mellitus. Diabetologia 34: 337-344. [DOI] [PubMed] [Google Scholar]
- LaPlaca MC, Zhang J, Raghupathi R, Li JH, Smith F, Bareyre FM, Snyder SH, Graham DI, McIntosh TK ( 2001) Pharmacologic inhibition of poly(ADP-ribose) polymerase is neuroprotective following traumatic brain injury in rats. J Neurotrauma 18: 369-376. [DOI] [PubMed] [Google Scholar]
- Love S, Barber R, Wilcock GK ( 1999) Neuronal accumulation of poly(ADP-ribose) after brain ischaemia. Neuropathol Appl Neurobiol 25: 98-103. [DOI] [PubMed] [Google Scholar]
- Mandir AS, Poitras MF, Berliner AR, Herring WJ, Guastella DB, Feldman A, Poirier GG, Wang ZQ, Dawson TM, Dawson VL ( 2000) NMDA but not non-NMDA excitotoxicity is mediated by poly(ADP-ribose) polymerase. J Neurosci 20: 8005-8011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris R ( 1984) Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods 11: 47-60. [DOI] [PubMed] [Google Scholar]
- Nellgard B, Wieloch T ( 1992) Cerebral protection by AMPA- and NMDA-receptor antagonists administered after severe insulin-induced hypoglycemia. Exp Brain Res 92: 259-266. [DOI] [PubMed] [Google Scholar]
- Olney JW, Labruyere J, Price MT ( 1989) Pathological changes induced in cerebrocortical neurons by phencyclidine and related drugs. Science 244: 1360-1362. [DOI] [PubMed] [Google Scholar]
- Purnell MR, Whish WJ ( 1980) Novel inhibitors of poly(ADP-ribose) synthetase. Biochem J 185: 775-777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmued LC, Hopkins KJ ( 2000) Fluoro-jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res 874: 123-130. [DOI] [PubMed] [Google Scholar]
- Southan GJ, Szabo C ( 2003) Poly(ADP-ribose) polymerase inhibitors. Curr Med Chem 10: 321-340. [DOI] [PubMed] [Google Scholar]
- Suh SW, Chen JW, Motamedi M, Bell B, Listiak K, Pons NF, Danscher G, Frederickson CJ ( 2000) Evidence that synaptically-released zinc contributes to neuronal injury after traumatic brain injury. Brain Res 852: 268-273. [DOI] [PubMed] [Google Scholar]
- Szabo C, Dawson VL ( 1998) Role of poly(ADP-ribose) synthetase in inflammation and ischaemia-reperfusion. Trends Pharmacol Sci 19: 287-298. [DOI] [PubMed] [Google Scholar]
- Szabo C, Zingarelli B, O'Connor M, Salzman AL ( 1996) DNA strand breakage, activation of poly (ADP-ribose) synthetase, and cellular energy depletion are involved in the cytotoxicity of macrophages and smooth muscle cells exposed to peroxynitrite. Proc Natl Acad Sci USA 93: 1753-1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokime T, Nozaki K, Sugino T, Kikuchi H, Hashimoto N, Ueda K ( 1998) Enhanced poly(ADP-ribosyl)ation after focal ischemia in rat brain. J Cereb Blood Flow Metab 18: 991-997. [DOI] [PubMed] [Google Scholar]
- Virag L, Szabo C ( 2002) The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev 54: 375-429. [DOI] [PubMed] [Google Scholar]
- Wang ZQ, Auer B, Stingl L, Berghammer H, Haidacher D, Schweiger M, Wagner EF ( 1995) Mice lacking ADPRT and poly(ADP-ribosyl)ation develop normally but are susceptible to skin disease. Genes Dev 9: 509-520. [DOI] [PubMed] [Google Scholar]
- Weise J, Isenmann S, Bahr M ( 2001) Increased expression and activation of poly(ADP-ribose) polymerase (PARP) contribute to retinal ganglion cell death following rat optic nerve transection. Cell Death Differ 8: 801-807. [DOI] [PubMed] [Google Scholar]
- Wieloch T ( 1985) Hypoglycemia-induced neuronal damage prevented by an N-methyl-d-aspartate antagonist. Science 230: 681-683. [DOI] [PubMed] [Google Scholar]
- Wieloch T, Harris RJ, Symon L, Siesjo BK ( 1984) Influence of severe hypoglycemia on brain extracellular calcium and potassium activities, energy, and phospholipid metabolism. J Neurochem 43: 160-168. [DOI] [PubMed] [Google Scholar]
- Wieloch T, Engelsen B, Westerberg E, Auer R ( 1985) Lesions of the glutamatergic cortico-striatal projections in the rat ameliorate hypoglycemic brain damage in the striatum. Neurosci Lett 58: 25-30. [DOI] [PubMed] [Google Scholar]
- Ying W, Han SK, Miller JW, Swanson RA ( 1999) Acidosis potentiates oxidative neuronal death by multiple mechanisms. J Neurochem 73: 1549-1556. [DOI] [PubMed] [Google Scholar]
- Ying W, Anderson CM, Chen Y, Stein BA, Fahlman CS, Copin JC, Chan PH, Swanson RA ( 2000) Differing effects of copper, zinc superoxide dismutase overexpression on neurotoxicity elicited by nitric oxide, reactive oxygen species, and excitotoxins. J Cereb Blood Flow Metab 20: 359-368. [DOI] [PubMed] [Google Scholar]
- Ying W, Sevigny MB, Chen Y, Swanson RA ( 2001) Poly(ADP-ribose) glycohydrolase mediates oxidative and excitotoxic neuronal death. Proc Natl Acad Sci USA 98: 12227-12232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying W, Chen Y, Alano CC, Swanson RA ( 2002) Tricarboxylic acid cycle substrates prevent PARP-mediated death of neurons and astrocytes. J Cereb Blood Flow Metab 22: 774-779. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Limmroth V, Irikura K, Moskowitz MA ( 1994) The NOS inhibitor, 7-nitroindazole, decreases focal infarct volume but not the response to topical acetylcholine in pial vessels. J Cereb Blood Flow Metab 14: 924-929. [DOI] [PubMed] [Google Scholar]
- Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, Poirier GG, Dawson TM, Dawson VL ( 2002) Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 297: 259-263. [DOI] [PubMed] [Google Scholar]
- Zhang J, Dawson VL, Dawson TM, Snyder SH ( 1994) Nitric oxide activation of poly(ADP-ribose) synthetase in neurotoxicity. Science 263: 687-689. [DOI] [PubMed] [Google Scholar]
- Zhang J, Lautar S, Huang S, Ramsey C, Cheung A, Li JH ( 2000) GPI 6150 prevents H(2)O(2) cytotoxicity by inhibiting poly(ADP-ribose) polymerase. Biochem Biophys Res Commun 278: 590-598. [DOI] [PubMed] [Google Scholar]